

A new section in MEJDD which is aimed to improve chinical decision making by presenting a real patient by an experienced clinician and who discuss the case and provide an evidence base decision making for diagnosis and treatment of patient in daily clinical practice.
Recurrent Cholestasis in a 13 Year-old Boy
A 13 year-old boy with generalized intermittent pruritis from three years preceding referred to Shariati Hospital for ERCP. His pruritis was aggravated at night and relieved by warm baths. It was associated with jaundice. During the attacks, he had acholic stool and dark urine. The patient had three attacks in the preceding years, each of which lasted for about 3-4 months. Associated symptoms were anorexia and mild weight loss (3kg /3 months). All complaints began after an attack of gastroenteritis. Between attacks, the patient was completely asymptomatic with no complaints.
History of pruritis has many differential diagnoses. Many of these are primary skin disorders such as xerosis, atopic dermatitis, lichen simplex chronicus, and psoriasis, to name a few; this patient had no history of any allergic reaction or skin lesions.1 Still, drug and allergic history are important and must be assessed. Likewise, some systemic diseases can cause pruritis, such as: renal and thyroid diseases, malignancies, multiple sclerosis, infection with human immune deficiency virus (HIV) and iron deficiency anemia. Therefore, it is imperative that a detailed history be taken and the necessary workup performed for patients. Another etiology is cholestasis, which looks more plausible as it is similarly associated with jaundice, acholic stool, dark urine, is aggravated at night and relieved with warm baths. Itching in cholestasis is typically generalized although it is most severe on the palms and soles. Dry skin, hot and humid weather, and wearing constricting clothes can worsen this type of itching. The pathogenesis of pruritis in cholestasis is incompletely understood, but accumulation of bile acids in the bloodstream can be a probable explanation. On the other hand, absence of pruritis in many patients with elevated bile acids and lack of correlation between pruritis and bile acid concentrations make this hypothesis a less approved culprit. Alternatively, leakage of pruritogens into the bloodstream can be another plausible etiology.2
Yet, another more acceptable theory is the increase in endogenous opioids.3 A history of gastroenteritis and generalized symptoms (anorexia and weight loss) can be features of viral, autoimmune or other cholestatic liver diseases. So, in this patient a comprehensive workup is recommended and should consist of a detailed drug history as well as laboratory analysis for metabolic and systemic diseases, including: complete blood count (CBC), thyroid stimulating hormone (TSH), renal tests, HIV antibody, blood sugar (BS), chest X-ray (CXR), liver enzymes and their pertinent tests.4
The patient’s drug history consisted of urso-deoxycholic acid (UDCA), hydroxyzine, zinc and loratadin, which had been prescribed during the attacks. No history of blood infusion, prolonged infancy jaundice, drug use prior to the attacks, or hepatitis was detected. His two brothers and parents had no history of any similar disease.
Absence of any drug consumption before an attack makes drug reaction less probable but we must presume that on many occasions patients consume drugs inadvertently or do not disclose this information to their physicians. Additionally, many people think herbal or over-the-counter (OTC) drugs or opiates are safe and may not mention them if not specifically asked. Many drugs can induce liver injury, among which are antibiotics such as amoxicillin-clavulanate, azithromycin, erythromycin and anti-tuberculosis drugs, among others. Non-steroidal anti-inflammatory drugs (NSAIDS) including ibuprofen, diclofenac, anti-epileptics and herbal drugs are among the most common drugs that cause liver damage.5 Older patients show a cholestatic pattern more frequently than other patterns of liver injury. Drugs taken by our patient were less likely to cause this kind of had begun taking these drugs after disease onset. However, risk factors for liver diseases must be evaluated in every patient with a similar history. Among these risk factors are a history of blood infusion, intravenous drug use (IDU), icter, any liver disease in infancy or childhood, or in parents and siblings.
Upon physical exam, he was not ill or toxic. His conjuctiva was not pale, sclera was mildly icteric, with no lymphadenopathy observed in the head, neck or axillary regions. Chest exam showed normal heart and lung sounds. Abdomen was soft with no tenderness or guarding, and liver and spleen were likewise normal. There were some excoriations on the trunk and limbs.
Icterus and excoriation in this patient make liver disease the most plausible explanation for his pruritis attacks. There were no signs or symptoms of liver insufficiency such as encephalopathy, ascites, or splenomegaly; but their absence does not rule out the possibility of chronic liver disease such as viral, autoimmune or cholestatic diseases. Therefore, obtaining general tests is imperative.
Such tests include a CBC, prothrombin time (PT), albumin and liver enzymes, in addition to a further workup. Moreover, metabolic diseases that include renal disease, diabetes mellitus (DM) and pertinent electrolytes must be examined.1
General laboratory data such as CBC, PT, partial thromboplastin time (PTT), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), lactate dehydrogenase (LDH), fasting blood sugar (FBS), blood urea nitrogen (BUN), creatinine (Cr), sodium, potassium, calcium, phosphorous, uric acid and urinalysis, in addition to a stool exam and culture were normal (Table 1).
Table 1 . Patient’s laboratory data.
| Test | Results |
| WBC | 7,400 (/ml) |
| Hb | 13 (g/dl) |
| MCV | 83.3 (fl) |
| PLT | 421,000 (/ml) |
| PT | 13 sec |
| PTT | 35 sec |
| INR | 1.2 |
| ESR | 10 (mm/hr) |
| CRP | Negative |
| LDH | 372 (IU/L) |
| FBS | 93 (mg/dl) |
| BUN | 7.9 (mg/dl) |
| Cr | 0.5 (mg/dl) |
| Uric acid | 3.9 (mg/dl) |
| S/E | Negative for OB and OP |
| S/C | Non-pathogen |
| Urinalysis | Normal |
Aspartate transminase (AST) and alanine transaminase (ALT) were about 2-3 times the upper limit of normal and alkaline phosphatase was markedly elevated during his three attacks, but all returned to normal during asymptomatic intervals.
Interestingly, gamma glutamyl transpeptidase (GGT) was normal during his attacks.
Bilirubin was only marginally elevated during the attacks and normal during asymptomatic periods (Table 2).
Table 2 . Patient’s subsequent laboratory data.
| Test | 1st attack | 2nd attack | 3rd attack | After 3rd attack |
| AST | 65 (IU/L) | 83 (IU/L) | 95 (IU/L) | 28 (IU/L) |
| ALT | 85 (IU/L) | 113 (IU/L) | 167 (IU/L) | 29 (IU/L) |
| ALP | 1162 (IU/L) | 968 (IU/L) | 1013 (IU/L) | 202 (IU/L) |
| GGT | 53 (<80) (IU/L) | |||
| Total bilirubin | 1.9 mg/dl | 1.4 mg/dl | 2.55 mg/dl | |
| Direct bilirubin | 0.2 mg/dl | 0.9 mg/dl | 1.7 mg/dl | |
| Amylase | 31 (IU/L) |
Normal platelets, PT, INR, electrolytes, FBS, BUN, Cr and uric acid rule out cirrhosis, DM, renal and metabolic diseases. Normal ESR, LDH and albumin make hemolysis, malignancy and liver insufficiency unlikely.
Elevated liver enzymes in this patient are typical of a cholestatic disease in which ALP is commonly more than 3-4 times the upper limits of normal, and ALT and AST are less than three times normal limits.6 Cholestasis has many different etiologies. The first step in evaluating cholestasis is to determine its extra- or intra-hepatic origin. History and physical exam were incomplete in this regard.
Transabdominal ultrasonography (TUS) is the best preliminary test for differentiating the origin of cholestasis.6
Absence of intra- and extra-hepatic biliary duct dilation raises the possibility of an intra-hepatic cholestatic liver disease. Still, false negative results occur due to cirrhosis, primary sclerosing cholestasis (PSC) in addition to partial biliary duct obstruction due to tumoral lesions, which cannot be ruled out with certainty by a normal ultrasound.7, 8
Abdominal ultrasonography was performed on this patient, which showed only fatty liver with normal gallbladder and normal diameter of the intra- and extra-hepatic ducts without splenomegaly.
TUS in this patient indicates an intrahepatic origin for his complaints. This syndrome has many etiologies resulting from acute and chronic hepatocellular diseases including: viral and autoimmune hepatitis, alcoholic and nonalcoholic steatohepatitis, drugs, PSC, primary biliary cirrhosis (PBC) and cirrhosis, which must be ruled out. Multifactorial etiologies can likewise present with this type of cholestasis and include: total parenteral nutrition (TPN), systemic infection, sickle cell disease, lysis, postoperative conditions, systemic infections and hypotension or hypoxemia due to congestive heart failure. On the contrary, there was no clue to these etiologies in the patient’s history.6 Lack of any history of alcohol intake rules out alcohol related diseases. Diseases such as celiac, Wilson, alfa-1 antityrpsin deficiency and immune deficiency syndromes (primary or acquired) may similarly present with cholestatic pattern in liver enzymes.6, 9
Inherited or infiltrative diseases must also be considered in this patient after ruling out the abovementioned causes. Therefore, a full battery of tests is imperative and may include screening for hepatitis B virus (HBV), hepatitis C virus (HCV), HIV, autoimmune markers, tein and immunoelectrophoresis. Examining ceruloplasmin, 24-hour urinary copper level, Kesier Flesier (KF) ring as well as alfa-1 anti-trypsin and glucose-6 phosphate dehydrogenase (G6PD) levels must be performed if all of the previous tests are negative.
Viral and autoimmune markers including HBsAg, HCV Ab, HIV Ab, ANA, ASMA, anti-LKM1, anti-dsDNA, and ANCA were negative. Complement levels, protein and immunoglubin electrophoresis were within normal ranges. Anti-TTG was negative and levels of alfa 1 antitrypsin, G6PD, ceruloplasmin and 24-hour urine copper were normal. Ophthalmologic exam showed no KF ring (Table 3).
Table 3 . Patient’s laboratory data (continued).
| Test | Result |
| Protein electrophoresis | Normal |
| Immunoelectrophoresis | Normal |
| Anti-TTG | 1.5 (IU/ml) (NL<12) |
| Alfa 1 antitrypsin | 200 (mg/dl) (NL: 63-170) |
| G6PD | 15 (min) (NL <60) |
| Ceruloplasmin | 41 (mg/dl) (NL: 18-45) |
| Urine 24 hr copper | 115 (micg/24h) (NL <150) |
| KF ring | Negative |
Negative results of viral, autoimmune, celiac and other aforementioned ailments show that the patient has probably a cholestatic disease such as PSC or infiltrative and/or inherited disease. Consequently, magnetic resonance cholangio-pancreatography (MRCP) and if unavailable, endoscopic retrograde cholangio-pancreatography (ERCP) are recommended to rule out PSC and probable partial obstruction due to malignancies.
Abdominal magnetic resonance imaging (MRI) and MRCP were normal. ERCP performed at another center was also normal (Figure 1).
Fig. 1 .

ERCP and MRCP views of the patient.
Normal MRCP and ERCP can rule out large duct PSC, small common bile stones, malignancies and diseases such as Caroli’s disease or Alagille syndrome, which can sometimes present with these symptoms. However, it can not preclude small duct PSC or infiltrative diseases.10-12
An important issue is that MRCP, by itself, can efficiently rule out these possible diagnoses. An ERCP does not have any additional benefit, but rather can have many complications. Thus, ERCP is not indicated. As a last resort, liver biopsy is recommended for evaluating bile duct injuries, deposition of amyloid proteins, malignant cells of amyloidosis or lymphomas, and the presence of granulomas or caseous necrosis as found in cases of tuberculosis or sarcoidosis.13, 14
In this case, liver biopsy showed only mild hydropic changes and intracanalicular cholestasis with no other findings.
Normal GGT and biopsy findings narrow the potential metabolic etiologies of a patient’s complaints to familial intrahepatic cholestasis disorders that range from mild to severe. Severe forms include progressive familial intrahepatic cholestasis (FIC 1 and 2). Mild forms include benign recurrent intrahepatic cholestasis (BRIC) types 1 and 2.15-17 It appears that the best diagnosis in this patient would be BRIC, which can be diagnosed with criteria initially presented by Tygstrup and Jensen.17
Their criteria included the following:1) several episodes of evident jaundice associated with severe pruritis, separated by asymptomatic intervals lasting for several months or years; 2) absence of any known etiology for intra- or extra-hepatic cholestasis (i.e., medications, pregnancy, etc); 3) signs of cholestasis in a biochemical profile contrasted with normal ERCP; and 4) bile plugs within the ducts as seen by liver histology.
Other disorders with low serum GGT activity during cholestasis include inborn errors of bile acid biosynthesis,18 familial hypercholanemia (FHC),19, 20 arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome21, Smith-Lemli-Opitz syndrome (SLOS)22, nonspecific failure of bile acid production23, and microvillus inclusion disease (MVID).24 However, these are less probable because many are associated with other organ involvement, and present during in infancy or early childhood with low bile acid levels. None were detected in this patient’s history.
Despite the evident criteria for correct diagnosis of BRIC, this disease may be erroneously labeled as cholecystitis, hepatitis or other intra-hepatic cholestatic diseases.25 In 75% of cases, BRIC manifests itself in subjects less than 20 years of age and the male to female ratio is 1.7:1.26 In a large case series, the age at disease presentation varies from 1 to 59 years.27 The majority of BRIC patients are diagnosed in adolescence or early adulthood,28 although in some cases it has been diagnosed in infancy16, 17and middle age.29
BRIC has not only been reported in individuals, it has also been detected within whole families30 and isolated communities.17 Wareham in 1985 and Lee in 1997 reported BRIC within first degree relatives.31 The majority of episodes of cholestasis began in winter.32
What are other symptoms and signs of BRIC?
As mentioned above, BRIC is characterized by several episodes of evident jaundice associated with severe pruritis, separated by asymptomatic intervals. Symptomatic episodes consist of two phases: pre-icteric and cholestatic.
The pre-icteric phase lasts from two to four weeks. During this phase, patients may suffer from several prodromal symptoms such as malaise, anorexia, abdominal pain, nausea, vomiting, fatigue and weight loss.15, 16, 33 However, pruritis is the main ultimate complaint of BRIC patients in the pre-icteric phase as reported by Williams et al. in 1964.34 The severity of pruritus is not proportionate to the moderately elevated parameters of cholestasis.35
In the cholestatic phase, pruritis will subsequently be followed by clinical jaundice. Dark colored urine and whitish defecation25, 26are other symptoms of BRIC in this phase. These attacks have not been reported to be induced by fatty foods.25 Other symptoms may include insomnia and frequent watery diarrhea.35
Episodes may also be associated with upper respiratory tract infections, influenza, otitis media, acute gastro-enteritis or abdominal pain.36 In a report by Chatila et al., attacks have been associated with intractable cough, which resolved when cholestasis subsided.37
The cholestatic phase may last from a few weeks to several months followed by complete clinical and biochemical resolution and may recur at intervals of several months to years.27 Recurrent attacks may resemble one another.38 As reported in previous studies, the number of episodes may range from 1 to as many as 27.31, 32
BRIC attacks may occur spontaneously, but De Pagter and colleagues (1976) have re ported that pregnancy, stress and surgeries may be triggers.38 In some patients, episodes may occur following oral contraceptive use.15 However, as intrahepatic cholestasis secondary to pregnancy or drug are separate diagnostic entities, their role in triggering BRIC is ruled out.
On examination, the typical patient is afebrile and presents with icterus without pallor.16, 31 There is usually no hepatomegaly, splenomegaly, or any sign of cirrhosis and liver failure such as spider angiomas, palmar erythema or thenar atrophy. Yet, the first reported cases of BRIC presented with enlarged and tender livers.16, 31 Signs of excoriation due to scratching and hyperemic pharynx may be noticed with physical examination.25, 39 No peripheral edema, clubbing, or signs of vitamin deficiencies are detected in BRIC patients.36 Hearing may be impaired and sweat glands may be dysfunctional.40 However, the rest of the systemic examination is usually normal and anthropometric parameters are within normal limits with average height and weight.
Is the pathogenesis of BRIC known?
The primary cause of cholestasis might be either a defect in hepatocellular bile acid transport or an intrinsic abnormality in hepatocyte bile acid secretion.15, 41 Attacks of BRIC seem to affect hepatic metabolism of organic anions. After attacks resolve, secretion of these anions either return to normal levels or remain slightly higher than seen in unaffected individuals.42
In BRIC it seems that altered bile acid metabolism is the underlying pathogenic mechanism. This hypothesis has been proposed, based on increased influx of sulphated lithocholoic acid conjugates from the intestine that have been observed in these patients.32 There is a decrease in primary bile acids such as cholate and chenodeoxycholate, and an increase in the secondary bile acids, lithocholate and deoxycholate, which are more cholestatic.
A high fat diet and colon infection can lead to increased secondary toxic bile acids due to fat malabsorption and bacterial metabolism in the colon. Environmental factors may additionally interfere with bile acid metabolism and bile secretion through an allergic or hypersensitivity reaction. Yet, the mechanism is still unclear.26 Recent studies in transgenic mice suggest that this finding may be secondary to suppression of apolipo-protein A-I gene transcription by the bile salt-binding nuclear receptor farensoid X-activated receptor (FXR).43 The aforementioned metabolic disorders as later proven by genetic studies, are the manifestations of mutations in certain genes.
In 1994-95 the gene for chromosome 18 was mapped using a genome search for a shared segment in three patients with BRIC from an isolated community.28, 44 Genetic studies mapped this defect in the long arm of chromosome 18q21-22.28Subsequently, the same region was reported to be the locus of the gene that caused progressive familial intrahepatic cholestasis type 1 (Byler disease).44 Both diseases were reported to be caused by mutations in a single gene which was later labeled as familial intrahepatic cholestasis 1 (FIC1).45 Studies have reported heterogeneities in this gene.46, 47 FIC1 appears to be a member of a p-type ATPase family that takes on the responsibility to transport aminophospholipid from the outer to the inner leaflet of a variety of cell membranes. FIC1 plays a critical role in the enterohepatic circulation of bile acids48and in maintaining fluidity of the cell membrane.49 FIC1 has been recently named ATP8B1.50, 51 Patients in whom a mutation in ATP8B1 is the underlying genetic abnormality of the disease are categorized as BRIC type 1.45, 50
The scheme of the BRIC gene is demonstrated in Figure 2.
Fig. 2 .
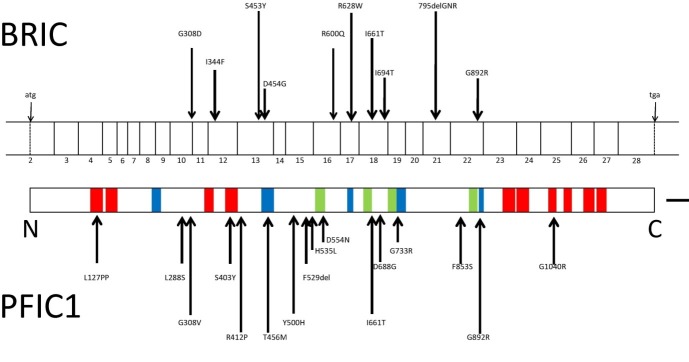
BRIC gene scheme.
Recently BRIC type 2 caused by another mutational change in ABCB11 gene located on chromosome 2 has been identified.49 The product of this gene is bile salt export pump (BSEP) which is the primary transporter responsible for canalicular bile salt secretion. This membrane transporter belongs to the ATP-binding cassette (ABC) superfamily of proteins that comprise one of the largest families of transporters within the human genome.52
BSEP plays an essential role in the physiologic maintenance of the enterohepatic circulation of bile acids.
Impairment in bile flow that results from genetic defects in this transporter leads to cholestatic liver disease, including progressive familial intrahepatic cholestasis type 2 (PFIC2), benign recurrent intrahepatic cholestasis type 2 (BRIC2), drug-induced cholestasis (DIC) and hormone-dependent intrahepatic cholestasis of pregnancy (ICP).53
Compared to patients with ATP8B1 mutations, patients with ABCB11 mutations lack extrahepatic symptoms such as pancreatitis and are more likely to exhibit cholstasis.54
BRIC types 1 and 2 are autosomal recessive hereditary diseases. A third type labeled as BRIC type 3 with autosomal dominant heritance has been recently identified, but is not related to mutations on chromosomes 18 or 2.55 BRIC3 is caused by the loss of MDR3 or ABCB4, which is a phosphatidyl choline floppase.56
Is there any other way to diagnose BRIC?
Liver biopsy is invaluable in excluding other icteric diseases.39 Results of light microscopic examination of a liver biopsy in the icteric period are normal or show centrilobular cholestasis with minimal reactive changes and mild inflammation of portal spaces infiltrated by mononuclear cells and occasional eosinophils and Kupffer cell hyperplasia.15, 28,32, 39 Cholestasis is secondary to accumulated bile pigment or bile plug in the hepatocytes and bile canals.17, 29 In some cases, mild fibrosis is observed in the peripheral and centrilobar zones (zones 1 and 3) and focal degeneration of hepatocytes can be noticed.31 There is typically no evidence of ductopenia, Mallory or Councilman bodies, siderosis or steatosis.39
On immunostaining, there is a slight deficiency in canalicular neutral endopeptidase (CD10) expression in some centrilobular regions. In addition, GGT expressions are considerably deficient throughout the lobule. However the transport protein BSEP and its homologue multi-drug resistance-associated protein 2 (MRP2) remain normally expressed along bile canaliculi throughout the lobule.57 In electron microscopy there is a marked alteration of bile canaliculi with distorted and reduced microvilli.58
What is the recommended treatment of this disease?
The main treatment for BRIC is reassurance. However, pruritis which is the major disturbing symptom of BRIC can be alleviated with several suggested medications that include: UDCA (13-15 mg/kg body weight/day), cholestyramine (12-16 g/day), rifampin (300-600 mg/day), phenobarbital28, 36, 39, and antihistamines (hydroxyzine and difenhydramine) especially for nocturnal pruritus.59
Rifampicin partly interferes with hepatic uptake of bile acids and decreases hepatocyte bile concentration.60 Rifampicin also induces 6-hydroxylation of secondary bile salts which facilitates their elimination.61
Corticosteroid therapy has been used to alleviate cholestasis. Bile secretion and intrahepatic cholestasis also involves cellular mechanisms such as cellular immunity, which is suppressed by corticosteroids and explains their mechanism of action.26
Simvastatin has also been proposed as an alternative medical therapy.35
Extracorporal albumin dialysis (MARS: Molecular Adsorbent Recycling System), is recently reported by some studies to be a successful treatment in resistant cases.62 MARS treatment shifts parts of the highly concentrated intracellular contents towards the extracellular compartments which results in decreased plasma concentrations of bile acids and bilirubin.63 MARS also improves transcription of apo A-1 gene which is suppressed in BRIC and therefore increases the plasma levels of apo A-1.35
Other plausible medications that may be able to reduce intrahepatic cholestasis are azathioprine, chlorambucil, cholchicine, cyclosporine, sadenosylmethionine, FK 506 and methotrexate.
However, the efficiency and safety of these medications need further investigation.25
Nasobiliary drainage via ERCP has been used in a single study from Holland for long-lasting relief from pruritis and jaundice.64
In case there is a known trigger for BRIC, the patient is advised to avoid risk factors. For example, if the disease is secondary to OCP, the patient will be advised to use other methods of contraception, such as IUD or sterilization.25
What is the natural history and prognosis of this disease?
As mentioned above, BRIC manifests by repeated cholestatic episodes.28, 33 The intensity and duration of episodes, and the length of the intervening periods are unpredictable.37
The number of attacks is variable in different patients, and ranges from 1 to 27. In a case-series by Tygstrup in 1999, the episodes recurred for 36 times in one patient. In their study, some patients who were followed for years continued to suffer from intermittent attacks, but with no progression to cirrhosis. It is worth noting however, that Van Ooteghem et al., in 2002, have reported the cases of four BRIC patients in whom liver histology worsened into fibrosis.51
The duration of attacks ranges from weeks to months before the disease resolves spontaneously.36 Improvement of the clinical profile of patients begins with improvement in appetite which is commonly followed by sudden and complete regression of pruritis and gradual disappearance of icter.17
Correct diagnosis is essential in the management of BRIC and prevents over-investigation of the patient either by ERCP or surgery.31 Surgery in the jaundiced patient is a predisposing factor that may increase risks and possibly aggravate the natural history.38
BRIC is a benign disease that does not progress to cirrhosis 65 or end stage liver disease, although in a few cases as reported by Ooteghem et al., the disease has been progressive.51 The episodes of jaundice and pruritis may be lengthened with debilitating symptoms as some patients are reported to have undergone liver transplantation for symptom relief.66
Commentary:
Our presented case had sufficient criteria for BRIC. Therefore, we reassured the patient and his parents and prescribed UDCA (400 mg TID) to alleviate his pruritis. During 3 years follow-up, there was no disease progression and no new attacks.
CONFLICT OF INTEREST
The authors declare no conflict of interest related to this work.
REFERENCES
- 1.Greco PJ, Ende J. Pruritus: a practical approach. J Gen Intern Med. 1992;7:340–9. doi: 10.1007/BF02598094. [DOI] [PubMed] [Google Scholar]
- 2.Ghent CN. Pruritus of cholestasis is related to effects of bile salts on the liver, not the skin. Am J Gastroenterol. 1987;82:117–8. [PubMed] [Google Scholar]
- 3.Thornton JR, Losowsky MS. Opioid peptides and primary biliary cirrhosis. BMJ. 1988;297:1501–4. doi: 10.1136/bmj.297.6662.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fazio SB. Pruritis. In: UpToDate. Rose BD (Ed) UpTo Date 2011, Waltham, 19.1.
- 5.Andrade RJ, Lucena MI, Fernández MC, Pelaez G, Pachkoria K, García-Ruiz E. et al. Drug-induced liver injury: an analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology. 2005;129:512–21. doi: 10.1016/j.gastro.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 6. Kaplan MM. Approach to the patient with abnormal liver function tests. In: UpToDate. Rose BD (Ed) UpTo Date 2011 , Waltham, 19.1.
- 7.Lapis JL, Orlando RC, Mittelstaedt CA, Staab EV. Ultrasonography in the diagnosis of obstructive jaundice. Ann Intern Med. 1978;89:61–3. doi: 10.7326/0003-4819-89-1-61. [DOI] [PubMed] [Google Scholar]
- 8.Pedersen OM, Nordgård K, Kvinnsland S. Pedersen OM, Nordgård K, Kvinnsland SValue of sonography in obstructive jaundiceLimitations of bile duct caliber as an index of obstruction. Scand J Gastroenterol. 1987;22:975–81. doi: 10.3109/00365528708991945. [DOI] [PubMed] [Google Scholar]
- 9.Abdo A, Meddings J, Swain M. Liver abnormalities in celiac disease. Clin Gastroenterol Hepatol. 2004;2:107–12. doi: 10.1016/s1542-3565(03)00313-6. [DOI] [PubMed] [Google Scholar]
- 10.Alagille D, Estrada A, Hadchouel M, Gautier M, Odièvre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110:195–200. doi: 10.1016/s0022-3476(87)80153-1. [DOI] [PubMed] [Google Scholar]
- 11.Dave M, Elmunzer BJ, Dwamena BA, Higgins PD. Primary sclerosing cholangitis: meta-analysis of diagnostic performance of MR cholangiopancreatography. Radiology. 2010;256:387–96. doi: 10.1148/radiol.10091953. [DOI] [PubMed] [Google Scholar]
- 12.Moff SL, Kamel IR, Eustace J, Lawler LP, Kantsevoy S, Kalloo AN. et al. Diagnosis of primary sclerosing cholangitis: a blinded comparative study using magnetic resonance cholangiography and endoscopic retrograde cholangiography. Gastrointest Endosc. 2006;64:219–23. doi: 10.1016/j.gie.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 13.Levy M, Fryd CH, Eliakim M. Intrahepatic obstructive jaundice due to amyloidosis of the liverA case report and review of the literature. Gastroenterology. 1971;61:234–8. [PubMed] [Google Scholar]
- 14.Wee A, Ludwig J. Pericholangitis in chronic ulcerative colitis: primary sclerosing cholangitis of the small bile ducts? Ann Intern Med. 1985;102:581–7. doi: 10.7326/0003-4819-102-5-581. [DOI] [PubMed] [Google Scholar]
- 15.Brenard R, Geubel AP, Benhamou JP. Benign recurrent intrahepatic cholestasisA report of 26 cases. J Clin Gastroenterol. 1989;11:546–51. doi: 10.1097/00004836-198910000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Summerskill WH, Walshe JM. Benign recurrent intrahepatic “obstructive” jaundice. Lancet. 1959;2:686–90. doi: 10.1016/s0140-6736(59)92128-2. [DOI] [PubMed] [Google Scholar]
- 17.Tygstrup N, Jensen B. Intermittent intrahepatic cholestasis of unknown etiology in five young males from the Faroe Islands. Acta Med Scand. 1969;185:523–30. doi: 10.1111/j.0954-6820.1969.tb07378.x. [DOI] [PubMed] [Google Scholar]
- 18.Bove KE, Heubi JE, Balistreri WF, Setchell KD. Bile acid synthetic defects and liver disease: a comprehensive review. Pediatr Dev Pathol. 2004;7:315–34. doi: 10.1007/s10024-002-1201-8. [DOI] [PubMed] [Google Scholar]
- 19.Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL. et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91–6. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 20.Zhu QS, Xing W, Qian B, von Dippe P, Shneider BL, Fox VL. et al. Inhibition of human m-epoxide hydrolase gene expression in a case of hypercholanemia. Biochim Biophys Acta. 2003;1638:208–16. doi: 10.1016/s0925-4439(03)00085-1. [DOI] [PubMed] [Google Scholar]
- 21.Eastham KM, McKiernan PJ, Milford DV, Ramani P, Wyllie J, van’t Hoff W. et al. ARC syndrome: an expanding range of phenotypes. Arch Dis Child. 2001;85:415–20. doi: 10.1136/adc.85.5.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grange DK, deMello DE, Hart MH, Kelley RI, Knisely AS, Nworkoro NA. Cholestatic liver disease in Smith-Lemli-Opitz syndrome(Abstract) Proc Greenwood Genet Center. 2002;21:48–9. [Google Scholar]
- 23.Kajiwara E, Akagi K, Tsuji H, Murai K, Fujishima M. Low activity of gamma-glutamyl transpeptidase in serum of acute intrahepatic cholestasis. Enzyme. 1991;45:39–46. doi: 10.1159/000468863. [DOI] [PubMed] [Google Scholar]
- 24.Peters J, Lacaille F, Horslen S. Microvillus inclusion disease treated by small bowel transplantation: Development of progressive intrahepatic cholestasis with low serum concentrations of gamma glutamyl transpeptidase activity. Hepatology. 2001;34:213A. [Google Scholar]
- 25.Akbar FN, Noer S, Lesmana L, Husodo UB, Marwoto W. Benign recurrent intrahepatic cholestasis. Ind J Gastro Hepat Dig Endos. 2001;2:41–4. [Google Scholar]
- 26. Sherlock S Cholestatsis, in Diseases of the liver and biliary system, Sherlock S and Dooley J, Editors., Blackwell Science Ltd: Oxford 1997. p. 230-2.
- 27.Summerfield JA, Scott J, Berman M, Ghent C, Bloomer JR, Berk PD. et al. Benign recurrent intrahepatic cholestasis: studies of bilirubin kinetics, bile acids, and cholangiography. Gut. 1980;21:154–60. doi: 10.1136/gut.21.2.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Houwen RH, Baharloo S, Blankenship K, Raeymaekers P, Juyn J, Sandkuijl LA. et al. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nat Genet. 1994;8:380–6. doi: 10.1038/ng1294-380. [DOI] [PubMed] [Google Scholar]
- 29.Summerskill WHJ. The syndrome of benign recurrent cholestasis. Am J Med. 1965;38:298–305. doi: 10.1016/0002-9343(65)90184-1. [DOI] [PubMed] [Google Scholar]
- 30.Goldberg DM, Hendry EB. Familial form of benign idiopathic recurrent cholestasis. Arch Intern Med. 1967;120:556–64. [PubMed] [Google Scholar]
- 31.Wareham NJ, Dickson CJ, Baskerville PA. Benign recurrent intrahepatic cholestasis. J R Soc Med. 1985;78:955–6. doi: 10.1177/014107688507801120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bijleveld CM, Vonk RJ, Kuipers F, Havinga R, Fernandes J. Benign recurrent intrahepatic cholestasis: a long-term follow-up study of two patients. Hepatology. 1989;9:532–7. doi: 10.1002/hep.1840090404. [DOI] [PubMed] [Google Scholar]
- 33.Liu CJ, Kao JH, Chen PJ, Lai MY, Mao TL, Wang TH. et al. Benign recurrent intrahepatic cholestasis. J Formos Med Assoc. 1997;96:370–3. [PubMed] [Google Scholar]
- 34.Williams R, Cartter MA, Sherlock S, Scheuer PJ, Hill KR. Idiopathic recueernt cholestasis: A study of the functional and pathologic lesion in four cases. Q J Med. 1964;33:387–99. [PubMed] [Google Scholar]
- 35.Sturm E, Franssen CF, Gouw A, Staels B, Boverhof R, De Knegt RJ. et al. Extracorporal albumin dialysis (MARS) improves cholestasis and normalizes low apo A-I levels in a patient with benign recurrent intrahepatic cholestasis (BRIC) Liver. 2002;22:72–5. doi: 10.1034/j.1600-0676.2002.00015.x. [DOI] [PubMed] [Google Scholar]
- 36.al Drees K, al Zaben A, al Amir A, Abdulla A. Benign recurrent intrahepatic cholestasis in a Saudi child. Ann Trop Paediatr. 1999;19:215–7. doi: 10.1080/02724939992563. [DOI] [PubMed] [Google Scholar]
- 37.Chatila R, Bergasa NV, Lagarde S, West AB. Intractable cough and abnormal pulmonary function in benign recurrent intrahepatic cholestasis. Am J Gastroenterol. 1996;91:2215–9. [PubMed] [Google Scholar]
- 38.de Pagter AG, van Berge Henegouwen GP, ten Bokkel Huinink JA, Brandt KH. Familial benign recurrent intrahepatic cholestasis. Interrelation with intrahepatic cholestasis of pregnancy and from oral contraceptives? Gastroenterology. 1976;71:202–7. [PubMed] [Google Scholar]
- 39.Ermis F, Oncu K, Ozel M, Yazgan Y, Gurbuz AK, Demirturk L. et al. Benign recurrent intrahepatic cholestasis: late initial diagnosis in adulthood. Ann Hepatol. 2010;9:207–10. [PubMed] [Google Scholar]
- 40.Oshima T, Ikeda K, Takasaka T. Sensorineural hearing loss associated with Byler disease. Tohoku J Exp Med. 1999;187:83–8. doi: 10.1620/tjem.187.83. [DOI] [PubMed] [Google Scholar]
- 41.Jacquemin E, Dumont M, Bernard O, Erlinger S, Hadchouel M. Evidence for defective primary bile acid secretion in children with progressive familial intrahepatic cholestasis (Byler disease) Eur J Pediatr. 1994;153:424–8. doi: 10.1007/BF01983406. [DOI] [PubMed] [Google Scholar]
- 42.Spiegel EL, Schubert W, Perrin E, Schiff L. Benign recurrent intrahepatic cholestasis , with response to cholestyramine. Am J Med. 1965;39:682–8. doi: 10.1016/0002-9343(65)90090-2. [DOI] [PubMed] [Google Scholar]
- 43.Claudel T, Sturm E, Duez H, Torra IP, Sirvent A, Kosykh V. et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109:961–71. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlton VE, Knisely AS, Freimer NB. Mapping of a locus for progressive familial intrahepatic cholestasis (Byler disease) to 18q21-q22, the benign recurrent intrahepatic cholestasis region. Hum Mol Genet. 1995;4:1049–53. doi: 10.1093/hmg/4.6.1049. [DOI] [PubMed] [Google Scholar]
- 45.Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M. et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–24. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 46.Floreani A, Molaro M, Mottes M, Sangalli A, Baragiotta A, Roda A. et al. Autosomal dominant benign recurrent intrahepatic cholestasis (BRIC) unlinked to 18q21 and 2q24. Am J Med Genet. 2000;95:450–3. doi: 10.1002/1096-8628(20001218)95:5<450::aid-ajmg8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 47.Sinke RJ, Carlton VE, Juijn JA, Delhaas T, Bull L, van Berge Henegouwen GP. et al. Benign recurrent intrahepatic cholestasis (BRIC): evidence of genetic heterogeneity and delimitation of the BRIC locus to a 7-cM interval between D18S69 and D18S64. Hum Genet. 1997;100:382–7. doi: 10.1007/s004390050520. [DOI] [PubMed] [Google Scholar]
- 48.Iyanagi T, Emi Y, Ikushiro S. Biochemical and molecular aspects of genetic disorders of bilirubin metabolism. Biochim Biophys Acta. 1998;1407:173–84. doi: 10.1016/s0925-4439(98)00044-1. [DOI] [PubMed] [Google Scholar]
- 49.Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clin Liver Dis. 2004;8:133–49. doi: 10.1016/S1089-3261(03)00133-8. [DOI] [PubMed] [Google Scholar]
- 50.Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ. et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27–38. doi: 10.1002/hep.20285. [DOI] [PubMed] [Google Scholar]
- 51.van Ooteghem NA, Klomp LW, van Berge-Henegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol. 2002;36:439–43. doi: 10.1016/s0168-8278(01)00299-9. [DOI] [PubMed] [Google Scholar]
- 52.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 53.Lam P, Soroka CJ, Boyer JL. The bile salt export pump: clinical and experimental aspects of genetic and acquired cholestatic liver disease. Semin Liver Dis. 2010;30:125–33. doi: 10.1055/s-0030-1253222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Mil SW, van Oort MM, van den Berg IE, Berger R, Houwen RH, Klomp LW. Fic1 is expressed at apical membranes of different epithelial cells in the digestive tract and is induced in the small intestine during postnatal development of mice. Pediatr Res. 2004;56:981–7. doi: 10.1203/01.PDR.0000145564.06791.D1. [DOI] [PubMed] [Google Scholar]
- 55.Liu C, Aronow BJ, Jegga AG, Wang N, Miethke A, Mourya R. et al. Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis. Gastroenterology. 2007;132:119–26. doi: 10.1053/j.gastro.2006.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. 2010;30:134–46. doi: 10.1055/s-0030-1253223. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y, Meng J, Wang J, Wang Z, Wang X, Linghu E. et al. Repeated rendezvous treatment of PTBD and ERCP in patients with recurrent obstructive jaundice. Hepatogastroenterology. 2010;57:1029–33. [PubMed] [Google Scholar]
- 58.Biempica L, Gutstein S, Arias IM. Morphological and biochemical studies of benign recurrent cholestasis. Gastroenterology. 1967;52:521–35. [PubMed] [Google Scholar]
- 59.Erlinger S. Molecular genetics of familial cholestasis. Gastroenterol Clin Biol. 1999;23:195–8. [PubMed] [Google Scholar]
- 60.Balsells F, Wyllie R, Steffen R, Kay M. Benign recurrent intrahepatic cholestasis: improvement of pruritus and shortening of the symptomatic phase with rifampin therapy: a case report. Clin Pediatr (Phila) 1997;36:483–5. doi: 10.1177/000992289703600809. [DOI] [PubMed] [Google Scholar]
- 61.Lachaux A, Loras-Duclaux I, Bouvier R, Dumontet C, Hermier M. Benign recurrent cholestasis with normal gamma-glutamyl-transpeptidase activity. J Pediatr. 1992;121:78–80. doi: 10.1016/s0022-3476(05)82546-6. [DOI] [PubMed] [Google Scholar]
- 62.Huster D, Schubert C, Achenbach H, Caca K, Mössner J, Berr F. Successful clinical application of extracorporal albumin dialysis in a patient with benign recurrent intrahepatic cholestasis (BRIC) Z Gastroenterol. 2001;39:13–4. doi: 10.1055/s-2001-919024. [DOI] [PubMed] [Google Scholar]
- 63.Stange J, Mitzner SR, Risler T, Erley CM, Lauchart W, Goehl H, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs. 1999;23:319–30. doi: 10.1046/j.1525-1594.1999.06122.x. [DOI] [PubMed] [Google Scholar]
- 64.Stapelbroek JM, van Erpecum KJ, Klomp LW, Venneman NG, Schwartz TP, van Berge Henegouwen GP. et al. Nasobiliary drainage induces long-lasting remission in benign recurrent intrahepatic cholestasis. Hepatology. 2006;43:51–3. doi: 10.1002/hep.20998. [DOI] [PubMed] [Google Scholar]
- 65.Nakamuta M, Sakamoto S, Miyata Y, Sato M, Nawata H. Benign recurrent intrahepatic cholestasis: a long-term follow-up. Hepatogastroenterology. 1994;41:287–9. [PubMed] [Google Scholar]
- 66.Cissarek T, Schumacher B, Schwöbel H, Sarbia M, Neuhaus H. Follow-up of benign recurrent intrahepatic cholestasis (Summerskill-Walshe-Tygstrup syndrome) over 46 years. Z Gastroenterol. 1998;36:379–83. [PubMed] [Google Scholar]