

Abstract
The Notch signaling pathway governs many distinct cellular processes by regulating transcriptional programs. The transcriptional response initiated by Notch is highly cell context dependent, indicating that multiple factors influence Notch target gene selection and activity. However, the mechanism by which Notch drives target gene transcription is not well understood. Herein, we identify and characterize a novel Notch-interacting protein, NACK, which acts as a Notch transcriptional co-activator. We show that NACK associates with the Notch transcriptional activation complex on DNA, mediates Notch transcriptional activity, and is required for Notch-mediated tumorigenesis. We demonstrate that Notch1 and NACK are co-expressed during mouse development and that homozygous loss of NACK is embryonic lethal. Finally, we show that NACK is also a Notch target gene, establishing a feed forward loop. Thus, our data indicate that NACK is a key component of the Notch transcriptional complex and is an essential regulator of Notch-mediated tumorigenesis and development.
Keywords: Notch, tumorigenesis, atypical kinase, esophageal adenocarcinoma, pancreatic ductal adenocarcinoma
Introduction
The Notch signaling pathway acts as a critical regulator during development by governing cell fate determination through direct regulation of transcriptional programs that drive cellular processes including proliferation, differentiation, self-renewal, and apoptosis. These same cellular processes are also driven by aberrant activation of Notch in tumorigenesis. Notch was first implicated in human cancer in T cell acute lymphoblastic leukemia (T-ALL), where activating mutations result in ligand-independent proteolytic cleavage of Notch1 and increased stability of the intracellular domain (NICD) (1). Evidence for genetic alterations in Notch genes can be found in solid tumors; however, the major mechanism for aberrant Notch activity in many non-hematological human malignancies appears to be elevated expression of Notch pathway components and/or loss of negative regulators (2–4).
This mechanism is relevant in solid tumors because Notch signaling is mediated through direct cell-to-cell contact between a Notch-expressing cell and a cell expressing a DSL (Delta, Serrate, Lag-2) ligand. This binding initiates a series of proteolytic cleavages ultimately leading to release of NICD from the plasma membrane and its translocation to the nucleus (5–7). What follows is a step-wise recruitment of Notch and co-activators of the Mastermind-like (Maml) family to the DNA (8–10). This complex binds to CSL (CBF1-Su(H)-LAG1), displacing the co-repressor complex and initiating transcription of target genes (11–13). Although a prevailing scheme for Notch signaling has been accepted, little is known regarding the regulation of transcription initiation by the Notch/Maml/CSL complex. In an effort to identify novel components that are involved in Notch-mediated transcription, we carried out a biochemical screen in Notch-dependent lymphomas and defined a new interacting partner of Notch. We identified and characterized NACK (Notch Activation Complex Kinase) as a new protein interacting with N1ICD and implicated in Notch transcriptional regulation.
Materials and Methods
Subcellular Fractionation
Subcellular fractionation was performed as described previously (14).
Cell Culture and Transfection
All cell culture reagents were purchased from Invitrogen, unless otherwise indicated. Mouse embryonic fibroblasts (MEFs) were prepared from wild-type C57Bl/6 embryos at day E13.5 following a standard MEF isolation protocol (15). Transfections were performed using Lipofectamine 2000.
Western Blotting
Cell lysates were resolved by SDS-PAGE, transferred onto Immobilon-P membranes (Millipore), blocked in 5% milk, and incubated with the appropriate primary antibody overnight. Primary antibodies were α-Flag (1:5000; Sigma), monoclonal α-NACK (1:1000; directed against aa209–287 and aa1051–1152; affinity purified using a NACK-GST fusion protein), polyclonal α-NACK (1:1000; directed against aa1051–1152; affinity purified using a NACK-GST fusion protein), α-CSL (1:1000; polyclonal), α-p27 (1:1000; Santa Cruz Biotechnology, Dallas, TX), α-β-actin (1:5000; Abcam), α-HeyL (1:1000; Abcam), and α-tubulin (1:15000; Sigma). After incubation with HRP-conjugated secondary antibodies, proteins were detected using an enhanced chemiluminescence reaction (Amersham Biosciences, Pittsburgh, PA) according to the manufacturer’s specification.
DNA Pull-down
Streptavidin agarose beads (Pierce, Rockford, IL) were incubated with previously annealed 47mer biotinylated dsDNA containing two high affinity CSL binding sites facing forward (2× CSL binding DNA) or 2 mutated CSL binding sites (Mut. DNA; for oligo sequences see Table S1). For the in vitro DNA pull-down, recombinant Flag-tagged proteins were incubated with DNA streptavidin beads and bound proteins were analyzed by western blot. For DNA pull-down experiments from cells, 293T cells were transfected with N1ICD, Maml1, and NACK and lysates were incubated with DNA streptavidin beads. Protein bound to the beads was analyzed by western blot.
Luciferase Reporter Assay
H1299 cells were transfected with 8× CSL luciferase reporter vector (10), SV40 β-galactosidase (internal transfection control; Clontech, Mountain View, CA), and N1ICD, Maml1, and NACK expression plasmids. Luciferase activity in the lysates was analyzed using the Luciferase Assay System (Promega, Madison, WI) according to the manufacturer’s instructions. Control siRNA, siRNA against human Maml1, and siRNA against human NACK were purchased from Dharmacon (Lafayette, CO).
N1ICD Lymphoma
N1ICD T cell lymphomas were generated as described previously (16).
Viral Infections
NACK shRNA and control (scrambled) shRNA were purchased in the pLKO vector from Open Biosystems (Thermo Scientific, Pittsburgh, PA). Lentivirus was packaged using psPAX2 packaging vector and pMD2.G envelope plasmid. Retrovirus was packaged using SV40 psi− packaging vector. Virus was collected 48 h post-transfection. Cells were infected overnight with virus-containing medium in the presence of 8 µg/mL hexadimethrine bromide (Polybrene, Sigma), and infected cells were selected with 2.5 µg/mL puromycin.
RT-PCR
RNA was isolated from cells using Trizol reagent (Invitrogen) following the manufacturer’s instructions. RNA was isolated from tumors using the RNeasy Mini Kit (Qiagen, Germantown, MD). cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) following the manufacturer’s directions. qPCR reactions were carried out in the Bio-Rad CFX96 thermal cycler using Sybr Green Master Mix (Bio-Rad, Hercules, CA). Gene expression in human and mouse was normalized to GAPDH and Hprt, respectively. Primer sequences are available upon request.
Chromatin Immunoprecipitation
OE33 cells were cross-linked with 1% formaldehyde and cross-linking was quenched by adding glycine to a final concentration of 0.125 M. Cells were resuspended in SDS lysis buffer and sonicated to yield chromatin fragments of approximately 300 to 800 bp. Lysates were immunoprecipitated with α-Notch 927 (polyclonal), α-Notch (ab27526, Abcam), or α-Pragmin (Bethyl Laboratories, Montgomery, TX) antibodies and were reverse cross-linked at 65°C in 200 mM NaCl for 4 h followed by incubation with RNase A and proteinase K. DNA was cleaned using PCR purification kit (Qiagen) and Hes1 and GAPDH were amplified by qPCR. Primer sequences are available upon request.
β-galactosidase Staining of Embryos
Whole embryos were extracted and washed in PBS at room temperature. Embryos were then fixed at 4°C in cold fixative for 60 min, then washed and stained for 24–36 h.
In situ Hybridization
In situ hybridization of Notch1 and NACK was performed as previously described (17,18). The Notch1 probe was designed in the ANK repeat domain and the NACK probe was designed in the kinase domain.
Immunohistochemistry
IHC was performed on 5 µm paraffin sections prepared from paraffin-embedded tissue arrays. Tissue sections were rehydrated, pre-treated with antigen unmasking solution (1:100 dilution; Vector Laboratories, Burlingame, CA), and then treated with 3% H2O2 and blocked with protein block serum-free (Dako, Carpinteria, CA). Sections were incubated with polyclonal antibodies against NACK (α-Pragmin, 1:50) or cleaved Notch1 (1:200 dilution; Abcam, ab-8925), then with biotinylated secondary antibodies (Vector Laboratories). Immunoreactivity was detected using the ABC Elite kit (Vector Laboratories) with AEC as the final chromogen and hematoxylin as the nuclear counterstain.
Soft Agar Experiments
HC11 cells were infected with shRNA against NACK and N1ICD and cells were plated in soft agar (base agar 0.5%, top agar 0.35%). Plates were incubated at 37°C until colonies were visible by eye and then colonies were stained with 1% MTT (Sigma).
Colony Formation
EAC cells were seeded in 6-well plates at a density of 10,000 cells/well and allowed to attach overnight. Cells were then infected with lentiviruses expressing control shRNA or shRNA against NACK. Seven days post-infection, colony formation was quantitated by staining cells with Crystal Violet (Millipore) and counting the number of colonies.
Xenografts
OE19 cells were infected with lentivirus expressing shRNA against NACK, and then were mixed 1:1 with Matrigel (BD Biosciences; 5 mg/mL) and injected into the flanks of nude mice (nu/nu, Jackson Laboratory, Bar Harbor, ME). Xenografts were measured weekly.
Knockout Mouse
Embryonic stem cells harboring a knock-out first (KOF) allele with a promoter-driven cassette inserted between exons 2–3 of the D8Ertd82e gene were generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (19). Chimeras were produced in C57Bl/6 mice by the Transgene Facility of Sylvester Comprehensive Cancer Center at the University of Miami (Miami, FL). Germline transmission was verified by PCR using primers specific to the common loxP site and the neomycin selection marker (Neo). NACKKOF denotes mice harboring the targeted D8Ertd82etm1a(KOMP)Mbp allele. To remove the KOF cassette, NACKKOF/+ mice were bred to heterozygous Tg(ACTFLPe)9205Dym mice (Jackson Laboratory), which have ubiquitous expression of FLPe recombinase and the resulting pups were heterozygous for the floxed allele (NACKflox/+). Primer sequences are available upon request. All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Miami.
Results
NACK is a novel protein with kinase fold that interacts with Notch1ICD
In order to gain a better understanding of the molecular machinery involved in Notch-mediated transcription, Notch complexes were affinity purified from Notch-induced T cell lymphomas and analyzed by liquid chromatography-mass spectrometry (LC-MS/MS) to identify novel binding partners of N1ICD (Figure S1A) (20). LC-MS/MS analysis revealed that all 3 Maml family proteins as well as CSL were co-purified with Notch, thereby authenticating our purification protocol (Figure S1B). Other proteins that were co-purified with Notch included an uncharacterized protein annotated as hypothetical protein D8Ertd82e. Since this protein was co-purified with N1ICD and core components of the transcriptional activation complex, and sequence analysis predicted the presence of a kinase domain at the C-terminus, we refer to it as Notch Activation Complex Kinase (NACK).
In order to validate the interaction between NACK and N1ICD, co-immunoprecipitation studies were performed from 293T cells co-expressing Myc-tagged NACK and Flag-tagged N1ICD (Figure S1C, lane 10). These studies recapitulated the purification results and therefore demonstrate that NACK is a novel interacting partner of N1ICD.
NACK is part of the Notch transcriptional activation complex
To determine the specificity of the relationship between Notch and NACK, we used a monoclonal NACK antibody to pull down NACK from lysates derived from the Notch-driven T cell lymphomas 4084 and NA1 (Ikaros-null), and the Myc-driven T cell lymphoma 6780 (16,21,22). Expression of NACK was robust in 4084 and NA1 cells compared to 6780 cells, demonstrating co-expression of Notch and NACK (Figure 1A). Subcellular fractionation of 4084 cells revealed that NACK and Notch co-localize to the nucleus (Figure 1B). To determine whether NACK interacts with the ternary Notch complex on DNA, we performed DNA pull-down experiments by incubating beads coupled to DNA containing 2 CSL sites (2× CSL) with nuclear lysate from 4084 lymphoma cells and wt mouse thymocytes. Beads coupled to DNA containing 2 mutated CSL sites (2× mut) were used as a control for specificity. We found that NACK is pulled down concomitantly with N1ICD and Maml1 in a CSL-dependent manner (Figure 1C). We achieved similar results when we applied the same technique to lysates from 293T cells transfected with different combinations of N1ICD, Maml1, and NACK (Figure 1D, lanes 9 and 16). Furthermore, the binding of NACK to the Notch complex on DNA appears to be Maml1-dependent, as Notch and CSL alone are not sufficient to recruit NACK to the DNA (Figure 1D, lanes 15 and 16).
Figure 1. NACK forms a complex with N1ICD and Maml1.
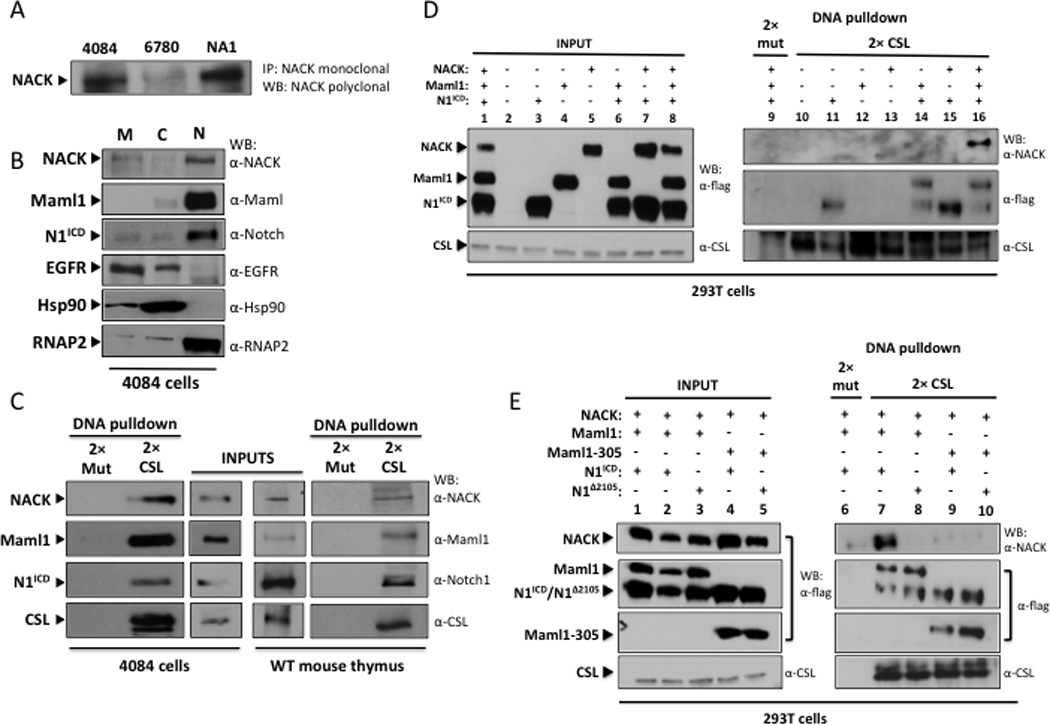
A. IP/western analysis of lysates from T-ALL cell lines. 4084 – Notch-induced lymphoma; 6780 – Myc-induced lymphoma; NA-1 – Ikaros-null lymphoma. B. Western analysis of subcellular fractions of 4084 cells. M – membrane; C – cytoplasm; N – nuclear. C. DNA pull-down and western analysis of nuclear lysates from 4084 cells and wt mouse thymocytes using beads conjugated to 2× CSL binding DNA or 2× mutant CSL binding DNA (2× mut). Input lanes represent 5% of total nuclear lysate. D. DNA pull-down and western analysis of lysates from 293T cells transfected with different combinations of N1ICD, Maml1, and NACK. Input lanes represent 10% of total lysate. E. DNA pull-down and western analysis of lysates from 293T cells transfected with different combinations of N1ICD, N1Δ2105, Maml1, Maml1–305, and NACK. Input lanes represent 10% of total lysate.
Previously, we reported mutations in Notch1 and Maml1 that inhibit the transcriptional activity of the Notch complex on a Notch-responsive promoter. Notch1Δ2105 has a deletion of amino acids 2105–2114 (which lie in the 7th Ank repeat) (23). Maml1–305 contains only the first 305 amino acids of Maml1, which include the Notch binding domain, and acts in a dominant negative manner (10). Gel filtration studies revealed that both mutated proteins can form the ternary complex with CSL on DNA; however, both the activity and the size of the complexes are dramatically reduced, suggesting that the mutated components fail to recruit a necessary co-activator (10,23). Based on our findings that NACK is a co-activator of Notch signaling, we hypothesized that these mutants of Notch and Maml might fail to recruit NACK to the ternary complex. We transfected cells with wt or mutant Notch1 and Maml1, plus NACK, and performed a DNA pull-down. The results show that mutation of either Notch1 (Notch1Δ2105) or Maml1 (Maml1–305) is sufficient to eliminate binding of NACK to the transcription activation complex on DNA (Figure 1D, lanes 8–10). This result suggests that these mutated Notch and Maml proteins are unable to activate transcription due to their failure to recruit NACK to the transcriptional activation complex.
NACK is a co-activator of Notch transcription
Given that NACK is part of the Notch transcriptional activation complex on DNA, we reasoned that NACK should play a role in Notch-mediated transcriptional activity. To address this, CSL-dependent transcriptional reporter assays were performed by transfecting different combinations of N1ICD, Maml1, and NACK into H1299 cells and assaying for luciferase driven by an artificial promoter containing 8 CSL binding sites (8× CSL). Transfection of N1ICD in H1299 cells increased CSL-directed transcription by about 26-fold when compared with vector alone (Figure 2A). When NACK was included, there was a marked increase in activity, similar to the increase seen when Maml1, a bona fide co-activator of Notch transcription, was transfected with N1ICD. Including both NACK and Maml1 in the transfection with N1ICD resulted in a robust cooperative effect on Notch activity, compared to Notch + Maml1 (Figure 2A), indicating that NACK, like Maml1, is a co-activator of Notch transcriptional activity. Titration of NACK suggests that NACK is a rate-limiting component of the transcription activation complex, because at every concentration of Maml, addition of NACK increases transcriptional activity (Figure 2B).
Figure 2. NACK is a co-activator of Notch signaling.
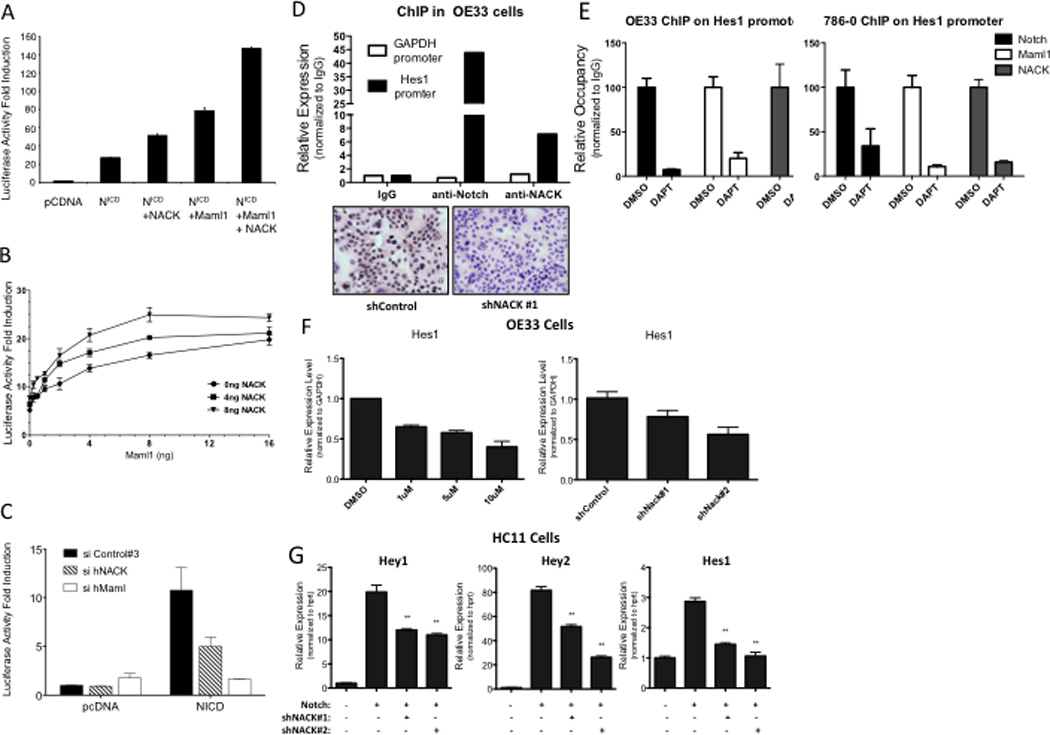
A–C. 8× CSL luciferase reporter assays in H1299 cells transfected with different combinations of plasmids. Experiments were performed in triplicate and bars represent mean (SEM). A. Histogram of luciferase activity. B. Titration curve showing luciferase activity with different quantities of Maml1 and/or NACK over a fixed quantity of N1ICD (2 ng). C. Histogram of luciferase activity after siRNA-mediated knockdown of Maml or NACK. D. ChIP of Notch and NACK on the Hes1 promoter in OE33 cells. IHC validates specificity of the α-NACK antibody. E. ChIP of Notch, Maml, and NACK on the Hes1 promoter in OE33 and 786-0 cells after treatment with DAPT. F. Hes1 expression in OE33 cells treated with DMSO or DAPT, or infected with shRNA against NACK. Bars represent mean (SEM) of 3 samples. **p<0.01 versus shControl. G. Expression of Notch target genes in HC11 cells infected with N1ICD and shRNA against NACK. Bars represent mean (SEM) of 3 samples. **p<0.01 versus Notch.
To assess the role of endogenous NACK on Notch-mediated transcriptional activity, we used targeted siRNA pools to reduce protein levels of hNACK in H1299 cells and examined the effects on luciferase reporter activity. Transfection of H1299 cells with hNACK siRNA resulted in a 74% knockdown of NACK mRNA expression (data not shown) and decreased the transcriptional activity of N1ICD (Figure 2C). Similarly, a siRNA-mediated decrease in hMaml1 mRNA expression (84% knockdown) also reduced N1ICD transcriptional activity, providing further evidence that NACK, like Maml, is a co-activator of Notch-mediated transcriptional activity.
The involvement of NACK in the Notch transcriptional complex suggests that NACK associates with N1ICD on chromatin. To explore this, we carried out chromatin immunoprecipitation (ChIP) experiments using chromatin extracts derived from OE33 esophageal adenocarcinoma cells, which are dependent on Notch activity (data not shown). Results demonstrate that NACK and N1ICD specifically co-localize on the promoter region of Hes1, a known Notch target gene. In contrast, there was no enrichment of either N1ICD or NACK on the GAPDH promoter, which is not a Notch target. (Figure 2D, upper panel). Immunohistochemistry on OE33 cells infected with shRNA targeting NACK shows the specificity of the NACK antibody used for ChIP (Figure 2D, lower panel). Additionally, treatment of OE33 cells or 786-0 renal adenocarcinoma cells with the γ-secretase inhibitor DAPT abrogated the binding of Notch1, Maml1, and NACK to the Hes1 promoter (Figure 2E), supporting the association of NACK with the Notch transcription complex on DNA. Further studies in these cells demonstrate that knockdown of NACK expression using shRNA results in a decrease in Hes1 expression similar to that induced by treatment with DAPT (Figure 2F), indicating that NACK is required for Notch-mediated transcription. To further explore the effect of NACK on transcription of Notch target genes, we showed that ectopic expression of Notch in HC11 mammary epithelial cells induced expression of the Notch target genes Hes1, Hey1, and Hey2, and knockdown of NACK in these cells attenuated the level of expression (Figure 2G). Taken together, these experiments indicate that NACK is an integral component of the Notch transcriptional activation complex on DNA, acting as a co-activator of Notch-mediated transcription.
NACK is a transcriptional target of Notch signaling
In the course of examining NACK gene expression in T-ALL, we found higher expression in tumor samples compared to normal thymus (Figure 3A). This led us to ask whether NACK is a target of Notch signaling. To test this, we used mouse embryonic fibroblasts (MEFs) as a cell culture system devoid of measurable Notch activity. MEFs were infected with retrovirus expressing N1Δ2444 or GFP as an infection control. Results show that NACK mRNA expression is dramatically increased when N1ICD is introduced into the system, as seen by RT-PCR (Figure 3B). As expected, mRNA expression of HeyL, a canonical target of Notch signaling, is also increased. In contrast, mRNA expression of p27, a gene down regulated by Notch signaling, is decreased. These data suggest that Notch1 can induce NACK expression.
Figure 3. NACK is a transcriptional target of Notch signaling.

A. mRNA expression in mouse lymphoma cells expressing exogenous N1ICD or wt mouse thymocytes. B. mRNA expression in MEFs infected with N1ICD. C. NACK expression in HC11 cells infected with N1ICD, N2ICD, N3ICD, or N4ICD. Histogram shows mean (SEM). D. Schematic representation showing architecture of the NACK promoter. Numbers denote genomic sequence from − to + with respect to the translation initiation codon ATG. TSS, transcription start site. E. ChIP of Notch on the NACK promoter in OE33 cells.
In order to determine whether other Notch family members drive NACK transcription, HC11 mouse mammary epithelial cells were infected with retroviruses expressing N1ICD, N2ICD, N3ICD, or N4ICD. Results show that all Notch family members increase NACK mRNA levels when compared to control (Mig) infection, as shown by qPCR analysis (Figure 3C).
To demonstrate that NACK is a direct target of Notch, we analyzed the DNA sequence of the NACK promoter and found two predicted CSL binding sites at −1981 bp and −1790 bp (Figure 3D), suggesting that Notch regulates NACK transcription by binding to its promoter. ChIP assay of the GAPDH and NACK promoters showed specific binding of Notch only to the NACK promoter (Figure 3E). Taken together, these results show NACK as a transcriptional target of Notch signaling.
NACK is co-expressed with Notch during development and in tumors
Since Notch is important during many stages of early development, we asked if NACK is also expressed during development, and whether the expression co-localizes with Notch. We generated a knock-in mouse by inserting the β-galactosidase gene under control of the NACK promoter and inhibitor methionine without disrupting the expression of the wt NACK protein. Staining of an E16.5 embryo showed widespread β-galactosidase expression throughout the embryo (Figure 4A). In situ hybridization demonstrated co-expression of NACK and Notch1 in the central nervous system of E12.5 and E16.5 wt mouse embryos (Figure 4B). These results indicate that NACK is expressed in the developing embryo and shares a high degree of expression overlap with Notch1.
Figure 4. NACK is co-expressed with Notch during development and in tumors.
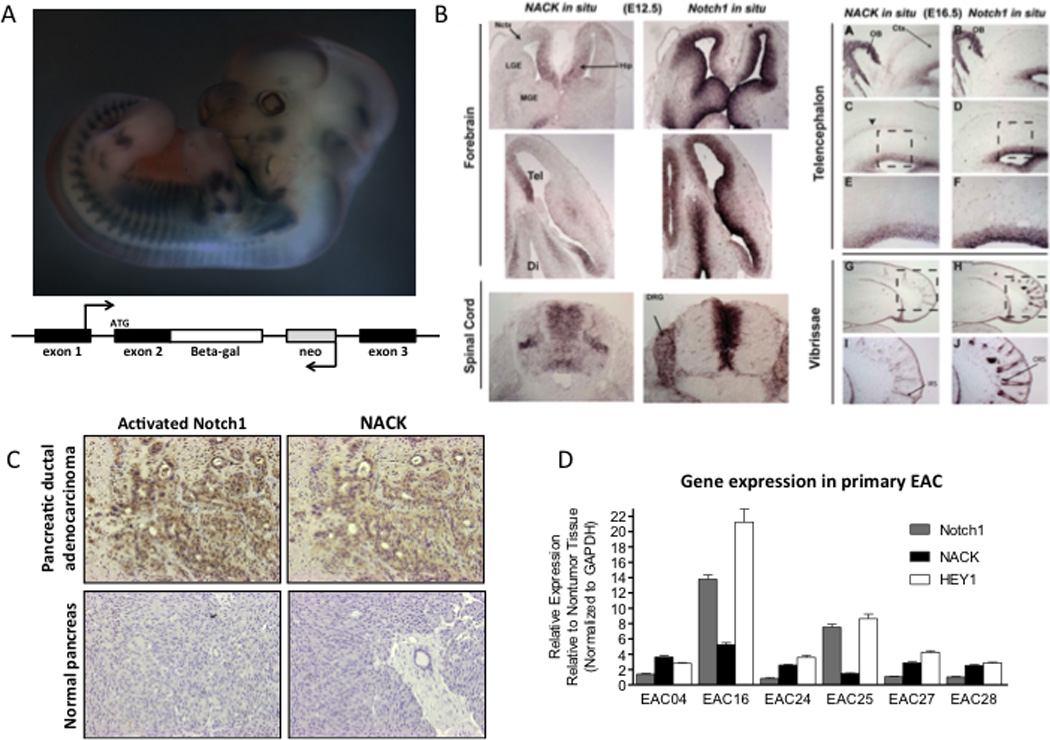
A. Upper panel, β-galactosidase staining of transgenic knock-in mouse E16.5 embryo. Bottom panel, schematic representation showing knock-in construct. B. In situ hybridization of NACK and Notch1 in mouse central nervous system during development. C. IHC of activated N1ICD and NACK in pancreatic ductal adenocarcinoma and normal pancreas (representative samples). D. Expression of Notch1, NACK, and Hey1 in esophageal adenocarcinoma and adjacent esophageal mucosa. Histogram shows mean (SEM).
Since many solid tumors display aberrant expression of Notch receptors and ligands, we hypothesized that NACK, as a specific co-activator, would be expressed at higher levels in cancer tissues compared to normal tissues. Using clinical samples derived from surgically resected pancreatic ductal adenocarcinoma and esophageal adenocarcinoma, we analyzed expression of N1ICD and NACK in tumor versus normal tissues by immunohistochemistry (IHC) and qPCR (Figure 4C and D). Higher levels of N1ICD and NACK protein expression where observed by IHC in pancreatic ductal adenocarcinoma tumor samples when compared to normal pancreas (Figure 4C, representative sample). Validation of the NACK antibody used for IHC is shown in Figure 2E. Expression of NACK, Notch1, and the Notch target gene Hey1 were all elevated in esophageal adenocarcinoma tissues compared to normal tissue (Figure 4D). These data demonstrate that expression of Notch and NACK are linked and that they are co-expressed in development and in tumors.
NACK is required for Notch1-mediated transformation and tumorigenesis
HC11 mammary epithelial cells have little to no endogenous Notch activity but can be transformed by the addition of exogenous N1ICD, as evidenced by the acquisition of anchorage-independent growth in soft agar. To determine if Notch-mediated transcription is dependent on NACK, we examined the effect of NACK on Notch-mediated transformation by knocking down endogenous expression of NACK in HC11 cells and analyzing the outcome by soft agar and qPCR assays. HC11 cells were infected with lentivirus expressing control shRNA or shRNA against NACK. After selection, cells were infected with N1ICD or empty control vector. While NACK knockdown had no effect on the growth of normal mouse mammary epithelial HC11 cells (Figure 5A, top panel), the number of soft agar colonies formed by HC11 cells infected with N1ICD was greatly reduced in the shRNA-infected samples (Figure 5A, bottom panel). Knockdown of NACK was confirmed by qPCR (Figure 5B). Moreover, the mRNA levels of the Notch target genes Hey1 and Cyclin D1 (24) were also decreased in the presence of shNACK, further supporting the role of NACK as a co-activator of Notch-mediated transcriptional activity (Figure 5B). These results demonstrate the requirement for NACK in Notch-mediated transformation.
Fig. 5. NACK is required for Notch-mediated transformation and tumorigenesis.
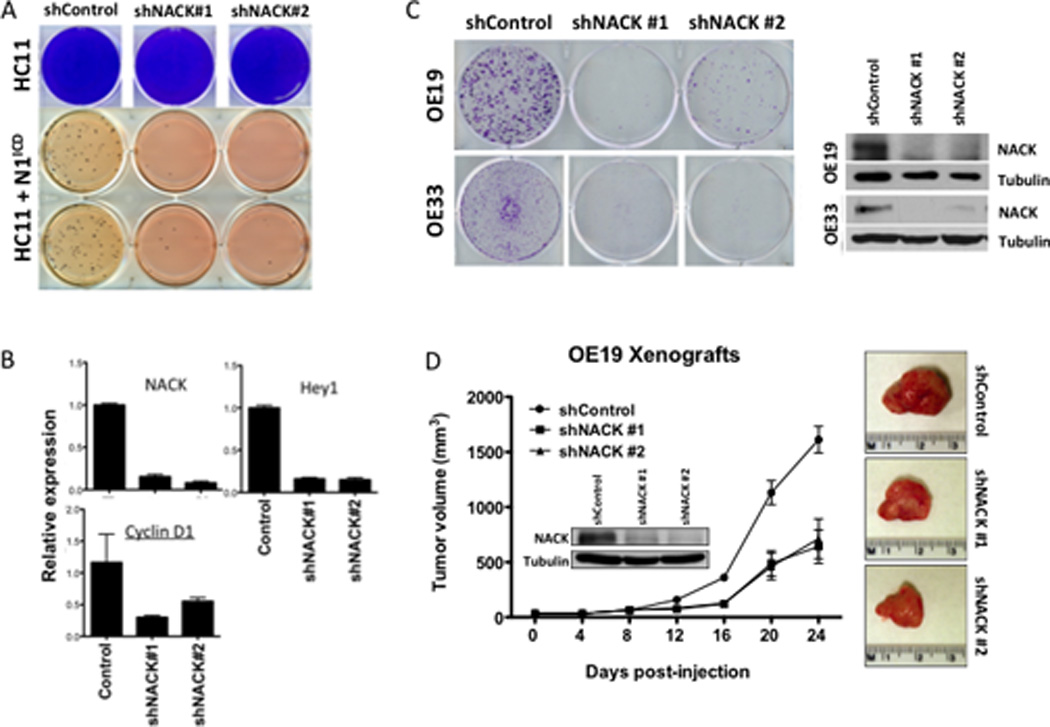
A. Upper panel, colony growth of HC11 cells infected with shRNA against NACK. Lower panel, growth of HC11 cells infected with N1ICD and shRNA against NACK in soft agar. B. Expression of NACK, Hey1, and Cyclin D1 in HC11 cells infected with shRNA against NACK. Bars represent mean (SEM) of fold-change relative to control shRNA. C. Colony formation in OE19 and OE33 cells infected with shRNA against NACK. NACK knockdown was verified by western blot. D. Xenograft formation from OE19 cells infected with shRNA against NACK. Tumor volume was measured weekly and error bars indicate SEM. n=8 per group. At 24 d post-injection, tumors were harvested and representative images are shown from each group.
We next examined the effect of NACK on Notch-mediated tumorigenesis. Knockdown of NACK in esophageal adenocarcinoma cells (OE19 and OE33), which was verified by western blot, resulted in dramatic inhibition of the clonogenic potential of these cells (Figure 5C). OE19 and OE33 cells are dependent on Notch activity for survival and have detectable levels of activated N1ICD protein and NACK (data not shown). To address the effect of NACK on tumor growth, OE19 cells infected with control shRNA or shNACK were injected subcutaneously into the flanks of nude mice and tumor size was measured every 4 days for 24 days. Knockdown of NACK, again verified by western blot, resulted in decreased tumor growth (Figure 5D), indicating a major role for NACK in Notch-mediated tumorigenesis.
NACK knockout is embryonic lethal
Embryonic stem (ES) cells harboring a targeted allele of D8Ertd82e were obtained from the International Knockout Mouse Consortium (KOMP). The targeted D8Ertd82etm1a(KOMP)Mbp allele (denoted in this manuscript as NACKKOF) carried a knockout first (KOF), promoter-driven cassette inserted between the second and third exons of the gene (Figure 6A, top panel). The presence of the splice acceptor site in the cassette is predicted to generate a non-functional truncated transcript. We determined that knockout of NACK is embryonic lethal based on the absence of homozygous NACKKOF/KOF pups among 114 live births from NACKKOF/+ breedings (Figure 6A, middle panel). In fact, we were unable to recover NACKKOF/KOF embryos even as early as E9.5. Representative genotyping results are shown in Figure 6A, bottom panel.
Figure 6. NACK knockout is embryonic lethal.
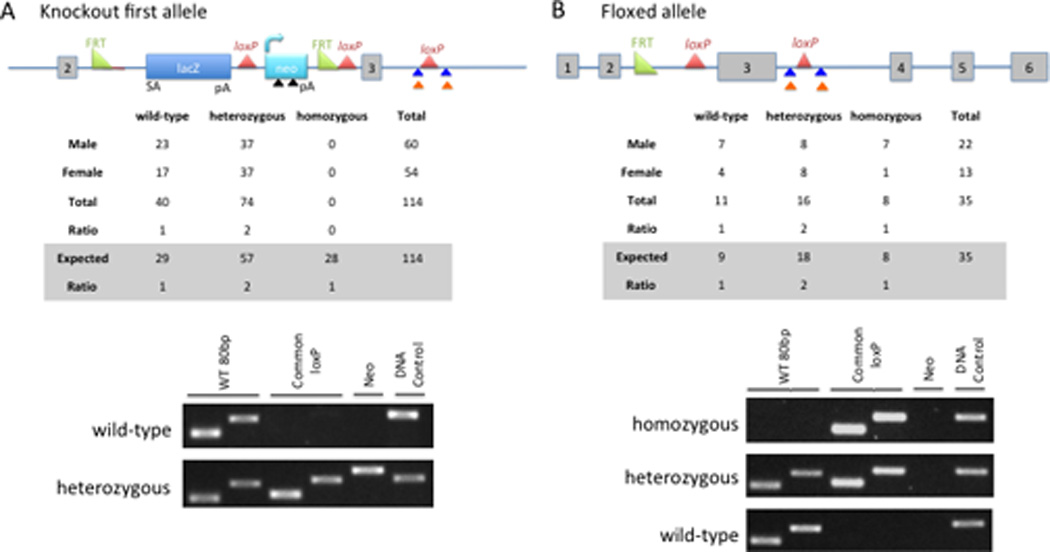
A. Upper panel, schematic of knockout first allele. Colored arrows mark the locations of the primers used for genotyping: black triangle, Neo primers; blue triangle, common loxP primers; orange triangle, WT 80bp primers. Middle panel, summary of live births achieved from mating two NACKKOF/+ mice. Bottom panel, genotyping results from wt and heterozygous pups. B. Top panel, schematic of floxed allele resulting from FLPe recombination. Middle panel, summary of live births achieved from mating two NACKflox/+ mice. Bottom panel, representative genotyping results from wt, heterozygous, and homozygous pups.
Crossing NACKKOF/+ mice with transgenic mice expressing FLPe (the enhanced version of the site-specific recombinase FLP) (25) resulted in recombination between the FRT sites, generating a floxed allele (Figure 6B, top panel). This recombination reverts the allele to the wild-type (but floxed) configuration. Genotyping results showed that the segregation of the floxed allele follows Mendelian inheritance, indicating that FLPe recombination rescues the embryonic lethality of the KOF allele (Figure 6B, middle panel). We confirmed the recombination by demonstrating the loss of Neo without loss of the common LoxP site (Figure 6B, bottom panel). Characterization of this NACK knockout first mouse model clearly demonstrates that NACK is an essential gene in development.
In conclusion, we have demonstrated that NACK is a novel integral component of the Notch transcriptional activation complex, acts as a co-activator of Notch signaling, and participates in a feed-forward loop whereby NACK increases the transcriptional activity of Notch, leading to increased transcription of NACK and other target genes (Figure 7A). Taken together, these data suggest that NACK is required for Notch-mediated transcription and that NACK knockout is embryonic lethal, highlighting the critical importance of this protein in both cancer and development.
Discussion
The importance of Notch signaling in hematologic malignancies and solid tumors has made Notch an attractive target for therapeutic intervention in cancer (2,4,26). Currently, therapeutic approaches have been limited to inhibition of NotchICD production either by blocking γ-secretase inhibitors or by antibody antagonists. A key feature of Notch signaling is rapid recruitment of pathway-specific co-activators to initiate a burst of target gene transcription and subsequent degradation and turnover of N1ICD transcription complexes. To better understand the mechanisms of Notch signaling during tumorigenesis and to identify novel druggable components of Notch transcription complexes, we performed a protein screen from Notch-induced lymphoma. Herein we report the identification of a novel binding partner of the Notch transcriptional activation complex, which we termed NACK. This protein has been previously identified as Pragmin and has been shown to interact with Rho family GTPases (27) and C-terminal Src kinase (28). Using both in vivo and in vitro approaches we demonstrated that NACK acts as a regulator of Notch transcription, is necessary for Notch-driven tumorigenesis, and is required for embryonic development. Taken together, this study demonstrates that NACK is critical for Notch function.
We propose that NACK is recruited to the Notch complex by interactions with Notch and Maml on chromatin. Two pieces of data suggest that the requirement for NACK in Notch transcriptional activation serves a role in the timing of Notch signaling: first, that NACK is rate limiting in transcription and second, that NACK is a target gene of Notch and thus establishes a feed-forward relationship.
Analysis of NACK expression in various cells and tissues reveals that its steady state level is low, but is exquisitely sensitive to activation of Notch. That is, in cells that lack activation of Notch we find low levels of NACK, but when Notch is activated, levels of NACK are rapidly induced. Therefore, we argue that NACK functions as a “reostat” for Notch transcriptional activity. Therefore, recruitment of NACK to create functional Notch transcriptional activation complexes is a mechanism to temporally regulate Notch signaling. Although we propose that NACK is required for basal Notch transcriptional activity, the possibility exists that NACK instead plays a role in modulating the strength and duration of the transcriptional activity of Notch complexes.
NACK is in a class of kinases annotated in the kinome as atypical kinases (29). Proteins in this class are predicted to have kinase folds, but they harbor substitutions in amino acid residues that are thought to be critical for catalysis. While it was originally thought that these substitutions rendered atypical kinases enzymatically inactive, there are several recent reports assigning kinase activity to previously identified “inactive” atypical kinases (30–32). NACK has several major protein kinase features, including the VAIK and HRD motifs; however, it lacks the conserved DFG motif that functions in metal binding. Substitution in the highly conserved Mg2+-binding DFG motif is also noted in the atypical kinase CASK, which has demonstrable kinase activity (33). These findings suggest that NACK might have kinase activity as well; however, no targets have yet been identified. Identification of proteins phosphorylated by NACK will provide further insight into the functional role of this protein and may highlight potential targets for therapeutic intervention.
Underscoring a role for NACK in Notch signaling, we observed a striking overlap in the expression of Notch1 and NACK during mouse development and in human tumor samples (Figure 4). Notch signaling acts as a central axis for cell proliferation and lineage specification during embryonic development, and also plays a pivotal role during neurogenesis (34). This supports our finding that NACK is co-expressed with Notch1 in the central nervous system of E12.5 and E16.5 wt mouse embryos (Figure 4B). Further evidence for the role of NACK during development comes from our NACK knockout-first mouse, which shows that loss of NACK is embryonic lethal (Figure 6). We were unable to detect NACKKOF/KOF embryos even as early as E9.5, suggesting that embryos with this genotype either do not implant in the uterine wall or are reabsorbed very early in gestation.
Knockout of either Notch1 or Notch2 is lethal by E11.5 (35–37), while knockout of Notch3 or Notch4 has no effect on development (38–40). Interestingly, CSL knockout results in death before E10.5 (41), likely due to the combined effect on all 4 Notch receptors. The severity of NACK knockout is similar to that of CSL knockout, and both are more severe than loss of a single Notch receptor, suggesting that loss of NACK might also impact signaling through all 4 Notch receptors. This is supported by our finding that each of the Notch receptors is able to induce NACK expression (Figure 3C). Alternatively, NACK could play a role in other signaling pathways that are important during development. This has been shown for Maml1, which is also a co-activator of the muscle-specific transcription factor MEF2C (42) as well as β-catenin signaling (43). Since it appears that NACK is recruited to the Notch ternary complex by Maml, it is plausible and perhaps likely that NACK is also participating in other developmental pathways.
Supplementary Material
Acknowledgments
The authors would like to thank members of the Capobianco laboratory for support and technical assistance, and J. Diez for assistance in generating the knockout mouse. NIH grants to Velocigene at Regeneron Inc. (U01HG004085) and the CSD Consortium (U01HG004080) funded the generation of gene-targeted ES cells for 8500 genes in the KOMP Program, archived and distributed by the KOMP Repository at UC Davis and CHORI (U42RR024244).
NCI RO1 CA 83736, RO1 CA 169805-01 to A.J.C.
Samuel Waxman Foundation for Cancer Research to A.J.C.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 2.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 3.Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 4.Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–8537. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 5.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 6.Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, et al. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- 7.Struhl G, Greenwald I. Presenilin-mediated transmembrane cleavage is required for Notch signal transduction in Drosophila. Proc Natl Acad Sci USA. 2001;98:229–234. doi: 10.1073/pnas.011530298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petcherski AG, Kimble J. Mastermind is a putative activator for Notch. Curr Biol. 2000;10:R471–R473. doi: 10.1016/s0960-9822(00)00577-7. [DOI] [PubMed] [Google Scholar]
- 9.Wu L, Aster JC, Blacklow SC, Lake R, Artavanis-Tsakonas S, Griffin JD. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat Genet. 2000;26:484–489. doi: 10.1038/82644. [DOI] [PubMed] [Google Scholar]
- 10.Jeffries S, Robbins DJ, Capobianco AJ. Characterization of a high-molecular-weight Notch complex in the nucleus of Notch(ic)-transformed RKE cells and in a human T-cell leukemia cell line. Mol Cell Biol. 2002;22:3927–3941. doi: 10.1128/MCB.22.11.3927-3941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carpio RRV-D, Kaplan FMF, Weaver KLK, VanWye JDJ, Alves-Guerra M-CM, Robbins DJD, et al. Assembly of a Notch transcriptional activation complex requires multimerization. Mol Cell Biol. 2011;31:1396–1408. doi: 10.1128/MCB.00360-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aster JC, Robertson ES, Hasserjian RP, Turner JR, Kieff E, Sklar J. Oncogenic forms of NOTCH1 lacking either the primary binding site for RBP-Jkappa or nuclear localization sequences retain the ability to associate with RBP-Jkappa and activate transcription. J Biol Chem. 1997;272:11336–11343. doi: 10.1074/jbc.272.17.11336. [DOI] [PubMed] [Google Scholar]
- 13.Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, et al. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H) Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- 14.Dignam JD, Martin PL, Shastry BS, Roeder RG. Eukaryotic gene transcription with purified components. Meth Enzymol. 1983;101:582–598. doi: 10.1016/0076-6879(83)01039-3. [DOI] [PubMed] [Google Scholar]
- 15.Hogan B, Beddington R, Constantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- 16.Beverly LJ, Felsher DW, Capobianco AJ. Suppression of p53 by Notch in lymphomagenesis: implications for initiation and regression. Cancer Res. 2005;65:7159–7168. doi: 10.1158/0008-5472.CAN-05-1664. [DOI] [PubMed] [Google Scholar]
- 17.Tiveron MC, Hirsch MR, Brunet JF. The expression pattern of the transcription factor Phox2 delineates synaptic pathways of the autonomic nervous system. J Neurosci. 1996;16:7649–7660. doi: 10.1523/JNEUROSCI.16-23-07649.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez C, Tatard VM, Bertrand N, Dahmane N. Differential modulation of Sonic-hedgehog-induced cerebellar granule cell precursor proliferation by the IGF signaling network. Dev Neurosci. 2010;32:59–70. doi: 10.1159/000274458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.University of California Davis, editor. [cited 2014 Jun 16];KOMP Repository [Internet] Available from: http://www.komp.org. [Google Scholar]
- 20.Demarest RM, Dahmane N, Capobianco AJ. Notch is oncogenic dominant in T-cell acute lymphoblastic leukemia. Blood. 2011 doi: 10.1182/blood-2010-05-286351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C-H, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci USA. 2007;104:13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gómez-del Arco P, Koipally J, Georgopoulos K. Ikaros SUMOylation: switching out of repression. Mol Cell Biol. 2005;25:2688–2697. doi: 10.1128/MCB.25.7.2688-2697.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeffries S, Capobianco AJ. Neoplastic transformation by Notch requires nuclear localization. Mol Cell Biol. 2000;20:3928–3941. doi: 10.1128/mcb.20.11.3928-3941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ronchini C, Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic) Mol Cell Biol. 2001;21:5925–5934. doi: 10.1128/MCB.21.17.5925-5934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. genesis. 2000;28:106–110. [PubMed] [Google Scholar]
- 26.La O, De J-P, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci USA. 2008;105:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka H, Katoh H, Negishi M. Pragmin, a novel effector of Rnd2 GTPase, stimulates RhoA activity. J Biol Chem. 2006;281:10355–10364. doi: 10.1074/jbc.M511314200. [DOI] [PubMed] [Google Scholar]
- 28.Safari F, Murata-Kamiya N, Saito Y, Hatakeyama M. Mammalian Pragmin regulates Src family kinases via the Glu-Pro-Ile-Tyr-Ala (EPIYA) motif that is exploited by bacterial effectors. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1107740108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manning GG, Whyte DBD, Martinez RR, Hunter TT, Sudarsanam SS. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 30.Brennan DF, Dar AC, Hertz NT, Chao WCH, Burlingame AL, Shokat KM, et al. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011;472:366–369. doi: 10.1038/nature09860. [DOI] [PubMed] [Google Scholar]
- 31.Ungureanu D, Wu J, Pekkala T, Niranjan Y, Young C, Jensen ON, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18:971–976. doi: 10.1038/nsmb.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS. Kinases and Pseudokinases: Lessons from RAF. Mol Cell Biol. 2014;34:1538–1546. doi: 10.1128/MCB.00057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukherjee K, Sharma M, Urlaub H, Bourenkov GP, Jahn R, Südhof TC, et al. CASK Functions as a Mg2+-independent neurexin kinase. Cell. 2008;133:328–339. doi: 10.1016/j.cell.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Louvi A, Artavanis-Tsakonas S. Notch signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:93–102. doi: 10.1038/nrn1847. [DOI] [PubMed] [Google Scholar]
- 35.Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–719. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 36.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- 37.Hamada Y, Kadokawa Y, Okabe M, Ikawa M, Coleman JR, Tsujimoto Y. Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development. 1999;126:3415–3424. doi: 10.1242/dev.126.15.3415. [DOI] [PubMed] [Google Scholar]
- 38.Krebs LT, Xue Y, Norton CR, Sundberg JP, Beatus P, Lendahl U, et al. Characterization of Notch3-deficient mice: normal embryonic development and absence of genetic interactions with a Notch1 mutation. genesis. 2003;37:139–143. doi: 10.1002/gene.10241. [DOI] [PubMed] [Google Scholar]
- 39.Kitamoto T, Takahashi K, Takimoto H, Tomizuka K, Hayasaka M, Tabira T, et al. Functional redundancy of the Notch gene family during mouse embryogenesis: analysis of Notch gene expression in Notch3-deficient mice. Biochemical and Biophysical Research Communications. 2005;331:1154–1162. doi: 10.1016/j.bbrc.2005.03.241. [DOI] [PubMed] [Google Scholar]
- 40.Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- 41.Oka C, Nakano T, Wakeham A, la Pompa de JL, Mori C, Sakai T, et al. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121:3291–3301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- 42.Shen H, McElhinny AS, Cao Y, Gao P, Liu J, Bronson R, et al. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes Dev. 2006;20:675–688. doi: 10.1101/gad.1383706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alves-Guerra M-C, Ronchini C, Capobianco AJ. Mastermind-like 1 Is a specific coactivator of beta-catenin transcription activation and is essential for colon carcinoma cell survival. Cancer Res. 2007;67:8690–8698. doi: 10.1158/0008-5472.CAN-07-1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.