

Abstract
Monocarboxylate transporter 8 (MCT8) deficiency is an X-linked disorder resulting from an impairment of the transcellular transportation of thyroid hormones. Within the central nervous system thyroid hormone transport is normally mediated by MCT8. Patients are described as affected by a static or slowly progressive clinical picture which consists of variable degrees of mental retardation, hypotonia, spasticity, ataxia and involuntary movements, occasionally paroxysmal. The authors describe the clinical and neuroradiological picture of 3 males patients with marked delayed brain myelination and in which the clinical picture was dominated by early onset nonparoxismal extrapyramidal symptoms. In one subject a novel mutation is described.
Keywords: MCT8, X-linked, leukodystrophy, hypomyelination, movement disorder, extrapyramidal, thyroid
Monocarboxylate transporter 8 deficiency (also known as Allan-Hernon-Dudley syndrome OMIM: #300523) is an X-linked disorder caused by an impairment of the transcellular transportation of thyroid hormones. In the brain, transcellular transport of thyroid hormones is mediated by MCT8, which is encoded by SLC16A2.1 The hallmark of the disease is a thyroid profile characterized by an increased free T3, normal to low free T4, low reverse T3, and normal to elevated TSH serum without any signs or symptoms of congenital hypothyroidism.2 Affected patients are described having a static or slowly progressive clinical picture, which consists of variable mental retardation, hypotonia, spasticity and pyramidal signs, facial/neck weakness, ataxia, and involuntary movements that may be paroxysmal.3–5 Since the first patient was published in 1944,6 a discrete number of patients have been reported describing the clinical and endocrinological picture,3,5–11 as well as the appearance of abnormal white matter signal on magnetic resonance imaging (MRI) early in the disease course.4,12,13 Here, the authors report the clinical and neuroradiological picture of 3 patients with markedly delayed brain myelination and in which the clinical picture was dominated by early onset extrapyramidal symptoms evident since the first months of life.
Case Reports
Patient 1
This patient is the first child of nonconsanguineous healthy parents of Albanian origin. The pregnancy and delivery were uneventful. At the age of 6 months, developmental delay became evident, accompanied by progressive dystonic posturing of upper limbs. At the age of 20 months, the first MRI demonstrated a delay of myelination (Figure 1A). At evaluation at 3 years and 3 months of age, the clinical picture was dominated by extrapyramidal symptoms (dystonic tetraparesis, fluctuation of muscle tone, startle reactions) associated with severe hypotonia (only partial head control) and pyramidal signs (bilateral Babinski reflexes, hypereflexia); concerning care, feeding, and motor independency, he needed constant assistance. Magnetic resonance repeated at 3 years and again at 5 years and 6 moths of age showed slow progression of myelination reaching almost a normal pattern (Figure 1B). Proton magnetic resonance spectroscopy (H-MRS) performed at 5 years of age showed normal N-acetylaspartate and myoinositol peak values and a choline peak values at the upper limits of normal. Computed tomography scans did not show calcifications. Brainstem auditory evoked potentials, visual evoked potentials, electroretinogram, and fundus oculi examination were all normal. An electroencephalogram did not show epileptic abnormalities. Laboratory and basic metabolic investigations performed (creatine kinase, lactic and pyruvic acid, ammonemia levels, uric acid, urinary organic acid, plasmatic and urinary amminoacid) were normal. Thyroid function tests demonstrated high free T3 level (10.2 pmol/L, normal values 3.80–6.80 pmol/L), low free T4 level (9 pmol/L, normal values 12–16 pmol/L) and normal TSH level. Molecular genetic analysis of SLC16A2 revealed the presence of an already described missense mutation (c.1412T>C [p.Leu471Pro]) (Figure 2).14,15
Figure 1.
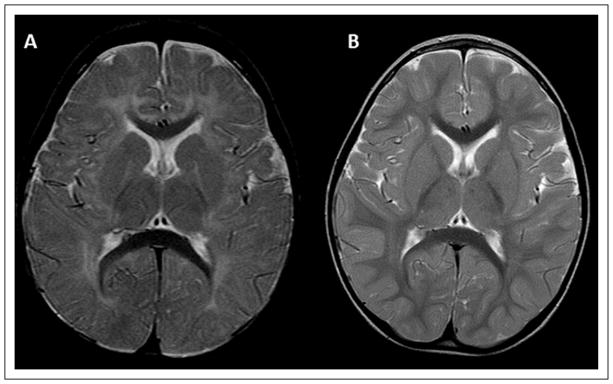
MR T2-weighted axial scan of patient 1. The first MRI (A) was performed when the patient was 20 months of age; the second one (B) was performed at 5 years and 6 months of age. The neuroradiological follow-up clearly shows the progression of the initially marked delayed myelination.
Figure 2.

SLC16A2 mutational analysis—chromatograms of the 2 different mutations identified in the reported patients.
Patient 2
This patient is the younger brother of patient 1. The pregnancy and delivery were uneventful. His development was delayed early on, and in view of his brother’s diagnosis, he was evaluated at 11 months. The clinical picture was dominated by symptoms suggestive of extrapyramidal system involvement (poor and slow movements, facial grimaces, persistent neonatal reflexes, fluctuating tone, exaggerated startle reactions, abnormal dystonic upper limb posture) associated with severe neuromotor delay (only partial head control, no use of hands) and pyramidal signs (bilateral Babinski reflexes, hypereflexia). Magnetic resonance showed a marked delay of myelination (Figure 3A–3B). H-MRS was not performed. Assessment of visual evoked potentials showed evidence of a conduction delay. Electroretinogram and fundus oculi examinations were unremarkable. Electroencephalogram showed no epileptiform features. Laboratory and metabolic investigations performed (creatine kinase, uric acid, lactic acid and ammonemia levels, prolactin, serum copper levels, urinary organic acid, plasmatic and urinary amminoacid) were normal. Thyroid function tests demonstrated high free T3 level (7.9 pmol/L, normal values 3.80–6.80 pmol/L), low free T4 level (8.7 pmol/L, normal values 12–16 pmol/L), and slight augmentation of TSH level (5.8 mU/L, normal values 0.27–4.20 mU/L). Molecular genetic analysis revealed the same hemizygous missense mutation of SLC16A2 as his brother (Figure 2).
Figure 3.

MR T1-weighted axial scan (A, C with gadolinium) and MR T2-weighted axial scan (B, D) images of patient 2 at 11 months (A, B) and patient 3 at 4 years (C, D) show delayed myelination and no basal ganglia abnormalities.
In addition, the mother of these 2 siblings has been shown to be a heterozygous carrier of the c.1412T>C [p.Leu471Pro] mutation.
Patient 3
This patient is the second of 4 healthy children born to noncon-sanguineous, healthy parents. The pregnancy and delivery were uneventful. From the age of 3 months, increased tone, abnormal upper limb posture (internal abducted rotation), and developmental delay were noted. At the age of 4 months, the first MRI was performed and considered normal. Progressively, he developed a clinical picture characterized by spastic-dystonic tetraplegia, poor spontaneous movement, facial grimaces, persistent neonatal reflexes, and severe cognitive delay. At 2.5 years, he was able to reach for and hold an object and be in a quadruped position when placed, but these skills were later lost. After 4 years of age he developed myoclonic seizures not clearly improved by levetiracetam 60 mg/kg/day. Because of extrapyramidal symptoms, he was treated by L-dopa/carbidopa (max dose 50 mg/12.5 mg twice a day) with a reported transient improvement (better head control and spontaneous movements). Concerning care, feeding, and motor independency, he needed constant assistance.
MRIs were repeated at the ages of 2, 3, and 4, years showing a slow progression of myelination that was still delayed at the last examination at 4 years of age (Figure 3C–3D). No basal ganglia abnormalities were evident on MRI (Figure 3C–3D). H-MRS was also performed at 3 years and showed markedly elevated levels of choline. Computed tomography scans did not show calcifications.
Metabolic and genetic investigations performed (biotinididase, very long chain fatty acids, phytanic acid, pristanic acid, urinary orgnic acids, urinary, free and esterified carnitine, aminoacids in plasma and spinal fluid, lactate and pyruvate, urinary mucopolysaccharides and oligosaccharides, succyniladenosine in spinal fluid, pyridoxal-5′ phosphate, neurotransmitters, pterins and folate, seric and sialic acid in spinal fluid, chromosome microarray, FMR1, PLP1, GJC2, LMNB1, AADC molecular analysis) were all normal. Thyroid function tests demonstrated normal free T3 level, low free T4 level (3.4 pmol/L, normal values 5.9–11.6 pmol/L), low rT3 (8 ng/dl, normal values 11–32 ng/dl) and normal TSH levels. Molecular genetic analysis of SLC16A2 gene revealed the presence of one novel nonsense mutation predicted to result in the replacement of the codon for the amino acid tryptophan with a premature termination codon at codon 219 in the SLC16A2 protein (c.656G>A [p.Trp219X]) (Figure 2). The mutation has never been reported, but is predicted to be pathogenic. Pathogenicity of this change is inferred as nonsense mutations may result in a truncated and usually nonfunctional protein product. In actuality, nonsense mutations are likely to be subjected to the nonsense-mediated mRNA decay pathway, in which mRNAs containing nonsense mutations are degraded before they are translated into a polypeptide. The production of a nonfunctional protein, or the absence of the protein, is considered in many cases to be causative of disease.
Discussion
The authors described the clinical and neuroradiological picture of 3 male patients affected by MCT8 deficiency. The clinical picture of these patients was dominated by an early onset predominantly dystonic tetraplegia with symptoms evident since the first months of life. Symptoms included extrapyramidal manifestations typically seen in young infants,16 including hypokinesia, facial grimaces, persistence of neonatal reflexes, abnormal upper limb postures, fluctuation of muscle tone, and exaggerated startle responses. In the second year of life, more classic patterns of a movement disorder, potentially exacerbated by stressful conditions or sensitive stimuli, were evident in these children.16 The presence extrapyramidal signs, even if almost always reported among symptoms, has not been underlined except in 2 recently described cases,5 and in 7 patients where the movement disorder was described as paroxysmal.3,5,7,8 It has not been stressed that in some cases extrapyramidal symptoms can dominate the clinical picture from the beginning. The age at onset of symptoms of extrapyramidal involvement is not always clear in the literature, maybe because these symptoms are not always easy to recognize. Semeiological aspects of extrapyramidal symptoms change in relation to age, as they are, indeed, age specific.16 These cases prompt us to emphasizes the possibility of an early onset, predominant, and nonparoxismal extrapyramidal picture. In patients, extrapyramidal symptoms were evident since the first months of life and were so predominant that the differential diagnosis initially included early onset X-linked extrapyramidal neurodegenerative syndromes such as Lesch-Nyhan disease.
In 2 cases with paroxysmal movement disorders,3,5,7,8 abnormal signal of the putamina on MRI has been observed.7,8 Conversely, in these cases, the MRIs did not show any abnormalities of basal ganglia.
White matter abnormalities on MRI in MCT8 deficiency have been described variably as delayed myelination13 or “hypomyelination.”4 According to the definition of hypomyelination, an unchanged pattern of deficient myelination on 2 MRIs at least 6 months a part in a child older than 1 year,17 van der Knaap and Wolf pointed out that white matter abnormalities observed in MCT8 deficiency should not be considered as true hypomyelination because of the progression of myelination, even if slow.12 The MRI follow-up in the first case (Figure 1) reinforces and confirms the idea of a marked delay in myelination and not hypomyelination as the prominent neuroradiological feature of the disease. This is also supported by the normality of H-MRS in this patient, performed when the slow myelination processes were almost finished. H-MRS in affected patients may show increased choline and myoinositol levels and decreased N-acetylaspartate9 when the myelination process is still delayed, as in earlier scans in patient 3.
In conclusion, even if a simple delay of myelination is itself a nonspecific feature,12 the authors think that the association of a marked delay of myelination on MRI together with an early onset clinical picture dominated by extrapyramidal symptoms in a male without history of perinatal asphyxia is very suggestive of MCT8 disorders. This association should prompt thyroid hormone testing, including reverse T3, which could be very helpful in achieving the final diagnosis when presented with the classical profile associated with MCT8 deficiency.
Acknowledgments
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Reprints and permission: sagepub.com/journalsPermissions.nav
Author Contributions
DT: study design, data collection, and analysis and manuscript and figure preparation. AV: data collection and analysis, figure preparation, manuscript review and corrections. AB: data collection and analysis, manuscript review and corrections. JLS: data collection and analysis, manuscript review and corrections. CDC: genetic analysis, manuscript review and corrections. FN: genetic analysis, manuscript review and corrections. ADG: data collection and analysis, manuscript review and corrections. AM: data collection and analysis, manuscript review and corrections. FT: neuroimaging evaluation, manuscript review and corrections. JEBH: data collection and analysis, manuscript review and corrections. OZ: genetic analysis, manuscript review and corrections. UB: data collection and analysis, manuscript review and corrections. SO: study design, data collection and analysis, manuscript review and corrections.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This work was performed in agreement with the ethical rules of IRCCS C. Mondino Institute of Neurology Foundation and Children’s National Medical Center. The authors received patient consent forms from the family of the patients.
References
- 1.Heuer H, Visser TJ. Minireview: pathophysiological importance of thyroid hormone transporters. Endocrinology. 2009;150(3):1078–1083. doi: 10.1210/en.2008-1518. [DOI] [PubMed] [Google Scholar]
- 2.Filho HC, Marui S, Manna TD, et al. Novel mutation in MCT8 gene in a Brazilian boy with thyroid hormone resistance and severe neurologic abnormalities. Arq Bras Endocrinol Metabol. 2011;55(1):60–66. doi: 10.1590/s0004-27302011000100008. [DOI] [PubMed] [Google Scholar]
- 3.Brockmann K, Dumitrescu AM, Best TT, Hanefeld F, Refetoff S. X-linked paroxysmal dyskinesia and severe global retardation caused by defective MCT8 gene. J Neurol. 2005;252(6):663–666. doi: 10.1007/s00415-005-0713-3. [DOI] [PubMed] [Google Scholar]
- 4.Vaurs-Barrière C, Deville M, Sarret C, et al. Pelizaeus-Merzbacher-like disease presentation of MCT8 mutated male subjects. Ann Neurol. 2009;65(1):114–118. doi: 10.1002/ana.21579. [DOI] [PubMed] [Google Scholar]
- 5.Gika AD, Siddiqui A, Hulse AJ, et al. White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med Child Neurol. 2010;52(5):475–482. doi: 10.1111/j.1469-8749.2009.03471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allan W, Herndon CN, Dudley FC. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Ment Defic. 1944;(48):325–334. [Google Scholar]
- 7.Kakinuma H, Itoh M, Takahashi H. A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid. J Pediatr. 2005;147(4):552–554. doi: 10.1016/j.jpeds.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs O, Pfarr N, Pohlenz J, Schmidt H. Elevated serum triiodothyronine and intellectual and motor disability with paroxysmal dyskinesia caused by a monocarboxylate transporter 8 gene mutation. Dev Med Child Neurol. 2009;51(3):240–244. doi: 10.1111/j.1469-8749.2008.03125.x. [DOI] [PubMed] [Google Scholar]
- 9.Sijens PE, Rodiger LA, Meiners LC, Lunsing RJ. 1H magnetic resonance spectroscopy in monocarboxylate transporter 8 gene deficiency. J Clin Endocrinol Metab. 2008;93(5):1854–1859. doi: 10.1210/jc.2007-2441. [DOI] [PubMed] [Google Scholar]
- 10.Holden KR, Zuniga OF, May MM, et al. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol. 2005;20(10):852–857. doi: 10.1177/08830738050200101601. [DOI] [PubMed] [Google Scholar]
- 11.Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A. Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150 V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol. 2005;153(3):359–366. doi: 10.1530/eje.1.01980. [DOI] [PubMed] [Google Scholar]
- 12.van der Knaap MS, Wolf NI. Hypomyelination versus delayed myelination. Ann Neurol. 2009;68(1):115. doi: 10.1002/ana.21751. [DOI] [PubMed] [Google Scholar]
- 13.Namba N, Etani Y, Kitaoka T, et al. Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur J Pediatr. 2007;167(7):785–791. doi: 10.1007/s00431-007-0589-6. [DOI] [PubMed] [Google Scholar]
- 14.Jansen J, Friesema EC, Kester MH, et al. Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab. 2007;92(6):2378–2381. doi: 10.1210/jc.2006-2570. [DOI] [PubMed] [Google Scholar]
- 15.Friesema EC, Grueters A, Biebermann H, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364(9443):1435–1437. doi: 10.1016/S0140-6736(04)17226-7. [DOI] [PubMed] [Google Scholar]
- 16.Krageloh-Mann I. Cerebral palsy. In: Aicardi J, editor. Diseases of the Nervous System in Childhood. 3. New York, NY: Wiley-Blackwell; 2009. pp. 221–222. [Google Scholar]
- 17.Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72(8):750–759. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]