

Abstract
Drug resistance and toxicity are major obstacles in cancer chemotherapy. Combination therapies can overcome resistance, and synergies can minimize dosing. Polymer nanocarriers are interesting vehicles for cancer therapeutics for their delivery and tumor targeting abilities. We synthesized a multilayered polymer nanoparticle (MLNP), comprising of poly(lactic-co-glycolic acid) with surface polyethyleneimine and functional peptides, for targeted drug and gene delivery. We confirmed the particle’s ability to inhibit tumor growth through synergistic action of the drug and gene product. MLNPs achieved transfection levels similar to lipofectamine, while maintaining minimal cytotoxicity. The particles delivered camptothecin (CPT), and plasmid encoding TNF related apoptosis inducing ligand (pTRAIL) (CT MLNPs), and synergistically inhibited growth of multiple cancer cells in vitro. The synergy of co-delivering CPT and pTRAIL via CT MLNPs was confirmed using the Chou-Talalay method: the combination index (CI) values at 50% inhibition ranged between 0.31–0.53 for all cell lines. Further, co-delivery with MLNPs resulted in a 3.1–15 fold reduction in CPT and 4.7–8.0 fold reduction in pTRAIL dosing. CT MLNPs obtained significant HCT116 growth inhibition in vivo compared to monotherapy. These results support our hypothesis that MLNPs can deliver both small molecules and genetic agents towards synergistically inhibiting tumor growth.
Keywords: PLGA, Nanoparticle, Gene therapy, Drug delivery
1. Introduction
Despite numerous discoveries in tumor biology and pathogenesis, cancer remains a highly prevalent and lethal disease. There are over 1.6 million new cases of cancer every year, and approximately 580,000 deaths each year[1]. Current chemotherapies are limited due to the development of tumor resistance and severe side effects. There is strong evidence that combination therapies can minimize drug resistance[2]. Yet side effects from the drugs can still limit treatment dosing, duration, and ultimately efficacy. Targeted therapies, utilizing multiple synergistic drugs can address these obstacles by decreasing dosing and avoiding non-neoplastic cells. Ongoing analysis of tumor biology has exposed the complexities of cancer pathogenesis, and it is increasingly clear that small drugs targeted to specific molecular pathways have limitations[3]. Gene therapy can potentially overcome this obstacle, since it has the potential to alter expression of any gene of interest[4]. Polymer nanoparticle (NP) technology provides an attractive vector for delivering multi-modal therapies against cancer in a targeted manner[5]. By allowing for co-delivery of two agents, nanoparticle systems consolidate these properties into one vehicle and ultimately ensure that the targeted tissue receives both agents at a ratiometric dose. Therefore, the optimal drug ratio can be tuned in vitro, and subsequently translated to the clinic effectively. Combining multiple agents into one carrier should streamline manufacturing and infusion, overcome batch to batch variability, and lower costs. The alternate approach of delivering the two agents in separate vehicles would add potential variability, since each vehicle could have independent pharmacokinetics, biodistribution, and clearance. Improvements in particle design are needed to enhance gene transfection capabilities, while maintaining the ideal biophysiochemical properties and functionality for in vivo delivery. Our work examines multi-functional, multi-layered polymer nanoparticles (MLNPs) for combination gene and small molecule therapy towards synergistically inhibiting tumor growth.
The optimal anti-neoplastic nanoparticle for combination drug and gene delivery should have several characteristics: 1) acceptable safety profile, 2) high transfection capabilities, 3) synergistic payload interactions, and 4) tumor targeting capabilities. Complexes formed with cationic lipids and cationic polymers have been shown to provide effective anti-tumor therapies, yet these systems continue to have issues with stability and toxicity[6–8]. The biophysiochemical properties of these cationic particles have resulted in high in vitro transfection efficiency; however, those same qualities can lead to increased clearance or toxicity in vivo. Surface modifications have improved their translation into in vivo applications, but often at the expense of the particle’s transfection potential. Further, many cationic systems are ideal for gene therapies, but do not lend themselves well for small molecule delivery, so co-delivery is difficult. On the other hand, solid polymeric nanoparticles—formed from materials such as poly(lactic-co-glycolic acid) (PLGA)—have many favorable characteristics including long term stability, low toxicity, and superior small molecule delivery[9–11]. Blends of PLGA, and cationic polymers including poly-L-lysine (PLL), polyethyleneimine (PEI), and poly(beta amino esters), have also been extensively studied[12–14]. Although nanoparticles can deliver combination therapies, improvements in transfection, synergy, and functionality are needed.
Here, we examine a delivery system for the co-delivery of agents that we hypothesized would act synergistically: camptothecin (CPT) and a plasmid encoding for TNF related apoptosis inducing ligand (TRAIL). These agents increase apoptosis synergistically through both the intrinsic and extrinsic pathways[15–18]. CPT is a hydrophobic, topoisomerase-1 inhibitor, which induces double stranded deoxyribonucleic acid (DNA) breaks, and subsequent apoptosis[19]. Since CPT is poorly soluble and readily hydrolyzed to an inactive form in water, polymer nanoparticles provide an ideal vector for delivery by improving plasma solubility and stability[20, 21]. Recombinant human TRAIL (rhTRAIL) protein has minimal systemic side-effects, yet has a short systemic half-life[22]. TRAIL induces apoptosis via the extrinsic pathway through binding of the surface death receptors (DR). Recent studies have shown that TRAIL can induce apoptosis in a variety of tumors with minimal systemic toxicities[23, 24]. Delivery of a plasmid encoding for the TRAIL gene (pTRAIL), can potentially address it’s short half-life, since transfected cells can continue producing TRAIL for extended periods, and inhibit tumor growth via the bystander effect[25].
Several previous studies examined the use of nanoparticles for combination drug and gene therapies for cancer[26–37], but significant problems remain. For instance, improving gene transfection capabilities and minimizing toxicity is necessary, while maintaining stable particle properties for in vivo therapies. While several of the systems described in the literature were effective in cell culture, this did not translate to promising results in vivo[27–29, 31, 32, 34]. Most combination delivery systems previously described have not demonstrated true synergism between the agents co-delivered, and therefore require delivery of high doses of both small molecules and genetic material. Here, we have improved on prior approaches by rationally designing a PLGA based, modular MLNP, capable of higher transfection and active tumor targeting. Further, we confirmed the synergism of our co-delivery system at a fixed payload ratio over multiple doses and on several cancer cell lines. These improvements allow for marked reduction in both small molecule and plasmid dosing, while maintaining significant tumor growth inhibition in vivo.
2. Materials and Methods
2.1. Materials and cell culture
All reagents were purchased from Sigma Aldrich unless otherwise noted. Plasmid DNA (pDNA) encoding luciferase (pGL4.13; pLuc), green fluorescent protein (GFP; pEGFP; pGFP), and TRAIL (pEGFP-TRAIL; pTRAIL) were purchased from Promega and Addgene respectively. Human embryonic kidney 293 (293T), human glioblastoma (U87), human colorectal adenocarcinoma (HCT116), and human breast adenocarcinoma (MDA-MB-231) cell lines were obtained from ATCC (American Type Culture Collection). All cells were grown in same culture media (CM), which consisted of DMEM medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies), 100 units/ml penicillin, and 100 mg/ml streptomycin (Life Technologies), in a 37C incubator containing 5% CO2. Fluorescence was determined using a UV-Vis spectrophotometer (Cary 50 Bio UV-Vis Spectrophotometer, Varian, Palo Alto, CA).
2.2. Nanoparticle fabrication
NPs were fabricated using established emulsion evaporation techniques. PLGA (50:50 PLGA Carboxylic Acid End Group; i.v. ~0.67 dL/g; Absorbable Polymers: Pelham, AL) was dissolved in organic solvent (ethyl acetate (EA) or dichloromethane (DCM)) at a ratio of 50 mg polymer to 1 mL organic solvent to form the oil phase (O). For drug loaded particles, CPT was dissolved at designated concentrations (5 mg/mL to 0.0001 mg/mL) in O. The polymer solution was then added dropwise to the outer aqueous phase (W2) which consisted of deionized water containing 2.5% (v/v) poly-vinyl alcohol (PVA) at a ratio of 1 mL O to 2 mL W2. The O/W2 emulsion was sonicated on ice three times for 10 s (Tekmar, 600 Watts 38%). When indicated, trans-1,2-Cyclohexanediol (TCHD) was incorporated by adding 12.5 uL of 200 mg/mL TCHD in deionized water as the inner aqueous phase (W1), and added dropwise to O. This W1/O emulsion was sonicated as above, and then added to W2 at the same ratio as above. The final O/W2 or W1/O/W2 emulsion was then diluted 25 fold into 0.3% (v/v) PVA in water. The diluted emulsion was left stirring at 400 rpm for at least 4 h. NPs were then centrifuged at 18,900 g for 15 min at 4C. All centrifugation steps were performed using these parameters. The supernatant was then decanted and the pellet was resuspended in 4 mL water by sonication for 10 sec in a water bath sonicator. Particles were always resuspended using these sonication parameters. Resuspended NPs were flash frozen in liquid nitrogen and lyophilized for long-term storage at −20C.
2.3. Surface modification
MLNP synthesis was adapted from previously reported protocols[11, 38]. Briefly, MLNPs were fabricated by forming PLGA nanoparticles as above with an outer aqueous phase containing 3.33 mg/mL PEI (25 kDa, branched). After spinning overnight, the particles were centrifuged and resuspended in 1 mL of pH 4.0 sodium acetate buffer containing 50 μg/mL of pDNA. The particle suspension was loaded on a rotisserie shaker for 15 min at room temperature (RT), and then centrifuged and resuspended in 1 mL of 10 mg/mL PEI in pH 4.0 sodium acetate buffer. The particle suspension was loaded on a rotisserie shaker for 15 min at RT, and then centrifuged and resuspended in 1 mL of pH 7.4 phosphate buffered saline (PBS) containing 1 mg/mL of a heterobifunctional PEG linker (Peirce Thermo Scientific). The PEG linker was functionalized with N-hydroxysuccinimide (NHS) and maleimide (MAL) and approximately 5 kDa (PEG5) or 1 kDa (PEG1) in size. The particle suspension was loaded on a rotisserie shaker for 30 min at RT, and then centrifuged and resuspended in 1 mL of pH 7.4 PBS containing 2 mg/mL modified antennapedia (mAP) (W. M. Keck Facility at Yale University). The particle suspension was loaded on a rotisserie shaker for 30 min at RT, and then centrifuged and resuspended in 4 mL water. The final MLNP suspension was flash frozen in liquid nitrogen and lyophilized for long-term storage at −20C. Step by step illustration of the conjugation steps are illustrated in Figure 1. Each surface modification step was performed with a molar excess of conjugating reagents.
Figure 1. Surface modification of MLNPs.

Schematic for the step-by-step fabrication and surface modifications of multi-layered nanoparticles (MLNPs). A–B. Camptothecin was encapsulated into poly(lactic-co-glycolic) (PLGA) nanoparticles. C–D. The PLGA nanoparticles were complexed with polyethyleneimine (PEI) which results in a net positive surface charge on the MLNPs. E–F. Plasmid DNA was then complexed onto the positive surface of the MLNPs. G–H. Another layer of PEI was subsequently complexed onto the MLNPs. I–J. A heterobifunctional polyethylene glycol (PEG) linker was then conjugated to the outer PEI layer. K–L. Finally, a cell penetrating peptide, specifically modified antennapedia, was conjugated to the PEG linker.
2.4. Cellular Transfection
For cell culture in 96 well plates, the culture volume was 100 μL and for 48 well plates, the culture volume was 250 μL. Cells were seeded at 50,000 cells/cm2, unless indicated otherwise, in CM without antibiotics, and transfected 24 h later. For polymer NP transfections, the CM was removed and replaced with NPs or MLNPs resuspended in CM containing antibiotics at designated particle concentrations. The particles were resuspended via brief sonication. After 24 h, the particles in CM were replaced with fresh CM. For lipofectamine transfections, 6.4 μL of Lipofectamine 2000 (Life Technologies) reagent was resuspended in 100 μL of OptiMEM (Life Technologies) at RT for 5 min. The lipofectamine solution was then added to an equivalent volume of pDNA in Optimem (11.6 ng/uL) for 20 min at RT. The lipid nanoparticle solution was then added to each cell culture well at 10% of the working volume. The CM containing lipid particles was aspirated and replaced with CM containing antibiotics as stated above after 5 h.
2.5. Cytotoxicity
Human embryonic kidney 293T cells were seeded at approximately 30,000 cells/cm2, and, 24 h later, transfected with 1 mg/mL of particles and lipofectamine containing an equivalent pDNA dose as described previously. Cell viability was determined 48 h post transfection by adding Celltiter Blue reagent to each well at 10% of the culture volume. After 2 h, 90 μL of the culture medium was transferred to a 96 well UV transparent plate (Corning) and assayed for fluorescence at Ex/Em 560/590 nm. The fluorescent signal for each treatment group was normalized to the no treatment (NT) group in order to determine viability. Toxicity was calculated as one minus viability.
2.6. Nanoparticle characterization
Scanning electron microscopy (SEM) was used to determine particle size and morphology. Dried nanoparticles were applied onto double-sided carbon tape and sputter coated with gold, under vacuum, for 30 s with a 40 mA current (Dynavac Mini Coater, Dynavac, USA). An XL-30 ESEM-FEG (FEI Company) having an acceleration voltage of 10–15 kV was used to visualize the nanoparticles. The average particle size was determined using available image analysis software (Image J, National Institute of Health). Particle hydrodynamic diameter and surface charge was determined by dynamic light scattering on a Zetasizer Nano ZS (Malvern Instruments, Ltd.).
2.7. Camptothecin loading and in vitro release
Polymer particles were dissolved at 1–10 mg/mL in dimethyl sulfoxide (DMSO) for 1 h at RT. Afterwards, the dissolved particle solution was diluted 10 fold in CPT extraction buffer (EB; PBS containing 1% sodium dodecyl sulfate and 1% 1 N HCl v/v) for 1 h at RT. CPT was dissolved in DMSO at 5mg/mL and diluted in CPT EB containing 10% DMSO (v/v) for the standard curve. Particle and CPT samples in EB were added to a 96 well UV transparent plate and evaluated for CPT fluorescence at Ex/Em 370/428 nm. Blank NP or MLNP fluorescence was subtracted from the drug sample signal and evaluated for loading against the standard curve.
To determine the CPT release characteristics, particles were suspended in 1 mL of PBS at 3–6 mg/mL, placed in a 10 kDa snake skin dialysis tubing, and sutured closed. The tubing was then placed into 15 mL conical tubes containing 10 mL PBS, which were loaded onto rotisserie shakers at 37C for 96 h. At designated time points, 1 mL of the supernatant was aliquoted for subsequent analysis and the remaining buffer was decanted. Ten mL of fresh PBS was added to each tube after each collection. Thirty μL of EB containing 3.33% DMSO (v/v) was added to 970 μL of sample. CPT was dissolved in DMSO at 5 mg/mL and diluted with EB containing 1% DMSO (v/v) for the standard curve. Samples and standards were analyzed for CPT content as described above.
2.8. Plasmid loading and in vitro release
MLNPs were dissolved at 2 mg/mL in DMSO for 1 h. The particle solution was diluted 10 fold in Tris-EDTA (TE) buffer containing 12.5 mg/mL heparin sulfate overnight at RT. Stock λ DNA was diluted in 12.5 mg/mL heparin sulfate in TE buffer for the standard curve. Particle samples and standards were transferred to a 96 well UV transparent plate, and combined with an equal volume of Quant-iT PicoGreen reagent (Life Technologies) diluted in TE buffer for 5 min at RT. Samples were assayed for fluorescence at Ex/Em 480/520 nm. Blank MLNP fluorescence was subtracted from the plasmid sample signal and evaluated for loading against the standard curve.
Plasmid release characteristics were determined by resuspending 0.5 mg/mL particles in a 15 mL conical tube containing 10 mL of PBS. The tubes were placed on a rotisserie shaker at 37C. At each time point, 1 mL of supernatant was aliquoted and replaced with 1 mL of fresh PBS. Each sample was diluted 10 fold in TE buffer containing 12.5 mg/mL heparin sulfate overnight at RT. Released plasmid content was measured using the Quant-iT PicoGreen reagent as previously described.
2.9. PEI Loading and in vitro release
MLNPs were dissolved at 2 mg/mL in 0.1 M sodium hydroxide (NaOH) overnight at RT. PEI was dissolved in 0.1 M NaOH at 1 mg/mL and diluted for the standard curve. Samples and standards were transferred to a 96 well UV transparent plate, and 10 μL of 7 mg/mL fluorescamine in acetone was added to each well (v/v 10:1) for 10 min at RT, and then assayed for fluorescence at Ex/Em 390/475 nm. Blank MLNP fluorescence was subtracted from the PEI sample signal and evaluated for loading against the standard curve.
PEI release characteristics were determined by resuspending 0.5 mg/mL particles in a 15 mL conical tube containing 10 mL of PBS. The tubes were placed on a rotisserie shaker at 37C. At each time point, 1 mL of supernatant was aliquoted and replaced with 1 mL of fresh PBS. Each sample was diluted 10 fold in 0.1 M NaOH overnight at RT. PEI content was measured using the fluorescamine reagent as previously described.
2.10. Transfection analysis
For the population transfection studies, MLNPs were loaded with pLuc (BL MLNP), and 293T cells were transfected with 1 mg/mL MLNPs and an equivalent plasmid dose using lipofectamine. Expression of luciferase was quantified using Luciferase Assay Reagent (LAR; Promega) 42 h post transfection. The CM was aspirated, and cells were lysed using lysis buffer. After one freeze thaw cycle, the lysates were spun down at 15,000 rpm for 3 min, 20 μL of supernatant was added to 100 μL of LAR, and luminescence was measured using a Glomax luminometer (Promega) over a 10 s integration period. Sample luminescence was normalized to total sample protein, which was determined using a bicinchoninic acid (BCA) protein assay (Thermo Scientific).
Percent cellular transfection was determined by loading MLNPs and lipid NPs with pGFP towards transfecting 293T cells as above. Cells were imaged at 48–72 h post transfection using a Leica SP5 confocal microscope. Images were analyzed for GFP expression by determining percent fluorescent area using ImageJ. Flow cytometry analysis was performed by resuspending the 293T cells in PBS, centrifuging once at 2,000 rpm for 3 min, and resuspending the pelleted cells in Fluorescence-activated cell sorting (FACS) buffer. The resuspended cells were then analyzed with a BD Biosciences FACScan to determine percent transfection.
2.11. in vitro cytotoxicity
The effect of CPT exposure and pTRAIL transfection timing on death kinetics was determined in vitro using free CPT and pTRAIL delivered with lipofectamine. Cells (U87, HCT116, and MDAMB231) were seeded in 96 well culture plates (Corning) at 2,500 cells/cm2. All treatment groups were transfected with pTRAIL 48 h after seeding, and exposed to CPT 24 h before (−24 h), during (0 h), or 24 h after (+24 h) CPT exposure. Treatment controls included NT, and CPT exposure alone at the respective time points. Treatment groups were analyzed for viability 48 h post transfection via Quant-iT Celltiter Blue reagent as described earlier. Figure S 1 illustrates the experimental design for the death kinetics experiments.
MLNPs were then fabricated to deliver CPT and pTRAIL (CT MLNPs), CPT and pLuc (CL MLNPs), and only pTRAIL (BT MLNPs) as described earlier. The initial loading of CPT in O1 ranged between 0.1 to 0.0001 mg/mL (CT1-3 or CL1-3, Figure 2H). U87, HCT116, and MDAMB231 cells were seeded in 96 well culture plates at 5,000 cells/cm2, and were transfected with particles 24 h later. The doses of particles ranged from 1.5 mg/mL to approximately 0.004 mg/mL, and cell viability was determined 72 h post transfection using the Quant-iT Celltiter reagent as described earlier. CT2 MLNPs, CL2 MLNPs, BT MLNPs, and CPT NPs were evaluated for efficacy in reducing tumor growth in vitro.
Figure 2. Characterization of MLNPs.

A–E. SEM images and size distributions of MLNPs after surface modifications (A – PLGA, B – PLGA-PD, C – PLGA-PDP, D – PLGA-PDP-PEG, E – PLGA-PDP-PEG-mAP). F. MLNP release profiles. CPT, PEI, and pDNA release from MLNPs. Approximately 80% of total CPT was released in 12–24 h, while approximately 80% of total pDNA was released between 24–48 h. G. Dry particle diameters as measured by SEM, surface charge, PEI loading, and pDNA loading for each particle formulation are provided. H. CPT encapsulation and loading efficiency for MLNPs. CPT - Camptothecin, CT – CPT and TRAIL encoding plasmid MLNPs, CL – CPT and luciferase encoding plasmid MLNPs, PEI - polyethyleneimine, pDNA – plasmid DNA, PEG - polyethylene glycol, PLGA - non-modified poly(lactic-co-glycolic) acid nanoparticles, PLGA-PD - PLGA nanoparticles with surface polyethyleneimine (PEI) and pDNA, PLGA-PDP - PLGA-PD particles modified with a second layer of PEI, PLGA-PDP particles modified with a heterobifunctional PEG linker (PLGA-PDP-PEG), PLGA-PDP-PEG-mAP - PLGA-PDP-PEG particles conjugated to modified antennapedia, MLNP – multi-layered nanoparticles, SEM – scanning electron microscopy, TRAIL – TNF related apoptosis inducing ligand.
2.12. Evaluating synergism
The dose effect data comparing the fraction of tumor cells affected (Fa) to drug or plasmid doses were analyzed using the Chou-Talalay analysis in order to evaluate for synergism[39]. Particle doses were converted to μg/mL of CPT or pTRAIL according to particle loading characteristics. The data were analyzed using the CompuSyn software in order to determine median effective dose (Dm), combination index (CI), and dose-reduction index (DRI) values.
2.13. Biodistribution
Coumarin-6 (C6) loaded MLNPs (C6 MLNPs, 1.7 μg/mg C6 loading; 87% loading efficiency) were created as described earlier, however, an equimolar concentration of both mAP (1 mg/mL) and internalizing RGD (iRGD, 0.25 mg/mL; Thermo Scientific) were used in the final conjugation step. C6 was loaded into O at 0.1 mg/mL in EA. Male athymic nude (NCr-nu/nu) mice were purchased from Taconic and maintained in a sterile environment. All animal procedures were approved by the Institutional Animal Care and Utilization Committee of Yale University. Xenografts were established by injecting mice (7 weeks) with 2e106 HCT116 tumor cells subcutaneously. Tumors were allowed to grow until they reached approximately 100 mm3. The mice were divided into three groups that received either PBS only (n=3), 1 dose of C6 MLNPs (n=3), or four doses of C6 MLNPs (n=4). Each dose was delivered via tail vein, as 100 μL of 20 mg/mL C6 MLNPs in PBS. The mice receiving PBS and 1 dose of C6 MLNPs were analyzed three hours after injection. The mice receiving four doses were given a dose of C6 MLNPs every day for four days, and analyzed 3 hours after the last injection. The methods for analysis were adapted from previous work[40]. Briefly, the mice were euthanized and blood was collected with cardiac puncture. Fifteen mL of PBS was then perfused through the left ventricle, and the organs were collected, flash frozen in liquid nitrogen, and lyophilized. Each organ sample was homogenized in 1 mL DMSO, shaken at 47C for at least four hours, and then homogenized again. The final homogenized solution was then spun on a benchtop centrifuge at 13,000 RPM for 10 minutes. One hundred μL of the supernatant was added to a 96 well plate and C6 fluorescence was quantified at Ex/Em 488/520 nm with a plate reader, and normalized to the weight of the analyzed tissue. To quantify the fluorophore in the blood and heart, 100 μL of the supernatant was added to 1 mL acetonitrile. The solution was spun on a benchtop centrifuge at 13,000 RPM for 10 min and then 800 μL supernatant was removed and added to an eppendorf tube. All the acetonitrile was evaporated with a SpeedVac and the C6 fluorescence was quantified as above.
2.14. Antitumor evaluation of MLNPs in vivo
CT2 MLNPs, BT MLNPs, CL2 MLNPs, and BL MLNPs were fabricated as described earlier, however, an equimolar concentration of both mAP (1 mg/mL) and iRGD (0.25 mg/mL) were used in the final conjugation step. HCT116 xenografts were established in male athymic nude mice (4 weeks) as stated above. Experiments were started when tumor volumes reached approximately 30–50 mm3. Mice were randomly divided into groups of three to six mice per treatment group as follows: group 1, PBS vehicle (n=3); group 2, BL MLNP (n=3); group 3, CL2 MLNP (n=6); group 4, BT MLNP (n=6); group 5, CT2 MLNP (n=6). Each mouse was injected with 2 mg of particles via tail vein three days a week until the end of the experiments. Each injection consisted of 100 μL of 20 mg/mL particles in PBS. Tumor size was measured using traceable digital vernier calipers (Fisher). The tumor length (l) and the width (w) measurements were obtained in order to calculate tumor volume (V =1/2*lw2). The growth curve was plotted with respect to tumor volumes. The animals were sacrificed after the tumors grew over 1 cm3, and the tissue was excised, and formalin-fixed for immunohistochemistry. Slides of serial sections were stained with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) for analysis of therapeutic effect (Yale Pathology Tissue Services).
2.15. Statistical and image analysis
For all in vitro studies, test groups were conducted in replicates of three, and compared using a two-sample heteroscedastic t-test. For all of the in vivo studies, two-sample heteroscedastic t-tests were used to determine significant differences between two specific groups. A p-value smaller than 0.05 was considered to be significant for all analyses. All error bars denote the standard error for each group. All summary data is presented as mean +/− standard deviation.
3. Results
3.1. Surface modifications and biotoxicity
Particle morphology and size were analyzed after each step in MLNP fabrication (Figure 2A–E). Sequential surface modification of PLGA particles with PEI, pDNA, PEG, and mAP did not alter particle size or morphology as all particles were spherical with average dry diameters between 101–117 nm (Figure 2G). The initial surface complexation of PLGA particles with PEI (PLGA-P) did increase particle surface charge from −23.3 mV to 32.3 mV with a total load of 3.7 μg of PEI per mg of particle (Figure 2G). A relative decrease in surface charge and a minimal drop in PEI content was observed with subsequent surface condensation of pDNA (PLGA-PD) (Figure 2G). The second layer of PEI increased the total content to 44.5 μg of PEI per mg particle with a concomitant increase in surface charge to 26.6 mV (PLGA-PDP). Conjugation of mAP to the outer PEI layer via a heterobifunctional PEG cross-linker (PLGA-PDP-PEG and PLGA-PDP-PEG-mAP) did not alter surface charge dramatically, however, there was a relative drop in total PEI content with each conjugation step, presumably due to shedding of surface PEI with incubation and sonication.
3.2. Loading and release studies
PEI and pDNA loading of the MLNPs were measured after each step in fabrication (Figure 2G): in all of these preparations, EA was used as the organic phase. Plasmid DNA content decreased with each fabrication step, and the final MLNP pDNA load was approximately 575 ng/mg of particle. CPT encapsulation within MLNPs was tunable relative to initial CPT loading (Figure 2H). Decreasing the initial CPT load from 0.01 mg CPT per mg PLGA (CT1 MLNP) to 0.001 mg CPT per mg PLGA (CT2 MLNP) resulted in 4–16 fold decrease in loading to approximately 0.018–0.071 μg/mg, with a modest increase in loading efficiency. Decreasing the initial CPT load even further to 0.0001 mg CPT per mg PLGA (CT3 MLNP) resulted in a decrease in loading to approximately 0.006–0.010 μg/mg while maintaining a similar loading efficiency. CPT release and PEI dissociation from CT2 MLNPs prepared with EA occurred readily, achieving 80–90% release within 24 hours (Figure 2F). Plasmid release lagged behind CPT release, and approximately 80% of the total pDNA amount was released within 48 hours (Figure 2F). MLNPs containing CPT and pLuc (CL2 MLNPs) were also produced with the same loading ratio as CT2 MLNPs for subsequent functional evaluation.
3.3. Particle cytotoxicity
Initial surface modification with PEI rendered particles toxic to cells (Figure 3A). But, after conjugation with PEG5-mAP, particle toxicity decreased significantly. Modifying MLNPs with PEG1 and co-encapsulation with TCHD did not alter particle toxicity. MLNPs with PEG, mAP, and TCHD were all significantly less toxic to cells than the lipofectamine control.
Figure 3. Functional evaluation of MLNPs for transfection and cytotoxicity.
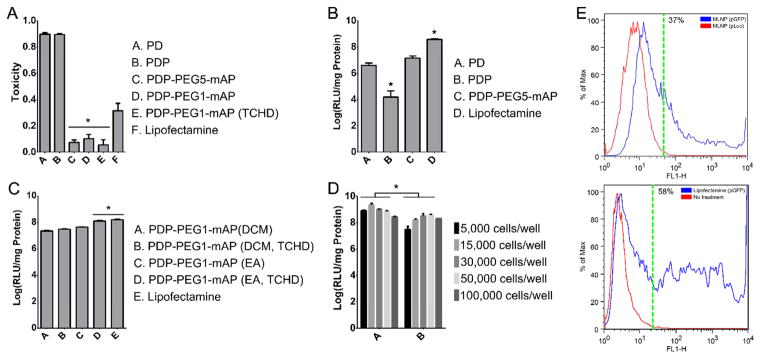
Population transfection optimization on 293T cells. MLNPs were loaded with pLuc. A. Toxicity of MLNPs after surface modification. B. The effect of surface modification on particle transfection. All MLNPs were synthesized with DCM. C. The effect of utilizing a smaller PEG1 linker, changing the organic solvent during fabrication to EA, and encapsulating TCHD within the PLGA core on particle transfection. D. The effect of initial seeding density on particle transfection. E. Percent cell transfection. FACS of 293T cells transfected with MLNPs or lipofectamine particles carrying pGFP. Fluorescence measured via the FL1-H channel detected transfected cells. After 72 h, approximately 37% of cells were transfected by MLNPs and 58% of cells were transfected by lipofectamine. The dotted green line indicates a gate containing at least 99% of the control population. *P<0.05. DCM – dichloromethane, EA – ethyl acetate, FACS - fluorescence-activated cell sorting, PEG - polyethylene glycol, PEG1 – 1 kDa PEG, PEG5 – 5 kDa PEG, PLGA - poly(lactic-co-glycolic) acid, PD - PLGA nanoparticles with surface polyethyleneimine (PEI) and plasmid DNA, PDP - PD particles modified with a second layer of PEI, PDP-PEG-mAP - PDP particles modified with a heterobifunctional PEG linker and modified antennapedia. MLNP – multi-layered nanoparticles, pLuc – luciferase encoding plasmid, pGFP – green fluorescent protein encoding plasmid, TCHD - trans-1,2-Cyclohexanediol.
3.4. Cellular transfection
Cellular transfection efficiency was determined by measuring luciferase activity in 293T cells 48 h after treatment with MLNPs or lipofectamine (Figure 3B–E). Surface modification with PEI and pLuc allowed for moderate transfection of 293T cells (Figure 3B). Further modification with a PEG5 cross-linker and mAP resulted in greater luciferase activity after transfection. However, in these initial formulations, lipofectamine still provided greater transfection than the MLNPs (Figure 3B). Transfection was subsequently improved by utilizing a lower molecular weight PEG1 and co-encapsulating TCHD (Figure 3C). In initial experiments, particles were prepared using DCM in the organic phase (Figure 3B–C). Subsequently, we discovered that preparing particles with EA instead of DCM in the organic phase improved MLNP transfection levels resulting in luciferase activity after transfection that was comparable to lipofectamine. The optimal MLNPs were those fabricated using EA, PEG1, and TCHD (Figure 3C). Subsequent transfection of 293T cells at lower seeding densities with the optimal MLNP formulation resulted in significantly greater luciferase activity than lipofectamine (Figure 3D).
To identify the fraction of cells that were successfully transfected, 293T cells were transfected with MLNPs loaded with pGFP and analyzed using flow cytometry after 72 hours. MLNP percent cellular transfection was measured at 37% compared to 58% obtained by lipofectamine (Figure 3E). The percent cell transfection was also determined by measuring GFP expression via confocal microscopy as well (Figure S 2). The fluorescent signal area fraction for the MLNP groups increased from 48–72 h from 2.1% to 2.4%, however, the signal from the lipofectamine groups decreased over this period from 6.6% to 5.3%.
3.5. in vitro cytotoxicity
Before evaluating MLNPs for effectiveness of combination drug and gene delivery, toxicity studies were conducted on U87, HCT116, and MDAMB231 cell lines to determine optimal timing of CPT exposure and pTRAIL transfection (Figure 4). Exposure to CPT concurrently with pTRAIL transfection resulted in the greatest relative decrease in viability for all cell lines, compared to exposure to CPT 24 hrs before or after transfection. Transfection with seven ng/well of pTRAIL resulted in significant decreases in cell viability with concomitant CPT exposure. HCT116 cells demonstrated the largest relative decrease in cell viability with CPT and pTRAIL transfection (Figure 4A). MLNPs loaded with both CPT and pTRAIL (CT MLNP) were tested for efficacy in reducing tumor cell proliferation in vitro (Figure 4). CT MLNPs were fabricated with decreasing CPT to pTRAIL loading ratios. All particles contained approximately 575 ng of pTRAIL per mg of PLGA, and were loaded with decreasing CPT content (CT1-3 and CL1-3; Figure 2H). After exposing cells to an effective 0.01 μM dose of CPT via MLNPs, CT2 MLNPs resulted in a significantly greater effect on reducing HCT116 cell growth in vitro (Figure 4B; A) CT1 MLNP, B) CT2 MLNP, C) CT3 MLNP). The CT2 MLNP formulation was then used for subsequent synergy analysis, which delivered a mass ratio of CPT to pTRAIL of 0.03.
Figure 4. Antitumor effects of MLNPs in vitro.

A. Tumor death kinetics relative to free CPT drug exposure and transfection with pTRAIL at multiple doses. U87, HCT116, and MDAMB231 cells were exposed to CPT 24 h before, during (0 h), or 24 h after transfection. B. HCT116 cell viability 72 h after exposure to MLNPs loaded with CPT to pTRAIL mass ratios of 5, 0.03, and 0.01 (CT1, CT2, and CT3 respectively). Treatment with CT2 MLNPs resulted in significantly lower cell proliferation. C–E. Dose effect curves. C. Tumor cells were exposed to CL2 MLNPs, and CT2 MLNPs. D. Tumor cells were exposed to CT2 MLNPs and CPT NPs. E. Tumor cells were exposed to BT MLNPs or CT2 MLNPs. CT2 and CL2 MLNPs were loaded with a CPT to pDNA mass ratio of 0.03. *P<0.05. BT MLNPs - MLNPs delivering only pTRAIL, CL2 MLNP - MLNPs delivering CPT and pLuc, CPT - camptothecin, CPT NP – PLGA nanoparticles encapsulating CPT, CT1-3 – CPT and TRAIL encoding plasmid MLNPs at different CPT to pTRAIL ratios, CT2 MLNP - MLNPs delivering CPT and pTRAIL, Fa – fraction effected, MLNP – multi-layered nanoparticles, PLGA - poly(lactic-co-glycolic), pLuc – luciferase encoding plasmid, pTRAIL – TRAIL encoding plasmid, TRAIL – TNF related apoptosis inducing ligand.
3.6. Evaluating synergism
Delivering combination CPT and pTRAIL via CT2 MLNPs shifted the dose effect curves to the left for all cell lines (Figure 4C–E), suggesting synergy between CPT and pTRAIL. CT2 MLNPs (containing both CPT and pTRAIL) were more effective than to CL2 MLNPs (containing CPT and pLUC, Figure 4C) and CPT NPs (containing only CPT, Figure 4D). CT2 MLNPs were also more effective than BT MLNPs (containing pTRAIL but no CPT, Figure 4E). To quantitate the degree of synergy, CompuSyn analysis was performed on these results, yielding CI values that were predominantly between 0.3–0.7 (Figure 5), confirming a synergistic interaction (CI < 1). The CPT Dm was markedly reduced when using the CT2 MLNP formulation for all cell lines (Table S 4). Chou-Talalay analysis of CT2 MLNPs compared to CL2 MLNPs and BT MLNPs (Figure 5A) as well as CPT NPs and BT MLNPs (Figure 5B) were performed, and the CI and DRI for both CPT and pTRAIL determined (Figure 5 and Figure S 3) for multiple Fa levels. The DRI at Fa0.5 ranged from 3.14–14.5 for CPT and 4.66–7.99 for pTRAIL. The linear regression coefficients for transforming the dose effect data to the median effect equation as determined by CompuSyn were above 0.90 for all dose affect curves (Table S 3).
Figure 5. Synergy analysis of MLNP therapy in vitro.

A. The Chou-Talalay analysis of the dose effect curves obtained in Figure 4. CI values for CT2 MLNPs are provided for all Fa levels on reducing U87, HCT116, and MDAMB231 cell growth. The CPT and pTRAIL DRI values of CT2 MLNPs relative to CL2 MLNPs and BT MLNPs are provided for multiple Fa levels. The theoretical values were those determined by the Chou-Talalay analysis model for all Fa levels, and the experimental values were those determined for actual CT2 MLNP doses. B. A similar synergy analysis as above is provided for comparing combination CPT and pTRAIL with CT2 MLNPs to monotherapy with CPT NPs and BT MLNPs. CI < 1 supports synergy with combination therapy. BT MLNPs - MLNPs delivering only pTRAIL, CI – combination index, CL2 MLNP - MLNPs delivering CPT and pLuc, CPT - camptothecin, CPT NP – PLGA nanoparticles encapsulating CPT, CT2 MLNP - MLNPs delivering CPT and pTRAIL, DRI – dose reduction index, Fa - fraction affected, MLNP – multi-layered nanoparticles, PLGA - poly(lactic-co-glycolic), pLuc – luciferase encoding plasmid, pTRAIL – TRAIL encoding plasmid, TRAIL – TNF related apoptosis inducing ligand.
3.7. Biodistribution
For the in vivo studies, the MLNPs were surface conjugated with both mAP and iRGD to improve tumor targeting[7, 11]. HCT116 xenografts were grown until tumors were approximately 103 +/− 11.8 mm3 in volume. C6 MLNPs accumulated primarily in the liver and tumor (Figure 6A). Three hours after a single dose, minimal C6 signal was detected, and the majority of particles were detected in the kidney. Another group of mice received four doses every 24 h. Three hours after the last dose, there was a significant signal increase in the liver and tumor, and minimal change in the remaining organ sites. The levels found in the liver and tumor were significantly greater than any other organ after four doses; however, the signal from the liver was not significantly different from the signal found in the tumor.
Figure 6. Evaluation of MLNPs in vivo.

A. Biodistribution of C6 MLNPs. MLNPs accumulated primarily in the liver and tumor. Three h after a single dose, there was minimal signal in most tissues. However, after four repeated doses over 48 h, there was significantly greater C6 signal in the tumor and liver. *P<0.5 comparing weight normalized C6 signal after four doses was significantly greater than the signal after one dose. **P<0.5 comparing weight normalized C6 signal between the tumor and liver is statistically similar, and significantly greater than signal from other tissue sites. B. Antitumor evaluation of MLNPs in vivo. Antitumor effects of MLNP treatment groups can be seen after five tail vein injections, and 16 days of treatment. Mice treated with CT2 MLNPs had significantly reduced tumor growth compared to all other treatment groups after 15 days. Gray arrows designate timing of tail vein injections. *P<0.5 compared to all other treatment groups. Representative gross tumor resections are displayed as images I–V (I. PBS, II. BL MLNP, III. CL2 MLNP, IV. BT MLNP, V CT2 MLNP). Scale bar set to 1 cm. C–E. TUNEL staining of representative tumor sections after five injections, and 16 days of growth at 10× magnification. CT2 MLNP treated mice displayed greater proportion of apoptotic cells. (C. BL MLNP, D. CL2 MLNP, E. BT MLNP, F. CT2 MLNP). BL MLNPs – MLNPs delivering only pLuc, BT MLNPs - MLNPs delivering only pTRAIL, CI – combination index, CL2 MLNP - MLNPs delivering CPT and pLuc, CPT - camptothecin, C6 – coumarin 6, CT2 MLNP - MLNPs delivering CPT and pTRAIL, MLNP – multi-layered nanoparticles, PBS – phosphate buffered saline, pLuc – luciferase encoding plasmid, pTRAIL – TRAIL encoding plasmid, TRAIL – TNF related apoptosis inducing ligand. TUNEL - terminal deoxynucleotidyl transferase dUTP nick end labeling.
3.8. Antitumor evaluation of MLNPs in vivo
Intravenous injection of optimized MLNPs co-delivering CPT and pTRAIL leads to substantial HCT116 antitumor effects (Figure 6B). Each treatment group received five injections at times indicated by the grey arrows. After 15–16 days, CT2 MLNP therapy resulted in a significantly greater inhibition of tumor volume (80–82%) when compared to all other treatment groups. CPT therapy alone via CL2 MLNPs resulted only in a modest effect on tumor volume (7–13%), whereas pTRAIL delivery via BT MLNPs also resulted in a greater inhibition of tumor growth (58–64%). Representative gross sections of the xenografts after 16 days of treatment revealed these same trends (Figure 6B). Mouse weights remained stable during the treatment period (Figure S 4). Representative TUNEL staining of tumor sections from each MLNP treatment group reveals significant induction of apoptosis in tumors treated with CT2 MLNPs (Figure 6).
4. Discussion
4.1. Nanoparticle fabrication and characterization
The modular design of MLNPs allowed for tuning of properties to improve synergy between drug and gene delivery to tumors. Evaluation of these particles began with analysis of basic particle characteristics at each fabrication step. Particle diameter remained constant at approximately 109 nm after surface modifications (Figure 2), which is beneficial for passive tumor targeting since the ideal particle size for utilizing the enhanced permeation and retention (EPR) effect is between 10 to 200 nm[41]. Surface attachment of PEI was successful, as indicated by a change in surface charge from −23 mV to 32 mV, and a PEI load of 3.7 ug/mg of particle (Figure 2G). Subsequent addition of another layer of PEI increased the surface potential again to values ranging from 27 to 31 mV. The bulk of the total particle PEI content appears to be located in the second layer as the total weight normalized PEI load increased substantially, from 3.7 ug/mg to 45 ug/mg, with the addition of the second layer (Figure 2G). PEI is a potent transfection agent, however, it is well known to be toxic to cells, which may be due to the interactions of surface PEI with the cellular or mitochondrial membrane[42]. The initial surface coating with PEI did render the particles highly toxic to 293T cells; however, subsequent modifications with PEG-mAP eliminated most of the cellular toxicity. Coating with PEG-mAP likely hindered these cytotoxic interactions, which significantly decreased toxicity to levels much lower than lipofectamine (Figure 3A).
PLGA particles effectively encapsulate the hydrophobic CPT molecule. Although the MLNPs were able to encapsulate CPT at high loading efficiencies, our goal was ultimately to deliver less CPT. In fact, delivering a greater ratio of pTRAIL to CPT was ultimately preferred. Particle loading of CPT was therefore decreased by reducing the initial CPT dissolved in the organic phase of the emulsion. Decreasing the initial CPT load per mg of PLGA resulted in reduced final MLNP loading (Figure 2H).
CPT and PEI were readily released from MLNPs, and there was a relative lag in plasmid release. This may suggest that the majority of the plasmid is complexed with the first PEI layer. The in vitro release studies were performed under conditions that do not accurately reflect either the tumor microenvironment or the endosomal compartment, which may influence particle release profiles. Importantly, heparin was required to accurately measure the total amount of released plasmid. DNA dissociated from the MLNPs as polyplexes with PEI. High concentrations of heparin were needed to facilitate the separation of DNA molecules from the PEI polyplex, allowing for accurate DNA detection. Plasmid dissociation in vivo will likely be more prolonged, which may be beneficial for protecting plasmids from both circulating and cytosolic endonucleases.
4.2. Transfection evaluation
The initial goal was to improve MLNP transfection capabilities. Population transfection studies were conducted by delivering pLuc to 293T cells via MLNPs. Optimization of particle fabrication parameters resulted in high 293T transfection levels that were ultimately comparable to lipofectamine (Figure 3). After coating the outer PEI layer with PEG-mAP, transfection levels measured by luciferase activity increased (Figure 3A–B), which was partly a result of the significant decrease in particle toxicity (Figure 3D). MLNPs fabricated with EA, TCHD, and 1 kDa PEG provided the greatest transfection levels, which were comparable to lipofectamine (Figure 3C). Decreasing PEG size may allow for a greater number of PEG units to bind to the PEI coating, and subsequently more maleimide groups for conjugating mAP. TCHD increases nuclear pore patency, which permits plasmid localization into the nucleus[11]. Incorporation of TCHD increased transfection levels by a factor of 2–5. Finally, switching the organic phase from DCM to EA improved particle transfection as well. Theoretically, this effect is due to the smaller size of the particles. Utilization of EA in the organic phase promotes formation of smaller particles due to its increased diffusion out of the PLGA organic solvent globule upon exposure to water, and its greater miscibility with the aqueous phase[43].
Figure 3D demonstrates that decreasing the seeding density can increase normalized population transfection levels obtained by MLNPs. This may be related to increased MLNP internalization due to the greater amount of particles per cell at lower seeding densities, and the higher mitotic activity of these cells. Percent cell transfection is higher with lipofectamine, however, population transfection levels obtained by MLNPs were similar if not greater than lipofectamine (Figure 3C–D). The decreased cell viability in the lipofectamine group may explain why the population transfection levels are similar to the MLNP group despite having a greater percent transfection. This property translates well for TRAIL gene therapy. Population transfection levels are likely a better predictor of pTRAIL transfection efficacy, since total TRAIL levels will determine the therapeutic effect rather than percent transfection.
4.3. in vitro cytotoxicity
There are multiple mechanisms for synergy between the apoptotic stimulus provided by CPT and TRAIL. Previous studies have reported nanoparticles for delivery of either CPT or TRAIL[7, 21]. However, no previous studies have examined the effectiveness of combination CPT and TRAIL therapy via a nanovector. Cancer cell lines have variable sensitivities to CPT and TRAIL. Here, in vitro tests evaluated the efficacy of combination therapies on U87, HCT116, and MDAMB231 cells. Previous studies analyzing the effect of combination CPT and the TRAIL peptide suggest that pretreatment with CPT sensitizes cancer cells to TRAIL mediated apoptosis by increasing DR expression[17]. Due to the lag between transfection and protein expression, the death kinetics of CPT and pTRAIL transfection were studied to determine the optimal timing for each therapy (Figure 4A). The resulting death kinetics suggest that CPT exposure during or before TRAIL transfection provides the greatest reduction in tumor cell growth. The most significant effect was seen with CPT delivery during transfection for all cell lines. These results add further support to the theory that CPT exposure sensitizes cells to TRAIL, by either up-regulating DR4 and DR5 expression, or inhibiting anti-apoptotic factors including Bcl-2[17, 44]. Furthermore, we designed particles that deliver CPT and pTRAIL at this optimal timing by using the MLNP delivery system (Figure 2F).
CT MLNPs were evaluated for delivering the most effective ratio of CPT to pTRAIL. In order to evaluate the efficacy of MLNPs in reducing tumor growth, pTRAIL was loaded onto CPT encapsulated MLNPs (CT MLNPs). CPT is a potent anti-cancer agent, and delivering a high ratio of CPT to pTRAIL should provide overwhelming toxicity primarily due to CPT activity. But our goal was to maximize synergy, and minimize potential toxicities to other tissues. Therefore, multiple MLNP formulations with decreasing CPT to pTRAIL loading ratios were fabricated (Figure 2H; Table S 2). Each formulation was evaluated for its effect on inhibiting HCT116 growth in vitro (Figure 4B). MLNPs with lower drug loading will therefore deliver a greater plasmid dose since more particles were delivered to meet the required CPT dose. The optimal formulation was CT2. Although CT3 MLNPs delivered more pTRAIL per molecule of CPT, they also provided more PEI, which could potentially reduce the efficacy of CPT due to lactone hydrolysis.
4.4. Analysis of synergism
The in vitro synergy analysis not only supports the efficacy of co-delivering CPT and pTRAIL for reducing tumor cell growth, but it also validates the use of MLNPs as a vehicle for co-delivery of the two agents (Figure 5). CT2 MLNPs provided a synergistic growth inhibition over a large dose range on multiple cancer cell lines (CI values between 0.3–0.7). CI values are ratios of the combination dose to the sum of individual doses at the same Fa level. Ratios less than one indicate synergism, whereas a ratio of one suggests an additive effect, and values greater than one demonstrate antagonism[39]. The resulting DRI values for the CT2 MLNP formulation indicates that we can deliver drug and DNA doses that are approximately an order of magnitude lower via combination delivery. The lower CPT DRI for HCT116 and U87s may be due to the increased sensitivity of this cell line to CPT compared to MDAMB231s. Decreased CPT dosing can potentially lead to a reduction in system toxicities and greater patient tolerance. Additionally, certain drugs have lower encapsulation efficiencies in PLGA nanoparticles. Combination therapies can potentially improve therapeutic efficacy for other cancer chemotherapeutics by decreasing the required dose for effectiveness in nanoparticle systems.
4.5. Antitumor evaluation of MLNPs in vivo
The in vitro evaluation demonstrates that MLNPs can effectively delivery both CPT and pTRAIL. In order to functionally translate these results into in vivo applications, iRGD was surface attached along with mAP using cysteine maleimide conjugation chemistries in order to improve tumor targeting capabilities. The iRGD peptide sequence recognizes the αvβ3/αvβ5 integrins up regulated on tumor endothelial cells[45]. Zhou et al. has shown that attaching both mAP and iRGD can improve tumor localization and transfection in vivo[11]. The biodistribution of C6 MLNPs preferentially accumulated in the tumor and liver when compared to other organs (Figure 6A). Repeated dosing for four days resulted in significantly greater particle accumulation in the liver and tumor, and the normalized fluorescence in the liver and tumor were not statistically different.
HCT116 was selected to create target xenografts for the in vivo antitumor studies. It was selected for its marked sensitivity to both CPT and TRAIL, and its promising synergistic growth inhibition by combination therapy in vitro (Figure 4). After 15–16 days, xenograft growth was significantly reduced by CT2 MLNP therapy when compared to all other treatment groups. Further, the effect of CT2 MLNP was greater than the combined effects of CL2 MLNPs and BT MLNPs. Tumors receiving both BT MLNP and CT2 MLNP therapy had lower cellular mass and greater increase in apoptotic cells, which can be visualized by the TUNEL stains of the resected tumors (Figure 6). Therapeutic efficacy was achieved by delivering very low concentrations of both CPT and pTRAIL, which can potentially minimize systemic toxicities. Each mouse received approximately 2.4 ng/kg of CPT and 46 ng/kg of pDNA with every injection. The low dose of CPT explains the modest effect of the CL2 MLNPs, although the amount delivered was adequate in improve the effect of concomitant pTRAIL transfection with CT2 MLNPs.
5. Conclusion
A PLGA-based MLNP delivery system can provide a multi-functional and efficient vehicle for targeted and multi-modal antitumor therapies. Tumors are highly heterogeneous, and so the necessity for personalized medicine and the ability to deliver multi-modal therapy is important. The MLNP design allowed for optimization of key particle properties necessary for effective combination delivery. The PLGA based particles have minimal cytotoxicity, while maintaining high transfection capabilities. Further they were able to provide synergistic combination drug and gene delivery, and tumor targeting capabilities. The step wise evaluation of these particles allowed for significantly greater tumor growth inhibition that was translated to in vivo therapy, while delivering extremely low amounts of drug and DNA. Future nanoparticle formulations for combination therapies should also be analyzed in vitro for synergism prior to in vivo evaluation. The MLNP’s modular, multifunctional design provides a robust system for efficiently delivering both small molecules and gene therapy, and creates an attractive vehicle for combination anti-cancer therapies.
Supplementary Material
Figure S 1: Experimental design for evaluating camptothecin and TRAIL plasmid transfection death kinetics.
The experimental design for determining death kinetics after CPT exposure and pTRAIL transfection. U87, HCT116, and MDAMB231 cells were seeded in 96 well culture plates at 2,500 cells/cm2. All treatment groups were transfected with pTRAIL 48 h after seeding, and exposed to CPT 24 h before (−24 h), during (0 h), or 24 h after (+24 h) CPT exposure. Treatment controls included NT, and CPT exposure alone at the respective time points. Treatment groups were analyzed for viability 48 h post transfection. Subtherapeutic doses of CPT and pTRAIL were used in order to appreciate the effect of combination delivery. U87 and MDAMB231 cells were exposed to 25 nM CPT. Since HCT116 cells are more sensitive, they were exposed to 2.5 nM CPT. Combination therapies were normalized to groups receiving a similarly timed CPT dose in order to determine the relative increase in tumor cell death. CPT – camptothecin, NT – no treatment, pTRAIL - TRAIL encoding plasmid, TRAIL – TNF related apoptosis inducing ligand.
Figure S 2: Green fluorescent protein expression after particle transfection.
Confocal fluorescent imaging of 293T cells transfected with MLNPs or lipofectamine particles carrying pGFP. A–B. 293T cells transfected with MLNPs after 48 h and 72 h respectively. C–D. 293T cells transfected with lipofectamine after 48 h and 72 h respectively. Fluorescent signal area was determined using ImageJ and provided as a percentage. MLNP – multi-layered nanoparticles, pGFP – green fluorescent protein encoding plasmid.
Figure S 3: Dose reduction index of camptothecin and TRAIL plasmid using CT2 MLNPs.
The dose-reduction index (DRI) for multi-layered nanoparticles (MLNPs) over all fraction affected (Fa) levels of tumor growth inhibition for U87, HCT116, and MDAMB231 cancer cell lines. A. DRI of MLNPs delivering camptothecin (CPT) and plasmid encoding TNF related apoptosis inducing ligand (pTRAIL) (CT2 MLNPs) relative to MLNPs delivering CPT and plasmid encoding for luciferase (CL2 MLNPs) and MLNPs containing only pTRAIL (BT MLNPs). B. DRI of CT2 MLNPs relative to poly(lactic-co-glycolic) (PLGA) nanoparticles encapsulating CPT only (CPT NPs) and BT MLNPs. All MLNPs were loaded with the same amount of plasmid and the same CPT to pTRAIL mass ratio of 0.03. CPT NPs were loaded with same amount of CPT as the CPT loaded MLNPs.
Figure S 4: Mouse weights during in vivo antitumor therapy.
Mouse weights remained stable during the 16 day study. Treatment groups included phosphate buffered saline (PBS), multi-layered nanoparticles (MLNPs) delivering luciferase encoding plasmid (pLuc) (BL MLNPs), MLNPs delivering camptothecin (CPT) and pLuc (CL2 MLNPs), MLNPs delivering TNF related apoptosis inducing ligand encoding plasmid (pTRAIL) (BT MLNPs), and MLNPs delivering both CPT and pTRAIL (CT2 MLNPs). All MLNPs were loaded with the same amount of plasmid and the same CPT to pTRAIL ratio of 0.03.
Table S 1: CPT to pTRAIL ratio for death kinetics experiments.
Table S 2: Camptothecin to pTRAIL ratio for MLNP formulations.
Table S 3: Goodness of fit of the Chou-Talalay analysis.
Table S 4: Median effect doses for CT2 MLNPs, CL2 MLNPs, and CPT NPs.
Acknowledgments
This work was supported by the Howard Hughes Medical Research Fellows grant, the Yale School of Medicine Office of Student Research, and a grant from the National Institutes of Health (R01 CA149128). We thank Elias Quijano, Young Seo, Jenny Sawyer, and Chris Cheng for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cancer Facts & Figures 2013. Atlanta: American Cancer Society; 2013. [Google Scholar]
- 2.De Vita VT, Jr, Young RC, Canellos GP. Combination versus single agent chemotherapy: a review of the basis for selection of drug treatment of cancer. Cancer. 1975;35:98–110. doi: 10.1002/1097-0142(197501)35:1<98::aid-cncr2820350115>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011;12:316–28. doi: 10.1038/nrg2971. [DOI] [PubMed] [Google Scholar]
- 5.Fahmy TM, Fong PM, Goyal A, Saltzman WM. Targeted for drug delivery. Mater Today. 2005;8:18–26. [Google Scholar]
- 6.Pirollo KF, Chang EH. Targeted delivery of small interfering RNA: approaching effective cancer therapies. Cancer Res. 2008;68:1247–50. doi: 10.1158/0008-5472.CAN-07-5810. [DOI] [PubMed] [Google Scholar]
- 7.Zhou J, Liu J, Cheng CJ, Patel TR, Weller CE, Piepmeier JM, et al. Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nat Mater. 2011;11:82–90. doi: 10.1038/nmat3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:185–98. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 9.Shea LD, Smiley E, Bonadio J, Mooney DJ. DNA delivery from polymer matrices for tissue engineering. Nat Biotechnol. 1999;17:551–4. doi: 10.1038/9853. [DOI] [PubMed] [Google Scholar]
- 10.Luo D, Woodrow-Mumford K, Belcheva N, Saltzman WM. Controlled DNA delivery systems. Pharm Res. 1999;16:1300–8. doi: 10.1023/a:1014870102295. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J, Patel TR, Fu M, Bertram JP, Saltzman WM. Octa-functional PLGA nanoparticles for targeted and efficient siRNA delivery to tumors. Biomaterials. 2011;33:583–91. doi: 10.1016/j.biomaterials.2011.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blum JS, Saltzman WM. High loading efficiency and tunable release of plasmid DNA encapsulated in submicron particles fabricated from PLGA conjugated with poly-L-lysine. J Control Release. 2008;129:66–72. doi: 10.1016/j.jconrel.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Son S, Kim WJ. Biodegradable nanoparticles modified by branched polyethylenimine for plasmid DNA delivery. Biomaterials. 2010;31:133–43. doi: 10.1016/j.biomaterials.2009.09.024. [DOI] [PubMed] [Google Scholar]
- 14.Fields RJ, Cheng CJ, Quijano E, Weller C, Kristofik N, Duong N, et al. Surface modified poly(beta amino ester)-containing nanoparticles for plasmid DNA delivery. J Control Release. 2012;164:41–8. doi: 10.1016/j.jconrel.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O’Dwyer PJ, Gordon MS, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol. 2010;28:2839–46. doi: 10.1200/JCO.2009.25.1991. [DOI] [PubMed] [Google Scholar]
- 16.Ciusani E, Croci D, Gelati M, Calatozzolo C, Sciacca F, Fumagalli L, et al. In vitro effects of topotecan and ionizing radiation on TRAIL/po2L-mediated apoptosis in malignant glioma. JNO. 2005;71:19–25. doi: 10.1007/s11060-004-9180-4. [DOI] [PubMed] [Google Scholar]
- 17.Singh TR, Shankar S, Chen X, Asim M, Srivastava RK. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 Ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res. 2003;63:5390–400. [PubMed] [Google Scholar]
- 18.Xiang H, Fox JA, Totpal K, Aikawa M, Dupree K, Sinicropi D, et al. Enhanced tumor killing by Apo2L/TRAIL and CPT-11 co-treatment is associated with p21 cleavage and differential regulation of Apo2L/TRAIL ligand and its receptors. Oncogene. 2002;21:3611–9. doi: 10.1038/sj.onc.1205449. [DOI] [PubMed] [Google Scholar]
- 19.Rothenberg ML. Topoisomerase I inhibitors: review and update. Ann Oncol. 1997;8:837–55. doi: 10.1023/a:1008270717294. [DOI] [PubMed] [Google Scholar]
- 20.Ertl B, Platzer P, Wirth M, Gabor F. Poly(D,L-lactic-co-glycolic acid) microspheres for sustained delivery and stabilization of camptothecin. J Control Release. 1999;61:305–17. doi: 10.1016/s0168-3659(99)00122-4. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Jiang Z, Zhang S, Saltzman WM. Poly(omega-pentadecalactone-co-butylene-co-succinate) nanoparticles as biodegradable carriers for camptothecin delivery. Biomaterials. 2009;30:5707–19. doi: 10.1016/j.biomaterials.2009.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelley SK, Harris LA, Xie D, DeForge L, Totpal K, Bussiere J, et al. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001;299:31–8. [PubMed] [Google Scholar]
- 23.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–63. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 25.Kagawa S, He C, Gu J, Koch P, Rha SJ, Roth JA, et al. Antitumor activity and bystander effects of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene. Cancer Res. 2001;61:3330–8. [PubMed] [Google Scholar]
- 26.Wang Y, Gao S, Ye WH, Yoon HS, Yang YY. Co-delivery of drugs and DNA from cationic core-shell nanoparticles self-assembled from a biodegradable copolymer. Nat Mater. 2006;5:791–6. doi: 10.1038/nmat1737. [DOI] [PubMed] [Google Scholar]
- 27.Saad M, Garbuzenko OB, Minko T. Co-delivery of siRNA and an anticancer drug for treatment of multidrug-resistant cancer. Nanomedicine (Lond) 2008;3:761–76. doi: 10.2217/17435889.3.6.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T, et al. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrugresistant cancer cells. Small. 2009;5:2673–7. doi: 10.1002/smll.200900621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaneshiro TL, Lu ZR. Targeted intracellular codelivery of chemotherapeutics and nucleic acid with a well-defined dendrimer-based nanoglobular carrier. Biomaterials. 2009;30:5660–6. doi: 10.1016/j.biomaterials.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 30.Hu C-MJ, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Deliv. 2010;1:323–34. doi: 10.4155/tde.10.13. [DOI] [PubMed] [Google Scholar]
- 31.Meng H, Liong M, Xia T, Li Z, Ji Z, Zink JI, et al. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano. 2010;4:4539–50. doi: 10.1021/nn100690m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu C, Jung S, Luo S, Meng F, Zhu X, Park TG, et al. Co-delivery of siRNA and paclitaxel into cancer cells by biodegradable cationic micelles based on PDMAEMA-PCL-PDMAEMA triblock copolymers. Biomaterials. 2010;31:2408–16. doi: 10.1016/j.biomaterials.2009.11.077. [DOI] [PubMed] [Google Scholar]
- 33.Sun T-M, Du J-Z, Yao Y-D, Mao C-Q, Dou S, Huang S-Y, et al. Simultaneous delivery of siRNA and paclitaxel via a “two-in-one” micelleplex promotes synergistic tumor suppression. ACS Nano. 2011;5:1483–94. doi: 10.1021/nn103349h. [DOI] [PubMed] [Google Scholar]
- 34.Zhao F, Yin H, Li J. Supramolecular self-assembly forming a multifunctional synergistic system for targeted co-delivery of gene and drug. Biomaterials. 2014;35:1050–62. doi: 10.1016/j.biomaterials.2013.10.044. [DOI] [PubMed] [Google Scholar]
- 35.Han L, Huang R, Li J, Liu S, Huang S, Jiang C. Plasmid pORF-hTRAIL and doxorubicin co-delivery targeting to tumor using peptide-conjugated polyamidoamine dendrimer. Biomaterials. 2011;32:1242– 52. doi: 10.1016/j.biomaterials.2010.09.070. [DOI] [PubMed] [Google Scholar]
- 36.Liu S, Guo Y, Huang R, Li J, Huang S, Kuang Y, et al. Gene and doxorubicin co-delivery system for targeting therapy of glioma. Biomaterials. 2012;33:4907–16. doi: 10.1016/j.biomaterials.2012.03.031. [DOI] [PubMed] [Google Scholar]
- 37.Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment. ACS Nano. 2013;7:9571–84. doi: 10.1021/nn4047925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shau MD, Shih MF, Lin CC, Chuang IC, Hung WC, Hennink WE, et al. A one-step process in preparation of cationic nanoparticles with poly(lactide-co-glycolide)-containing polyethylenimine gives efficient gene delivery. Eur J Pharm Sci. 2012;46:522–9. doi: 10.1016/j.ejps.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Chou T-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 40.Deng Y, Saucier-Sawyer JK, Hoimes CJ, Zhang J, Seo Y-E, Andrejecsk JW, et al. The effect of hyperbranched polyglycerol coatings on drug delivery using degradable polymer nanoparticles. Biomaterials. 2014;35:6595–602. doi: 10.1016/j.biomaterials.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–46. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 42.Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005;11:990–5. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 43.Choi S-W, Kwon H-Y, Kim W-S, Kim J-H. Thermodynamic parameters on poly(d,l-lactide-co-glycolide) particle size in emulsification–diffusion process. Colloids Surf Physicochem Eng Aspects. 2002;201:283–9. [Google Scholar]
- 44.Hellwig CT, Rehm M. TRAIL signaling and synergy mechanisms ssed in TRAIL-based combination therapies. Mol Cancer Ther. 2012;11:3–13. doi: 10.1158/1535-7163.MCT-11-0434. [DOI] [PubMed] [Google Scholar]
- 45.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–20. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S 1: Experimental design for evaluating camptothecin and TRAIL plasmid transfection death kinetics.
The experimental design for determining death kinetics after CPT exposure and pTRAIL transfection. U87, HCT116, and MDAMB231 cells were seeded in 96 well culture plates at 2,500 cells/cm2. All treatment groups were transfected with pTRAIL 48 h after seeding, and exposed to CPT 24 h before (−24 h), during (0 h), or 24 h after (+24 h) CPT exposure. Treatment controls included NT, and CPT exposure alone at the respective time points. Treatment groups were analyzed for viability 48 h post transfection. Subtherapeutic doses of CPT and pTRAIL were used in order to appreciate the effect of combination delivery. U87 and MDAMB231 cells were exposed to 25 nM CPT. Since HCT116 cells are more sensitive, they were exposed to 2.5 nM CPT. Combination therapies were normalized to groups receiving a similarly timed CPT dose in order to determine the relative increase in tumor cell death. CPT – camptothecin, NT – no treatment, pTRAIL - TRAIL encoding plasmid, TRAIL – TNF related apoptosis inducing ligand.
Figure S 2: Green fluorescent protein expression after particle transfection.
Confocal fluorescent imaging of 293T cells transfected with MLNPs or lipofectamine particles carrying pGFP. A–B. 293T cells transfected with MLNPs after 48 h and 72 h respectively. C–D. 293T cells transfected with lipofectamine after 48 h and 72 h respectively. Fluorescent signal area was determined using ImageJ and provided as a percentage. MLNP – multi-layered nanoparticles, pGFP – green fluorescent protein encoding plasmid.
Figure S 3: Dose reduction index of camptothecin and TRAIL plasmid using CT2 MLNPs.
The dose-reduction index (DRI) for multi-layered nanoparticles (MLNPs) over all fraction affected (Fa) levels of tumor growth inhibition for U87, HCT116, and MDAMB231 cancer cell lines. A. DRI of MLNPs delivering camptothecin (CPT) and plasmid encoding TNF related apoptosis inducing ligand (pTRAIL) (CT2 MLNPs) relative to MLNPs delivering CPT and plasmid encoding for luciferase (CL2 MLNPs) and MLNPs containing only pTRAIL (BT MLNPs). B. DRI of CT2 MLNPs relative to poly(lactic-co-glycolic) (PLGA) nanoparticles encapsulating CPT only (CPT NPs) and BT MLNPs. All MLNPs were loaded with the same amount of plasmid and the same CPT to pTRAIL mass ratio of 0.03. CPT NPs were loaded with same amount of CPT as the CPT loaded MLNPs.
Figure S 4: Mouse weights during in vivo antitumor therapy.
Mouse weights remained stable during the 16 day study. Treatment groups included phosphate buffered saline (PBS), multi-layered nanoparticles (MLNPs) delivering luciferase encoding plasmid (pLuc) (BL MLNPs), MLNPs delivering camptothecin (CPT) and pLuc (CL2 MLNPs), MLNPs delivering TNF related apoptosis inducing ligand encoding plasmid (pTRAIL) (BT MLNPs), and MLNPs delivering both CPT and pTRAIL (CT2 MLNPs). All MLNPs were loaded with the same amount of plasmid and the same CPT to pTRAIL ratio of 0.03.
Table S 1: CPT to pTRAIL ratio for death kinetics experiments.
Table S 2: Camptothecin to pTRAIL ratio for MLNP formulations.
Table S 3: Goodness of fit of the Chou-Talalay analysis.
Table S 4: Median effect doses for CT2 MLNPs, CL2 MLNPs, and CPT NPs.