

Abstract
Objectives:
To investigate clinical, imaging, and pathologic associations of the cingulate island sign (CIS) in dementia with Lewy bodies (DLB).
Methods:
We retrospectively identified and compared patients with a clinical diagnosis of DLB (n = 39); patients with Alzheimer disease (AD) matched by age, sex, and education (n = 39); and cognitively normal controls (n = 78) who underwent 18F-fluorodeoxyglucose (FDG) and C11 Pittsburgh compound B (PiB)-PET scans. Among these patients, we studied those who came to autopsy and underwent Braak neurofibrillary tangle (NFT) staging (n = 10).
Results:
Patients with a clinical diagnosis of DLB had a higher ratio of posterior cingulate to precuneus plus cuneus metabolism, cingulate island sign (CIS), on FDG-PET than patients with AD (p < 0.001), a finding independent of β-amyloid load on PiB-PET (p = 0.56). Patients with CIS positivity on visual assessment of FDG-PET fit into the group of high- or intermediate-probability DLB pathology and received clinical diagnosis of DLB, not AD. Higher CIS ratio correlated with lower Braak NFT stage (r = −0.96; p < 0.001).
Conclusions:
Our study found that CIS on FDG-PET is not associated with fibrillar β-amyloid deposition but indicates lower Braak NFT stage in patients with DLB. Identifying biomarkers that measure relative contributions of underlying pathologies to dementia is critical as neurotherapeutics move toward targeted treatments.
Dementia with Lewy bodies (DLB) is the second most common cause of dementia.1 Despite revised criteria,2 clinically distinguishing DLB from other neurodegenerative diseases continues to be problematic. In fact, the National Alzheimer's Coordinating Center registry reported the sensitivity of clinical diagnoses of DLB compared with subsequent pathologic examination to be 32.1%.3
Complicating the inaccuracies of clinical diagnosis, the majority of dementia cases have multiple pathologies at autopsy.4,5 Coexisting Alzheimer disease (AD) pathology occurs in 51% to 66% of those with pathologically confirmed DLB.1,6
Accurate antemortem prediction of underlying causes of dementia is important for determining a prognosis,7,8 predicting treatment response,9 and planning for clinical trials. Additional biomarkers are required to better distinguish DLB from AD and to predict relative contributions of different underlying pathologies to DLB. By measuring β-amyloid (Aβ) deposition with C11 Pittsburgh compound B (PiB) binding, investigators have demonstrated that more than half of patients with DLB have Aβ deposition in the brain.10,11
Antemortem 18F-fluorodeoxyglucose (FDG)-PET hypometabolism correlates with neurofibrillary tangle (NFT)-tau burden, rather than Aβ, at autopsy.12 The cingulate island sign (CIS), a term referring to sparing of the posterior cingulate relative to the precuneus and cuneus, has been proposed as an FDG-PET imaging feature of DLB.13,14 Although the CIS performed well in differentiating clinically diagnosed cases of AD from DLB, few cases underwent autopsy.14 Our objective was to investigate the clinical and pathologic associations of the CIS using PiB-PET for the assessment of Aβ pathology and Braak staging of the NFT pathology at autopsy.
METHODS
Study patients.
We performed a case-control study with patients recruited from the Mayo Clinic Alzheimer's Disease Research Center, where participants undergo annual clinical evaluation, MRI, and neuropsychometric evaluation.6,11,15 Diagnosis is made at a consensus meeting of behavioral neurologists, neuropsychologists, and nurses. We retrospectively identified patients who met consensus clinical diagnosis of probable DLB (n = 39) on the basis of published criteria2 and had antemortem FDG- and PiB-PET scans. We also identified cognitively normal controls who were matched for age, sex, and education (n = 78) without evidence of Aβ deposition (PiB-negative) and patients with AD dementia matched for age, sex, and education who met the National Institute of Neurological Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria for probable AD (n = 39) for comparison.16
Clinical DLB features were recorded through the following measures: (1) visual hallucinations were fully formed, occurring more than once, and were not attributable to other medical factors, including medications and advanced dementia; (2) parkinsonism was rated with the Unified Parkinson's Disease Rating Scale (UPDRS); (3) probable REM sleep behavior disorder was recorded if patients met the International Classification of Sleep Disorders II diagnostic criteria B; and (4) a score of 3 or 4 on the Mayo Fluctuations Questionnaire was required for fluctuations to be considered present.17
Standard protocol approvals, registrations, and patient consents.
The present study was approved by the Mayo Clinic Institutional Review Board. Informed consent for participation was obtained from every patient or an appropriate surrogate.
Neuropathologic assessment.
Ten patients in the study underwent autopsy. Sampling was done according to the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) protocol18 and the Third Report of the DLB Consortium.2 NFTs and corresponding Braak stage were detected with thioflavin-S microscopy or Bielschowsky silver stain and classified in accordance with National Institute on Aging–Reagan Institute criteria.19 A polyclonal antibody to α-synuclein was used to categorize regional involvement of Lewy bodies as brainstem, limbic, and neocortical. The neuropathologic diagnosis of DLB was made in accordance with the Third Report of the DLB Consortium criteria without consideration of clinical presentation.2
MRI, PiB-PET, and FDG-PET acquisitions.
MRI examinations were performed at 3 tesla with an 8-channel phased array coil (GE Healthcare; Waukesha, WI). A 3-dimensional high-resolution magnetization-prepared rapid-acquisition gradient echo acquisition with repetition time/echo time/inversion time of 7/3/900 milliseconds, a flip angle of 8°, an in-plane resolution of 1.0 mm, and a slice thickness of 1.2 mm was performed for anatomical segmentation and labeling. PET images were obtained with a LYSO PET/CT scanner (DRX; GE Healthcare) functioning in 3-dimensional mode. Attenuation correction was achieved by obtaining a CT image. Patients received PiB (average [range], 596 [292–729] MBq) and FDG (average [range], 540 [366–399] MBq) injections. FDG-PET and C11 PiB images were acquired 1 hour apart on the same day. After a 40-minute PiB uptake period, a 20-minute PiB scan was acquired. PiB-PET was obtained with four 5-minute dynamic frames, acquired between 40 and 60 minutes postinjection. An 8-minute FDG scan was obtained, after a 30-minute FDG uptake period. Image acquisition included four 2-minute dynamic frames, acquired from 30 to 38 minutes after injection. Standard corrections were performed.
PiB-PET and FDG-PET analysis.
The methodology of PiB-PET analysis has been described previously.11 Values greater than 1.5 were considered PiB-positive.20 FDG-PET image volumes of each patient were coregistered to the patient's own T1-weighted MRI scan with the modified automated anatomical labeling atlas,21 using an affine registration of 6 degrees of freedom with mutual information cost function. In the patient's T1-weighted MRI space, atlas-based parcellation of FDG images into regions of interest (ROIs) was performed. Partial volume correction of CSF and tissue compartments was applied through the 2-compartment model22 to remove atrophy effects on the FDG uptake on PET images. FDG-PET ratio images were derived using pons uptake as the internal reference. Group differences in voxel-wise FDG uptake were assessed with 2-sided t test within the general linear model framework of Statistical Parametric Mapping (SPM)5. Statistical maps displaying differences between groups were shown at a significance level of p < 0.05, using family-wise error correction for multiple comparisons. Modified automated anatomical labeling atlas ROIs were used to extract statistics on image voxel values. The median value in the posterior cingulate gyrus ROI was divided by the median value in the precuneus plus cuneus ROI to derive the CIS ratio from the FDG-PET images.14 Figure e-1 on the Neurology® Web site at Neurology.org shows the ROIs analyzed in this study.
Statistical analysis.
When comparing the matched groups, we used Friedman test for continuous variables and χ2 tests from conditional logistic regressions stratifying on the matched sets for binary variables. For comparison of groups in unmatched data subsets, Kruskal-Wallis and Wilcoxon rank sum tests were performed for continuous variables and χ2 tests for categorical variables. Spearman rank correlation was used for comparing imaging with Braak stage, parkinsonism, and global cognitive function. All analyses were performed with SAS version 9.3 (SAS Institute Inc., Cary, NC) and R statistical software version 2.14.0 (R Project for Statistical Computing).
RESULTS
Study patients.
Demographic features were similar among the clinical groups (table 1). Dementia severity using Mattis Dementia Rating Scale (DRS) and Clinical Dementia Rating scores did not distinguish DLB from AD groups.
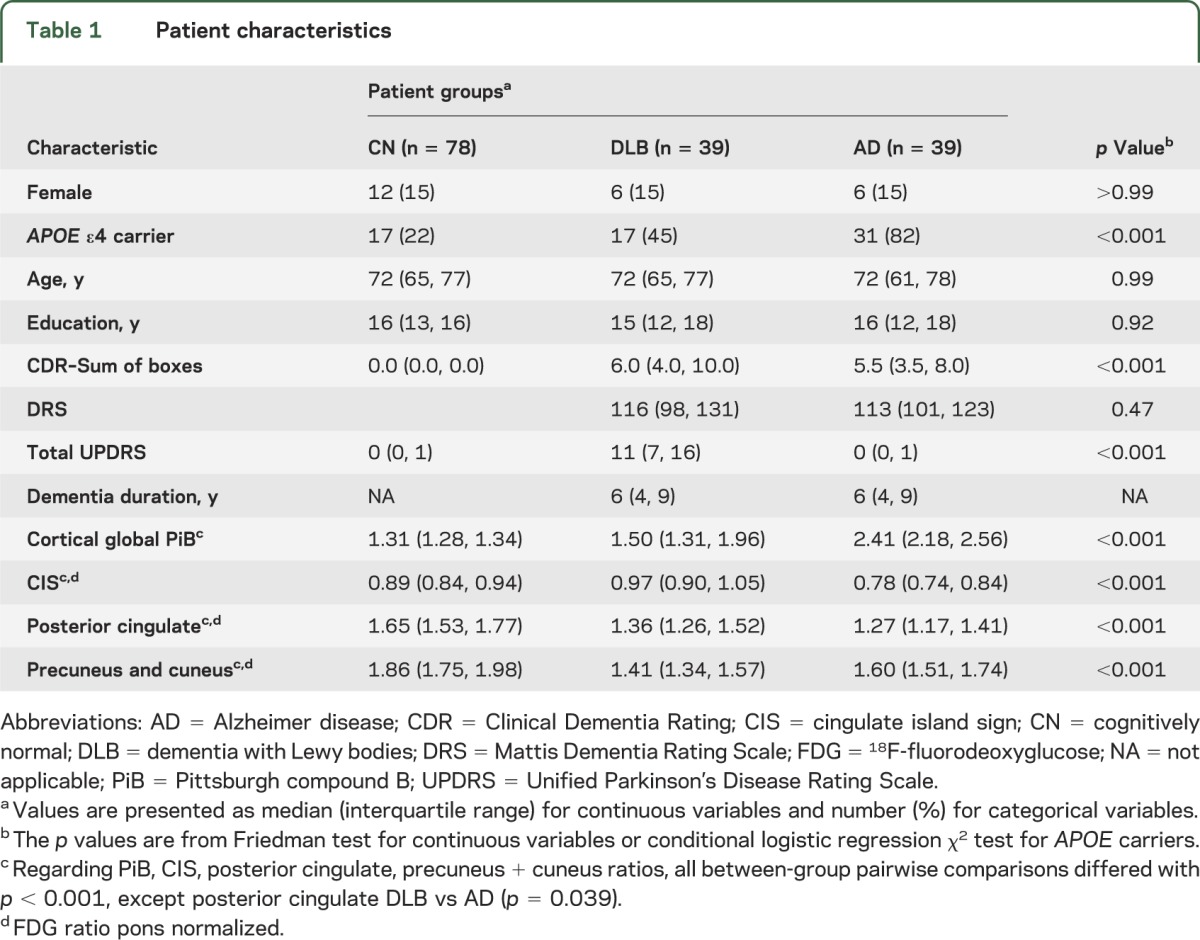
Table 1.
Patient characteristics
CIS in patients with DLB, AD, and normal cognitive function.
Table 1 shows the global cortical PiB ratios and FDG-PET values for the pons-normalized ratios of the CIS, posterior cingulate, and precuneus plus cuneus among patients with clinically diagnosed DLB, with clinically diagnosed AD, and with normal cognitive function. The CIS ratio was higher in patients with a clinical diagnosis of DLB than in patients with a clinical diagnosis of AD (p < 0.001). Disease duration did not correlate with the CIS in DLB (r = −0.043; p = 0.79).
CIS in PiB-positive and PiB-negative patients with DLB.
Table 2 divides the DLB group into PiB-positive and PiB-negative. No differences in demographic and clinical characteristics were observed among the PiB-positive and PiB-negative patients with DLB. Figure 1 plots the FDG-PET values of the CIS among DLB PiB-negative, DLB PiB-positive, cognitively normal, and AD patients showing a higher CIS in DLB than AD, regardless of PiB status (p < 0.001). No differences in CIS were observed between DLB PiB-negative patients and DLB PiB-positive patients (p = 0.56). DLB PiB-negative and DLB PiB-positive patients had a higher CIS compared with controls (p < 0.01). The lowest CIS ratio in the PiB-negative clinical DLB cases was 0.87. The 2 lowest CIS ratios (0.67 and 0.80) in the PiB-positive clinically diagnosed DLB cases showed a high likelihood of AD pathology without Lewy body pathology at autopsy. There was no relationship between PiB-PET as a continuous variable and FDG uptake in any components of the island sign (p > 0.38).
Table 2.
Characteristics of PiB-positive and PiB-negative patients with DLB
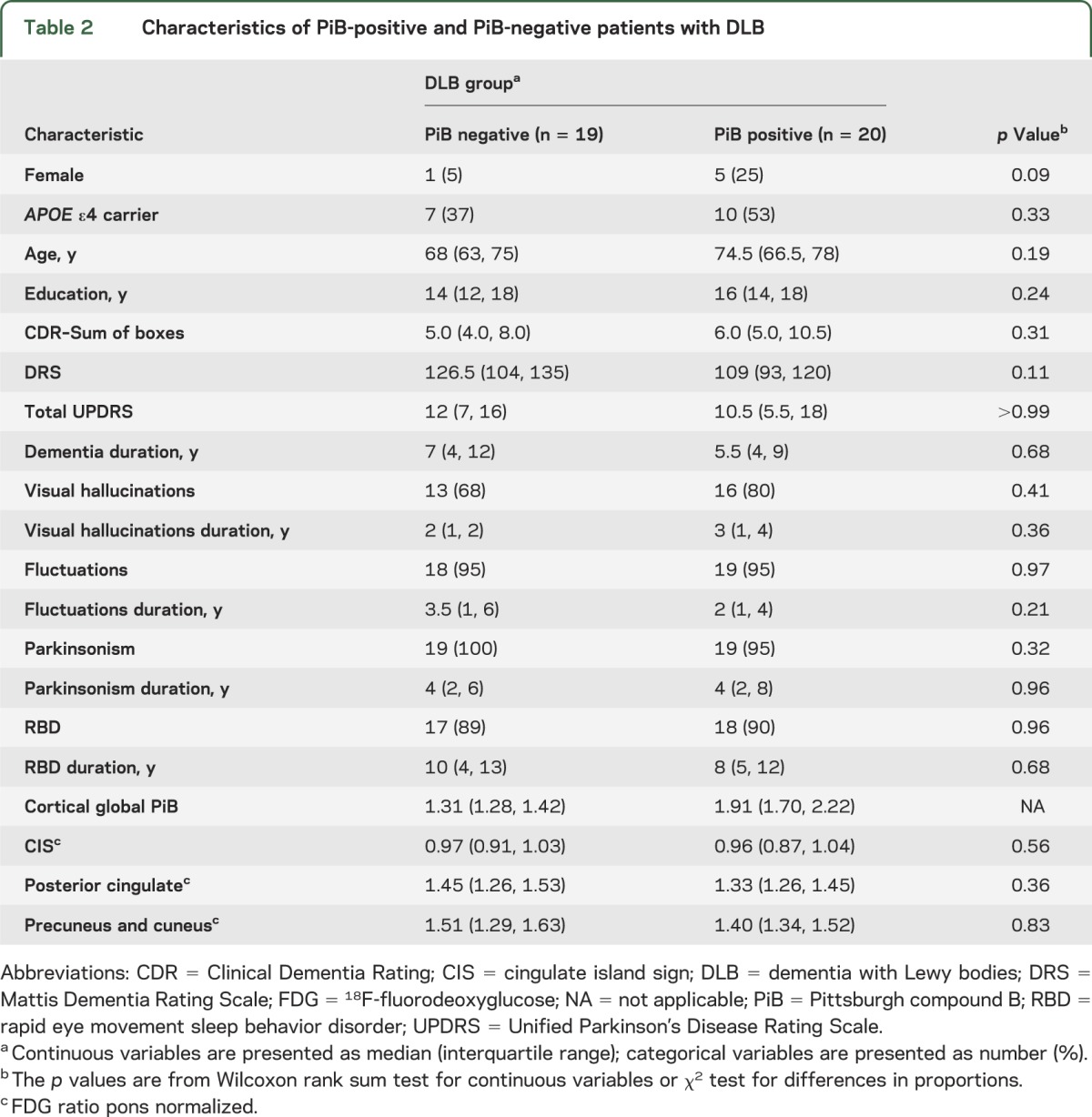
Figure 1. Box plots of the cingulate island sign ratio among the patient groups.
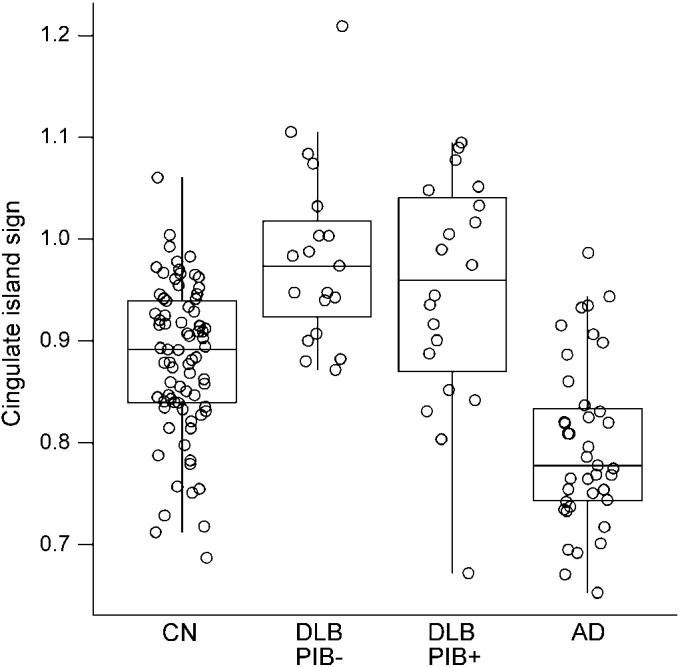
AD = Alzheimer disease; CN = cognitively normal; DLB = dementia with Lewy bodies; PiB = Pittsburgh compound B; PiB− = PiB negative; PiB+ = PiB positive.
Figure 2 is an SPM analysis of FDG-PET in PiB-positive and PiB-negative patients with DLB compared with controls. It shows relative preservation of the posterior cingulate and inferior precuneus metabolism, regardless of Aβ status (p > 0.05; family-wise error corrected). Voxel-based analysis of FDG-PET did not show any differences between PIB-positive and PiB-negative DLB groups (p > 0.05; family-wise error corrected).
Figure 2. SPM5 analysis of FDG-PET in patients with DLB compared with controls.
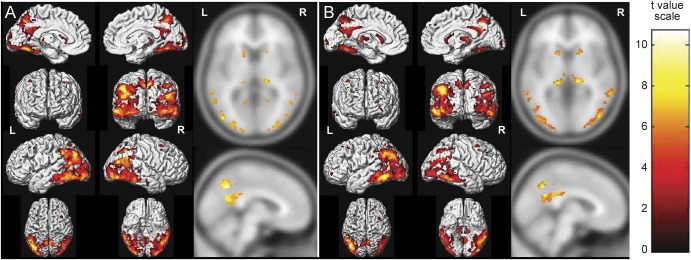
(A) Hypometabolism in PiB-positive DLB. (B) Hypometabolism in PiB-negative DLB. Patients with DLB compared with controls (p < 0.05; family-wise error corrected for multiple comparisons). The pattern of hypometabolism is similar among PiB-positive and PiB-negative groups and involved the posterior temporal, parietal, and occipital association cortices and the retrosplenial cortex but sparing the posterior and middle cingulate gyrus and inferior precuneus, visually identified as the cingulate island sign on FDG-PET images. Pulvinar of thalamus and head of the caudate nucleus are also involved. DLB = dementia with Lewy bodies; FDG = 18F-fluorodeoxyglucose; PiB = Pittsburgh compound B.
FDG correlates of parkinsonism and global cognitive function.
No associations of CIS were found with UPDRS and DRS in DLB (p > 0.05). We further investigated the relationship of UPDRS and DRS with the numerator and denominator components of the CIS ratio. Whereas lower precuneus plus cuneus hypometabolism correlated with higher UPDRS (r = −0.41; p = 0.01) and lower DRS (r = 0.44; p = 0.039), lower posterior cingulate hypometabolism correlated with lower DRS (r = 0.42; p = 0.045) but not UPDRS (p > 0.05).
Neuropathology.
Ten patients in the study came to autopsy; 8 had a clinical diagnosis of DLB and 2 a clinical diagnosis of AD. Of the 8 patients with diagnosed DLB, 6 had an intermediate or high likelihood of DLB and 2 had a high likelihood of AD with no Lewy body pathology at autopsy. All 6 patients with autopsy confirmation of DLB had CIS positivity on visual inspection of FDG-PET. The 2 patients who had clinical diagnosis of DLB but were found to have AD pathology at autopsy had a negative CIS. In addition, the 2 patients with a clinical diagnosis of AD had a high likelihood of AD at autopsy, and neither had CIS positivity. The CIS ratio was 0.81 or greater in all autopsy-confirmed DLB cases; the 3 autopsy-confirmed AD cases had a ratio less than 0.81. Figure 3 shows the relation between the CIS, posterior cingulate, and precuneus plus cuneus FDG-PET ratios with Braak NFT stage in the 10 cases that came to autopsy. A strong relation was present between a higher CIS and a lower Braak NFT stage in patients with DLB (r = −0.96 [95% confidence interval, −0.99, −0.83]; p < 0.001). Figure e-2 provides enlarged versions of FDG-PET scans in patients diagnosed with DLB from figure 3. Cases 1 and 6 had none and sparse amyloid neuritic plaques. Case 1 was PiB-negative and case 6 was borderline PiB-positive. Cases 2, 3, 4, and 5 had moderate neuritic plaques. Only case 2 was PiB-negative. Cases 7, 8, 9, and 10 had frequent neuritic plaques. All were PiB-positive. Cases 1, 2, and 6 had transitional Lewy body–type pathology, while cases 3, 4, 5, and 7 had diffuse Lewy body–type pathology.
Figure 3. Pathology-confirmed clinical cases arranged by DLB/AD likelihood according to pathology and cingulate island sign.

(A) Corresponding PET scan and Braak NFT stage in rows below. Cases 1 through 6 and cases 8 and 10 had clinically diagnosed DLB, and all but cases 8 and 10 had intermediate or high likelihood of DLB at autopsy. Cases 7 and 9 were diagnosed with AD clinically. (B) Numbers represent the corresponding cases shown above; blue numbers are DLB at autopsy and red numbers are AD at autopsy. AD = Alzheimer disease; DLB = dementia with Lewy bodies; NFT = neurofibrillary tangle.
DISCUSSION
The main finding of this study was that the preservation of the posterior cingulate metabolism on FDG-PET relative to the cuneus and precuneus (the CIS) is not associated with Aβ load but does predict lower Braak NFT stage in clinically diagnosed DLB cases. In the small group of patients who came to autopsy, the CIS distinguished patients with pathologic DLB from AD with a ratio of 0.81, similar to a prior study of clinically diagnosed cases.14 Furthermore, all PiB-negative patients with DLB had a CIS ratio of 0.87 or more. Notably, the presence of Aβ pathology on PiB-PET scans did not influence the CIS. In addition, our study confirmed that the CIS distinguishes clinically diagnosed patients with DLB from patients with AD. Although an overlap was found between the DLB and AD groups, the 2 lowest island signs in the clinically diagnosed DLB PiB-positive group showed AD pathology. Finally, our study showed that clinical symptoms of DLB (parkinsonism and global cognitive function) correlate with precuneus plus cuneus hypometabolism but not the CIS.
The majority of probable DLB cases have coexisting AD pathology. Recognition of the degree of AD pathology through biomarkers in DLB cases is important because imaging biomarkers of coexisting AD pathology predict worse treatment response to acetylcholinesterase inhibitors in DLB.9 Furthermore, coexisting AD pathology in DLB cases may be a predictor of survival23 and therefore may have implications when patients are enrolled for disease-modifying therapies that target AD pathology. Many patients with DLB have diffuse Aβ plaques, but others have neuritic Aβ plaques and associated NFT-tau pathology. Although the ability to predict Aβ deposition in DLB is accomplished with PiB-PET scans, coexisting Braak NFT stage has to be identified with surrogate markers until the imaging ligands of tau become widely available. Aβ pathology is a hallmark of AD; however, Braak NFT stage correlates better with the clinical symptoms in AD dementia, which may also be true for DLB.24 In clinically diagnosed DLB cases, the most frequent Braak NFT stage at autopsy is 3–4, with 5–6 being the second most frequent.4 As the neurodegenerative field moves toward targeting specific proteinopathies, antemortem prediction of relative contributions of underlying pathologies will become more important. Therefore, the CIS on FDG-PET may be used in conjunction with Aβ PET scan in the differential diagnosis of probable DLB to determine the presence and extent of coexisting AD pathology.
The CIS in DLB often occurred in the presence of a negative Aβ PET scan, with all patients who were PiB-negative having a CIS, with the lowest ratio being 0.87. Only a subset of the PiB-positive patients had a CIS ratio of less than 0.87, representing either patients with AD clinically mimicking DLB (patients 8 and 10 on figure 3) or patients with DLB with high Braak stage, although figure 1 shows that CIS ratios overlap in the clinically diagnosed cases. PiB positivity may not always point to high likelihood of AD because a positive PiB-PET scan has been observed in patients with Lewy body disease who had frequent diffuse plaques but sparse neuritic plaques and low to intermediate Braak NFT stage, which would classify them as having high likelihood of DLB.25 Therefore, additional information from the CIS may be useful.
Prior PET studies have shown greater occipital hypometabolism in patients with DLB than in controls.11,13,26 Occipital hypometabolism also differentiates clinical and autopsy-proven DLB from AD cases.13,26,27 In DLB, occipital hypometabolism correlates with visual hallucinations28 and improves with acetylcholinesterase inhibitor treatment.29 The occipital hypometabolism in DLB is independent of Aβ load11 and is thought to be related to impaired cholinergic transmission or synaptic dysfunction associated with α-synuclein.30–32 In addition, occipital hypometabolism is characteristic of posterior cortical atrophy (atypical AD) and advanced AD and thus is not specific for DLB.33,34 Although hippocampal atrophy is also associated with the Braak NFT stage in DLB,15 hippocampal preservation in posterior cortical atrophy can add further diagnostic confusion. An illustrative example is patient 8, who met diagnostic criteria for DLB (i.e., parkinsonism, early hallucinations, and visual spatial dysfunction) but was found to have posterior cortical atrophy (atypical AD) at autopsy. In this patient, the CIS would have provided important ancillary diagnostic information of Braak NFT stage.
The posterior cingulate hypometabolism on FDG-PET serves as a useful marker of AD pathology because metabolism is severely reduced early in AD, even compared with temporal and parietal cortices.35 Posterior cingulate abnormalities are independent of atrophy or partial volume-averaging effects, and they precede decreases in hippocampal metabolism or hippocampal atrophy in cognitively normal APOE ε4 carriers.36 In AD, the posterior cingulate gyrus shows marked neurodegeneration and volume loss.37
Of the 3 indices—the CIS, posterior cingulate hypometabolism, and precuneus plus cuneus hypometabolism—the precuneus plus cuneus hypometabolism correlated best with overall cognitive impairment and severity of parkinsonism in patients with probable DLB, likely reflecting its important interconnections with other structures directly affected by DLB pathology, rather than a direct role in the development of global cognitive dysfunction and parkinsonism.38 The precuneus has been implicated as part of the attention network that is abnormal in patients with DLB. For example, patients with DLB have increased connectivity between the precuneus and components of the dorsal attention network.39 Of interest, SPM analysis comparing patients with DLB to controls in the present study showed hypometabolism not only in the precuneus, but also in the pulvinar, a key component of the dorsal attention network.40 Patients with DLB who had PiB positivity had slightly greater posterior cingulate gyrus hypometabolism than those who had PiB negativity. However, the CIS persisted in the PiB-positive patients with DLB because the precuneus and cuneus were more hypometabolic than in AD or PiB-negative DLB, possibly because of synergistic effects of having both DLB and AD pathologies on the precuneus and cuneus regions.
The study has several limitations. Because the AD pathology is the driver of the difference between patients with DLB and those with AD, significant overlap exists between the subjects with DLB and controls. Therefore, the utility of the CIS is when a diagnosis of dementia already exists. Also, given the limited number of autopsied cases, we did not evaluate the CIS as a diagnostic biomarker with receiver operating characteristic curves. Because these cases were sampled according to the CERAD protocol, targeted investigation of the pathologic burden in posterior cingulate and precuneus was not possible in all subjects. Furthermore, correlations between CIS and neuropsychometric measures of episodic memory were not possible because of missing data. Our study confirms that the CIS can help distinguish DLB from AD. It extends previous work through showing that the presence of the CIS predicts a lower Braak NFT stage at autopsy, but the small number of autopsy-confirmed cases limits the generalizability of the CIS as an antemortem biomarker of Braak NFT stage in probable DLB. Although the relationship observed between Braak NFT stage and the CIS was strong, only 10 cases came to autopsy. Further pathologic confirmation of DLB cases with FDG-PET scans should be performed in order to establish a reliable cutoff of Braak NFT stage at which the CIS disappears. Future studies are needed to determine whether the CIS can serve as a marker of acetylcholinesterase inhibitor response or can predict prognosis in patients with DLB.
Supplementary Material
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- CERAD
Consortium to Establish a Registry for Alzheimer's Disease
- CIS
cingulate island sign
- DLB
dementia with Lewy bodies
- DRS
Mattis Dementia Rating Scale
- FDG
18F-fluorodeoxyglucose
- NFT
neurofibrillary tangle
- PiB
Pittsburgh compound B
- ROI
region of interest
- SPM
Statistical Parametric Mapping
- UPDRS
Unified Parkinson's Disease Rating Scale
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
J. Graff-Radford and M.E. Murray: study concept and design, acquisition of data, analysis and interpretation of the data, drafting the manuscript. V.J. Lowe: acquisition of data, analysis and interpretation of the data, drafting the manuscript, critical revision of the manuscript for important intellectual content. B.F. Boeve: acquisition of data, critical revision of the manuscript for important intellectual content. T.J. Ferman: acquisition of data, analysis and interpretation of the data, drafting the manuscript. S.A. Przybelski and T.G. Lesnick: study concept and design, analysis and interpretation of the data, critical revision of the manuscript for important intellectual content. M.L. Senjem, J.L. Gunter, G.E. Smith, and D.S. Knopman: acquisition of data, critical revision of the manuscript for important intellectual content. C.R. Jack Jr: acquisition of data, analysis and interpretation of the data, critical revision of the manuscript for important intellectual content. D.W. Dickson: study concept and design, acquisition of data, analysis and interpretation of the data, drafting the manuscript. R.C. Petersen: acquisition of data, critical revision of the manuscript for important intellectual content. K. Kantarci: study concept and design, acquisition of data, analysis and interpretation of the data, drafting the manuscript.
STUDY FUNDING
Supported by grant UL1 TR000135 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Funded by NIH (K23 AG030935, R01 AG040042, R01 AG11378, P50 AG16574, U01 AG06786, C06 RR018898), Mangurian Foundation, the Elsie and Marvin Dekelboum Family Foundation, and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program.
DISCLOSURE
J. Graff-Radford and M. Murray report no disclosures relevant to the manuscript. V. Lowe is a consultant for Bayer Schering Pharma AG and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, Inc., the NIH, the Elsie and Marvin Dekelboum Family Foundation, the Minnesota Partnership for Biotechnology and Medical Genomics, and the Leukemia and Lymphoma Society. B. Boeve has served as an investigator for clinical trials sponsored by Cephalon, Inc., Allon Pharmaceuticals Inc., and GE Healthcare. He receives royalties from the publication of a book titled The Behavioral Neurology of Dementia (Cambridge University Press, 2009). He has received honoraria from the American Academy of Neurology. He serves on the scientific advisory board of the Tau Consortium. He receives research support from the National Institute on Aging (NIA) (P50 AG016574, U01 AG006786, R01 AG032306, and R01 AG041797) and the Mangurian Foundation. T. Ferman is funded by the NIH (Mayo Clinic Alzheimer's Disease Research Center/Project 1-P50 AG16574/P1 [Co-I]). S. Przybelski, T. Lesnick, M. Senjem, and J. Gunter report no disclosures relevant to the manuscript. G. Smith is funded by the NIH (P50 AG16574). D. Knopman serves as Deputy Editor for Neurology®; has served on a data safety monitoring board for Lilly Pharmaceuticals; serves on a data safety monitoring board for Lundbeck Pharmaceuticals and for the Dominantly Inherited Alzheimer Network study; has served as a consultant to TauRx Pharmaceuticals Ltd.; was an investigator in clinical trials sponsored by Baxter International Inc. and Elan Corp.; is an investigator in a clinical trial sponsored by TauRx; and receives research support from the NIH. C. Jack Jr serves as a consultant for Janssen Pharmaceuticals, Inc.; Bristol-Myers Squibb Co.; General Electric; and Johnson & Johnson. He is involved in clinical trials sponsored by Allon and Baxter International, Inc., and receives research support from Pfizer Inc., the NIA (AG11378 [PI], P50 AG16574 [Co-I], and U01 AG024904-01 [Co-I]), and the Alexander Family Professor of Alzheimer's Disease Research fund of Mayo Foundation. D. Dickson is funded by the NIH (P50 AG16574/Neuropathology Core [PI], P01 AG017216 [PI], P50 NS072187 [PI], and R01 AG040042 [Co-I]). R. Petersen serves on scientific advisory boards for Elan Corp., Wyeth Pharmaceuticals, Inc., and GE Healthcare and receives research support from the NIH (P50 AG16574 [PI], U01 AG06786 [PI], R01 AG11378 [Co-I], and U01-24904 [Co-I]). K. Kantarci serves on the data safety monitoring board of Takeda Global Research and Development Center, Inc., and the data monitoring boards of Pfizer Inc. and Janssen Alzheimer Immunotherapy and is funded by the NIH (R01 AG040042 [PI], R21 NS066147 [PI], P50 AG44170/Project 2 [PI], P50 AG16574/Project 1 [PI], and R01 AG11378 [Co-I]). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Barker WW, Luis CA, Kashuba A, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord 2002;16:203–212 [DOI] [PubMed] [Google Scholar]
- 2.McKeith IG, Dickson DW, Lowe J, et al. ; Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872 [DOI] [PubMed] [Google Scholar]
- 3.Nelson PT, Jicha GA, Kryscio RJ, et al. Low sensitivity in clinical diagnoses of dementia with Lewy bodies. J Neurol 2010;257:359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujishiro H, Iseki E, Higashi S, et al. Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett 2010;486:19–23 [DOI] [PubMed] [Google Scholar]
- 5.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69:2197–2204 [DOI] [PubMed] [Google Scholar]
- 6.Ferman TJ, Boeve BF, Smith GE, et al. Inclusion of RBD improves the diagnostic classification of dementia with Lewy bodies. Neurology 2011;77:875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rongve A, Vossius C, Nore S, Testad I, Aarsland D. Time until nursing home admission in people with mild dementia: comparison of dementia with Lewy bodies and Alzheimer's dementia. Int J Geriatr Psychiatry 2014;29:392–398 [DOI] [PubMed] [Google Scholar]
- 8.Williams MM, Xiong C, Morris JC, Galvin JE. Survival and mortality differences between dementia with Lewy bodies vs Alzheimer disease. Neurology 2006;67:1935–1941 [DOI] [PubMed] [Google Scholar]
- 9.Graff-Radford J, Boeve BF, Pedraza O, et al. Imaging and acetylcholinesterase inhibitor response in dementia with Lewy bodies. Brain 2012;135:2470–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 2008;79:1331–1338 [DOI] [PubMed] [Google Scholar]
- 11.Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging 2012;33:2091–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imamura T, Ishii K, Sasaki M, et al. Regional cerebral glucose metabolism in dementia with Lewy bodies and Alzheimer's disease: a comparative study using positron emission tomography. Neurosci Lett 1997;235:49–52 [DOI] [PubMed] [Google Scholar]
- 14.Lim SM, Katsifis A, Villemagne VL, et al. The 18F-FDG PET cingulate island sign and comparison to 123I-beta-CIT SPECT for diagnosis of dementia with Lewy bodies. J Nucl Med 2009;50:1638–1645 [DOI] [PubMed] [Google Scholar]
- 15.Kantarci K, Ferman TJ, Boeve BF, et al. Focal atrophy on MRI and neuropathologic classification of dementia with Lewy bodies. Neurology 2012;79:553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944 [DOI] [PubMed] [Google Scholar]
- 17.Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology 2004;62:181–187 [DOI] [PubMed] [Google Scholar]
- 18.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486 [DOI] [PubMed] [Google Scholar]
- 19.Newell KL, Hyman BT, Growdon JH, Hedley-Whyte ET. Application of the National Institute on Aging (NIA)–Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol 1999;58:1147–1155 [DOI] [PubMed] [Google Scholar]
- 20.Jack CR, Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 2002;15:273–289 [DOI] [PubMed] [Google Scholar]
- 22.Meltzer CC, Kinahan PE, Greer PJ, et al. Comparative evaluation of MR-based partial-volume correction schemes for PET. J Nucl Med 1999;40:2053–2065 [PubMed] [Google Scholar]
- 23.Jellinger KA, Wenning GK, Seppi K. Predictors of survival in dementia with Lewy bodies and Parkinson dementia. Neurodegener Dis 2007;4:428–430 [DOI] [PubMed] [Google Scholar]
- 24.Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 1995;16:285–298 [DOI] [PubMed] [Google Scholar]
- 25.Kantarci K, Yang C, Schneider JA, et al. Antemortem amyloid imaging and β-amyloid pathology in a case with dementia with Lewy bodies. Neurobiol Aging 2012;33:878–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albin RL, Minoshima S, D'Amato CJ, Frey KA, Kuhl DA, Sima AA. Fluoro-deoxyglucose positron emission tomography in diffuse Lewy body disease. Neurology 1996;47:462–466 [DOI] [PubMed] [Google Scholar]
- 27.Minoshima S, Foster NL, Sima AA, Frey KA, Albin RL, Kuhl DE. Alzheimer's disease versus dementia with Lewy bodies: cerebral metabolic distinction with autopsy confirmation. Ann Neurol 2001;50:358–365 [DOI] [PubMed] [Google Scholar]
- 28.Perneczky R, Drzezga A, Boecker H, Forstl H, Kurz A, Haussermann P. Cerebral metabolic dysfunction in patients with dementia with Lewy bodies and visual hallucinations. Dement Geriatr Cogn Disord 2008;25:531–538 [DOI] [PubMed] [Google Scholar]
- 29.Satoh M, Ishikawa H, Meguro K, Kasuya M, Ishii H, Yamaguchi S. Improved visual hallucination by donepezil and occipital glucose metabolism in dementia with Lewy bodies: the Osaki-Tajiri project. Eur Neurol 2010;64:337–344 [DOI] [PubMed] [Google Scholar]
- 30.Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 2007;27:1405–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 2009;73:273–278 [DOI] [PubMed] [Google Scholar]
- 32.Zaja-Milatovic S, Keene CD, Montine KS, Leverenz JB, Tsuang D, Montine TJ. Selective dendritic degeneration of medium spiny neurons in dementia with Lewy bodies. Neurology 2006;66:1591–1593 [DOI] [PubMed] [Google Scholar]
- 33.Ishii K, Sasaki M, Kitagaki H, et al. Reduction of cerebellar glucose metabolism in advanced Alzheimer's disease. J Nucl Med 1997;38:925–928 [PubMed] [Google Scholar]
- 34.Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004;63:1168–1174 [DOI] [PubMed] [Google Scholar]
- 35.Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol 1997;42:85–94 [DOI] [PubMed] [Google Scholar]
- 36.Reiman EM, Chen K, Alexander GE, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci USA 2004;101:284–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brun A, Gustafson L. Distribution of cerebral degeneration in Alzheimer's disease: a clinico-pathological study. Arch Psychiatr Nervenkr 1976;223:15–33 [DOI] [PubMed] [Google Scholar]
- 38.Cavanna AE, Trimble MR. The precuneus: a review of its functional anatomy and behavioural correlates. Brain 2006;129:564–583 [DOI] [PubMed] [Google Scholar]
- 39.Galvin JE, Price JL, Yan Z, Morris JC, Sheline YI. Resting bold fMRI differentiates dementia with Lewy bodies vs Alzheimer disease. Neurology 2011;76:1797–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller EK, Buschman TJ. Cortical circuits for the control of attention. Curr Opin Neurobiol 2013;23:216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.