

Abstract
Objectives:
Prospective cohort study to characterize the clinical features and course of spinal muscular atrophy type I (SMA-I).
Methods:
Patients were enrolled at 3 study sites and followed for up to 36 months with serial clinical, motor function, laboratory, and electrophysiologic outcome assessments. Intervention was determined by published standard of care guidelines. Palliative care options were offered.
Results:
Thirty-four of 54 eligible subjects with SMA-I (63%) enrolled and 50% of these completed at least 12 months of follow-up. The median age at reaching the combined endpoint of death or requiring at least 16 hours/day of ventilation support was 13.5 months (interquartile range 8.1–22.0 months). Requirement for nutritional support preceded that for ventilation support. The distribution of age at reaching the combined endpoint was similar for subjects with SMA-I who had symptom onset before 3 months and after 3 months of age (p = 0.58). Having 2 SMN2 copies was associated with greater morbidity and mortality than having 3 copies. Baseline electrophysiologic measures indicated substantial motor neuron loss. By comparison, subjects with SMA-II who lost sitting ability (n = 10) had higher motor function, motor unit number estimate and compound motor action potential, longer survival, and later age when feeding or ventilation support was required. The mean rate of decline in The Children's Hospital of Philadelphia Infant Test for Neuromuscular Disorders motor function scale was 1.27 points/year (95% confidence interval 0.21–2.33, p = 0.02).
Conclusions:
Infants with SMA-I can be effectively enrolled and retained in a 12-month natural history study until a majority reach the combined endpoint. These outcome data can be used for clinical trial design.
Proximal spinal muscular atrophy (SMA) is an autosomal recessive motor neuron disorder with an incidence of 1:11,000 live births.1 Mutations in the survival of motor neuron 1, telomeric (SMN1) gene cause SMA.2 A nearly identical gene, SMN2, harbors an exonic splicing enhancer mutation that limits inclusion of exon 7.3 This results in a reduced amount of functional full-length SMN protein. The SMN2 copy number is inversely related to clinical severity.4 SMN protein functions within a spliceosomal complex and is important in RNA processing.5 SMA is a clinical continuum, divided into 4 phenotypes based on maximal motor function achieved: type I (SMA-I, onset by 6 months, nonsitter), type II (SMA-II, onset 6–18 months, sitter), ambulatory type III children, and type IV adults.6,7 Prior natural history studies have demonstrated shortened lifespan for SMA-I, with 68% succumbing within 2 years and 82% by 4 years of age.6,7 SMA-I has been further subdivided into 3 groups: type IA (or zero, in some reports8), presentation at birth with joint contractures and respiratory compromise; type IB, onset of symptoms before age 3 months; and type IC, onset after 3 months of age.9,10 Recent utilization of nutritional and respiratory support has altered the natural history of SMA-I, reducing mortality to approximately 30% at age 2 years, with approximately half of these survivors fully reliant on noninvasive ventilation.11
This prospective study was designed to describe the current natural history of SMA-I and guide planning of clinical trials for SMA-I.
METHODS
This study was performed by the Pediatric Neuromuscular Clinical Research Network for SMA, with clinical sites at Columbia University Medical Center (New York), University of Pennsylvania (The Children's Hospital of Philadelphia), and Harvard University (Boston Children's Hospital). Data management and statistical analysis were performed by the Biostatistics Center of the Muscle Study Group at the University of Rochester (New York).
The overall study design and data on subjects with SMA types II and III have been reported separately.12,13 The diagnosis of SMA and subtype classification were made by the principal investigator at each site (R.F., P.K., B.D.). Confirmation of the SMN1 exon 7/8 common deletion was performed by PCR amplification and restriction digest of DNA using primers flanking SMN1 and SMN2 exon 7, and SMN2 copy number was determined as previously described.12 Only patients with homozygous deletions were included.
Previously identified patients followed in our clinics and newly diagnosed patients were enrolled. All eligible patients were offered participation. Study visits were scheduled at baseline and at 2, 4, 6, 9, and 12 months and every 6 months thereafter. The SMA standard of care guidelines published in 2007 were used as a basis for providing uniform care among the study sites.14 For purposes of this study, sitting (for SMA-II) was defined as being able to sit independently for >10 seconds. To permit an analysis on the basis of function, rather than SMA type, subjects with type II (n = 45) were included and divided into type IIA, having lost the ability to sit (n = 10), and type IIB, having maintained the ability to sit (n = 35).
Demographic information included age, sex, and ethnicity. SMA history included age at symptom onset (by parental recollection and by verification of medical records when available), age at clinical diagnosis, means of diagnosis (clinical impression, molecular genetic confirmation), history of motor developmental milestones gained and/or lost, and family history of SMA. Other relevant medical and surgical history was captured, with attention to feeding/nutrition and respiratory function, hospitalizations, therapy received, and medication/supplements taken (none were disallowed). The primary outcomes of interest were age at death and age at reaching the combined endpoint of either death or requiring at least 16 hours/day of noninvasive ventilation support for at least 14 days in the absence of an acute reversible illness or perioperatively (as a surrogate for death).10 Physical examination findings included weight, length/height, head and chest circumference, vital signs, motor function, scoliosis, and joint contractures. Measurement of the serum comprehensive metabolic panel and complete blood count was performed at each study visit. Laboratory abnormalities were determined based on each hospital laboratory's normal values for age.
Motor function testing utilized The Children's Hospital of Philadelphia Infant Test for Neuromuscular Disorders (CHOP INTEND), a validated 16-item, 64-point scale shown to be reliable and sensitive to change over time for SMA-I.15,16 The CHOP INTEND was performed on all subjects with SMA-I and, for comparison, 5 subjects with SMA-II. Motor function testing was performed by clinical evaluators after a training and reliability session and reliability was re-established annually.12
Electrophysiologic measurements, the compound motor action potential (CMAP) and motor unit number estimate (MUNE, multipoint method), were obtained as previously described.12
Standard protocol approvals, registrations, and patient consents.
All guardians of participants provided written informed consent approved by the individual institutional review boards at the participating sites.
Statistical analysis.
For purposes of analysis, subjects with SMA-I were subdivided into types IB and IC, and classified as “recent” if enrolled within 3 months of diagnosis or “chronic” if beyond 3 months. The distributions of age at death and age at reaching the combined endpoint of either death or requiring at least 16 hours/day of noninvasive ventilation support for at least 2 weeks were described using Kaplan-Meier curves. The distributions of age at reaching the combined endpoint were compared between subjects with type IB and type IC, boys and girls, and those with 2 and 3 SMN2 copies using log-rank tests. The mean rate of change over time (slope) in CHOP INTEND score was estimated using a mixed-effects linear regression model with random slopes and intercepts. The mean slopes were compared between subjects who were recently diagnosed and those with chronic SMA-I, as well as between subjects with type IB and type IC, by adding the subgroup variable and its interaction with time to the mixed-effects model and testing for significance of the interaction term. The distributions of electrophysiologic variables (CMAP and MUNE) at baseline were compared between various subgroups (type IB vs type IC; recently diagnosed vs chronic SMA-I) using Wilcoxon rank sum tests. SMA-I subjects with 2 vs 3 copies of SMN2 were compared regarding baseline variables using Wilcoxon rank sum tests or χ2 tests, as appropriate. Fisher exact tests were used to compare SMA-I and SMA-II subgroups regarding the percentage of subjects with abnormal laboratory values.
RESULTS
All ages are listed in months. Descriptive results for several variables are expressed as median and interquartile range (IQR). Seventy-nine subjects (34 SMA-I and 45 SMA-II) were enrolled between May 2005 and April 2009. The baseline subject characteristics are summarized in table 1, and longitudinal data are summarized in table 2. Figure e-1 (available on the Neurology® Web site at Neurology.org) depicts the relationship between age at symptom onset and time between symptom onset and enrollment for subjects with SMA-I, and indicates the type of supportive care received at baseline. Nutritional support (nasogastric tube or gastrostomy tube; median 8.0, IQR 6–13) was initiated approximately 3 months earlier than ventilation support (noninvasive ventilation or intubation leading to tracheostomy; median 11.0, IQR 5–19). At baseline, all subjects with SMA-I older than 12 months required feeding or combined feeding and ventilation support, and 14 required 16+ hours/day of ventilation support (11 with bilevel positive airway pressure by mask, 3 with tracheostomy). For the entire SMA-I cohort, the age at symptom onset (median 3.0, IQR 2–4) was approximately 3 months before the age at diagnosis (median 6.0, IQR 4–7) and the interval from diagnosis to enrollment (median 9.0, range 0.3–252) reflects that many chronic patients were included.
Table 1.
Baseline demographic and clinical characteristics of study participants
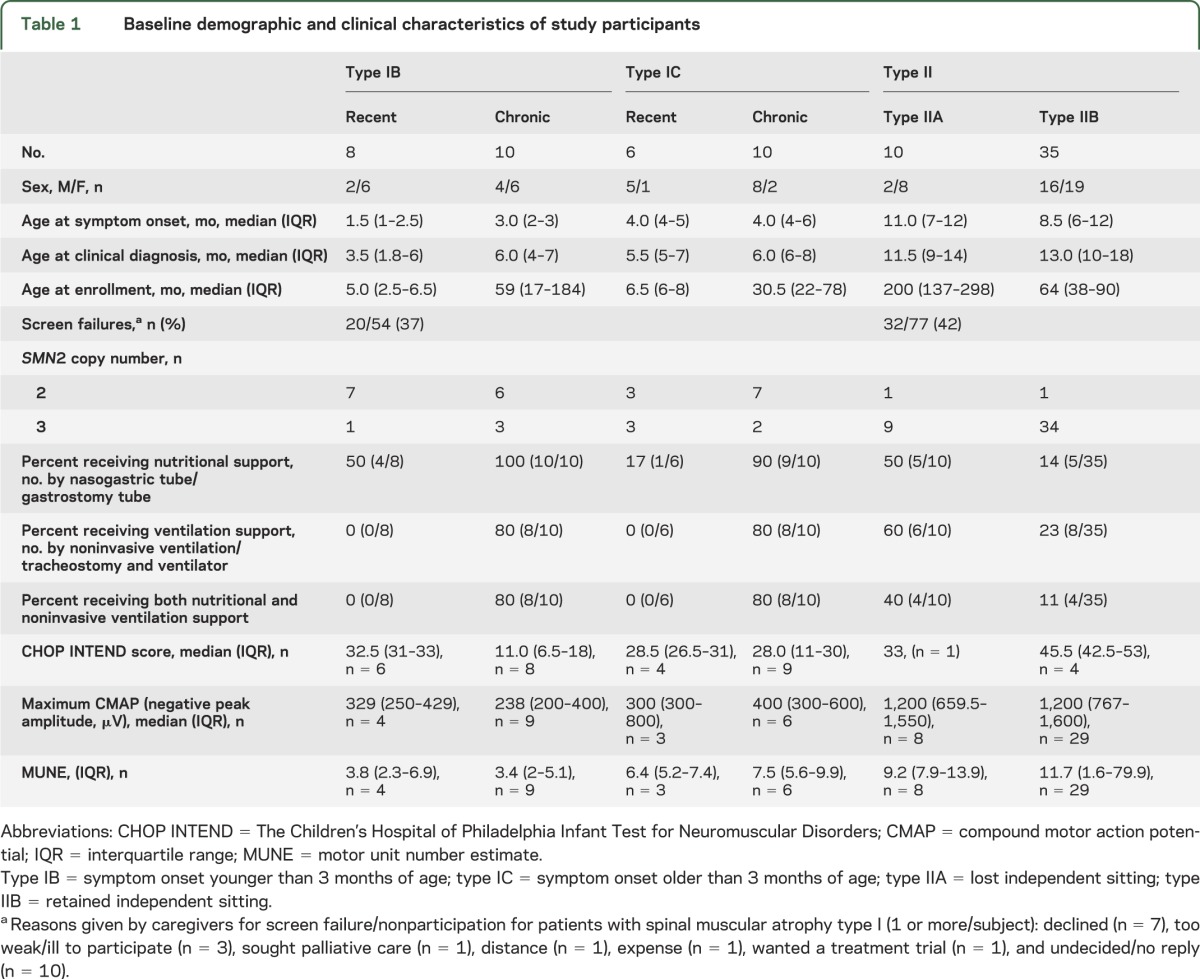
Table 2.
Follow-up data for subjects with SMA-I by subtype (IB and IC) and by recent/chronic status (see text)
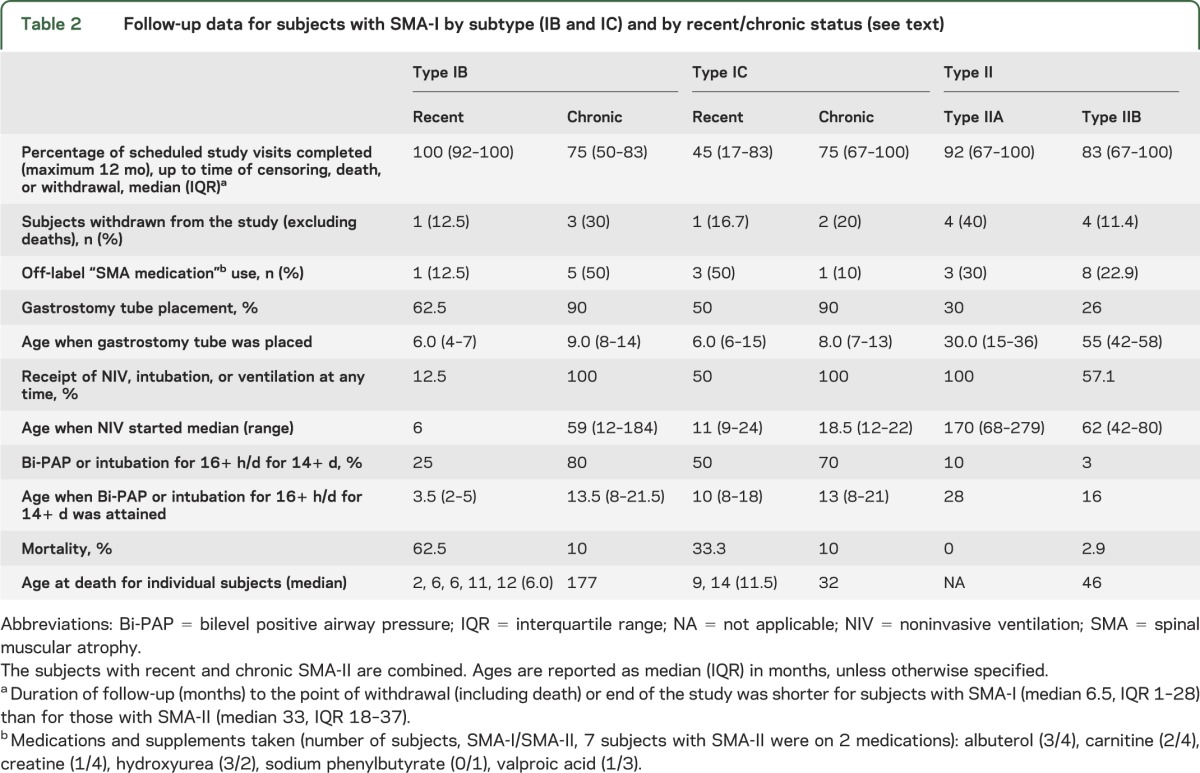
Mortality and combined endpoint data are presented in figure 1 and ventilation support data are detailed in table 1. Nine subjects with SMA-I died during follow-up: 6 with type IB and 3 with type IC. Causes of death were acute pulmonary infection (n = 6), airway obstruction (n = 2), and bradycardic arrest (n = 1). The median age at reaching the combined endpoint in subjects with SMA-I was 13.5 (IQR 8.1–22.0). This distribution did not differ significantly by sex (p = 0.19) or SMA-I subtype (figure 1B, p = 0.58): the median age at the combined endpoint for subjects with type IB was 11.9 (IQR 7.0–22.0) and for subjects with type IC was 13.6 (IQR 8.8–20.1). SMN2 copy number data were obtained in 31 of 34 subjects and are summarized in table 1. Nine subjects had an SMN2 copy number of 3; none died and 4 required 16+ hours of ventilation for at least 2 weeks during the course of this study. The distribution of the age at reaching the combined endpoint differed by SMN2 copy number (figure 1C, p = 0.002): the median age at the combined endpoint for subjects with 2 SMN2 copies was 10.5 (IQR 8.1–13.6), but the 25th percentile was 22.0 for those with 3 SMN2 copies (not enough events occurred to permit estimation of the median age).
Figure 1. Time-to-event curves for SMA-I.

Kaplan-Meier curves for SMA-I. (A) Probability of survival with advancing age by SMA-I subtype (type IB, n = 18; type IC, n = 16). (B) Probability of not reaching the combined endpoint of death or the need for a minimum of 16 hours/day of noninvasive ventilation support for a minimum of 14 continuous days, in the absence of an acute reversible illness or perioperatively, with advancing age by SMA-I subtype. (C) Probability of not reaching the combined endpoint with advancing age by SMN2 copy number (2 copies, n = 23; 3 copies, n = 9). SMA-I = spinal muscular atrophy type I.
Motor function as measured by the CHOP INTEND is summarized in table 1. CHOP INTEND scores did not differ significantly at baseline whether or not nutritional support was required, while the age was older and the scores were notably lower when both nutritional and ventilation support were required (table e-1). Seventeen subjects with SMA-I (4 recent, 13 chronic) were evaluated on at least 2 occasions using the CHOP INTEND (figure 2 by study visit and figure e-2 by age). The mean rate of change over time in the CHOP INTEND score overall was −1.27 points/year (95% confidence interval [CI] −2.33 to −0.21, p = 0.02). The mean rate of change was similar (p = 0.90) for the recently diagnosed subgroup (−1.24 points/year, 95% CI −2.39 to −0.09, p = 0.03) and the chronic subgroup (−1.07 points/year, 95% CI −3.53 to 1.39, p = 0.39). The mean decline was slightly worse among type IB subjects (−1.83 points/year, 95% CI −3.35 to −0.32, p = 0.02) than among type IC subjects (−0.83 points/year, 95% CI −2.18 to 0.52, p = 0.22), but the group difference in mean rate of change was not significant (p = 0.32).
Figure 2. CHOP INTEND motor function in SMA-I: Longitudinal data.

The CHOP INTEND longitudinal data are represented for each subject with 2 or more assessments. Those subjects enrolled within 3 months of diagnosis (“recent”) are shown with a blue line and those enrolled more than 3 months after diagnosis (“chronic”) are shown with a red line. CHOP INTEND = The Children's Hospital of Philadelphia Infant Test for Neuromuscular Disorders; SMA-I = spinal muscular atrophy type I.
Of the 54 eligible subjects with SMA-I screened, 34 (63%) were enrolled, with an average of 1.20 subjects with SMA-I screened/month and 0.74 subjects enrolled/month among the 3 study sites. Overall, 50% of subjects with SMA-I and 84% of subjects with SMA-II completed at least 12 months of follow-up. Of those who lived for at least 12 months after enrollment, 5 of 25 (20%) completed all 6 scheduled study visits and 15 of 25 (60%) completed at least 4 of the 6 visits for the initial 12 months. Missed visits were attributable to illness, surgery, and travel difficulties. Early withdrawal occurred in 16 of the 34 subjects (47%): 9 because of death (all within the first 12 months) and 7 for other reasons (1 illness, 2 noncompliant, 2 enrolled in a treatment study, 1 time constraints for the caregivers, 1 withdrew consent).
Study procedures were well tolerated. Of the 34 adverse events that were reported, 65% were related to the pulmonary system (respiratory syncytial virus in 3), 21% to gastrointestinal issues, and 6% were perioperative in nature. All were determined to be related to progression of disease or to scheduled hospital admissions for surgery or testing; none were attributed to the study assessments or travel to the study site. No primary cardiac events were identified. The duration of study visits was approximately 4 hours, including breaks.
Most laboratory values were within normal ranges or were slightly outside the range of normal (alanine aminotransferase, hemoglobin, creatinine, glucose; table e-2).
CMAP and MUNE responses were obtained at the baseline visit in 20 of 34 subjects with SMA-I (59%) and were omitted otherwise upon parental request. The ulnar CMAP amplitude (median = 300 μV, range 41–1,100 μV) and MUNE (median 5, range 1–18) responses were substantially reduced relative to reference data for the lower limit of normal in infants from neonate to 2 years of age (CMAP: 1,800–5,000 μV; MUNE: 100–250). Normative CMAP values rise with development before plateauing at adult levels by the end of the first decade. Cross-sectional MUNE norms have been reported in infants and some age groups, although the trajectory of MUNE values during normal development has not been as well charted.17 There were no significant differences in CMAP and MUNE between the recently diagnosed and chronic SMA-I subgroups (CMAP: p = 0.80; MUNE: p = 0.97), nor was there a significant difference in CMAP between the type IB and type IC subgroups (p = 0.35), but the MUNE values tended to be higher in the type IC subgroup than in the type IB subgroup (p = 0.04, table 1).
DISCUSSION
We have demonstrated that a prospective, longitudinal natural history study of SMA-I can be performed effectively among 3 clinical sites. We found that subjects with type IB and type IC fare similarly regarding time to the combined endpoint. The need for gastrostomy tube placement and noninvasive ventilation was not significantly different in these SMA-I subgroups. Thus, it would be sensible to include both subgroups in a clinical trial that used time to reach the combined endpoint as the primary outcome variable, and there would appear to be no advantage of stratifying by subgroup in the randomization or statistical analysis. In a different study, time to the combined endpoint in patients with SMA-I did not differ between those receiving supportive nutritional care only vs proactive respiratory support.18
For purposes of clinical trial planning, our data suggest that the probability of reaching the combined endpoint at age 12 months (i.e., after 6–9 months of trial participation) is approximately 50%. A sample size of 63 subjects per group (126 total) would be required in a 2-group trial to provide 80% power to detect a relatively large group difference of 50% vs 75% regarding event-free survival (not reaching the combined endpoint), corresponding to a hazard ratio of 0.42, using a log-rank test and a 5% significance level (2-tailed), assuming a 10% dropout rate. An additional 6 months of follow-up would increase the probability of reaching the combined endpoint to approximately 65%, possibly allowing for detection of a smaller treatment effect, but one would have to consider the increased risk of dropout with longer-term follow-up in sample-size planning.
Motor status, as measured by the CHOP INTEND total score, declined over time in subjects with SMA-I but the mean rate of decline was not significantly different between the type IB and type IC subjects or between the recently diagnosed and chronic subgroups.
Despite advanced denervating disease, CMAP and MUNE could be obtained at baseline in the majority of subjects in this cohort. CMAP amplitudes, however, were similar across groups in patients with SMA-I and therefore less informative, likely because they were very low at the time of baseline evaluation. The baseline MUNE, however, while still quite low, was relatively higher in the type IC than in the type IB subgroup. CMAP amplitude and MUNE from the distal ulnar nerve may serve as useful markers in early-phase trials if an intervention causes some rescue of motor neurons, hastens deterioration, or promotes collateral reinnervation. However, the absence of change in these measures does not necessarily indicate a lack of benefit because they do not reflect motor neuron pools that control bulbar or respiratory function and may be limited because of very advanced denervation.
SMN2 copy number was associated with severity of disease and with the time to reach the combined endpoint in our cohort. Limiting a clinical trial to subjects with a copy number of 2 will yield a higher number of participants who reach the combined endpoint; however, this could make recruitment more difficult. It would seem useful, however, to stratify by SMN2 copy number in the randomization of participants in future trials.
The elevated alanine aminotransferase findings may reflect muscle atrophy (creatine kinase levels were not evaluated here, but have been reported to be elevated in SMA19). Reduced serum bicarbonate may reflect an underlying metabolic acidosis, as has been described previously.20 Reduced serum creatinine values reflect reduced muscle mass; these tended to occur more frequently in subjects with SMA-II than in subjects with SMA-I (p = 0.08). The use of serum cystatin C may be better than serum creatinine for evaluation of renal function. Random blood glucose levels were abnormally reduced or elevated in 8 of 26 subjects with SMA-I (30.8%) and in 23 of 43 subjects with SMA-II (53.5%). Both hypo- and hyperglycemia warrant close monitoring, especially in time of illness or perioperatively, as has been demonstrated.21
Limitations to the interpretation of these data include the relatively small sample size, incomplete retention, and having a mixture of recently diagnosed and more chronic subjects. The latter group (20 of 34 subjects, 59%) is more likely to have milder disease and to have longer-term survival. While all subjects with SMA were included, focusing on the recently diagnosed group may give a clearer picture of the natural history of SMA-I using contemporary support options.
Patients with SMA-I can be effectively enrolled in a longitudinal study. Subjects should be enrolled in trials shortly after diagnosis, before age 8 months, by which time about half of the subjects already need feeding support. Enrollment within 1 month of diagnosis is feasible but will be a challenge. The subjects tolerated extensive testing with appropriate rest periods. Retention was variable for the subjects with SMA-I, who missed many study visits because of illness. The frequency of study visits was challenging for many subjects with SMA-I and their parents. To maximize retention, study visits should be minimized; the use of remote assessments in lieu of some study visits should be considered. Multiple pulmonary-related adverse events and minor laboratory abnormalities should be anticipated in an intervention study. SMN2 copy number serves as a predictive biomarker potentially useful for stratification, in contrast to SMA-I subtype (IB and IC). The composite endpoint of death or the need for at least 16 hours/day of noninvasive ventilation support for at least 2 weeks accurately captures the milestone of sustained respiratory failure, a surrogate for death. The CHOP INTEND may serve as a useful secondary outcome measure.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to the patients and their parents who participated in this study, without whom this effort would not have succeeded. The generous support from the Spinal Muscular Atrophy Foundation to the PNCR made this possible and the suggestions from Loren Eng, Karen Chen, Cynthia Joyce, and Dione Kobayashi have been particularly helpful. The authors thank Rabi Tawil, Erica Sanborn, Jason Caracciolo, Hailly Butler, Christine Annis, Elizabeth Luebbe, Carrie Irvine, and Benjamin Koo for their contributions to this research.
GLOSSARY
- CHOP INTEND
The Children's Hospital of Philadelphia Infant Test for Neuromuscular Disorders
- CI
confidence interval
- CMAP
compound motor action potential
- IQR
interquartile range
- MUNE
motor unit number estimate
- SMA
spinal muscular atrophy
- SMN1
survival of motor neuron 1, telomeric
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Finkel's contributions include drafting/revising the manuscript for content, study concept and design, analysis and interpretation of data, acquisition of data, study supervision or coordination, and obtaining funding. Dr. McDermott's contributions include drafting/revising the manuscript for content and analysis or interpretation of data. Dr. Kaufmann's contributions include drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, study supervision or coordination, and obtaining funding. Dr. Darras' contributions include drafting/revising the manuscript for content, study concept or design, acquisition of data, study supervision or coordination, and obtaining funding. Dr. Chung's contributions include revising the manuscript for content and data acquisition. Dr. Sproule's contributions include drafting/revising the manuscript for content, study concept or design, analysis, interpretation of data, acquisition of data, and study supervision or coordination. Dr. Kang's contributions include data acquisition and revision of the manuscript for content. Dr. Foley's contributions include review/revision of the manuscript and data acquisition. Dr. Yang's contributions include review/revision of the manuscript and data acquisition. William Martens' contributions include revising the manuscript for content, analysis or interpretation of data, and data acquisition. Dr. Oskoui's contributions include revising the manuscript for content and data acquisition. Allan Glanzman's contributions include review/revision of the manuscript and data acquisition. Jean Flickinger's contributions include drafting/revising the manuscript for content and data acquisition. Jacqueline Montes' contributions include drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, and study supervision or coordination. Sally Dunaway's contributions include revising the manuscript for content and data acquisition. Jessica O’Hagen's contributions include revising the manuscript for content, data acquisition, and study supervision or coordination. Janet Quigley's contributions include review/revision of the manuscript and data acquisition. Susan Riley's contributions include review/revision of the manuscript and data acquisition. Maryjane Benton's contributions include review/revision of the manuscript, analysis or interpretation of data, data acquisition, and study supervision or coordination. Patricia Ryan's contributions include revising the manuscript for content and data acquisition. Megan Montgomery's contributions include review/revision of the manuscript for content, data acquisition, and study supervision or coordination. Jonathan Marra's contributions include review/revision of the manuscript and data acquisition. Dr. Gooch's contributions include drafting/revising the manuscript for content, study concept or design, acquisition of data, and study supervision or coordination. Dr. De Vivo's contributions include drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, study supervision or coordination, and obtaining funding.
STUDY FUNDING
This study was sponsored by the SMA Foundation (New York) through funding to the Pediatric Neuromuscular Clinical Research Network. Additional clinical research support was provided through CTSA 1 NIH UL1 RR024134 (The Children's Hospital of Philadelphia), CTSA NIH 1 UL1 RR024156 and the NSADA K12 program (Columbia University), and NIH 1 UL1 RR025755 from the National Center for Research Resources, NIH, to the Harvard Catalyst Clinical & Translational Science Center (Harvard Catalyst).
DISCLOSURE
R. Finkel served as a Data and Safety Monitoring Board (DSMB) member for a trial sponsored by Sarepta Therapeutics and now serves on a DSMB for an scAAV9 gene therapy clinical trial at Nationwide Children's Hospital, serves on advisory boards for DuchenneConnect, Families of SMA, SMA-REACH, PTC Therapeutics, Inc., Isis Pharmaceuticals, Inc; has received honoraria from Novartis and Roche, has received travel expenses for lectures not funded by industry; serves on the editorial board of Neuromuscular Disorders and Journal of Neuromuscular Disorders; receives research support from Isis Pharmaceuticals, Inc., PTC Therapeutics, Inc. (PTC124-016 study, site PI), Santhera Pharmaceuticals (DELOS Study in Duchenne MD, site PI), the NIH (5R21-NS058926 [PI], U54 AR0526446-03 [Co-I], 1U54 NS065712-01 [Co-I]), RO1-AR056973 [Co-I]), the SMA Foundation (PNCR network for SMA, site PI), the Muscular Dystrophy Association, Genzyme Corporation, and the Charcot-Marie-Tooth Association; and his spouse serves on the editorial board of Arthritis Research and Therapy, holds and has received license fees for numerous patents related to T-cell activation and HIV, and receives research support from the Gates Foundation, Merck Serono, and the NIH in the field of T-cell activation, HIV, and genomics of juvenile arthritis. M. McDermott serves as a DSMB member for trials sponsored by Isis Pharmaceuticals, Biogen Idec, Inc., The ALS Association/FDA, and the Muscular Dystrophy Association. He serves on the editorial board for Movement Disorders and has been a consultant for the New York State Department of Health, Teva Pharmaceutical Industries, Ltd., Synosia, Inc., Smith & Nephew, Inc., Impax Pharmaceuticals, Bioness, Inc., and Asubio Pharmaceuticals, Inc. He receives research support from the Michael J. Fox Foundation, Spinal Muscular Atrophy Foundation, Muscular Dystrophy Association, American Dental Association, the FDA, and NIH. P. Kaufmann reports no disclosures relevant to the manuscript. B. Darras has received publishing royalties from UpToDate and consulting fees from Isis Pharmaceuticals, Quest Diagnostics, Guidepoint Global Consultation, Easton Associates, Clearview Healthcare Partners, and Gerson Lehrman Group. He has received honoraria from the American Academy of Neurology and the Muscular Dystrophy Association. He has received research support from PTC Therapeutics, Isis Pharmaceuticals, NIH/NIAMS 2P01NS-40828-6A11, NIH/National Institute of Neurological Disorders and Stroke (NINDS) 1U10NS077269; NIH/NIAMS 1R01AR060850, SMA Foundation, Muscular Dystrophy Association, and Slaney Family Fund for SMA. He serves on the editorial boards of Neurology and Pediatric Neurology. W. Chung reports no disclosures relevant to the manuscript. D. Sproule receives research funding from PTC Therapeutics, the SMA Foundation, and NINDS-sponsored Neurological Sciences Academic Development Award (K12 NS01698). P. Kang received travel funding from Isis Pharmaceuticals and various nonprofit entities during the study period. He previously served as an occasional consultant for LEK consulting, Gerson Lehrman Group, and Leerink Swann, all commercial consulting firms. He has received honoraria for grant reviews from nonprofit entities and federal government agencies. He previously performed medico-legal consulting for Gross, Minsky & Mogul and currently does so for a federal government agency. He performs EMGs (15% effort). Dr. Kang receives honoraria for serving as an officer of the Massachusetts Medical Society, a nonprofit entity, and has received occasional honoraria from other nonprofit entities. He has received compensation from Oakstone Publishing and Springer, both commercial publishing firms. Dr. Kang receives research support from the NIH and the Muscular Dystrophy Association, and previously received research support from Harvard University and Boston Children's Hospital. His spouse receives research support from the NIH and Boston Children's Hospital, and receives royalties from a gene therapy patent. A. Foley receives research support from the Muscular Dystrophy Campaign (UK). M. Yang and W. Martens report no disclosures relevant to the manuscript. M. Oskoui received research support from the Public Health Agency of Canada, project RT736230, for 2 years and the Spinal Muscular Atrophy Foundation (2004–2007, fellowship). Dr. Oskoui receives research support from SickKids Foundation–CIHR over 3 years. Dr. Oskoui is a Clinician Research Scholar of the Fonds de recherche du Québec–Santé (FRQS). A. Glanzman received funding for travel and training from GlaxoSmithKline, PTC Therapeutics, and Eli Lilly, and receives research support from the SMA Foundation. J. Flickinger reports no disclosures relevant to the manuscript. J. Montes is a consultant for Isis Pharmaceuticals Inc. and is funded in part by Department of Defense grant USAMRAA 09131005. S. Dunaway is funded in part by Department of Defense grant USAMRAA 09131005. J. O'Hagen and J. Quigley report no disclosures relevant to the manuscript. S. Riley received honoraria from the SMA Foundation for speaking at an SMA/Scoliosis conference at the Children's Hospital of Philadelphia. M. Benton, P. Ryan, M. Montgomery, and J. Marra report no disclosures relevant to the manuscript. C. Gooch is a consultant for NeuralStem, a medical advisory board member of the GBS/CIDP Foundation International, and an employee of the FDA. He is an editorial board member of the Journal of Clinical Neuromuscular Disease and Neurology (journal of the American Academy of Neurology). He receives travel funding from the NIH as chair of the DSMB (IVIg in Autonomic Neuropathy). D. De Vivo serves on the scientific advisory boards for Colleen Giblin Foundation, SMA Foundation, Canavan Foundation, Pediatric Neurotransmitter Disease Association, Milestones for Children, Will Foundation, Glut1 Deficiency Foundation, and Isis Pharmaceuticals. He has received compensation as a consultant for Isis Pharmaceuticals and Ultragenyx Pharmaceutical Inc. Dr. De Vivo receives research support from the NIH/NICHD and NINDS, Department of Defense, SMA Foundation, Colleen Giblin Foundation, Milestones for Children, Glut1 Deficiency Foundation, and the Will Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet 2012;20:27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–165 [DOI] [PubMed] [Google Scholar]
- 3.Lorson C, Hahnen E, Androphy E, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 1999;96:6307–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feldkötter M, Schwarzer V, Wirth R, Wienker T, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002;70:358–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gabanella F, Carissimi C, Usiello A, Pellizzoni L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum Mol Genet 2005;14:3629–3642 [DOI] [PubMed] [Google Scholar]
- 6.Zerres K, Rudnik-Schöneborn S. Natural history in proximal spinal muscular atrophy: clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol 1995;52:518–523 [DOI] [PubMed] [Google Scholar]
- 7.Munsat T, Davies K. Spinal muscular atrophy. 32nd ENMC International Workshop. Naarden, The Netherlands, 10–12 March, 1995. Neuromuscul Disord 1996;6:125–127 [DOI] [PubMed] [Google Scholar]
- 8.Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol 1999;3:49–51 [DOI] [PubMed] [Google Scholar]
- 9.Thomas NH, Dubowitz V. The natural history of type I (severe) spinal muscular atrophy. Neuromuscul Disord 1994;4:497–502 [DOI] [PubMed] [Google Scholar]
- 10.Bertini E, Burghes A, Bushby K, et al. 134th ENMC International Workshop: outcome measures and treatment of spinal muscular atrophy, 11–13 February, 2005, Naarden, The Netherlands. Neuromuscul Disord 2005;15:802–816 [DOI] [PubMed] [Google Scholar]
- 11.Oskoui M, Levy G, Garland C, et al. The changing natural history of spinal muscular atrophy type 1. Neurology 2007;69:1931–1936 [DOI] [PubMed] [Google Scholar]
- 12.Kaufmann P, McDermott MP, Darras BT, et al. Observational study of spinal muscular atrophy type 2 and 3: functional outcomes over 1 year. Arch Neurol 2011;68:779–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaufmann P, McDermott MP, Darras BT, et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 2012;79:1889–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, Finkel R, Bertini E, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol 2007;22:1027–1049 [DOI] [PubMed] [Google Scholar]
- 15.Glanzman A, Mazzone E, Main M, et al. The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord 2010;20:155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glanzman AM, O'Hagen JM, McDermott MP, et al. Validation of the Expanded Hammersmith Functional Motor Scale in spinal muscular atrophy type II and III. J Child Neurol 2011;26:1499–1507 [DOI] [PubMed] [Google Scholar]
- 17.Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol 2005;57:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemoine TJ, Swoboda KJ, Bratton SL, Holubkov R, Mundorff M, Srivastava R. Spinal muscular atrophy type 1: are proactive respiratory interventions associated with longer survival? Pediatr Crit Care Med 2012;13:e161–e165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudnik-Schoneborn S, Lutzenrath S, Borkowska J, Karwanska A, Hausmanowa-Petrusewicz I, Zerres K. Analysis of creatine kinase activity in 504 patients with proximal spinal muscular atrophy types I–III from the point of view of progression and severity. Eur Neurol 1998;39:154–162 [DOI] [PubMed] [Google Scholar]
- 20.Crawford TO, Sladky JT, Hurko O, Besner-Johnston A, Kelley RI. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann Neurol 1999;45:337–343 [DOI] [PubMed] [Google Scholar]
- 21.Bowerman M, Swoboda KJ, Michalski JP, et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol 2012;72:256–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.