

Abstract
Hepatitis B virus (HBV) has killed countless lives in human history. The invention of HBV vaccines in the 20th century has reduced significantly the rate of the viral infection. However, currently there is no effective treatment for chronic HBV carriers. Newly emerging vaccine escape mutants and drug resistant strains have complicated the viral eradication program. The entire world is now facing a new threat of HBV and human immunodeficiency virus co-infection. Could phage display provide solutions to these life-threatening problems? This article reviews critically and comprehensively the innovative and potential applications of phage display in the development of vaccines, therapeutic agents, diagnostic reagents, as well as gene and drug delivery systems to combat HBV. The application of phage display in epitope mapping of HBV antigens is also discussed in detail. Although this review mainly focuses on HBV, the innovative applications of phage display could also be extended to other infectious diseases.
Keywords: Phage display, Hepatitis B virus, Epitope mapping, Drug delivery, Gene delivery, Antiviral drug, Therapeutics, Diagnosis, Hepatocellular carcinoma, Virus-like particle, Vaccine
Core tip: Hepatitis B virus (HBV) poses a major health problem worldwide and currently there is no effective treatment for HBV infection. Treatments of patients with nucleoside analogues have resulted in the selection of vaccine escape and drug resistant mutants. Most frightening, HBV is now linking arms with human immunodeficiency virus to threaten the world. Phage display has been employed extensively to solve these life-threatening problems. This article reviews critically the innovative applications of phage display in epitope mapping and the development of vaccines, therapeutic agents, diagnostic reagents, as well as gene and drug delivery systems.
INTRODUCTION
Hepatitis B virus (HBV), which causes liver cirrhosis and hepatocellular carcinoma (HCC), is one of the greatest killers in human history. It is 200 times more infectious than human immunodeficiency virus (HIV) and so far has killed more people than HIV. HBV poses a serious global health problem, about one third of the world’s population have been infected by this virus, of which 370 million are chronic carriers and about one million people die each year[1]. The virus is commonly present in human population, at least 7% of the people in South East Asia, China and Africa are chronically infected by HBV. The virus is now classified in the family of Hepadnaviridae and it has a narrow host range, infecting only human and other higher primates such as chimpanzees and orangutans. The modes of HBV transmission include blood transfusion, unprotected sexual contact, contaminated needles and syringes, and via perinatal transmission from an infected mother to her baby during childbirth.
Hepatitis B surface antigen (HBsAg) was accidentally discovered in 1960’s by an American doctor, Baruch Blumberg, in the blood of an Australian aborigine, when he was studying the inherited variations in human beings[2]. In 1970, for the first time, Dane et al[3] reported the observation of the virus particles under a transmission electron microscope. The infectious particle or known as the Dane particle, is roughly spherical with a diameter about 42 nm (Figure 1). This particle can be found in the blood of a chronically infected patient and its three-dimensional structure has been determined by cryo-electron microscopy[4]. The virion is enveloped by a lipid bilayer derived from the host cell membranes[5]. Associated with the lipid are three distinct but related forms of surface protein (HBsAg): S- (small), M- (middle) and L- (large) HBsAg.
Figure 1.
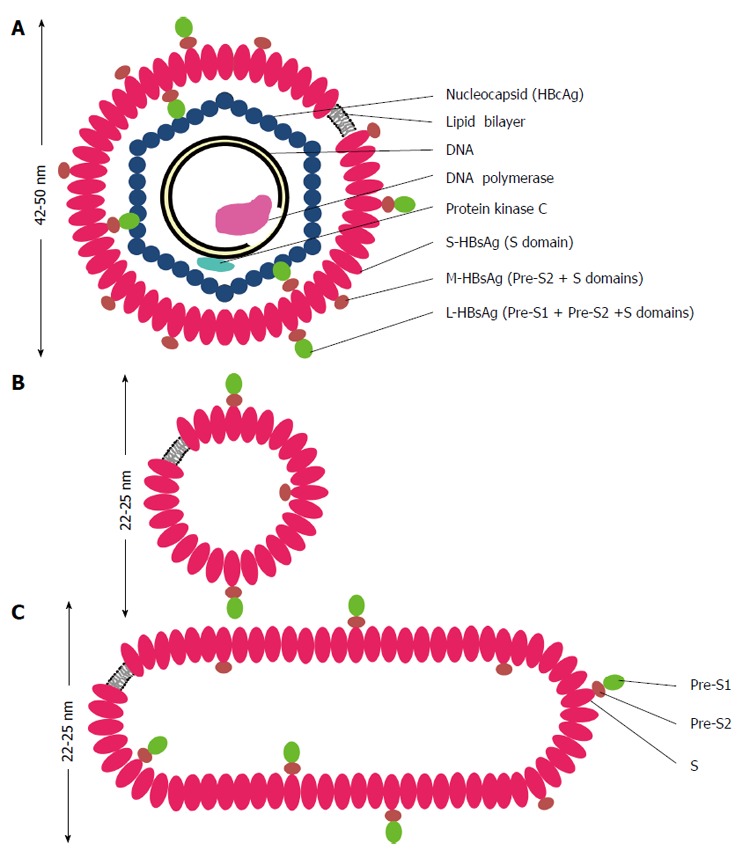
Schematic representations of hepatitis B virus virion (A), spherical (B) and filamentous (C) particles. The envelope of hepatitis B virus (HBV) virion or the Dane particle (A) contains three forms of hepatitis B surface antigen (HBsAg): large (L-) HBsAg has the PreS1, PreS2 and S domains; middle (M-) HBsAg contains the PreS2 and S domains; and small (S-) HBsAg has only the S domain. The representations of the L-, M-, and S-HBsAg have no quantitative or positional significance. The L-HBsAg interacts with the viral capsid which is made of many copies of the core protein (HBcAg). The capsid encapsidates a partially double stranded DNA molecule, a DNA polymerase containing the primase and reverse transcriptase activities. The protein kinase C phosphorylates the capsid protein. The diameter of HBV virion is about 42 nm when it is stained negatively and observed under a transmission electron microscope, but it appears bigger in cryo-electron microscopy. The spherical (B) and filamentous (C) particles have a diameter about 22 nm in negative staining and appear bigger in cryo-electron microscopy. The length of the filamentous particle varies. Both the non-infectious particles contain the L-, M-, S-HBsAg and lipid.
Inside the envelope is the viral nucleocapsid which is made of many copies of core protein or commonly known as core antigen (HBcAg). Within the capsid is a partially double-stranded DNA genome. The polymerase protein (P) which has reverse transcriptase and DNA-dependent DNA polymerase activities is covalently linked to a partially double-stranded circular DNA genome of about 3.2 kb.
Apart from the virion, another two distinct forms of non-infectious particles are also observed in the serum of a chronic carrier. They appear as spheres or filaments with a diameter of about 22 nm when observed under an electron microscope (Figure 1). The filamentous particles have different length. The amount of these non-infectious particles is about 1000- to 100000-fold in excess compared to the virion[6] and it is thought to serve as decoys to fool human’s immune system.
On the other hand, not all viruses are harmful to human beings. Viruses that infect and replicate in bacteria or known as bacteriophages (phages) are believed to control bacterial populations on this planet. They are distributed in oceans, rivers, soils, animals, insects and places populated by bacteria. These useful viruses were discovered separately by Frederick Twort and Félix d’Hérelle in 1910’s[7], about 50 years earlier before HBV was discovered. They occur in a wide variety of different sizes and shapes with DNA or RNA as their genomes. One may not be aware of phages’ impacts on the global economy and the advancement of science and technology. In fact, they were used widely as anti-bacterial agents in the early and mid-20th century before antibiotics were discovered. Phages also contributed significantly in understanding the fundamentals of DNA replication, transcription, translation, recombination and regulation. Some modification enzymes used in molecular biology, including ligase and RNA polymerase are isolated from these viruses. Phages, such as M13 and lambda, are employed as DNA cloning vectors for protein productions and determination of nucleotide sequences in organisms, including the human being[8].
On June 14, 1985, the Science, published an article by George P. Smith, describing an innovative method to display peptides on a filamentous phage by fusing their coding sequences to the phage’s genome[9]. This article marks the beginning of phage display, so simple that it links a genotype and a phenotype physically in a single phage particle, and so powerful that it allows the amplification of these two elements in billion folds in bacteria. Immediately, phage display ignited a spark of interest among scientists around the world, who incorporated, refined and applied it to almost every field of biological sciences, which include drug discovery, antibody engineering, epitope mapping, vaccine development, gene and drug delivery, development of diagnostic reagents, organ targeting, enzyme technology, protein-protein interactions and protein-DNA interactions. At the time of writing this manuscript, there are more than 20000 scientific articles about phage display on Google Scholar, demonstrating the impact of this method on current global economy, science and technology. Phage display has now developed into one of the most important tools in biological research, and of course it has also been employed extensively to study HBV particularly its innovative applications to control this virus. However, to the best of our knowledge, so far there is no review paper summarizing the applications of phage display on HBV research. Therefore, the main aim of this paper is to provide a comprehensive and critical review on the innovative applications of phage display to combat HBV. In order to achieve this aim, this article is divided into five sections based upon the main strategies that have been used to control and manage HBV.
VACCINE DEVELOPMENT
Edward Jenner’s brilliant idea about small pox vaccination in 18th century, also worked effectively for HBV vaccination in 20th and 21st centuries to prevent the spread of HBV, and of course this has saved countless lives worldwide. The first HBV vaccine was prepared by Baruch Blumberg and Irving Millman by using the non-infectious HBV particles (Figure 1) purified from infected sera[10]. This invention was patented by Fox Chase Cancer Center, licensed to Merck and Company, Inc. and approved by the U.S. Food and Drug Administration (FDA)[10]. In 1981, Merck marketed the first hepatitis B vaccine, Heptavax, but was discontinued in 1990 and replaced by a recombinant DNA vaccine[11].
The invention of recombinant DNA technology in 1970s allows the genetic materials from different organisms to be joined together, artificially introduced into an organism and expressed in the host. This innovative technology was quickly employed by Kenneth Murray, from the University of Edinburgh and also a co-founder of Biogen Inc., to insert the HBV DNA in a bacterial plasmid and to produce the recombinant proteins in Escherichia coli (E. coli)[12,13]. The experiment was successful and the first patent applications were filed in December 1978[14]. In early 1980s, the HBsAg was successfully produced in yeast by using recombinant DNA technology[15-17] and was demonstrated to protect chimpanzees from HBV infection[18,19]. This second generation vaccine is safer compared to the non-infectious particles purified from the sera of infected patients as the latter may be contaminated with other human pathogens. Production of this recombinant vaccine, when compared with the plasma-derived vaccine, is less laborious and less time-consuming. From the commercial aspect, Biogen Inc. licensed the patent to SmithKline Beecham, and almost at the same time Chiron Corporation, founded by William Rutter[20], worked with Merck to commercialize the vaccine. The recombinant vaccine manufactured by Merck (Recombivax HB®) and SmithKline Beecham (Engerix-B®) was eventually approved by the United States FDA in late 1980s[21]. The mass immunization of infants worldwide as recommended by the WHO has dramatically reduced the rate of HBsAg carrier in many countries[22]. For instance, in China the prevalence of HBsAg carriers dropped from 14.6% to 1.4%[23]. In addition, the incidence of HCC also declined by 4-folds in children after the implementation of mass immunization in Taiwan[24], indicating that HBV vaccine could prevent liver cancer.
Currently, there are effective vaccines in the market to prevent HBV infection. Is there a need to invent and formulate new vaccines? Yes, or at least until the virus is totally eradicated from this planet. Immune-escape mutants with amino acid substitutions, deletions and insertions across the immunodominant region or the “a” determinant of HBsAg have been reported widely[25-28]. Besides, a prolonged treatment of chronic hepatitis B patients with nucleotide or nucleoside analogs have resulted in the selection of vaccine escape mutants harboring nucleotide substitutions in their polymerase (pol) and S genes[29]. Most frightening, millions of people worldwide, particularly sub-Saharan Africa and East Asia, are co-infected by HBV and human immunodeficiency virus (HIV)[30]. These people have a higher rate of liver-related mortality compared to those only infected by HIV-1 or HBV alone[31]. In addition, only 20%-70% of HIV-infected patients developed an anti-HBsAg response after a standard HBV vaccination compared to 90%-95% in healthy adult individuals[32]. All the above scientific and medical evidences as well as the viral genetic diversity, justify strongly a continuing need for the development of new HBV vaccines. We believe that phage display can provide alternative solutions to these life-threatening problems. In general, up till now, phage display has been employed extensively to address these problems and can be grouped into three categories based on its applications: (1) display of immunogens on phage particles; (2) identification of epitope or mimotope mimics from sera; (3) phage-delivered DNA vaccine. These applications are summarized in Table 1 and discussed separately in the following sections.
Table 1.
Phage display in vaccine discovery
| Strategy | Phage (carrier) | Vector (target) | Epitope/mimotope/gene | Vaccination | Results | Ref. |
| Display of immunogens on phage particles | T7 (10B) | T7 Select 415-B | “a” determinant of HBsAg (residues 111-156) | Rabbit (subcutaneous) Freund’s complete adjuvant | Anti-HBsAg antibody induced | Tan et al[78] |
| M13 (pVIII) | pC89 with helper phage VASM13 | HBsAg28-39 | Mice (intradermal and intraperitoneal) no adjuvant added | MHC class I restricted HBsAg specific CTL response | Wan et al[40] | |
| M13 (pIII) | pCANTAB5E with helper phage M13KO7 | PreS1 | NA | NA | Kok et al[41] | |
| M13 (pIII) | pCANTAB5E with helper phage M13KO7 | HBcAg | Mice (intraperitoneal) incomplete Freund’s adjuvant | Anti-HBcAg antibody induced | Bahadir et al[42] | |
| Identification of epitope or mimotope mimics | M13 nonapeptide library (pVIII) | (Selected from HBV-infected sera) | Φ13 CRTCAHPGEHA Φ17 CIPFYLSAPQC Φ30 CGPFFLSPTSC Φ41 CGPFFLAASVC | Mice (intraperitoneal) no adjuvant added | Anti-HBsAg antibody induced | Folgori et al[54] |
| M13 (pVIII and pIII) | (Selected from HBV-infected sera) | Φ13, Φ35 (CVTCDTPPTY) ΦIII/17, ΦIII41 and ΦpIII/13 | Mice (intranasal/oral) cholera toxin as adjuvant | Anti-HBsAg antibody induced | Delmastro et al[56] | |
| Phage-delivered DNA vaccine | λ | λ-gt11 | HBsAg gene | Mice (subcutaneous) | Anti-HBsAg antibody induced | Clark et al[59] |
| λ | λ-gt11 | HBsAg gene | Rabbits (Intramuscular) | Anti-HBsAg antibody induced | Clark et al[61] |
NA: Not applicable; HBV: Hepatitis B virus; HBsAg: Hepatitis B surface antigen; MHC: Major histocompatibility complex; HBcAg: Hepatitis B core antigen; CTL: Cytotoxic T lymphocyte.
Display of immunogens on phage particles
Several polypeptide carriers have been developed to display or serve as a fusion partner for HBV immunogenic regions. These include HBV capsid (reviewed by[33,34]), ice nucleation protein (INP) located on the outer membrane of Pseudomonas syringae[35], heat shock protein 70 from Mycobacterium tuberculosis (TBhsp70)[36] and heat shock protein 65[37]. Apart from these potential carriers, phage particles provide an alternative means to display HBV epitopes and mimotopes.
Tan et al[38] demonstrated that the T7 phage is an efficient carrier for the highly conformational immunodominant region or denoted as “a” determinant of HBsAg (residues 111-156). This region was fused to the C-terminal end of the 10B capsid protein of T7 phage and about 1016 copies of reasonably pure immunodominant region can be produced and purified from 1 L culture within 6 h. The recombinant phage, namely T7-HBsAg111-156, was demonstrated to be highly immunogenic in rabbits and the immune response was as good as that of human-derived HBsAg, illustrating the potential of the whole recombinant phage particle as a vaccine candidate. The biological and physical features of T7 phage make it an excellent choice to display polypeptides: (1) the robust structure of the phage particle with an icosahedral head allows it to survive in extreme conditions; (2) the icosahedral head is composed of 415 copies of 10B proteins, enabling a high copy number of polypeptides, up to 50 residues, to be displayed on the surface of the phage particle[39]; and (3) T7 phage propagated faster than filamentous bacteriophages and its progenies assembled in the cytoplasm of E. coli cells and are released by breaking the host cells, thus the displayed peptides do not have to possess the capability to secrete through the periplasm and the cell membrane, as required by filamentous phages.
Several HBV immunogenic epitopes have also been displayed on filamentous phages as reported by Wan et al[40], Kok et al[41] and Bahadir et al[42]. In the first article, Wan et al[40], fused a 12-mer epitope, HBsAg28-39, to the pVIII coat protein of M13 phage. The vector, phagemid pC89, was used in the cloning of the coding sequence and the hybrid phage particles were produced by co-infecting with a helper phage, VASM13. The recombinant phage particles were inoculated into BALB/c (H-2d) mice in the absence of an adjuvant. Interestingly an MHC class I restricted HBsAg specific cytotoxic T lymphocytes (CTL) response was observed after 8 days of inoculation, demonstrating the potential of the recombinant M13 phage particles as potent immunogens without an adjuvant. The pVIII protein allows a very high copy number of display because an M13 phage particle is made up of about 2700 copies of this major coat protein. On the other hand, the pIII protein can only display up to 5 copies of foreign peptides at the tip of a filamentous phage. Kok et al[41] displayed the PreS region (163 residues) on M13 phage by using the pCANTAB5E vector with the help of the M13KO7 helper phage. The expression level of the fusion protein, PreS-g3P, was very low, most likely due to the low copy number of the pIII protein. Nevertheless, the fusion protein was shown to be antigenic. Recently, Bahadir et al[42] fused a larger protein, a full-length HBcAg (about 180 residues), to the pIII protein of M13 phage, by using the vector and helper phage as described by Kok et al[41]. The recombinant phage particles were shown to be highly immunogenic in BALB/c mice.
The filamentous phage is an excellent immunogenic carrier because: (1) its DNA can be manipulated easily and the viral particle can be conjugated chemically; (2) it does not lyse its host cells and these two major components can be separated easily by a simple centrifugation or any separation methods; (3) it elicits strong antibody responses at low doses and in the absence of an adjuvant[43-45]; (4) its immunogenicity is also enhanced by host lipopolysaccharide (LPS) associated with the purified phage preparation[46,47]; and (5) it elicits T cell responses[48]. The high immunogenicity of the phage particle itself may mask the immunogenicity of the displayed peptides. In order to reduce the phage’s immunogenicity, van Houten et al[49] deleted the immunodominant region of the pIII protein which enhanced and focused the antibody response against the chemically-conjugated synthetic peptide. This approach plus other mutagenesis techniques, amino acid substitution and insertion, can be employed to reduce the immunogenicity of phage particles, be it λ, MS2, P4, T4 or T7.
The use of T4 phage to display HBV epitopes has yet to be reported. Interestingly, the ability of this phage to co-display two immunogenic components on a single phage particle by exploiting two different capsid proteins, Hoc and Soc[50], would be an advantage to display HBV and HIV epitopes together, to prevent the co-infection by these two life-threatening viruses. The limitation of phage display is that the displayed peptide is not post-translationally modified as in eukaryotic systems, but this can be overcome by identification of epitope or mimotope mimics from sera as described below.
Identification of epitope or mimotope mimics
HBsAg are glycoproteins; the S-HBsAg is either glycosylated or un-glycosylated at Asn-146 of the S region (Figure 2) and the M-HBsAg has an additional glycosylation site at Asn-4 of the PreS2 region[51]. A myristyl group is linked to the glycine residue at the N-terminus of L-HBsAg[51], which is required for the virus infectivity[52]. The phosphorylation of HBcAg, the sole monomer of the viral capsid, is believed to facilitate the transport of the viral genome into host’s nucleus[53]. All these post-translational processes plus the formation of cysteine bonds and others have yet to be discovered processes, may play significant roles in stimulating both humoral and cellular immune responses. This poses a major challenge to the current recombinant DNA technology and chemical synthesis technology. Our understanding about the diversity and complexity of the cellular and humoral responses of HBV infection is still limited. For a start, the complexity of a random peptide library displayed on phages (about 1 billion different peptide sequences for a 6-mer peptide library), provides a practical approach to dissect the immune response of HBV infection. Both monoclonal and polyclonal antibodies from human sera can be used as substrates for the selection of epitopes or mimotopes that mimic antigen-antibody contact sites (molecular mimicry). For instance, Folgori et al[54] successfully identified mimotopes of HBsAg using HBV-infected sera. Some of the selected peptides share similarity with the amino acids located at positions 121-127 of HBsAg. Subsequently, immunization of mice and rabbits with the selected phages harboring the mimotopes mounted cellular[54] and humoral[55] responses resembling those induced by HBsAg. This discovery provides a method to map an immune response by using phage displayed peptide library, without any information about the infected agent. Delmastro et al[56] further demonstrated that the mimotope mimics induced specific antibodies when administered orally to BALB/c mice, suggesting the potential of phage display in oral vaccine development.
Figure 2.
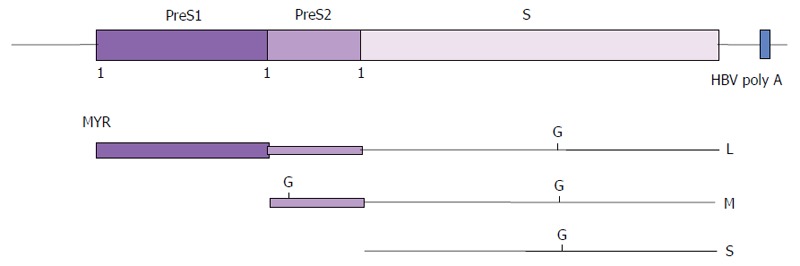
Envelope proteins of hepatitis B virus. The translation products of the HBsAg gene are shown as lines of different thickness. Small (S-), middle (M-) and large (L-) HBsAg are translated from a common open reading frame of the HBsAg gene by the use of three in-frame initiation codons (1) at the N-termini of PreS1, PreS2 and S. G: Glycosylation; MYR: Myristic acid; HBV: Hepatitis B virus; HBsAg: Hepatitis B surface antigen.
Phage-delivered DNA vaccine
A direct injection of a plasmid DNA encoding the HBsAg into mouse muscles has led to the production of the viral surface antigens in the tissues which eventually induced both humoral and cell-mediated immune responses[57]. This finding provides an alternative means for HBV vaccine development, apart from the well established virus-based and protein-based vaccines. The application of this DNA-based vaccine was further extended by Mancini et al[58], who demonstrated that the HBsAg transgenic mice representing HBV chronic carriers, mounted an immune response after a single intramuscular injection which resulted in a complete clearance of circulating HBsAg. This result opens new inroads in designing more effective ways in treating HBV chronic carriers. A clinical trial of the DNA vaccine in hepatitis-naive human volunteers using a gene gun (PowderJect™ system) as a delivery means into skin cells showed that the vaccine induced protective antibody titers as well as humoral and cell-mediated immune responses. However, up till now, there is no DNA vaccine available in the market to prevent HBV infection or treatment of chronic carriers. Perhaps, a longer time is needed, to overcome all the shortcomings of a DNA vaccine, before it can be used practically for mass inoculation in human. It is hope that phage display could play some roles in leading us towards this ultimate aim.
A novel concept to deliver DNA vaccines by using phage particles was introduced by Clark and March[59]. The HBsAg gene was inserted into a phage λ vector containing a DNA vaccine expression cassette and the cytomegalovirus promoter (PCMV). The whole phage particles harboring the recombinant DNA, rather than the naked DNA, were used to immunize mice and rabbits. Interestingly, the anti-HBsAg antibody levels were found to be higher than the standard plasmid-based DNA immunization in these animals[59,60]. Recently, the researchers have compared the phage-delivered HBsAg DNA vaccine with commercially available protein-based vaccine, Engerix B (GlaxoSmithKline Inc.). Amazingly, the results showed that phage-mediated DNA vaccination gave rise to anti-HBsAg antibody levels that were higher than the commercially available recombinant protein vaccine in rabbits immunized intramuscularly[61].
The advantages of phage DNA vaccines include: (1) the DNA is encapsidated by phage coat proteins, thus it is protected from degradation by nucleases; (2) phages are stable at room temperature, which are convenient for storage and transportation; (3) phages grow rapidly in bacterial hosts, hence production and purification of phages are relatively faster, simpler, and cheaper compared to protein- and virus-based vaccines; (4) they can be delivered orally as demonstrated by Delmastro et al[56]; (5) the genomes of certain bacteriophages including phage λ can carry a large foreign DNA fragment (up to 15 kb), which allow several genes from different etiological agents to be packaged in single phage particle; and (6) potential vaccine candidates for newly emerging diseases and mutants, particularly those of veterinary importance, could be developed easily and rapidly. Nevertheless, in spite of these advantages and promising results of phage-delivered DNA vaccine in small animals, it is of utmost importance to investigate the distribution, cellular uptake and expression of the DNA at cellular lever as well as their subsequent mechanisms in stimulating cellular and humoral responses. A potential HBV DNA vaccine should also be tested and validated in primates closely related to human, followed by thorough clinical trials.
DEVELOPMENT OF DIAGNOSTIC REAGENTS AND ASSAYS
The life-threatening complications posed by HBV and its global economic impact have always been the driving force for the development of streamlined diagnostic assays. From past to present, and perhaps in the future, the assays for detecting HBV infection focus on the viral antigens, nucleic acids and the antibodies produced by the human body. The appearance of these serological and genetic markers during acute, chronic and occult infections is summarized in Figure 3. The serological markers used routinely in clinical practice are HBsAg, anti-HBsAg, HBeAg, anti-HBeAg and anti-HBcAg (including total anti-HBcAg antibodies and anti-HBcAg IgM). In an acute HBV infection, the HBsAg is the first detectable viral antigen to appear during infection, followed by HBeAg. About three months after the viral infection, the anti-HBcAg is the first antibody to appear, followed by anti-HBeAg and anti-HBsAg[14]. The primary markers for the identification of acute HBV infection are HBsAg and anti-HBcAg. For some lucky people (about 90%-95%), the infection is utterly controlled and resolved by their immune systems. This is observed by the disappearance of the HBsAg, but the anti-HBcAg and anti-HBsAg remain positive which protect the body from further infection. If the infection is not resolved completely, these individuals become chronic carriers with the presence of HBsAg and anti-HBcAg. A repeat HBsAg test after six months will determine whether the infection is resolved or chronic. For chronic carriers, HBeAg and anti-HBeAg are tested to measure the level of viral infectivity and seroconversion status[62]. Anti-HBcAg (total) normally indicates a prior infection and it also provides evidence of an occult HBV infection, in which the viral DNA is present without detectable HBsAg[63]. Apart from detecting occult infection, the amount of viral DNA is commonly used to monitor HBV replication, disease progression and also to investigate the responses to drug treatment[62].
Figure 3.
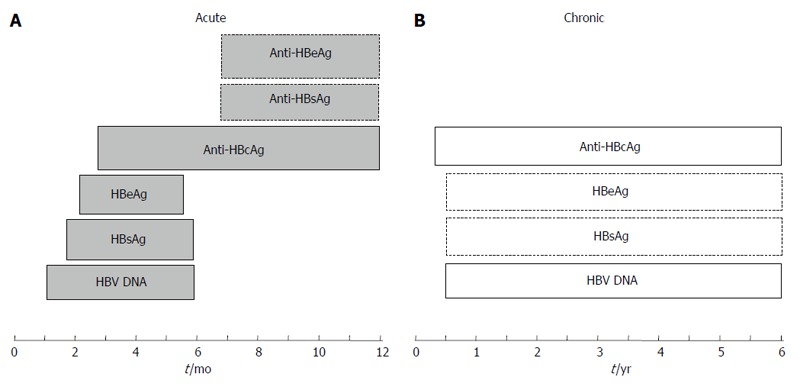
Hepatitis B virus markers in sera during acute (A) and chronic (B) infections. The periods in months (acute infection) and years (chronic infection) are indicated below the boxes containing the markers. The presence of the markers indicated in boxes with broken lines varies in individuals. HBV: Hepatitis B virus; HBsAg: Hepatitis B surface antigen; HBeAg: Hepatitis B e antigen; HBcAg: Hepatitis B core antigen.
A highly sensitive and specific radioimmunoassay (RIA) involving antigen-antibody interaction on a solid phase and radioisotopes was developed in early 1970s for the detection of HBsAg and anti-HBsAg[64-66]. Due to the drawbacks of RIA which involves radioisotopes, special precautions, licensing and equipment are required, therefore it was quickly replaced by ELISA for detecting HBsAg[67], HBeAg[68,69], anti-HBcAg IgM[70], HBcAg[71] and anti-HBeAg[72]. Today, the quantitative real-time PCR is able to monitor the replication of HBV DNA and quantify its amount in different stages of the viral infection[73]. In the advancement of these methods, phage display has demonstrated its potential in generating antibodies and diagnostic reagents. There is no doubt in the importance of phage display in the development of diagnostic assays in the 21st century to combat HBV.
Phage displayed antibodies as diagnostic reagents
Polyclonal antibodies produced in animals have been used widely for the detection of HBV antigens before the introduction of hybridoma technology for the production of monoclonal antibodies. This revolutionary technology involves the fusion of antibody producing spleen cells from mice with immortal myeloma cell lines[74]. The use of monoclonal antibody that interacts with a single epitope on an antigen has increased the specificity of a diagnostic assay and thus reduced cross-reactivity with other antigens. However, the production of monoclonal antibody by using hybridoma technology is time consuming and laborious. In addition, the application of mouse monoclonal antibodies as therapeutic agents is limited by the fact that they are highly immunogenic and cleared rapidly by the human immune system. All these limitations can now be overcome by the rapid advancement of recombinant DNA technology, in vitro isolation of antibodies from a combinatorial library and large scale production of antibodies in bacteria. Phage display fulfills these criteria and it is commonly used to display single chain fragment variable (scFv[75]), fragment antigen binding (Fab[76]), the variable heavy domain of camels (VHH) and humans (dAb[77]). These antibody fragments which are smaller than the immunoglobulin (IgG) are illustrated in Figure 4.
Figure 4.
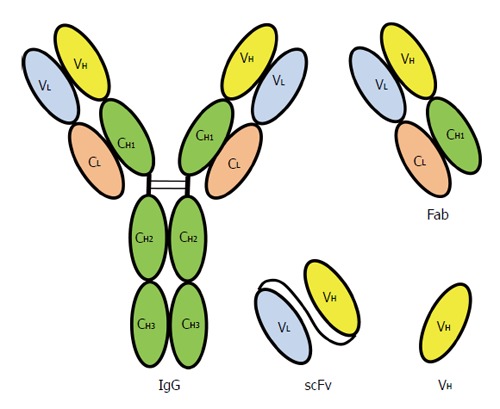
Recombinant antibodies displayed on bacteriophage compared with an immunoglobulin G molecule. Chain structure of a human immunoglobulin G (IgG) molecule. V: Variable region; C: Constant region; H: Heavy chain; L: Light chain; VH: Variable region of heavy chain; Fab: Fragment antigen binding; scFv: Single chain fragment variable, the VL and VH are linked by a peptide linker.
A phage-displayed scFv that interacts specifically with HBcAg was constructed by Tan et al[78] and the whole phage particle was used to establish a phage-ELISA for detecting HBcAg in human sera. The HBcAg is one of the markers of HBV infection because it is correlated with the infectious particles and also proportional to the level of HBV genome in a serum[71,79]. For the construction of a scFv library, mice were first inoculated with purified HBcAg and total RNA molecules were extracted from their spleens. The genes encoding heavy (VH) and light (VL) chains were amplified by PCR and linked by a linker to generate scFv coding regions. These coding regions were then inserted into the pComb3x vector and introduced into E. coli. The resultant phage displayed scFv library was panned against HBcAg immobilized on a solid phase and a phage clone that interacted tightly with the immunodominant region of HBcAg was isolated. The selected phage was used to establish a phage-enzyme-linked immuno sorbent assay (ELISA) for detecting HBcAg in human serum samples. This assay was able to detect as low as 10 ng of HBcAg and it did not cross react with HBsAg and HBeAg[78].
A phage displayed Fab that interacts tightly with the PreS1 (residues 37-45) of HBsAg was developed by Kim et al[80]. The library was generated by immunizing mice with a PreS1 epitope fused to the N-terminal domain of human thrombopoietin. Phagemid vector pC3-na was used in the construction of the cDNA library. A phage clone harboring the Fab was selected from a biopanning process and demonstrated to bind tightly to the PreS1 region with a KD of 1.2 nmol/L. The recombinant phage was also shown to interact with HBV particle by an immunoprecipitation assay.
Up till now, the phage displayed antibodies against HBeAg and HBxAg have yet to be reported. In principle, the methods described above can be employed to generate specific recombinant phage antibodies against these two important antigens of HBV. The phage-based diagnostic assays that can be developed are not limited to ELISA, the phage antibodies can also be incorporated into other diagnostic technology such as lateral flow strip assays, proximity ligation assay and flow cytometry. Production of monoclonal antibodies and their related fragments via phage display technology are without doubt, simpler, cheaper and faster compared with hybridoma technology.
Phage displayed peptides as diagnostic reagents
The key feature of a diagnostic assay is molecular interactions. In most cases, particularly ELISA, the main interaction involved is between antibody and antigen. To be more precise, the paratope of an antibody interacts with an epitope or a mimotope (discontinuous epitope) on the surface of an antigen. The contact region in an antibody-antigen interaction could be less than 10 residues, therefore, in principle the antibody can be replaced by a peptide which can be selected from a random phage displayed peptide library. This principle was proven correct by Tan et al[81] who successfully isolated a phage-displayed cyclic peptide bearing the sequence ETGAKPH that interacts tightly with the immunodominant region of HBsAg. The paratope mimic, ETGAKPH, was selected from a disulfide constrained random heptapeptide library displayed on the pIII of phage M13 by panning against HBsAg purified from human plasma. The phage harboring the peptide was shown to bind tightly to HBsAg with a KD of 2.9 nmol/L. The whole phage was then employed as a diagnostic reagent to establish a direct and an indirect phage-ELISA to detect HBsAg in HBV infected patients (Figure 5). Both assays were able to detect HBsAg down to 1 pg/mL. Most recently, the three-dimensional structure of this paratope mimic was elucidated by nuclear magnetic resonance (NMR) and demonstrated to interact with the “a” determinant of HBsAg[82]. These cumulative data prove that an antibody can be replaced by a short peptide and a phage harboring this peptide can be used as a sensitive diagnostic reagent.
Figure 5.
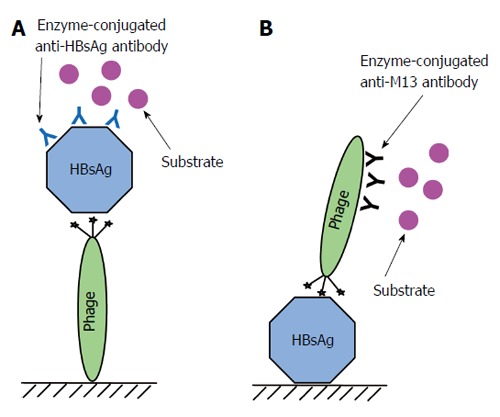
Phage-ELISA for detecting hepatitis B virus surface antigen. A: Major steps used in an assay for detecting hepatitis B virus surface antigen (HBsAg) by immobilizing phage carrying the peptide on a solid support. The phage harboring the peptide is immobilized on a solid support. The unsaturated area of the solid phase is blocked by 10% milk diluent. The sample containing HBsAg is added to interact with the peptide. Mouse anti-HBsAg antibody is added to interact with the captured HBsAg. Then anti-mouse antibody conjugated to an enzyme is added to interact with the anti-HBsAg antibody. Finally, substrates for the enzyme are added to produce a measurable signal; B: Major steps used in an assay for detecting HBsAg immobilized on a solid support using a phage carrying the peptide. A sample containing HBsAg is immobilized on a solid support. The unsaturated area of the solid phase is blocked by 10% milk diluent. The phage harboring the peptide is added to interact with immobilized HBsAg. Anti-M13 phage antibody conjugated to an enzyme is added to interact with the bound phage and substrates for the enzyme are added to produce a measurable signal. ELISA: Enzyme-Linked Immunosorbent Assay.
Along the same lines, an M13 phage displaying a paratope mimic that interacts tightly with HBcAg was used as a diagnostic reagent to develop a phage-ELISA, dot blot assay and immunoprecipitation assay[83]. The paratope mimic with the sequence WSFFSNI was isolated from a disulfide constrained random heptapeptide library by panning against the purified recombinant HBV capsid immobilized on a solid phage[84]. The recombinant phage with a KD of 2.5 nmol/L, was able to detect a minimum amount of 10 ng HBcAg in both the phage-ELISA and dot blot assay. These assays can also be used to detect HBcAg released from the virion in HBV positive serum samples[83]. The sensitivity of the phage-based diagnostic assay was further improved by Monjezi et al[85] by combining TaqMan based real-time PCR and ELISA. The resultant phage display mediated immuno-PCR (PD-IPCR) is 10000 times more sensitive than the phage-ELISA.
The vast diversity of a combinatorial phage displayed peptide library also allows one to select epitopes by panning against antibodies, be it monoclonal antibodies or polyclonal antibodies from sera. The selected epitopes, on the other hand, can be used to diagnose antibodies in sera. The advantage of this strategy is that prior knowledge about a newly emerging etiological agent inducing disease-specific antibodies is not required.
Compared with phage displayed antibodies, the isolation of phage born peptides is faster, cheaper and less tedious as no immunization of animals is involved. The affinity of a selected peptide towards an antigen or antibody can be increased by displaying the peptide on the major coat protein of a bacteriophage, such as the pVIII in the filamentous phage. Apart from HBV, phage born peptides have also been employed extensively as diagnostic reagents for cucumber mosaic virus[86], Newcastle disease virus[87,88], Salmonella enerica Serovar Typhi[89,90], Nipah virus[91] and HIV-1[92]. All these remarkable examples demonstrate the potential of phage displayed peptides as diagnostic reagents for detecting etiological reagents and of course this can be further extended to detect an unknown microorganism.
DRUG DISCOVERY
HBV has killed millions of innocent people in this century and the previous one. Currently, there are about 370 million chronic HBV carriers worldwide, and their major hope is a rapid discovery of an effective treatment for HBV. Therefore, searching for a cure is no doubt will change human history. The introduction of mass vaccination of infants worldwide with HBV vaccines has significantly reduced the incident of liver cancer and chronic infection. Despite of effective vaccines against HBV, mutations of the viral genome have resulted in vaccine escape mutants. Treatments of HBV chronic carrier with nucleoside or nucleotide analogs have also given rise to mutants that are resistant to anti-viral drugs. Most frightening, HBV is now associated with HIV to threat the world. Treatments of patients co-infected with these two viruses, either by using drugs against one or both viruses, have led to the selection of resistance mutations. Therefore, there is always a continuing need for the discoveries of a new therapeutic agent to control HBV infection particularly for the newly emerging mutants.
Traditionally, drugs and therapeutic agents are discovered through isolation of active ingredients from medicinal plants or microorganisms following effective treatments of the illnesses with the crude extract of these sources. In modern drug discovery, structural bioinformatics allow in silico screening of chemical compounds that ‘fit’ the binding pocket of a targeted protein from virtual chemical libraries. The actual dynamic and physical properties of the in silico selected compounds always end in disappointment due to the difficulties in chemical synthesis, poor solubility, affinity and specificity, although, in some cases, the binding mechanism can be explained by structural analysis using X-ray crystallography and NMR. The emergence of phage display technology has offered a powerful and rapid method to identify novel peptides and antibodies from phage fusion peptide/antibody libraries that bind to wide range of antigens ranging from whole cells such as bacteria, parasites and viruses to macromolecules for instances proteins, carbohydrates and lipids. DNA recombinant technology enables peptides to be fused either to the major or the minor fusion coat proteins and eventually displayed on the surface of various bacteriophages[93]. Vast peptide libraries can be screened for specific ligands that bind a targeted protein through a biopanning process[94]. The isolated phages can be easily propagated by infecting E. coli and the peptide sequence can be deduced by nucleotide sequencing. The corresponding peptide can be chemically synthesized or produced using bacterial expression systems. Owing to the stringency multiple cycles of selection, the selected peptides always display high specific and affinity towards their targets, however for some peptides, stability and solubility are the main problems. Nowadays, peptide binding mechanism can be studied by structural approaches and binding assay using ELISA.
For the past two decades, phage-displayed combinatorial peptide libraries have been used widely to select anti-viral peptides against many viruses (see review in[95]). Similar to other anti-viral discoveries, the major processes in HBV life cycle are the potential targets for drug discovery. Specific ligands that bind the key players of the viral life cycle would interfere with HBV replication intracellularly or extracellularly. The potential extracellular targets include (1) neutralization of matured infectious virion; and (2) viral-host cell receptor interaction. The infectious viral particles can be neutralized easily by specific antibodies. By screening combinatorial phage display Fab fragment libraries against HBsAg, several groups have successfully isolated Fab fragments that neutralize infectious HBV particles[96-103]. In addition, the PreS1 region which possesses a dual function in the viral assembly and infectivity has also been targeted for anti-viral drug discovery by panning with phage display peptide libraries[81,104-107]. Intracellularly, HBcAg forms the viral nucleocapsid and interacts with the PreS region during the viral assembly, therefore, HBcAg is a common target for the discovery of HBV inhibitors[84,108]. In addition to the intracellular inhibition of viral assembly, HBV release from the infected hepatocytes can be blocked by Fab fragments of intrabodies that could be selected easily from phage displayed antibody libraries. Intrabodies are intracellular antibody fragments that are expressed and functional inside a cell[109]. Several studies have shown that these phage derived intrabodies fragments bind immature HBV particles or proteins and potentially inhibit the viral secretion in vivo[78,110-112]. Targeting the extracellular steps of the viral cycle is relatively simpler and more straightforward compared to those of the intracellular steps, in which the anti-viral compounds must enter the membranous barrier of the liver cells. Nevertheless, selection of peptides against the association of HBcAg and L-HBsAg has been studied extensively and intensively, however, in vivo anti-viral activity has yet to be characterized in depth.
Intracellular targeting
HBcAg is an attractive target for the discovery of novel therapeutic agents. In the process of nucleocapsid formation, one copy of the viral pre-genomic RNA (pgRNA) is encapsidated in the cytoplasm[5]. Following the conversion of pgRNA to a mature viral genome (partially double stranded DNA), the nucleocapsid moves to the endoplasmic reticulum (ER) where it interacts with the ER membrane embedded with HBsAg which results in the envelopment of nucleocapsid. The mature virion is either secreted into the bloodstream or moved back to cytoplasm. This process is followed by disassembly of the nucleocapsid into individual HBcAg at the nuclear pore complex and the viral DNA is released back to the nucleus[113,114]. HBcAg has been shown to function as a transcriptional activator which enhances the viral replication[115]. The enrichment of HBcAg in this manner correlates with the high level of viral replication. Several anti-viral agents such as aptamers[116], heteroaryldihydropyrimidines[117], and single-chain variable fragment intrabodies[118] have been developed to target HBcAg. Using phage display technology, peptides were successfully selected from phage display peptide libraries to prevent its interaction with HBsAg[84,108] and Fab fragments of intrabodies neutralize HBcAg inside liver cells were also selected from phage display antibody libraries[78,110-112].
HBV was the first virus used in the selection of peptide inhibitors from a 6-mer linear phage display peptide library[108]. A phage bearing amino acid sequence LLGRMK that interacts specifically with the immobilized HBcAg particles with a KD of 0.17 μmol/L was selected and the corresponding synthetic peptide ALLGRMK was shown to block the association of L-HBsAg and HBV nucleocapsid in vitro. Interestingly, neither peptide 21LLTRIL27 of S-HBsAg nor GRMKG (C-terminal part of the selected peptide) inhibited the binding of L-HBsAg to HBcAg but this interaction was partially inhibited by the PreS1 amino acids 19LDPAFR24, the epitope of monoclonal antibody 18/7[108]. This indicates that the peptide mimics the internal region of HBsAg comprising distal amino acids which are brought into close proximity upon protein folding. Cell culture experiment also proved that the synthetic peptide was able to reduce HBV replication in vivo and the peptide binding site was later located to the tips of the spike that formed by amino acids 78-82 (78DPASR82) of HBcAg[119]. Mutations of D78 and E77 to an Alanine, in turn, dramatically reduced the binding affinity. Cross-linking study also proved that these residues are essential for the peptide-HBcAg interaction[120]. Although the 3-dimensional structure of the complex of HBcA-peptide was solved by cryo-electron microscopy, the detailed binding mechanism remains elusive due to its poor resolution[119]. In 2003, a modified biopanning approach using a disulfide-constrained heptapeptide library was employed. This approach has led to the isolation of cyclic peptide CWSFFSNIC and CWPFWGPWC exhibiting a higher affinity (> 10-fold higher than LLGRMK) towards HBcAg and similarly, the synthetic peptides blocked the association of HBcAg and L-HBsAg or monoclonal antibody C1-5, which has an epitope located at amino acids 78-83 of HBcAg, from interacting with its epitope[84]. These findings indicate that the linear and cyclic peptides bind different set of amino acids close to the tip of the spikes. The conformational constraint cyclic peptides were composed of mainly hydrophobic amino acids and it is believed that they interact with the hydrophobic amino acids located at the immunodominant region of HBcAg. Interestingly, the N-terminal half of peptide LLGRMK also contains hydrophobic amino acids, however it is unclear whether these residues interact with the same amino acids as the cyclic peptides. The 3-dimensional structure of the cyclic peptides in complex with HBcAg has yet to be identified. Further characterizations with the synthetic cyclic peptides were hampered by their hydrophobicity. Therefore these peptides need to be further modified in order to improve the solubility and bioavailability.
In 2009, Serruys et al[111] demonstrated for the first time that viral secretion in mammalian cells can be inhibited by intrabody-mediated inhibition. They constructed a single-domain antibody (VHH)-phage display peptide library and biopanned it against human plasma derived HBsAg. Up to 85% of the isolated phages are associated with S-HBsAg in relative to only 45% of the soluble VHHs expressed intracellularly. These intrabodies were shown to reduce HBsAg particles in plasma when they were expressed in the ER and shown to reduce the viral load up to 100-fold in a mouse model[111]. In the following year, similar approach was used by the same research group to isolate VHH intrabodies that bind HBcAg of ayw and adw subtypes[110]. As demonstrated in HBV transfected HepG2 cells, the nucleotropic targeted intrabodies reduced intracellular HBcAg, but interestingly, two out of six isolates increased the detection levels of intracellular HBeAg, which is likely due to HBeAg antigenicity displayed by the non-particulate HBcAg or retention of HBeAg inside the cells as a result of interaction with these intrabodies[110]. This hypothesis is in good agreement with that of Walsh et al[112], who isolated intrabodies by screening a naive single variables domain (VNAR) display phage library against HBV precore antigen (preHBcAg) and proved that these intrabodies reduced the extracellular amount of HBeAg and intracellular preHBcAg[112]. HBeAg is believed to function as an immune tolerogen that could lead to the establishment of chronic infection[121]. Therefore, down-regulation of HBeAg should improve clinical outcome. In an earlier study, Tan et al[78] isolated single-chain variable fragment (scFv) against HBcAg from a phage display scFv library. Although the initial aim of this study was to develop an antibody system to detect HBcAg, expression of the ScFv inside liver cells particularly in the ER membrane could possibly convert the antibody fragments into potent therapeutic agents that could neutralize and inhibit viral secretion in vivo.
Extracellular targeting
The first step of HBV replication is the attachment of an infectious HBV particle with a receptor on the surface of hepatocyte. Molecules that mimic the structural elements of a host cell receptor could block subsequent infection steps. The major problems to study HBV infection are attributed to its narrow host range and the absence of a susceptible cell line for virus infection[122]. However, biochemical studies with purified components of duck HBV (DHBV) have revealed several candidates that involved in DHBV infection[123]. Several glycoproteins have been identified as potential DHBV receptors such as glycoproteins gp180/p170 that bind DHBV and E. coli derived GST-PreS polypeptides. These glycoproteins are later classified as the prototype members of membrane bound carboxypeptidase D (CPD)[124,125]. Urban and coworkers showed that DHBV PreS region binds duck CPD with a KD of 0.46 nmol/L[126] and also demonstrated that CPD plays pivotal roles for the virus entry and attachment via its carboxypeptidase-like (A and B) and non-enzymatic (C) domains, respectively[127]. Although these host proteins have been identified as the cellular receptor of HBV, identification of a promising inhibitor has yet to be reported. On the other hand, the PreS1 region that plays pivotal roles in the viral infectivity and assembly has been the main focus of many anti-viral studies using phage display technology[81,104,107].
Tan et al[81] biopanned the HBsAg (subtype ad) purified from human plasma with a disulfide constrained phage display peptide library and identified a predominant M13 pIII fusion heptapeptide CETGAKPHC that binds to the immunodominant region of HBsAg (amino acids 111-156) with dissociation constants 2.9 ± 0.9 nmol/L and 0.83 ± 0.63 nmol/L[81]. NMR study indicated that the peptide adopts cis/trans conformations due to Pro rotamerization and in silico analysis revealed two binding sites which correspond to the first (amino acids 107-137) and second (amino acids 138-149) loops of the immunodominant region[82]. This finding indicates that the peptide could be further developed as a therapeutic agent that blocks the entry of HBV into hepatocytes.
Instead of using the purified HBsAg from an infected human serum as a substrate for affinity selection, Deng et al[104] biopanned the thioredoxin tagged-PreS using an M13 pVIII 12-mer fusion cyclic peptide library. The recombinant PreS1 is more soluble and stable compared with the PreS region alone[105]. Following 5 rounds of biopanning, Deng et al[104] isolated peptides with a common sequence motif of W/FTXWW/F that interact with amino acids 21 to 47 of the PreS1 region. Biotinylated peptide bearing an amino acid sequence SGSGWTNWWST (amino acids SGSG is a linker) was able to precipitate the HBV particles produced by HepG2 cells[104]. Several other HepG2 proteins were also bound to the same PreS1 region[128,129]. Among others, homology search has identified a lipoprotein lipase; a key enzyme involved in the lipoprotein metabolism; which contains amino acid sequence SWSDDWWS at its C-terminal region and this sequence was conclusively shown to interact with the PreS1 amino acids 21-47 and HBV particles. Alanine mutation scanning of the tryptophan residues of this peptide abolished the binding of the PreS1 and HBV particles; however, these activities were retained if tryptophan was replaced with phenylalanine. The authors speculated that the tryptophan/phenylalanine may mediate interaction of protein and lipid/aqueous interface during the viral attachment due to its amphipathic character. Overall, this PreS1 binding peptide displays anti-viral activities by blocking the viral attachment to a hepatocyte and potentially reduces the viral infectivity. Independently, Wang et al[107] and Deng et al[106] isolated a dodecapeptide (KHMHWHPPALNT) and a disulfide-constraint heptapeptide (SRLLYGW), respectively, against the PreS1 region from phage display peptide libraries[106,107]. Although both peptides were shown to bind the PreS1 region and capture HBV virion produced by HepG2 cells, no sequence consensus was found between these peptides and also those isolated by Tan et al[81] and Deng et al[104]. Variability in the construction of phage display library and biopanning procedures may lead to identification of different peptides that might potently block viral-host cell interactions. Diversity of the selected peptide sequences indicates that various cellular proteins are involved during the viral attachment. Targeting these proteins particularly the cellular receptor could potentially reduce the viral infectivity in vivo.
EPITOPE MAPPING
HBV can be easily transmitted from asymptomatic carrier mothers to their newborn babies. Such perinatal transmission is commonly caused by an exposure to HBV contaminated blood during birth. To reduce the risk of such mother to baby transmission, passive immunization has been introduced in which babies born to HBsAg positive mothers are immunized at birth with hepatitis B immunoglobulin containing antibodies against HBsAg. These antibodies are effective in preventing HBV infection in infants who do not have sufficient time to develop their own immune response. Apart from preventing perinatal transmission, these antibodies could also be used in acute exposure to blood containing HBsAg, sexual exposure to HBsAg, positive carriers and household exposure to people with acute HBV infection. After intramuscularly administration, the antibodies interact specifically with HBsAg epitopes and neutralize the virus. In other words, the paratopes of the antibodies interact with the epitopes on HBV. Antigenic epitopes are the region of an antigen that interacts specifically with an antibody or a T-cell receptor. In general, epitopes are divided into linear (continuous) and conformational (discontinuous) epitopes. A linear epitope is defined as a minimum stretch of continuous amino acids that are recognized by an antibody while the conformational epitope or mimotope are composed of discontinuous amino acids that brought into proximity as a result of 3-dimensional protein folding. A detailed study of epitope is the fundamental basis for the development of epitope-based vaccines, therapeutics, viral inhibitors and diagnostic reagents.
Various methods have been used to map the epitope of a specific antibody. Most of these methods rely on the ability of the antibody to recognize a linear or conformational epitope displayed on the surface of an antigen. The earliest method for epitope mapping known as the “pepscan method” was developed by Mario Geysen and coworkers in 1984[130]. With this method, a peptide library consisting of different lengths of overlapping oligopeptides covering an epitope is scanned over immobilized antibodies. Peptide array that interacts with the coated antibodies corresponds to certain aspects of the epitope. The major limitation of the pepscan method is that it only allows the identification of linear epitopes instead of conformational ones. X-ray analysis of antigen-antibody complexes is capable to identify the precise location of epitopes up to atomic resolutions, however, not all antigen-antibody complexes are readily crystallizable[131]. To date, about 70 co-crystals of antigen-antibodies have been solved by X-ray crystallography, demonstrating an exceptionally low efficiency of this method[132]. On the other hand, NMR is an alternative approach to achieve similar purpose but this method is limited to antigen-antibody complexes with molecular weight less than 30 kDa[133]. Nevertheless, NMR provides the thermodynamic characteristics of antigen-antibody complexes in solution. Overall, the accuracy and precision of the above mentioned biophysical approaches towards epitope mapping are restricted by the tedious and laborious techniques and the requirement of advanced instrumentation and expertise.
In 1990, Scott and Smith[94] demonstrated that epitope mapping can be accomplished easily by screening a random peptide library against sera or monoclonal antibodies that immobilized on a solid surface. Epitope mapping using phage display technology offers the advantages of (1) identifying both continuous and discontinuous epitopes; (2) independent of advanced instrumentations; (3) cost effective and simple experimental protocol; and (4) exploring the variety of binding sequences by screening with phage display peptide libraries containing fusion peptides with different lengths or conformations such as linear or disulfide-constrained fusion peptides. Precise epitope mapping of sequential or conformational epitopes recognized by monoclonal or polyclonal antibodies is important to understand the mechanism of immune response, host-virus interactions and development of vaccines and diagnostic tools. Such mapping requires the identification of shortest amino acid stretches within the polypeptide that is still capable to bind to the target antibody.
Generally, immunological studies have identified three HBV antigens to be clinically relevant: (1) HBsAg; (2) HBcAg; and (3) HBeAg. HBsAg has been extensively used to induce protective immune response in immunized individuals while HBcAg and HBeAg have served widely as diagnostic markers. Due to the vaccination value, be it an active or passive immunization, most of the early epitope mapping of HBV proteins was focused on the PreS regions particularly the PreS1 region that associates with the viral host cell receptors. With the development of phage display technology, epitopes of various antibodies raised against the PreS1[96,134,135], PreS2[136] and S regions[137-140] have been mapped by screening phage display peptide libraries. The strategies and outcomes of the epitope mapping are summarized in Table 2, and reviewed critically in the following sections.
Table 2.
Epitope mapping of anti-hepatitis B virus antibodies
| Antibodies against | Phage library used | Epitopes | Remarks | Ref. |
| PreS1 region | ||||
| MA 18/7 | M13 pIII 6-mer | ALDPAY, SLDPGF, PDPGFN, QLDPGF | The dipeptide DP was conserved from rounds 2 to 4 among all the analyzed phagotopes. Selected peptides were mapped to amino acids 19-23 of the PreS1 region | Germaschewski et al[134] |
| MA 18/7 | M13 15-mer liner | SDTRGDPVFNLPFQ APVDSVFDRAFSAYL | Isolated peptides contain motif similar to LDPAF | D'Mello et al[135] |
| MA BX-182 | M13 pIII 12-mer linear | SVPPPHTRSASG | Peptide matched amino acids 17SVPNP21 of the PreS1 | Zhang et al[144] |
| M13 pIII 7-mer disulfide constrained | CTNPVLRSC | Peptide spatially matched some amino acids within the same region but in a C- to N-terminal direction | ||
| PreS2 region | ||||
| HBsAg polyclonal sera | M13 pIII 12-mer linear | TANGFYRLPSGS | Peptide sequence mapped to amino acids 135-146 of the PreS2 region | Zhang et al[136] |
| S-region | ||||
| MA RFHBs6 | M13 pIII 15-mer linear | TSNTHAC(R/K)TCSNPSR | The peptide sequence is similar to amino acids 115-129 of the S region | Motti et al[140] |
| MA BA1 | M13 pIII 15-mer linear | PHDGNRAFPRTKVTM HMPRDANRHHQHPST SSLGSDHNARWVKRF | Low sequence homology with the S region | |
| MA 6H6B6 | M13 pIII 12-mer linear | WPHNWWPHFKVK | Peptide sequence homology not found | Jolivet-Reynaud et al[137] |
| M13 pIII 6-mer linear | QGFLPQ | Peptide sequence mapped to amino acids 101-105 of the S-region | ||
| MA H166, H5, H35, H53 | M13 pIII 30-mer linear | 1Peptide Xn-CXTC-Xn | Sequence motif of CXTC was mapped to amino acids 121-124 of the S-region | Chen et al[138] |
| IgG derived from S-HBsAg immunized chimpanzee | M13 pIII 6-mer linear | TRVPRR, SRLPLR, SRLPKR | Sequence motif SxxPxR was mapped to amino acids 117-122 of the S-HBsAg | Germaschewski et al[139] |
xn represents random flanking amino acid sequences. Position X is constituted either by arginine, lysine or valine. MA: Monoclonal antibody; HBsAg: Hepatitis B surface antigen; IgG: Immunoglobulin G.
Epitope mapping of the PreS1 region
Germaschewski and Murray[134] biopanned the first established monoclonal antibody MA18/7 that interacts specifically with the PreS1 region. Following four rounds of biopanning, phage bearing pIII fusion hexapeptides with amino acid sequence motif LDPX (F/Y), in which, X represents small side-chain amino acids such as alanine, glycine or valine were selected. The dipeptide DP was conserved from rounds 2 to 4 among all the analyzed phagotopes. This sequence motif mimics three or four residues of 19LDPAF23 of the PreS1. Recently, the 3-dimensional structure of monoclonal antibody MA18/7 in complex with epitope 19LDPAF23 fused to HBcAg immunodominant region was determined by cryoelectron microscopy[141]. Interestingly, this short amino acid sequence was also identified as the receptor-binding domain of L-HBsAg although similar motif was also found in some bacteria and cellular adhesion molecules[133].
This conserved motif was also reported when a 15-mer phage display peptide library was biopanned against the monoclonal antibody MA18/7 that had been immobilized on polystyrene beads[135]. Diversity of the longer flanking amino acid sequences around the tetramer [DPX(F/Y)] suggests that these adjacent amino acids do not contribute to specific binding. Alignment of the selected sequences with the PreS1 amino acids 28-35 (28HQLDPAFGAN35) indicates that all the phagotopes contained the core motif DX1X2F, with X1 constituted of either proline or arginine although leucine and serine occurred once each and X2 encompassed by small side-chain residues such as valine, alanine, glycine and serine, which is consistent with Germaschewski and Murray’s[134] earlier finding. The biopanning results either using 6- or 15-mer phage display peptide libraries are consistent with those of epitope mapping by using the bidirectional shortening of the PreS1 with exonuclease digestion, which conclusively defined the minimal epitope of the PreS1 to be DPAF[142]. Searching through the Protein Databank (PDB) revealed that the amino acid sequence DPAF adopts either a β-turn or as part of an α-helical structure. The main-chain carbonyl of the aspartate is always hydrogen bonded to the main-chain nitrogen of phenylalanine[143]. Therefore, it is not surprising that the alanine cannot be replaced with amino acids bearing bulkier side-chains.
In a separate study conducted by Zhang et al[144], a monoclonal antibody raised against HBsAg of adw subtype (monoclonal antibody BX-182) was used to select phagotopes from two different random peptide libraries. An early study demonstrated that a monoclonal antibody preferentially neutralized infectivity of HBV adw subtype in a chimpanzee model and this prompted the authors to map the neutralization epitope of adw HBsAg. Initial affinity selection using a disulfide-constrained heptapeptide library successfully identified phage bearing a cyclic heptapeptide CTNPVLRSC. In a separate experiment, affinity selection with a 12-mer phage display peptide library identified a linear dodecapeptide SVPPPHTRSASG. Sequence homology search showed that part of the dodecapeptide (SVPPP) matched the N-terminal region of the PreS1 between amino acids 17SVPNP21 with a replacement of proline with asparagine. On the other hand, the disulfide-constrained loop of CTNPVLRSC also spatially matched some amino acids within the same region but in a C- to N-terminal direction, indicating that 18VP19 are the core amino acids for monoclonal antibody BX-182 binding as they are conserved in both isolated sequences. Sequence homology search also revealed that the Val/Pro motif is conserved among genotypes A, B, C, F and H of ad subtype but it is absent in most genotypes of ay subtypes or in genotypes D, E, and G of ad subtype, in which these antigenic residues are replaced with Thr/Ser, Thr/Thr or Ala/Ser, respectively. This result indicates that the binding of monoclonal antibody BX-182 is subtype-specific and the isolated peptide can be used to discriminate ad/ay subtypes in the diagnosis of potential HBV escape mutants.
Epitope mapping of the PreS2 region
The PreS2 region possesses significant clinical importance and it contains T-cell and B-cell epitopes which confer protection by inducing strong immune response in immunized chimpanzees[145,146]. To our knowledge, epitope mapping against the antibodies that recognize the PreS2 region was scarcely reported particularly with phage display peptide library. This is most probably due to its short amino acid sequence comprises only 55 amino acids. Nevertheless, Zhang et al[136] biopanned a phage display library of dodecapeptides against the HBsAg polyclonal human sera. Following 3 rounds of affinity selection, phage bearing the fusion pIII peptide TANGFYRLPSGS; which mapped to amino acids 135-146 (135RVRGLYLPAGGS146) of the PreS2 region; was selected. This region was previously proved to be the antigenic epitope of monoclonal antibody 25-19, which has been shown to display neutralizing and protective activities[147,148]. Several epitope mappings of the PreS2 region were conducted by using the pepscan method[149-152].
Epitope mapping of the S region
Two antibodies namely monoclonal antibodies RFHBs6 and BA1, were pooled from HBV chronic carriers and used as targets for biopanning using a 15-mer phage display peptide library[140]. Single round of affinity screening against the monoclonal antibody RFHBs6 successfully isolated phage carrying pIII fusion peptide TSNTHAC(R/K)TCSNPSR, which is strikingly similar to amino acids 115-129 of the S region (115TTSTGPCKTCTTPAQ129). The two cysteine residues of the peptide are believed to be disulfide bonded and antigenically mimicking the natural epitope. On the other hand, similar approach was applied to isolate phagotopes that recognize monoclonal antibody BA1. Three peptides bearing the amino acid sequences PHDGNRAFPRTKVTM, HMPRDANRHHQHPST and SSLGSDHNARWVKRF were identified. Multiple sequence alignment indicated a low sequence similarity among these sequences with amino acids 127-135 at the N-terminal region of S-HBsAg (127PAQGNSMFP135). Interestingly, phagotopes selected against monoclonal antibodies RFHBs6 and BA1 did not cross-react with their counterpart’s target, indicating that the antigenic determinants are distinct.
Epitope mapping of monoclonal antibody 6H6B6 which recognizes specifically the S-HBsAg was carried out by biopanning the 6-mer and 12-mer phage display peptide libraries[137]. A selected phage bearing the fusion dodecapeptide WPHNWWPHFKVK did not share similarities with the primary sequence of the S-HBsAg as well as the PreS2 sequence but reacted with the monoclonal antibody 6H6B6, indicating that the selected phagotope mimics the epitope conformation. In contrast, biopanning with a 6-mer phage display peptide library had selected a pIII fusion hexapeptide QGFLPQ that shared 4 identical amino acids localized between amino acids 101-105 (101QGMLP105) of S-HBsAg. Overlapping peptides (99DYQGMLPVCPLI110 and 102GMLPVCPLIP111) corresponding to this region blocked the binding of monoclonal antibody 6H6B6 to the phage bearing pIII fusion peptide QGFLPQ. Furthermore, peptides coated on an ELISA plate were not recognized by monoclonal antibody 6H6B6, suggesting that amino acids 101-105 is part of a conformational epitope that corresponds to the N-terminal region of the major hydrophilic domain of the S-HBsAg. Peptide affinity selection against monoclonal antibodies 27E7F10 and 2G2G10 isolated highly hydrophobic peptides which were localized to amino acids 214-219 and 199-208, respectively, of the S-HBsAg. Antibody-peptide competition assay, however, was not carried out due to hydrophobicity of the flanking peptide sequence.
Affinity selection using a 30-mer phage display peptide library against monoclonal antibody H166 identified phagotope bearing a sequence motif CXTC, which localized to residues 121-124 of the S-HBsAg[138]. Coincidently, in the same year, Germaschewski and Murray[139] selected hexapeptides that bound to serum IgG derived from the S-HBsAg immunized chimpanzee. This ‘shotgun’ approach selected 20 different pIII fusion hexapeptides with some common motifs that match groups of 3 or 4 amino acids along the S-HBsAg. In an additional round of selection (micropanning using a phage sandwich ELISA), the number of tight binders had been reduced to 7 with pIII fusion peptide bearing the motif SxxPxR, which corresponds to amino acids 117-122 of the S-HBsAg (117STGPCR122)[139]. It is believed that the two cysteine residues within this region cyclized to form a disulfide-constrained loop conformation that resembles the native structure of HBsAg. Biopanning with other monoclonal antibodies (H5, H35 and H53) also identified fusion peptides flanking by cysteine residues that are separated by 2 to 15 amino acids[138]. Some selected peptide contained two separate regions that matched the S-HBsAg sequence, indicating that these segments are brought together into a close proximity as a result of protein folding.
DRUG AND GENE DELIVERY
HBV carriers have an increased risk of developing liver cancer or HCC. Worldwide, HCC is the fifth most common cancer that contributes to 5.6% of all human cancers and ranks third among cancer-related mortalities[153]. Recently, gene therapy has become a promising approach to treat cancers and virus-based nanoparticles, particularly bacteriophages, have been proposed as gene and drug delivery vectors.
Filamentous bacteriophages have been at the cutting edge of new developments in nanotechnology which create innovative applications in gene and drug delivery to target specific cell types. The M13 phage particle has a diameter of about 6 nm and about 900 nm long, which looks like a long thread under an electron microscope. This flexible nano-scale filament has been exploited as a template for the synthesis of semiconducting nanowires and lithium ion battery electrodes[154]. Several studies have shown that filamentous single-stranded bacteriophages can transfer genetic materials into mammalian cells in the presence of DEAE dextran and lipopolyamine[155]. In 1994, Hart et al[156] demonstrated that filamentous bacteriophages can be targeted to the cell surface and shown to be internalized with peptides. Larocca et al[157] showed that bacteriophage harboring the gene for growth factors are capable of delivering the gene into mammalian cells. Phages are potential gene and drug delivery systems for several main reasons. They lack tropism for eukaryotic cells; therefore phage vectors are safer than animal viruses. They are also physically stable and can be produced in large amount at lower cost. Besides, they can be cultured easily up to industrial scale, their coat proteins protect internal genetic materials from cellular degradation and they are physically stable within a wide range of temperature and pH[158]. Before a phage can be used as a drug or gene delivery vehicle for human, the specificity of the phages towards bacteria in human must be studies in depth. This could prevent or reduce the change of microbiome patterns in a healthy individual.
An interaction between HBV envelope protein, particularly the PreS regions, and a specific cell surface receptor is believed to be the initial step of HBV infection through attachment to hepatocytes. In order to develop a gene delivery system, the PreS regions were displayed on the pIII protein of phage M13[41] and used to transfect human hepatocarcinoma cell line HepG2[159]. The recombinant phage, namely M13-PreS, was shown to transfect HepG2 cells but the efficiency was very low, which could be attributed to the low valency of the displayed ligands. In order to improve the level of transfection, two fragments of the PreS1 region, residues 1-47 and 60-108, were fused separately to the 10B protein and 415 copies of these fragments were successfully displayed on the surface of phage T7. The recombinant phage displaying the amino acids 60-108 was shown to be the most effective in transfecting and internalizing into HepG2 cells in a dose- and time-dependent manner[160]. The capability of the phage to transfect HepG2 cells indicates the potential of the phage display system as a gene or drug delivery system for HCC. In an in vivo study, Zhang et al[161] further demonstrated that a filamentous phage bearing the amino acid sequence FQHPSFI bound to tumor cells following intravenous injections into BALB/c mice. This reaffirms the potential of phage display as a drug or gene delivery system to target HCC.
A combination of phages and other virus-like particles, for instance HBV capsid, has broadened their applications in gene and drug delivery. HBV capsid is made of 240 or 180 copies of HBcAg which form large and small icosahedral structures with triangulation number T = 4 or T = 3, respectively[162]. For the past 30 years, HBV capsid has been used extensively as molecular carrier for foreign epitopes in the development of multi-component vaccines (for reviews, see[33,34,163]). In addition, Lee et al[164] demonstrated that HBV capsid produced in E. coli can be dissociated into dimers by urea and guanidine hydrochloride (GdnHCl) and reassembled when the denaturing agents were removed by dialysis. This feature allows the capsid to package molecules in its empty inner cavity and can be further developed into a vehicle for delivering therapeutic molecules into a targeted organ. Cryo-electron microscopy[162] and X-ray crystallography[165,166] revealed that both the large and small HBV capsids are spiky and each spike is formed by four α-helix bundles, two from each HBcAg monomer. Affinity selection of HBV capsid with a hexapeptide phage display library, has successfully isolated peptides with the core motif LLGRMK[108]. Subsequently, the binding site of peptides containing this motif sequence was located precisely at the tip of the spike of HBV capsid by using cryoelectron microscopy[119] and mass-spectrometry[120]. A 12-mer peptide GGGSLLGRMKGA containing this motif sequence was designed as a ‘‘nanoglue’’ to display cell-internalizing peptides (CIP) of HeLa cells on the surface of HBV capsid[167]. Astonishingly, the HBV capsid delivered its cargo, be it oligonucleotides or fluorescein molecules into HeLa cells specifically[167]. Thus, a combination of phage display, viral-like particles (VLPs) and the nanoglue concept creates innovative applications, not only to combat HBV, but other diseases.
CONCLUSION
In general, mass immunization of world population with the recombinant HBV vaccine produced in yeast is an enormous success. HBV prevalence has now dropped dramatically and of course countless lives have been saved. For pharmaceutical industry, the recombinant vaccine has grown into a multi-billion-dollar business. Initially the market was monopoly by two giant pharmaceutical companies, but now many biotechnology companies are mushrooming and penetrating into this industry. Are these newly established companies employed the conventional technology to produce the same vaccine or are they going to invest and introduce a novel vaccine which is cheaper and more effective? If innovation is the only solution, phage display provides an alternative means for scientists and entrepreneurs to invent and introduce a novel HBV vaccine into the market. The strategies that can be used to achieve this aim are not only limited to; (1) display of immunogens on phage particles; (2) identification of epitope or mimotope mimics from sera; and (3) phage-delivered DNA vaccine. Although, the phage-based immunogens yielded promising results in animal immunizations, could they survive the stringent test of the time (about 10 years) and cost of clinical trials as well as product development, which would at least amount to about US$ 300 million? Economically, can they compete with the existing effective recombinant vaccine?
HBV is commonly used as a proof-of-concept target for novel diagnostic technologies due to its threat on global public health. The ultimate aims of these new technologies are: (1) to attain the highest sensitivity and specificity in the shortest time; (2) to obtain as much information as possible in a single test with the cheapest cost; and (3) to achieve simplicity. For the detection of HBV antigens in sera by using ELISA, the type of antibody mostly used is the IgG molecule, either raised in animals or produced via hybridoma technology. The IgG is a relative large and complex molecule with disulfide bonds and glycosylation which reduce its stability during storage and transportation. In addition, its production is laborious, time consuming and costly. These limitations can be overcome by phage display technology which allows the antigen recognition fragments of antibodies to be engineered and selected from a combinatorial library. Many studies have demonstrated the potential of phage display antibodies and peptides as diagnostic reagents for detecting HBV serological markers. Most recently, we have proven that phage-ELISA can be combined with real-time PCR for the detection of HBV antigen.
Phage display technology offers the opportunities to display vast number of peptide sequences on the surface of the minor or major coat proteins of bacteriophages. Affinity selection through biopanning has led to the identification of ligands that interact with the target molecules. Using liver cell lines, anti-viral activities of the isolated peptides/antibody fragments were demonstrated to inhibit intracellular and extracellular steps of HBV life cycle and subsequently reduced the HBV replication rate significantly. To date, most of the isolated therapeutic agents and their derivatives are targeting the viral proteins particularly the PreS1 region and HBcAg instead of the host cell proteins, although several authentic host proteins were discovered. These host cell proteins involved directly or indirectly in the viral attachment and penetration. It is believed that a set of periplasmic and cytoplamic proteins is involved in a cascade of reactions that lead to the internalization of the viral particles. In addition to current anti-viral therapeutics, discovery of therapeutic agents targeting the host cell proteins involved in the viral infection process is another potential strategy to combat HBV replication and infection.
The most heavily mapped HBV protein is the PreS1 region due to its clinical importance in vaccine design, diagnostic and therapeutic purposes. The S-HBsAg was also mapped considerably due to its abundances in the serum of HBV infected individuals. Although HBcAg is important for diagnostic purposes, its epitope has not been mapped extensively particularly with phage display technology. This is partly due to the availability of the 3-dimensional structure of HBcAg capsid which provides structural basis for the understanding of antibody-antigen binding. The epitopes of HBcAg capsid have been mapped extensively and precisely by examining the complex of core shell with antibodies using cryo-electron microscopy[168-171]. In contrast, the 3-dimensional structure of HBsAg has not been elucidated, probably due to its heterogeneity owing to high lipid content and extensive disulfide linkages. Thus, biophysical determination of epitope with other means is essential to map the epitope of HBsAg. Without the ‘golden standard’ mapping approaches such as X-ray crystallography, cryo-electron microscopy and NMR analysis, the pre-defined peptide epitopes have to be validated by various means which include mutagenesis, peptide scanning, cell culture and animal immunization. Furthermore, interaction between the phages and the antibody provides a means in terms of physical dynamics, which is greatly affected by physical parameters such as pH, temperature, ionic strength, and the peptide sequence polarity. It is common that a synthetic peptide alone behaves differently from the corresponding phage fusion peptide. Optimization of the internal or flanking sequence, sometime, is required to improve the binding efficacy.
The ultimate aim of drug or gene delivery is efficient conveyance of bioactive compounds to a specific target. The activity of the delivered substance is maintained for a desired length of time. The delivery vehicle must be biologically stable under physiological conditions. The packaging materials must be carefully chosen in order to protect the cargo. HBcAg with its unique capability to self-assemble into VLPs under permissive conditions is an excellent packaging material for foreign substances. Denaturing and renaturing of HBcAg VLPs allow therapeutic molecules to be packaged and protected inside the VLPs. These therapeutics molecules can be targeted specifically to an organ or a tissue by displaying CIPs on VLPs via the nanoglue concept. Nevertheless, the stability, solubility, specificity, and bioavailability of the nano-delivery vehicles and their therapeutic compounds must be studied thoroughly.
The innovative applications of phage display in epitope mapping and the development of vaccines, therapeutic agents, diagnostic reagents, as well as gene and drug delivery systems are not limited to HBV, they can be further extended to other microorganisms.
ACKNOWLEDGMENTS
This article is dedicated to Prof. Sir Kenneth Murray (1930-2013) who invented the first effective recombinant vaccine against HBV and saved countless lives worldwide. We thank him most sincerely for his care and love. His passion in experiments, science, commercialization and charity always inspires creativity and innovation in every aspect of our lives.
Footnotes
P- Reviewer: Arai M, Sabahi F, Song MJ S- Editor: Ma YJ L- Editor: A E- Editor: Wang CH
References
- 1.Michel ML, Tiollais P. Hepatitis B vaccines: protective efficacy and therapeutic potential. Pathol Biol (Paris) 2010;58:288–295. doi: 10.1016/j.patbio.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Blumberg BS. Polymorphisms of the serum proteins and the development of iso-precipitins in transfused patients. Bull N Y Acad Med. 1964;40:377–386. [PMC free article] [PubMed] [Google Scholar]
- 3.Dane DS, Cameron CH, Briggs M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet. 1970;1:695–698. doi: 10.1016/s0140-6736(70)90926-8. [DOI] [PubMed] [Google Scholar]
- 4.Seitz S, Urban S, Antoni C, Böttcher B. Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J. 2007;26:4160–4167. doi: 10.1038/sj.emboj.7601841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruss V. Envelopment of the hepatitis B virus nucleocapsid. Virus Res. 2004;106:199–209. doi: 10.1016/j.virusres.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 6.Chai N, Chang HE, Nicolas E, Han Z, Jarnik M, Taylor J. Properties of subviral particles of hepatitis B virus. J Virol. 2008;82:7812–7817. doi: 10.1128/JVI.00561-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shasha SM, Sharon N, Inbar M. [Bacteriophages as antibacterial agents] Harefuah. 2004;143:121–125, 166. [PubMed] [Google Scholar]
- 8.Voet D, Voet JG, Pratt CW. Fundamentals of Biochemistry. 2nd ed. Asia: John Wiley and Sons (Asia) Pte Ltd; 2006. [Google Scholar]
- 9.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 10.Baruch S. Blumberg - Biographical. 2013. Available from: http://www.nobelprize.org/nobel_prizes/medicine/laureates/1976/blumberg-bio.html.
- 11.Muraskin W. The war against hepatitis B: a history of the international task force on hepatitis B immunization. Philadelphia: University of Pennsylvania Press; 1995. [Google Scholar]
- 12.Burrell CJ, Mackay P, Greenaway PJ, Hofschneider PH, Murray K. Expression in Escherichia coli of hepatitis B virus DNA sequences cloned in plasmid pBR322. Nature. 1979;279:43–47. doi: 10.1038/279043a0. [DOI] [PubMed] [Google Scholar]
- 13.Pasek M, Goto T, Gilbert W, Zink B, Schaller H, MacKay P, Leadbetter G, Murray K. Hepatitis B virus genes and their expression in E. coli. Nature. 1979;282:575–579. doi: 10.1038/282575a0. [DOI] [PubMed] [Google Scholar]
- 14.Hofschneider P, Murray K. Combining science and business: from recombinant DNA to vaccines against hepatitis B virus. In: Buckel P, editor. Recombinant protein drugs. Basel, Switzerland: Birkhäuser Verlag; 2001. pp. 43–64. [Google Scholar]
- 15.Valenzuela P, Medina A, Rutter WJ, Ammerer G, Hall BD. Synthesis and assembly of hepatitis B virus surface antigen particles in yeast. Nature. 1982;298:347–350. doi: 10.1038/298347a0. [DOI] [PubMed] [Google Scholar]
- 16.Hitzeman RA, Chen CY, Hagie FE, Patzer EJ, Liu CC, Estell DA, Miller JV, Yaffe A, Kleid DG, Levinson AD, et al. Expression of hepatitis B virus surface antigen in yeast. Nucleic Acids Res. 1983;11:2745–2763. doi: 10.1093/nar/11.9.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyanohara A, Toh-e A, Nozaki C, Hamada F, Ohtomo N, Matsubara K. Expression of hepatitis B surface antigen gene in yeast. Proc Natl Acad Sci USA. 1983;80:1–5. doi: 10.1073/pnas.80.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. Human hepatitis B vaccine from recombinant yeast. Nature. 1984;307:178–180. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- 19.Murray K, Bruce SA, Hinnen A, Wingfield P, van Erd PM, de Reus A, Schellekens H. Hepatitis B virus antigens made in microbial cells immunise against viral infection. EMBO J. 1984;3:645–650. doi: 10.1002/j.1460-2075.1984.tb01861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.William J. Rutter - A history of UCSF. 2014. Available from: http://history.library.ucsf.edu/rutter.html.
- 21.Frost LJ, Reich MR. Access: how do good health technologies get to poor people in poor countries? 1st ed. Cambridge, MA: Harvard Center for Population and Development Studies; 2009. pp. 68–90. [Google Scholar]
- 22.Chen DS. Toward elimination and eradication of hepatitis B. J Gastroenterol Hepatol. 2010;25:19–25. doi: 10.1111/j.1440-1746.2009.06165.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen DS. Hepatitis B vaccination: The key towards elimination and eradication of hepatitis B. J Hepatol. 2009;50:805–816. doi: 10.1016/j.jhep.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Chang MH, Chen CJ, Lai MS, Hsu HM, Wu TC, Kong MS, Liang DC, Shau WY, Chen DS. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336:1855–1859. doi: 10.1056/NEJM199706263362602. [DOI] [PubMed] [Google Scholar]
- 25.Zanetti AR, Tanzi E, Manzillo G, Maio G, Sbreglia C, Caporaso N, Thomas H, Zuckerman AJ. Hepatitis B variant in Europe. Lancet. 1988;2:1132–1133. doi: 10.1016/s0140-6736(88)90541-7. [DOI] [PubMed] [Google Scholar]
- 26.Coleman PF. Detecting hepatitis B surface antigen mutants. Emerg Infect Dis. 2006;12:198–203. doi: 10.3201/eid1202.050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuckerman JN, Zuckerman AJ. Mutations of the surface protein of hepatitis B virus. Antiviral Res. 2003;60:75–78. doi: 10.1016/j.antiviral.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Pawlotsky JM. The concept of hepatitis B virus mutant escape. J Clin Virol. 2005;34 Suppl 1:S125–S129. doi: 10.1016/s1386-6532(05)80021-6. [DOI] [PubMed] [Google Scholar]
- 29.Lapiński TW, Pogorzelska J, Flisiak R. HBV mutations and their clinical significance. Adv Med Sci. 2012;57:18–22. doi: 10.2478/v10039-012-0006-x. [DOI] [PubMed] [Google Scholar]
- 30.van den Berg R, van Hoogstraten I, van Agtmael M. Non-responsiveness to hepatitis B vaccination in HIV seropositive patients; possible causes and solutions. AIDS Rev. 2009;11:157–164. [PubMed] [Google Scholar]
- 31.Thio CL, Seaberg EC, Skolasky R, Phair J, Visscher B, Muñoz A, Thomas DL. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS) Lancet. 2002;360:1921–1926. doi: 10.1016/s0140-6736(02)11913-1. [DOI] [PubMed] [Google Scholar]
- 32.Laurence JC. Hepatitis A and B immunizations of individuals infected with human immunodeficiency virus. Am J Med. 2005;118 Suppl 10A:75S–83S. doi: 10.1016/j.amjmed.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 33.Murray K, Shiau AL. The core antigen of hepatitis B virus as a carrier for immunogenic peptides. Biol Chem. 1999;380:277–283. doi: 10.1515/BC.1999.038. [DOI] [PubMed] [Google Scholar]
- 34.Pumpens P, Grens E. HBV core particles as a carrier for B cell/T cell epitopes. Intervirology. 2001;44:98–114. doi: 10.1159/000050037. [DOI] [PubMed] [Google Scholar]
- 35.Kim EJ, Yoo SK. Cell surface display of hepatitis B virus surface antigen by using Pseudomonas syringae ice nucleation protein. Lett Appl Microbiol. 1999;29:292–297. doi: 10.1046/j.1365-2672.1999.00612.x. [DOI] [PubMed] [Google Scholar]
- 36.Peng ML, Chen M, Ren H. [Immune adjuvant effect of TB.HSP70 to its accompanying cytotoxic T lymphocytes epitope elicits HBV specific immune response] Zhonghua Gan Zang Bing Za Zhi. 2006;14:406–409. [PubMed] [Google Scholar]
- 37.Yang BF, Zhao HL, Xue C, Xiong XH, Zhang W, Yao XQ, Liu ZM. Recombinant heat shock protein 65 carrying hepatitis B core antigen induces HBcAg-specific CTL response. Vaccine. 2007;25:4478–4486. doi: 10.1016/j.vaccine.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 38.Tan GH, Yusoff K, Seow HF, Tan WS. Antigenicity and immunogenicity of the immunodominant region of hepatitis B surface antigen displayed on bacteriophage T7. J Med Virol. 2005;77:475–480. doi: 10.1002/jmv.20479. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg A, Griffin K, Studier W, McCormick M, Berg J, Novy R, Mierendorf R. T7 Select phage display system: A powerful new protein display system based on bacteriophage T7. InNovations. 1996;6:1–6. [Google Scholar]
- 40.Wan Y, Wu Y, Bian J, Wang XZ, Zhou W, Jia ZC, Tan Y, Zhou L. Induction of hepatitis B virus-specific cytotoxic T lymphocytes response in vivo by filamentous phage display vaccine. Vaccine. 2001;19:2918–2923. doi: 10.1016/s0264-410x(00)00561-2. [DOI] [PubMed] [Google Scholar]
- 41.Kok WL, Yusoff K, Nathan S, Tan WS. Cloning, expression and display of the PreS domain of hepatitis B virus on filamentous bacteriophage M13. J Biochem Mol Biol Biophys. 2002;6:55–58. doi: 10.1080/10258140290010241. [DOI] [PubMed] [Google Scholar]
- 42.Bahadir AO, Balcioglu BK, Uzyol KS, Hatipoglu I, Sogut I, Basalp A, Erdag B. Phage displayed HBV core antigen with immunogenic activity. Appl Biochem Biotechnol. 2011;165:1437–1447. doi: 10.1007/s12010-011-9365-1. [DOI] [PubMed] [Google Scholar]
- 43.Greenwood J, Willis AE, Perham RN. Multiple display of foreign peptides on a filamentous bacteriophage. Peptides from Plasmodium falciparum circumsporozoite protein as antigens. J Mol Biol. 1991;220:821–827. doi: 10.1016/0022-2836(91)90354-9. [DOI] [PubMed] [Google Scholar]
- 44.van Houten NE, Zwick MB, Menendez A, Scott JK. Filamentous phage as an immunogenic carrier to elicit focused antibody responses against a synthetic peptide. Vaccine. 2006;24:4188–4200. doi: 10.1016/j.vaccine.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Houten NE, Scott JK. Phage libraries for developing antibody targeted diagnostics and vaccines. In: Sidhu SS, editor. Phage display in biotechnology and drug discovery. Boca Raton: CRC Press/Taylor & Francis; 2005. pp. 165–254. [Google Scholar]
- 46.Eriksson F, Tsagozis P, Lundberg K, Parsa R, Mangsbo SM, Persson MA, Harris RA, Pisa P. Tumor-specific bacteriophages induce tumor destruction through activation of tumor-associated macrophages. J Immunol. 2009;182:3105–3111. doi: 10.4049/jimmunol.0800224. [DOI] [PubMed] [Google Scholar]
- 47.Grabowska AM, Jennings R, Laing P, Darsley M, Jameson CL, Swift L, Irving WL. Immunisation with phage displaying peptides representing single epitopes of the glycoprotein G can give rise to partial protective immunity to HSV-2. Virology. 2000;269:47–53. doi: 10.1006/viro.2000.0185. [DOI] [PubMed] [Google Scholar]
- 48.Willis AE, Perham RN, Wraith D. Immunological properties of foreign peptides in multiple display on a filamentous bacteriophage. Gene. 1993;128:79–83. doi: 10.1016/0378-1119(93)90156-w. [DOI] [PubMed] [Google Scholar]
- 49.van Houten NE, Henry KA, Smith GP, Scott JK. Engineering filamentous phage carriers to improve focusing of antibody responses against peptides. Vaccine. 2010;28:2174–2185. doi: 10.1016/j.vaccine.2009.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shivachandra SB, Li Q, Peachman KK, Matyas GR, Leppla SH, Alving CR, Rao M, Rao VB. Multicomponent anthrax toxin display and delivery using bacteriophage T4. Vaccine. 2007;25:1225–1235. doi: 10.1016/j.vaccine.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Persing DH, Varmus HE, Ganem D. The preS1 protein of hepatitis B virus is acylated at its amino terminus with myristic acid. J Virol. 1987;61:1672–1677. doi: 10.1128/jvi.61.5.1672-1677.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bruss V, Hagelstein J, Gerhardt E, Galle PR. Myristylation of the large surface protein is required for hepatitis B virus in vitro infectivity. Virology. 1996;218:396–399. doi: 10.1006/viro.1996.0209. [DOI] [PubMed] [Google Scholar]
- 53.Duclos-Vallée JC, Capel F, Mabit H, Petit MA. Phosphorylation of the hepatitis B virus core protein by glyceraldehyde-3-phosphate dehydrogenase protein kinase activity. J Gen Virol. 1998;79(Pt 7):1665–1670. doi: 10.1099/0022-1317-79-7-1665. [DOI] [PubMed] [Google Scholar]
- 54.Folgori A, Tafi R, Meola A, Felici F, Galfré G, Cortese R, Monaci P, Nicosia A. A general strategy to identify mimotopes of pathological antigens using only random peptide libraries and human sera. EMBO J. 1994;13:2236–2243. doi: 10.1002/j.1460-2075.1994.tb06501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meola A, Delmastro P, Monaci P, Luzzago A, Nicosia A, Felici F, Cortese R, Galfrè G. Derivation of vaccines from mimotopes. Immunologic properties of human hepatitis B virus surface antigen mimotopes displayed on filamentous phage. J Immunol. 1995;154:3162–3172. [PubMed] [Google Scholar]
- 56.Delmastro P, Meola A, Monaci P, Cortese R, Galfrè G. Immunogenicity of filamentous phage displaying peptide mimotopes after oral administration. Vaccine. 1997;15:1276–1285. doi: 10.1016/s0264-410x(97)00072-8. [DOI] [PubMed] [Google Scholar]
- 57.Davis HL, Michel ML, Whalen RG. DNA-based immunization induces continuous secretion of hepatitis B surface antigen and high levels of circulating antibody. Hum Mol Genet. 1993;2:1847–1851. doi: 10.1093/hmg/2.11.1847. [DOI] [PubMed] [Google Scholar]
- 58.Mancini M, Hadchouel M, Davis HL, Whalen RG, Tiollais P, Michel ML. DNA-mediated immunization in a transgenic mouse model of the hepatitis B surface antigen chronic carrier state. Proc Natl Acad Sci USA. 1996;93:12496–12501. doi: 10.1073/pnas.93.22.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark JR, March JB. Bacteriophage-mediated nucleic acid immunisation. FEMS Immunol Med Microbiol. 2004;40:21–26. doi: 10.1016/S0928-8244(03)00344-4. [DOI] [PubMed] [Google Scholar]
- 60.March JB, Clark JR, Jepson CD. Genetic immunisation against hepatitis B using whole bacteriophage lambda particles. Vaccine. 2004;22:1666–1671. doi: 10.1016/j.vaccine.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 61.Clark JR, Bartley K, Jepson CD, Craik V, March JB. Comparison of a bacteriophage-delivered DNA vaccine and a commercially available recombinant protein vaccine against hepatitis B. FEMS Immunol Med Microbiol. 2011;61:197–204. doi: 10.1111/j.1574-695X.2010.00763.x. [DOI] [PubMed] [Google Scholar]
- 62.Hatzakis A, Magiorkinis E, Haida C. HBV virological assessment. J Hepatol. 2006;44:S71–S76. doi: 10.1016/j.jhep.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 63.Said ZN. An overview of occult hepatitis B virus infection. World J Gastroenterol. 2011;17:1927–1938. doi: 10.3748/wjg.v17.i15.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lander JJ, Alter HJ, Purcell RH. Frequency of antibody to hepatitis-associated antigen as measured by a new radioimmunoassay technique. J Immunol. 1971;106:1166–1171. [PubMed] [Google Scholar]
- 65.Ling CM, Overby LR. Prevalence of hepatitis B virus antigen as revealed by direct radioimmune assay with 125 I-antibody. J Immunol. 1972;109:834–841. [PubMed] [Google Scholar]
- 66.Purcell RH, Wong DC, Alter HJ, Holland PV. Microtiter solid-phase radioimmunoassay for hepatitis B antigen. Appl Microbiol. 1973;26:478–484. doi: 10.1128/am.26.4.478-484.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolters G, Kuijpers L, Kacaki J, Schuurs A. Solid-phase enzyme-immunoassay for detection of hepatitis B surface antigen. J Clin Pathol. 1976;29:873–879. doi: 10.1136/jcp.29.10.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Waart M, Snelting A, Cichy J, Niermeijer P, Gips CH, Huizenga JR, Schuurs A. The hepatitis B-related ‘e’ antigen-antibody system as measured by enzyme-immunoassay [proceedings] Antonie Van Leeuwenhoek. 1978;44:461–462. doi: 10.1007/BF00394327. [DOI] [PubMed] [Google Scholar]
- 69.von der Waart M, Snelting A, Cichy J, Wolters G, Schuurs A. Enzyme-immunoassay in diagnosis of hepatitis with emphasis on the detection of “e” antigen (HBeAg) J Med Virol. 1978;3:43–49. doi: 10.1002/jmv.1890030111. [DOI] [PubMed] [Google Scholar]
- 70.Gerlich WH, Lüer W. Selective detection of IgM-antibody against core antigen of the hepatitis B virus by a modified enzyme immune assay. J Med Virol. 1979;4:227–238. doi: 10.1002/jmv.1890040308. [DOI] [PubMed] [Google Scholar]
- 71.Bredehorst R, von Wulffen H, Granato C. Quantitation of hepatitis B virus (HBV) core antigen in serum in the presence of antibodies to HBV core antigen: comparison with assays of serum HBV DNA, DNA polymerase, and HBV e antigen. J Clin Microbiol. 1985;21:593–598. doi: 10.1128/jcm.21.4.593-598.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korec E, Dostálová V, Korcová J, Mancal P, König J, Borisova G, Cibinogen V, Pumpen P, Gren E, Hlozánek I. Monoclonal antibodies against hepatitis B e antigen: production, characterization, and use for diagnosis. J Virol Methods. 1990;28:165–169. doi: 10.1016/0166-0934(90)90031-a. [DOI] [PubMed] [Google Scholar]
- 73.Jardi R, Rodriguez F, Buti M, Costa X, Cotrina M, Valdes A, Galimany R, Esteban R, Guardia J. Quantitative detection of hepatitis B virus DNA in serum by a new rapid real-time fluorescence PCR assay. J Viral Hepat. 2001;8:465–471. doi: 10.1046/j.1365-2893.2001.00322.x. [DOI] [PubMed] [Google Scholar]
- 74.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 75.Hust M, Dübel S. Phage display vectors for the in vitro generation of human antibody fragments. Methods Mol Biol. 2005;295:71–96. doi: 10.1385/1-59259-873-0:071. [DOI] [PubMed] [Google Scholar]
- 76.Hoet RM, Cohen EH, Kent RB, Rookey K, Schoonbroodt S, Hogan S, Rem L, Frans N, Daukandt M, Pieters H, et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat Biotechnol. 2005;23:344–348. doi: 10.1038/nbt1067. [DOI] [PubMed] [Google Scholar]
- 77.Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003;21:484–490. doi: 10.1016/j.tibtech.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 78.Tan GH, Yusoff K, Seow HF, Tan WS. A phage-displayed single chain variable fragment that interacts with hepatitis B core antigen: library construction, selection and diagnosis. J Clin Virol. 2007;38:49–56. doi: 10.1016/j.jcv.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 79.Kimura T, Rokuhara A, Matsumoto A, Yagi S, Tanaka E, Kiyosawa K, Maki N. New enzyme immunoassay for detection of hepatitis B virus core antigen (HBcAg) and relation between levels of HBcAg and HBV DNA. J Clin Microbiol. 2003;41:1901–1906. doi: 10.1128/JCM.41.5.1901-1906.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim SJ, Jang MH, Ahn HJ, Kim JH, Lim JH, Ryu CJ, Lim NK, Kim KS, Park MJ, Park I, et al. Selection of an affinity-matured antibody against a defined epitope by phage display of an immune antibody library. J Immunol Methods. 2008;329:176–183. doi: 10.1016/j.jim.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 81.Tan WS, Tan GH, Yusoff K, Seow HF. A phage-displayed cyclic peptide that interacts tightly with the immunodominant region of hepatitis B surface antigen. J Clin Virol. 2005;34:35–41. doi: 10.1016/j.jcv.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 82.Muhamad A, Ho KL, Rahman MB, Uhrín D, Tan WS. Solution structure and in silico binding of a cyclic peptide with hepatitis B surface antigen. Chem Biol Drug Des. 2013;81:784–794. doi: 10.1111/cbdd.12120. [DOI] [PubMed] [Google Scholar]
- 83.Hasmoni SS, Yusoff K, Tan WS. Detection and precipitation of hepatitis B core antigen using a fusion bacteriophage. J Gen Appl Microbiol. 2005;51:125–131. doi: 10.2323/jgam.51.125. [DOI] [PubMed] [Google Scholar]
- 84.Ho KL, Yusoff K, Seow HF, Tan WS. Selection of high affinity ligands to hepatitis B core antigen from a phage-displayed cyclic peptide library. J Med Virol. 2003;69:27–32. doi: 10.1002/jmv.10266. [DOI] [PubMed] [Google Scholar]
- 85.Monjezi R, Tan SW, Tey BT, Sieo CC, Tan WS. Detection of hepatitis B virus core antigen by phage display mediated TaqMan real-time immuno-PCR. J Virol Methods. 2013;187:121–126. doi: 10.1016/j.jviromet.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 86.Gough KC, Cockburn W, Whitelam GC. Selection of phage-display peptides that bind to cucumber mosaic virus coat protein. J Virol Methods. 1999;79:169–180. doi: 10.1016/s0166-0934(99)00014-2. [DOI] [PubMed] [Google Scholar]
- 87.Ramanujam P, Tan WS, Nathan S, Yusoff K. Pathotyping of Newcastle disease virus with a filamentous bacteriophage. Biotechniques. 2004;36:296–300, 302. doi: 10.2144/04362RR04. [DOI] [PubMed] [Google Scholar]
- 88.Lee TC, Yusoff K, Nathan S, Tan WS. Detection of virulent Newcastle disease virus using a phage-capturing dot blot assay. J Virol Methods. 2006;136:224–229. doi: 10.1016/j.jviromet.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 89.Tang SS, Tan WS, Devi S, Wang LF, Pang T, Thong KL. Mimotopes of the Vi antigen of Salmonella enterica serovar typhi identified from phage display peptide library. Clin Diagn Lab Immunol. 2003;10:1078–1084. doi: 10.1128/CDLI.10.6.1078-1084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thong KL, Tang SS, Tan WS, Devi S. Peptide mimotopes of complex carbohydrates in Salmonella enterica serovar typhi which react with both carbohydrate-specific monoclonal antibody and polyclonal sera from typhoid patients. Microbiol Immunol. 2007;51:1045–1052. doi: 10.1111/j.1348-0421.2007.tb03997.x. [DOI] [PubMed] [Google Scholar]
- 91.Eshaghi M, Tan WS, Yusoff K. Identification of epitopes in the nucleocapsid protein of Nipah virus using a linear phage-displayed random peptide library. J Med Virol. 2005;75:147–152. doi: 10.1002/jmv.20249. [DOI] [PubMed] [Google Scholar]
- 92.Palacios-Rodríguez Y, Gazarian T, Rowley M, Majluf-Cruz A, Gazarian K. Collection of phage-peptide probes for HIV-1 immunodominant loop-epitope. J Microbiol Methods. 2007;68:225–235. doi: 10.1016/j.mimet.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 93.Smith GP, Petrenko VA. Phage Display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 94.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 95.Castel G, Chtéoui M, Heyd B, Tordo N. Phage display of combinatorial peptide libraries: application to antiviral research. Molecules. 2011;16:3499–3518. doi: 10.3390/molecules16053499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang JL, Gou JJ, Zhang ZY, Jing YX, Zhang L, Guo R, Yan P, Cheng NL, Niu B, Xie J. Screening and evaluation of human single-chain fragment variable antibody against hepatitis B virus surface antigen. Hepatobiliary Pancreat Dis Int. 2006;5:237–241. [PubMed] [Google Scholar]
- 97.Zhang ZC, Hu XJ, Yang Q. Generation of high affinity human single-chain antibody against PreS1 of hepatitis B virus from immune phage-display antibody library. Hepatobiliary Pancreat Dis Int. 2004;3:77–81. [PubMed] [Google Scholar]
- 98.Park SG, Jeong YJ, Lee YY, Kim IJ, Seo SK, Kim EJ, Jung HC, Pan JG, Park SJ, Lee YJ, et al. Hepatitis B virus-neutralizing anti-pre-S1 human antibody fragments from large naïve antibody phage library. Antiviral Res. 2005;68:109–115. doi: 10.1016/j.antiviral.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 99.Yang GH, Yoon SO, Jang MH, Hong HJ. Affinity maturation of an anti-hepatitis B virus PreS1 humanized antibody by phage display. J Microbiol. 2007;45:528–533. [PubMed] [Google Scholar]
- 100.Jia L, Yu J, Song H, Liu X, Ma W, Xu Y, Zhang C, Dong S, Li Q. Screening of human antibody Fab fragment against HBsAg and the construction of its dsFv form. Int J Biol Sci. 2008;4:103–110. doi: 10.7150/ijbs.4.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tiwari A, Dutta D, Khanna N, Acharya SK, Sinha S. Generation and characterization of high affinity humanized fab against hepatitis B surface antigen. Mol Biotechnol. 2009;43:29–40. doi: 10.1007/s12033-009-9165-9. [DOI] [PubMed] [Google Scholar]
- 102.Kim SH, Park SY. Selection and characterization of human antibodies against hepatitis B virus surface antigen (HBsAg) by phage-display. Hybrid Hybridomics. 2002;21:385–392. doi: 10.1089/153685902761022742. [DOI] [PubMed] [Google Scholar]
- 103.Bose B, Khanna N, Acharya SK, Sinha S. High affinity mouse-human chimeric Fab against hepatitis B surface antigen. World J Gastroenterol. 2005;11:7569–7578. doi: 10.3748/wjg.v11.i48.7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deng Q, Zhai JW, Michel ML, Zhang J, Qin J, Kong YY, Zhang XX, Budkowska A, Tiollais P, Wang Y, et al. Identification and characterization of peptides that interact with hepatitis B virus via the putative receptor binding site. J Virol. 2007;81:4244–4254. doi: 10.1128/JVI.01270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deng Q, Kong YY, Xie YH, Wang Y. Expression and purification of the complete PreS region of hepatitis B Virus. World J Gastroenterol. 2005;11:3060–3064. doi: 10.3748/wjg.v11.i20.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Deng Q, Zhuang M, Kong YY, Xie YH, Wang Y. Screening for PreS specific binding ligands with a phage displayed peptides library. World J Gastroenterol. 2005;11:4018–4023. doi: 10.3748/wjg.v11.i26.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang W, Liu Y, Zu X, Jin R, Xiao G. Blocking peptides against HBV: preS1 protein selected from a phage display library. Biochem Biophys Res Commun. 2011;412:633–637. doi: 10.1016/j.bbrc.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 108.Dyson MR, Murray K. Selection of peptide inhibitors of interactions involved in complex protein assemblies: association of the core and surface antigens of hepatitis B virus. Proc Natl Acad Sci USA. 1995;92:2194–2198. doi: 10.1073/pnas.92.6.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lobato MN, Rabbitts TH. Intracellular antibodies and challenges facing their use as therapeutic agents. Trends Mol Med. 2003;9:390–396. doi: 10.1016/s1471-4914(03)00163-1. [DOI] [PubMed] [Google Scholar]
- 110.Serruys B, Van Houtte F, Farhoudi-Moghadam A, Leroux-Roels G, Vanlandschoot P. Production, characterization and in vitro testing of HBcAg-specific VHH intrabodies. J Gen Virol. 2010;91:643–652. doi: 10.1099/vir.0.016063-0. [DOI] [PubMed] [Google Scholar]
- 111.Serruys B, Van Houtte F, Verbrugghe P, Leroux-Roels G, Vanlandschoot P. Llama-derived single-domain intrabodies inhibit secretion of hepatitis B virions in mice. Hepatology. 2009;49:39–49. doi: 10.1002/hep.22609. [DOI] [PubMed] [Google Scholar]
- 112.Walsh R, Nuttall S, Revill P, Colledge D, Cabuang L, Soppe S, Dolezal O, Griffiths K, Bartholomeusz A, Locarnini S. Targeting the hepatitis B virus precore antigen with a novel IgNAR single variable domain intrabody. Virology. 2011;411:132–141. doi: 10.1016/j.virol.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 113.Kann M, Schmitz A, Rabe B. Intracellular transport of hepatitis B virus. World J Gastroenterol. 2007;13:39–47. doi: 10.3748/wjg.v13.i1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rabe B, Vlachou A, Panté N, Helenius A, Kann M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci USA. 2003;100:9849–9854. doi: 10.1073/pnas.1730940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nassal M. Hepatitis B virus replication: novel roles for virus-host interactions. Intervirology. 1999;42:100–116. doi: 10.1159/000024970. [DOI] [PubMed] [Google Scholar]
- 116.Butz K, Denk C, Fitscher B, Crnkovic-Mertens I, Ullmann A, Schröder CH, Hoppe-Seyler F. Peptide aptamers targeting the hepatitis B virus core protein: a new class of molecules with antiviral activity. Oncogene. 2001;20:6579–6586. doi: 10.1038/sj.onc.1204805. [DOI] [PubMed] [Google Scholar]
- 117.Deres K, Schröder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Krämer T, Niewöhner U, Pleiss U, Stoltefuss J, et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299:893–896. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- 118.Yamamoto M, Hayashi N, Takehara T, Ueda K, Mita E, Tatsumi T, Sasaki Y, Kasahara A, Hori M. Intracellular single-chain antibody against hepatitis B virus core protein inhibits the replication of hepatitis B virus in cultured cells. Hepatology. 1999;30:300–307. doi: 10.1002/hep.510300105. [DOI] [PubMed] [Google Scholar]
- 119.Böttcher B, Tsuji N, Takahashi H, Dyson MR, Zhao S, Crowther RA, Murray K. Peptides that block hepatitis B virus assembly: analysis by cryomicroscopy, mutagenesis and transfection. EMBO J. 1998;17:6839–6845. doi: 10.1093/emboj/17.23.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tang KF, Abdullah MP, Yusoff K, Tan WS. Interactions of hepatitis B core antigen and peptide inhibitors. J Med Chem. 2007;50:5620–5626. doi: 10.1021/jm070468d. [DOI] [PubMed] [Google Scholar]
- 121.Chen M, Sällberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, Billaud JN, Milich DR. Immune tolerance split between hepatitis B virus precore and core proteins. J Virol. 2005;79:3016–3027. doi: 10.1128/JVI.79.5.3016-3027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Beck J, Nassal M. Hepatitis B virus replication. World J Gastroenterol. 2007;13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schultz U, Grgacic E, Nassal M. Duck hepatitis B virus: an invaluable model system for HBV infection. Adv Virus Res. 2004;63:1–70. doi: 10.1016/S0065-3527(04)63001-6. [DOI] [PubMed] [Google Scholar]
- 124.Tong S, Li J, Wands JR. Interaction between duck hepatitis B virus and a 170-kilodalton cellular protein is mediated through a neutralizing epitope of the pre-S region and occurs during viral infection. J Virol. 1995;69:7106–7112. doi: 10.1128/jvi.69.11.7106-7112.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kuroki K, Eng F, Ishikawa T, Turck C, Harada F, Ganem D. gp180, a host cell glycoprotein that binds duck hepatitis B virus particles, is encoded by a member of the carboxypeptidase gene family. J Biol Chem. 1995;270:15022–15028. doi: 10.1074/jbc.270.25.15022. [DOI] [PubMed] [Google Scholar]
- 126.Urban S, Kruse C, Multhaup G. A soluble form of the avian hepatitis B virus receptor. Biochemical characterization and functional analysis of the receptor ligand complex. J Biol Chem. 1999;274:5707–5715. doi: 10.1074/jbc.274.9.5707. [DOI] [PubMed] [Google Scholar]
- 127.Urban S, Schwarz C, Marx UC, Zentgraf H, Schaller H, Multhaup G. Receptor recognition by a hepatitis B virus reveals a novel mode of high affinity virus-receptor interaction. EMBO J. 2000;19:1217–1227. doi: 10.1093/emboj/19.6.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dash S, Rao KV, Panda SK. Receptor for pre-S1(21-47) component of hepatitis B virus on the liver cell: role in virus cell interaction. J Med Virol. 1992;37:116–121. doi: 10.1002/jmv.1890370208. [DOI] [PubMed] [Google Scholar]
- 129.De Falco S, Ruvoletto MG, Verdoliva A, Ruvo M, Raucci A, Marino M, Senatore S, Cassani G, Alberti A, Pontisso P, et al. Cloning and expression of a novel hepatitis B virus-binding protein from HepG2 cells. J Biol Chem. 2001;276:36613–36623. doi: 10.1074/jbc.M102377200. [DOI] [PubMed] [Google Scholar]
- 130.Geysen HM, Meloen RH, Barteling SJ. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc Natl Acad Sci USA. 1984;81:3998–4002. doi: 10.1073/pnas.81.13.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 132.Gershoni JM, Roitburd-Berman A, Siman-Tov DD, Tarnovitski Freund N, Weiss Y. Epitope mapping: the first step in developing epitope-based vaccines. BioDrugs. 2007;21:145–156. doi: 10.2165/00063030-200721030-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wüthrich K. Protein structure determination in solution by NMR spectroscopy. J Biol Chem. 1990;265:22059–22062. [PubMed] [Google Scholar]
- 134.Germaschewski V, Murray K. Screening a monoclonal antibody with a fusion-phage display library shows a discontinuity in a linear epitope within PreS1 of hepatitis B virus. J Med Virol. 1995;45:300–305. doi: 10.1002/jmv.1890450311. [DOI] [PubMed] [Google Scholar]
- 135.D’Mello F, Partidos CD, Steward MW, Howard CR. Definition of the primary structure of hepatitis B virus (HBV) pre-S hepatocyte binding domain using random peptide libraries. Virology. 1997;237:319–326. doi: 10.1006/viro.1997.8774. [DOI] [PubMed] [Google Scholar]
- 136.Zhang WY, Wan Y, Li DG, Tang Y, Zhou W. A mimotope of pre-S2 region of surface antigen of viral hepatitis B screened by phage display. Cell Res. 2001;11:203–208. doi: 10.1038/sj.cr.7290087. [DOI] [PubMed] [Google Scholar]
- 137.Jolivet-Reynaud C, Lésenéchal M, O’Donnell B, Becquart L, Foussadier A, Forge F, Battail-Poirot N, Lacoux X, Carman W, Jolivet M. Localization of hepatitis B surface antigen epitopes present on variants and specifically recognised by anti-hepatitis B surface antigen monoclonal antibodies. J Med Virol. 2001;65:241–249. doi: 10.1002/jmv.2026. [DOI] [PubMed] [Google Scholar]
- 138.Chen YC, Delbrook K, Dealwis C, Mimms L, Mushahwar IK, Mandecki W. Discontinuous epitopes of hepatitis B surface antigen derived from a filamentous phage peptide library. Proc Natl Acad Sci USA. 1996;93:1997–2001. doi: 10.1073/pnas.93.5.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Germaschewski V, Murray K. Identification of polyclonal serum specificities with phage-display libraries. J Virol Methods. 1996;58:21–32. doi: 10.1016/0166-0934(95)01980-4. [DOI] [PubMed] [Google Scholar]
- 140.Motti C, Nuzzo M, Meola A, Galfré G, Felici F, Cortese R, Nicosia A, Monaci P. Recognition by human sera and immunogenicity of HBsAg mimotopes selected from an M13 phage display library. Gene. 1994;146:191–198. doi: 10.1016/0378-1119(94)90292-5. [DOI] [PubMed] [Google Scholar]
- 141.Roseman AM, Borschukova O, Berriman JA, Wynne SA, Pumpens P, Crowther RA. Structures of hepatitis B virus cores presenting a model epitope and their complexes with antibodies. J Mol Biol. 2012;423:63–78. doi: 10.1016/j.jmb.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beck J, Bartos H, Nassal M. Experimental confirmation of a hepatitis B virus (HBV) epsilon-like bulge-and-loop structure in avian HBV RNA encapsidation signals. Virology. 1997;227:500–504. doi: 10.1006/viro.1996.8329. [DOI] [PubMed] [Google Scholar]
- 143.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang P, Yu MY, Venable R, Alter HJ, Shih JW. Neutralization epitope responsible for the hepatitis B virus subtype-specific protection in chimpanzees. Proc Natl Acad Sci USA. 2006;103:9214–9219. doi: 10.1073/pnas.0603316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Itoh Y, Takai E, Ohnuma H, Kitajima K, Tsuda F, Machida A, Mishiro S, Nakamura T, Miyakawa Y, Mayumi M. A synthetic peptide vaccine involving the product of the pre-S(2) region of hepatitis B virus DNA: protective efficacy in chimpanzees. Proc Natl Acad Sci USA. 1986;83:9174–9178. doi: 10.1073/pnas.83.23.9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cupps TR, Tibbles J, Hurni WM, Miller WJ, Ellis RW, Milich D, Wetter N. In vitro T cell immune responses to the preS2 antigen of the hepatitis B virus envelope protein in preS2 + S vaccine recipients. Absence of cross-reactivity of subtypes at a major T cell recognition site. J Immunol. 1993;151:3353–3360. [PubMed] [Google Scholar]
- 147.Mimms LT, Floreani M, Tyner J, Whitters E, Rosenlof R, Wray L, Goetze A, Sarin V, Eble K. Discrimination of hepatitis B virus (HBV) subtypes using monoclonal antibodies to the PreS1 and PreS2 domains of the viral envelope. Virology. 1990;176:604–619. doi: 10.1016/0042-6822(90)90031-l. [DOI] [PubMed] [Google Scholar]
- 148.Heijtink RA, de Wilde GA, van Hattum J, Schalm SW. Long-term immune reactivity to pre-S(2)-antigen after acute hepatitis B infection. J Med Virol. 1989;27:95–99. doi: 10.1002/jmv.1890270205. [DOI] [PubMed] [Google Scholar]
- 149.Sominskaya I, Bichko V, Pushko P, Dreimane A, Snikere D, Pumpens P. Tetrapeptide QDPR is a minimal immunodominant epitope within the preS2 domain of hepatitis B virus. Immunol Lett. 1992;33:169–172. doi: 10.1016/0165-2478(92)90043-n. [DOI] [PubMed] [Google Scholar]
- 150.Sominskaya I, Paulij W, Jansons J, Sobotta D, Dreilina D, Sunnen C, Meisel H, Gerlich WH, Pumpens P. Fine-mapping of the B-cell epitope domain at the N-terminus of the preS2 region of the hepatitis B surface antigen. J Immunol Methods. 2002;260:251–261. doi: 10.1016/s0022-1759(01)00551-8. [DOI] [PubMed] [Google Scholar]
- 151.Lee MK, Kim KL, Hahm KS. Epitope mapping of preS2 of the hepatitis B virus surface antigen against a conformation-dependent monoclonal antibody using synthetic peptides. Biochem Mol Biol Int. 1996;40:1077–1085. doi: 10.1080/15216549600201713. [DOI] [PubMed] [Google Scholar]
- 152.Meisel H, Sominskaya I, Pumpens P, Pushko P, Borisova G, Deepen R, Lu X, Spiller GH, Krüger DH, Grens E. Fine mapping and functional characterization of two immuno-dominant regions from the preS2 sequence of hepatitis B virus. Intervirology. 1994;37:330–339. doi: 10.1159/000150397. [DOI] [PubMed] [Google Scholar]
- 153.Thomas MB, Jaffe D, Choti MM, Belghiti J, Curley S, Fong Y, Gores G, Kerlan R, Merle P, O’Neil B, et al. Hepatocellular carcinoma: consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol. 2010;28:3994–4005. doi: 10.1200/JCO.2010.28.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nam KT, Kim DW, Yoo PJ, Chiang CY, Meethong N, Hammond PT, Chiang YM, Belcher AM. Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes. Science. 2006;312:885–888. doi: 10.1126/science.1122716. [DOI] [PubMed] [Google Scholar]
- 155.Yokoyama-Kobayashi M, Kato S. Recombinant f1 phage-mediated transfection of mammalian cells using lipopolyamine technique. Anal Biochem. 1994;223:130–134. doi: 10.1006/abio.1994.1557. [DOI] [PubMed] [Google Scholar]
- 156.Hart SL, Knight AM, Harbottle RP, Mistry A, Hunger HD, Cutler DF, Williamson R, Coutelle C. Cell binding and internalization by filamentous phage displaying a cyclic Arg-Gly-Asp-containing peptide. J Biol Chem. 1994;269:12468–12474. [PubMed] [Google Scholar]
- 157.Larocca D, Kassner PD, Witte A, Ladner RC, Pierce GF, Baird A. Gene transfer to mammalian cells using genetically targeted filamentous bacteriophage. FASEB J. 1999;13:727–734. doi: 10.1096/fasebj.13.6.727. [DOI] [PubMed] [Google Scholar]
- 158.Jepson CD, March JB. Bacteriophage lambda is a highly stable DNA vaccine delivery vehicle. Vaccine. 2004;22:2413–2419. doi: 10.1016/j.vaccine.2003.11.065. [DOI] [PubMed] [Google Scholar]
- 159.Tang KH. Transfection of HepG2 cells with bacteriophages T7 and M13 displaying regions of hepatitis B surface antigens. Department of Microbiology. Universiti Putra Malaysia: Universiti Putra Malaysia; 2008. [Google Scholar]
- 160.Tang KH, Yusoff K, Tan WS. Display of hepatitis B virus PreS1 peptide on bacteriophage T7 and its potential in gene delivery into HepG2 cells. J Virol Methods. 2009;159:194–199. doi: 10.1016/j.jviromet.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 161.Zhang B, Zhang Y, Wang J, Chen J, Pan Y, Ren L, Hu Z, Zhao J, Liao M, Wang S. Screening and identification of a targeting peptide to hepatocarcinoma from a phage display peptide library. Mol Med. 2007;13:246–254. doi: 10.2119/2006-00115.Zhang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Crowther RA, Kiselev NA, Böttcher B, Berriman JA, Borisova GP, Ose V, Pumpens P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell. 1994;77:943–950. doi: 10.1016/0092-8674(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 163.Whitacre DC, Lee BO, Milich DR. Use of hepadnavirus core proteins as vaccine platforms. Expert Rev Vaccines. 2009;8:1565–1573. doi: 10.1586/erv.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lee KW, Tan WS. Recombinant hepatitis B virus core particles: association, dissociation and encapsidation of green fluorescent protein. J Virol Methods. 2008;151:172–180. doi: 10.1016/j.jviromet.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 165.Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. 1999;3:771–780. doi: 10.1016/s1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 166.Tan WS, McNae IW, Ho KL, Walkinshaw MD. Crystallization and X-ray analysis of the T = 4 particle of hepatitis B capsid protein with an N-terminal extension. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:642–647. doi: 10.1107/S1744309107033726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lee KW, Tey BT, Ho KL, Tejo BA, Tan WS. Nanoglue: an alternative way to display cell-internalizing peptide at the spikes of hepatitis B virus core nanoparticles for cell-targeting delivery. Mol Pharm. 2012;9:2415–2423. doi: 10.1021/mp200389t. [DOI] [PubMed] [Google Scholar]
- 168.Conway JF, Cheng N, Zlotnick A, Stahl SJ, Wingfield PT, Belnap DM, Kanngiesser U, Noah M, Steven AC. Hepatitis B virus capsid: localization of the putative immunodominant loop (residues 78 to 83) on the capsid surface, and implications for the distinction between c and e-antigens. J Mol Biol. 1998;279:1111–1121. doi: 10.1006/jmbi.1998.1845. [DOI] [PubMed] [Google Scholar]
- 169.Conway JF, Watts NR, Belnap DM, Cheng N, Stahl SJ, Wingfield PT, Steven AC. Characterization of a conformational epitope on hepatitis B virus core antigen and quasiequivalent variations in antibody binding. J Virol. 2003;77:6466–6473. doi: 10.1128/JVI.77.11.6466-6473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Harris A, Belnap DM, Watts NR, Conway JF, Cheng N, Stahl SJ, Vethanayagam JG, Wingfield PT, Steven AC. Epitope diversity of hepatitis B virus capsids: quasi-equivalent variations in spike epitopes and binding of different antibodies to the same epitope. J Mol Biol. 2006;355:562–576. doi: 10.1016/j.jmb.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 171.Belnap DM, Watts NR, Conway JF, Cheng N, Stahl SJ, Wingfield PT, Steven AC. Diversity of core antigen epitopes of hepatitis B virus. Proc Natl Acad Sci USA. 2003;100:10884–10889. doi: 10.1073/pnas.1834404100. [DOI] [PMC free article] [PubMed] [Google Scholar]