

Synopsis
Primary abnormalities of the erythrocyte membrane, including the hereditary spherocytosis and hereditary elliptocytosis syndromes, are an important group of inherited hemolytic anemias. Classified by distinctive morphology on peripheral blood smear, these disorders are characterized by clinical, laboratory, and genetic heterogeneity. Among this group, hereditary spherocytosis patients are more likely to experience symptomatic anemia. Treatment of hereditary spherocytosis with splenectomy is curative in most patients. Once considered routine, growing recognition of the longterm risks of splenectomy, including cardiovascular disease, thrombotic disorders, and pulmonary hypertension, as well as the emergence of penicillin-resistant pneumococci, a concern for infection in overwhelming postsplenectomy infection, have led to re-evaluation of the role of splenectomy. Current management guidelines acknowledge these important considerations when entertaining splenectomy and recommend detailed discussion between health care providers, patient, and family. The hereditary elliptocytosis syndromes are the most common primary disorders of erythrocyte membrane proteins. However, most elliptocytosis patients are asymptomatic and do not require therapy.
Keywords: Hereditary spherocytosis, hereditary elliptocytosis, hereditary pyropoikilocytosis, erythrocyte membrane, anemia, splenectomy
Introduction
Primary abnormalities of the erythrocyte membrane lead to a variety of clinical syndromes including hereditary spherocytosis, hereditary elliptocytosis, and related disorders.1 Clinical and laboratory manifestations, as well as associated molecular defects, of these disorders vary widely. Abnormalities of erythrocyte shape on peripheral blood smear often provide clues to the underlying pathobiology and clinical diagnosis of the underlying disorder.
Hereditary Spherocytosis
The hereditary spherocytosis (HS) syndromes are a group of disorders associated with a primary defect in erythrocyte membrane proteins.2 HS was first described based on the finding of spherocytes, characteristic erythrocytes lacking central pallor, on peripheral blood smear. HS occurs worldwide in all racial and ethnic groups. It is the most common inherited anemia in individuals of northern European ancestry, affecting ∼ 1:2500 individuals in the United States. Clinical, laboratory, and molecular heterogeneity characterize the HS syndromes.3
The principal abnormality in HS erythrocytes is loss of membrane surface area relative to intracellular volume, which leads to spherically shaped erythrocytes with decreased deformability.4 The loss of surface area results from increased membrane fragility due to primary and secondary abnormalities in erythrocyte membrane proteins, particularly ankyrin, α- and β-spectrin, band 3, and protein 4.2 (Figure 1).2 Increased erythrocyte fragility leads to vesiculation and membrane loss. Splenic destruction of poorly deformable HS erythrocytes is the primary cause of hemolysis experienced by HS patients.5, 6
Figure 1. The Erythrocyte Membrane.
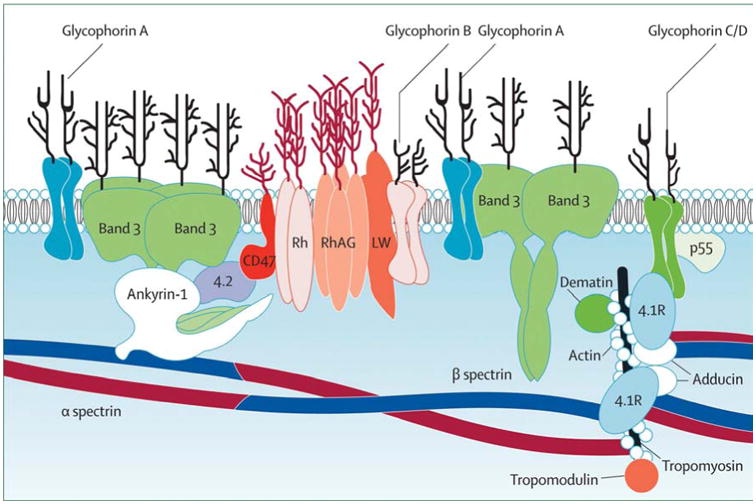
A model of the major proteins of the erythrocyte membrane is shown: α- and β-spectrin, ankyrin, band 3 (the anion exchanger), 4.1 (protein 4.1) and 4.2 (protein 4.2), actin and glycophorin. From Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet 2008;372:1411-1426; with permission.
HS is inherited as in autosomal dominant manner in ∼two thirds of patients, associated with mutations in the ankyrin, β-spectrin, or band 3 genes.2 In the remaining patients, inheritance is non-dominant due to autosomal recessive inheritance or a de novo mutation.3, 7 Autosomal recessive inheritance is associated with mutations of either the α-spectrin or protein 4.2 genes. A number of de novo mutations have been reported in the HS genes.8, 9
Clinical Manifestations and Classification
Clinical manifestations of the spherocytosis syndromes vary widely. Typical HS is associated with pallor, jaundice, splenomegaly, anemia, reticulocytosis, spherocytes on peripheral blood smear, positive osmotic fragility or flow cytometric analysis of eosin-5-maleimide-labeled erythrocytes (EMA binding) (see page 6, below), and a positive family history. Mild, moderate, and severe forms of HS have been defined according to the severity of anemia and the degree of compensation for the hemolysis (Table 1).10
Table 1. Classification of hereditary spherocytosisa.
| Carrier | Mild spherocytosis | Moderate spherocytosis | Severe spherocytosisb | |
|---|---|---|---|---|
| Hemoglobin (g/dl) | Normal | 11–15 | 8–12 | 6–8 |
| Reticulocytes (%) | ≤3 | 3–6 | ≥6 | ≥10 |
| Bilirubin (mg/dl) | 0–1 | 1–2 | ≥2 | ≥2 |
| Spectrin content (% of normal) | 100 | 80–100 | 50–80 | 40–60 |
| Peripheral smear | Normal | Mild spherocytosis | Spherocytosis | Spherocytosis |
| Osmotic fragility fresh blood | Normal | Normal or slightly increased | Distinctly increased | Distinctly increased |
| Incubated blood | Slightly increased | Distinctly increased | Distinctly increased | Distinctly increased |
From Eber SW, Armbrust R, Schroter W. Variable clinical severity of hereditary spherocytosis: relation to erythrocytic spectrin concentration, fragility, and autohemolysis. J Pediatr 1990;117:409-416.
Values in untransfused patients.
HS may present at any age, but typically it presents in childhood. Anemia is the most frequent finding at presentation (50%), followed by splenomegaly, jaundice, or a positive family history.3 The majority of HS patients have incompletely compensated hemolysis with mild to moderate anemia that is asymptomatic except for fatigue and pallor. Jaundice is visible at some time in over half of HS patients, usually in association with viral infection or other stress. The jaundice is typically acholuric, i.e. unconjugated hyperbilirubinemia without detectable bilirubinuria. By late childhood, palpable splenomegaly is found in most HS patients. Approximately a quarter of HS patients have compensated hemolysis, i.e. erythrocyte production and destruction are balanced.11 These patients are not anemic and are usually asymptomatic. The remaining 5-10% of HS patients experience moderate to severe anemia. This category includes patients with both dominant and recessive HS. The most severely affected patients are transfusion-dependent and almost always have recessive HS.12-14 Chronically transfused patients are at risk for developing complications of recurrent transfusion and iron overload.
HS may present in the neonatal period. Some patients present with significant neonatal jaundice requiring phototherapy or even exchange transfusion.15, 16 Others present with significant anemia presenting in the first few weeks of life and may require several transfusions in infancy. Most of these patients become transfusion-independent during the first year of life. A subset of patients presents with severe anemia in utero or immediately after birth and require red blood cell transfusion.17-19 These patients frequently remain transfusion dependent and suffer from severe HS.
Initial Assessment/Physical Examination
Initial assessment of a patient with suspected HS includes a detailed family history and questions about a history of pallor, jaundice, anemia, gallstones, and splenectomy. Physical examination includes attention to:
pallor
scleral icterus
splenomegaly
After diagnosing a patient with HS, family members should be examined for the presence of HS.
Laboratory Findings
Laboratory findings in HS are heterogeneous. Initial studies in a patient with suspected HS include:
complete blood count/erythrocyte indices
peripheral blood smear
reticulocyte count
bilirubin
flow cytometric analysis of eosin-5-maleimide-labeled erythrocytes (EMA binding) or incubated osmotic fragility
Erythrocyte indices. The majority of HS patients have some degree of anemia with reticulocytosis.11, 20 The mean corpuscular volume (MCV) is normal or slightly decreased in most patients except in severe cases, when it is decreased despite reticulocytosis, reflecting membrane loss and cellular dehydration.21 The mean corpuscular hemoglobin concentration (MCHC) is increased (≥34.5g/dL) due to relative cellular dehydration in >50% of patients.22 The red cell distribution width (RDW) is increased (>14) in most patients. Combining the MCHC and red cell distribution width (>35.4g/dL and >14, respectively) or combining the MCHC with histograms of hyperdense erythrocytes (MCHC>40g/dL) obtained from laser-based cell counters, have been utilized to rapidly identify HS patients.21, 23
Peripheral blood smear. Typical HS patients have blood smears with easily detectable spherocytes, i.e. erythrocytes lacking central pallor, which are distinctive but not diagnostic (Figure 2A). Occasionally, only a few spherocytes are seen on peripheral smear, or, in contrast, numerous small, dense spherocytes and bizarre erythrocyte morphology with anisocytosis and poikilocytosis may be observed in severely affected patients (Figure 2B). Specific findings have been identified in some patients with specific membrane protein abnormalities such as pincered erythrocytes (band 3) or spherocytic acanthocytes (β spectrin).24, 25
Figure 2. Peripheral Blood Smears in Hereditary Spherocytosis.
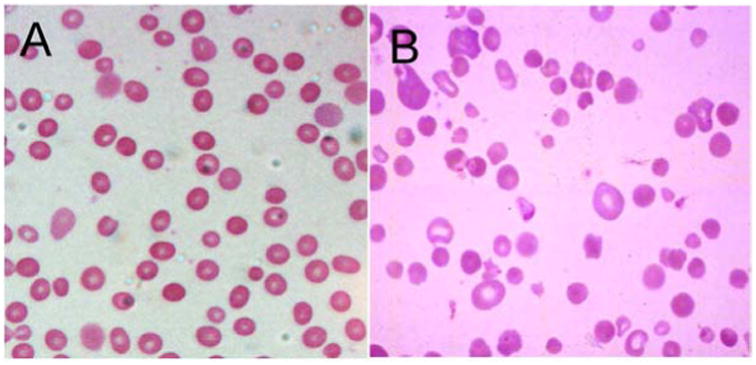
A. Typical hereditary spherocytosis. Characteristic spherocytes lacking central pallor are seen. B. Severe, recessively-inherited spherocytosis. Numerous small, dense spherocytes and bizarre erythrocyte morphology with anisocytosis and poikilocytosis associated with severe hemolysis are seen.
EMA binding and osmotic fragility. The EMA (eosin-5-maleimide) binding test is a flow cytometry-based analysis of the relative amounts of fluorescence reflecting the amount EMA binding to band 3 and Rh-related proteins in the erythrocyte membrane.26, 27 The basis of the test is the reduction in band 3 and related membrane proteins leading to a decrease in fluorescence intensity, typically to ∼65% of normal (Figure 3A).28 Even though defects of band 3 protein are only found in ∼25% of typical HS patients, decreased fluorescence intensity is also observed in HS erythrocytes with defects in ankyrin and spectrin, thought to be due to transmission of long range effects of varying protein defects across the membrane thereby influencing EMA binding to band 3. In laboratories with the ability to perform FACS-based studies, EMA binding has high sensitivity and specificity and is simple and rapidly performed.29 EMA-binding results are not influenced by shipping or storage for several days, and may be suitable for study of patients who have been recently transfused.
Figure 3. Testing in Hereditary Spherocytosis.
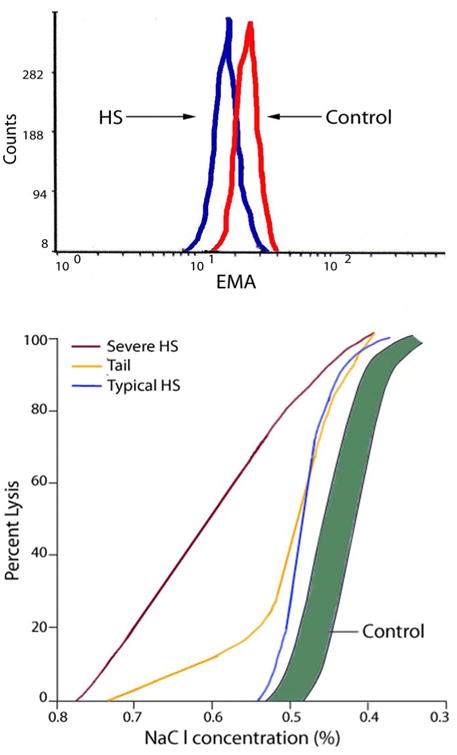
A. Eosin-5-maleimide (EMA) binding. Histogram of fluorescence of EMA-labeled erythrocytes from a normal control and a patient with typical hereditary spherocytosis. Decreased fluorescence in observed from HS erythrocytes. B. Osmotic fragility curves in hereditary spherocytosis. The shaded region is the normal range. Results representative of both typical and severe spherocytosis are shown. A tail, representing fragile erythrocytes conditioned by the spleen, is common in spherocytosis patients prior to splenectomy.
The osmotic fragility (OF) test, which measures the in vitro lysis of erythrocytes suspended in solutions of decreasing osmolarity, is frequently utilized in the diagnosis of HS. Due to the loss of membrane surface area relative to intracellular volume, HS erythrocytes are unable to withstand the introduction of small amounts of free water that occurs when they are placed in increasingly hypotonic saline solutions. As a consequence, HS erythrocytes hemolyze more readily than normal erythrocytes at any saline concentration. Hemolysis is determined by measuring the amount of hemoglobin released from red cells into the extracellular fluid. Approximately 25% of HS patients have a normal osmotic fragility on freshly drawn blood, with the osmotic fragility curve approximating the number of spherocytes on peripheral smear. After incubation at 37°C for 24 h, HS erythrocytes lose membrane surface area more readily than normal because their membranes are leaky and unstable, revealing the membrane defect in osmotic fragility testing. When the spleen is present, a subpopulation of fragile erythrocytes which have been conditioned by the spleen, form the ‘tail’ of the osmotic fragility curve that disappears after splenectomy (Figure 3B).
Neither EMA binding nor OF testing detect 100% of HS patients. Both tests struggle in the identification of cases of mild HS, with OF suffering from poorer sensitivity.30 Other conditions such as congenital dyserythropoietic anemia type II, Southeast Asian ovalocytosis and hereditary pyropoikilocytosis, may yield reduced fluorescence in EMA binding.31 OF testing is unreliable in patients who have small numbers of spherocytes, including those who have been recently transfused, and it is abnormal in other conditions where spherocytes are present. Although some laboratories in Europe combine EMA binding studies with the acidified glycerol lysis test to improve diagnostic sensitivity,27 this test is not available in most clinical laboratories in North America.
Specialized testing and molecular studies. Ektacytometry is a highly sensitive test of membrane deformability. It is available only in a few specialized centers. Similarly, quantitation of major erythrocyte membrane proteins via sodium dodecyl sulfate-polyacrylamide gel electrophoresis has limited availability. Mutation detection in the major erythrocyte membrane protein genes is now available commercially in the United States. It will be of use in diagnosing difficult cases and in cases where a molecular diagnosis is desired.
Other laboratory findings. Nonspecific markers of ongoing hemolysis, increased bilirubin, increased lactate dehydrogenase, increased urinary and fecal urobilinogen, and decreased haptoglobin, may be found, reflecting increased erythrocyte production and/or destruction.
Diagnosis of HS in the neonate
The diagnosis of HS in the neonate requires careful consideration of clinical and laboratory findings.15 In the neonatal period, findings on peripheral smear are variable, with anemia and hyperbilirubinemia typically present. Eleveated MCHC is often a clue to underlying HS, especially when >36g/dl.16 If specific diagnostic testing is pursued, the diagnosis can be established in the neonatal period. However, EMA binding or osmotic fragility testing must utilize appropriate age-matched controls, and then, the results interpreted in concert with clinical course, family history, and detailed examination of the peripheral smear and complete blood cell indices.
Imaging Studies
Splenomegaly is typically detected on abdominal ultrasound examination or radionuclide scanning. Cholelithiasis may be detected on abdominal ultrasound.
Differential Diagnosis
Hemolytic anemia with spherocytes on peripheral blood smear is found in a number of conditions including autoimmune hemolytic anemia, liver disease, thermal injury, micro- and macroangiopathic hemolytic anemias, Clostridial sepsis, transfusion reaction with hemolysis, severe hypophosphatemia, ABO incompatibility, and poisoning with certain snake, spider, and hymenoptera venoms. It is usually not difficult to distinguish HS from other disorders by additional diagnostic testing, such as autoimmune hemolytic anemia via a Coombs' test (the direct anti-globulin test or DAT), or by viewing the condition in the appropriate clinical context.
Complications
gallbladder disease
hemolytic crisis
aplastic crisis
megaloblastic crisis
uncommon: chronic leg ulceration, cardiomyopathy, neuromuscular abnormalities, “tumors” due to extramedullary erythropoiesis32, 33
Gallbladder disease. Chronic hemolysis may lead to the formation of bilirubinate gallstones, the most complication in HS patients. In most HS patients, gallstones become apparent in childhood and young adulthood. Interval ultrasonography to detect gallstones is recommended to allow timely diagnosis and treatment, thereby prevent complications of symptomatic biliary tract disease such as biliary obstruction, cholecystitis, and cholangitis.
Crises. Similar to other chronic hemolytic diseases, HS patients suffer from a variety of crises. Hemolytic crises present with jaundice, increased splenomegaly, decreased hematocrit, and reticulocytosis. They are generally mild and are associated with viral infection. Rarely, in severe cases, there is marked jaundice, lethargy, abdominal pain, tender splenomegaly, and significant anemia that require hospitalization and erythrocyte transfusion. Aplastic crises are typically due to viral bone marrow suppression by parvovirus B19 infection, which selectively infects erythropoietic progenitor cells and inhibits their growth.34 During the aplastic phase, the hemoglobin and reticulocyte count fall, paralleling a decrease in production of new red blood cells. Aplastic crises usually last 7-10 days, about half the life span of typical HS erythrocytes, with the hemoglobin falling to about ∼50% of its normal level before recovery. In severe HS patients, the anemia may be severe due to extremely short life span of erythrocytes, requiring hospitalization and transfusion. Parvovirus infection may be the first manifestation of HS, and multiple HS family members infected with parvovirus have developed aplastic crises at the same time, leading to descriptions of “epidemics” of HS.35 Megaloblastic crises with exaggeration of anemia occur due to exhaustion of folate reserves by the sustained increase in net DNA synthesis associated with increased folate demands,36 such as during pregnancy, during periods of rapid growth in childhood, and during the recovery phase of an aplastic crisis.
Treatment
Treatment of HS encompasses supportive care and when appropriate, splenectomy.30 HS patients should be followed regularly to assess the degree of anemia, development of complications such as gallstones, and in children, to monitor growth and development. Folate supplementation should be provided to patients with moderate and severe HS. Counseling of the patient regarding HS and its complications as well as investigation of other family members should be provided where appropriate. There should be close observation for hematologic decompensation during acute illnesses or stress. Many care providers obtain serial parvovirus B19 antibody titers until positive to assess risk for parvovirus-associated aplastic crisis.
Splenectomy. Splenic sequestration and destruction is the primary determinant of erythrocyte survival in HS patients. Thus in most HS patients, splenectomy cures the anemia, and decreases the incidence of cholelithiasis.37 Even severe HS patients experience marked improvement in their anemia postsplenectomy.12 Early complications of splenectomy include local infection, bleeding, and pancreatitis due to injury to the tail of the pancreas during surgery. Overwhelming postsplenectomy infection (OPSI), typically from encapsulated organisms, is an uncommon but significant late complication of splenectomy, especially in the first few years of life.38 Immunization with pneumococcal vaccine and promotion of early antibiotic therapy for asplenic febrile children have led to decreases in the incidence of OPSI. However, increasing rates of penicillin-resistant pneumococci has raised concerns about the potential increases in infection with this virulent organism. Another postsplenectomy complication is an increased risk of cardiovascular disease, particularly thrombosis and pulmonary hypertension.39-41 Finally, as global travel increases, increasing consideration has been given to the contribution of the spleen in protection from parasitic diseases such as malaria and babesiosis.
For many years, splenectomy was considered a routine procedure in HS patients. However, the risk of OPSI with penicillin-resistant pneumococci, increased recognition of postsplenectomy cardiovascular disease, and increased international travel, have all contributed to a re-evaluation of the role of splenectomy in the treatment of HS.42 Health care providers, patients, and family members should review both the risks and benefits when considering splenectomy.42, 43 Most experts agree that is reasonable to splenectomize all patients with severe HS and all patients with significant anemia suffering from growth failure, skeletal changes, extramedullary hematopoietic tumors, and leg ulcers.30 Whether patients with moderate HS and compensated, asymptomatic anemia should have a splenectomy remains controversial. In this group, splenectomy should be considered on a case-by-case basis. Patients with mild HS and well compensated hemolysis can be managed expectantly, deferring splenectomy unless clinically indicated.10 Management of patients with mild to moderate HS and gallstones is also controversial, as new treatments for cholelithiasis, including laparoscopic cholecystectomy, endoscopic sphincterotomy, and extracorporal choletripsy, lower the risk of treating this complication.
When splenectomy is indicated, laparoscopic splenectomy is the method of choice, resulting in less postoperative discomfort, shorter hospitalization, and decreased costs.44, 45 Partial splenectomy has been advocated for infants and young children with significant anemia associated with HS and it may be of benefit in typical HS patients.46, 47 In young children, the goal of partial splenectomy is to ameliorate the hemolytic anemia while maintaining residual splenic immune function. Prior to splenectomy, patients should be immunized with vaccines against pneumococcus, Haemophilus influenzae type b, and meningococcus.30, 48
Hereditary Elliptocytosis, Hereditary Pyropoikilocytosis, and Related Disorders
Introduction
The hereditary elliptocytosis (HE) syndromes are a heterogeneous group of disorders characterized by the presence of elliptical-shaped erythrocytes on peripheral blood smear.49, 50 Clinical manifestations range from the asymptomatic carrier state to severe, transfusion-dependent hemolytic anemia. HE occurs worldwide in all racial and ethnic groups. It is much more common in areas of endemic malaria, particularly in people of African and Mediterranean ancestry, presumably because elliptocytes confer some resistance to malaria.51 The world-wide incidence of HE has been estimated to be 1 in 2000 to 4000 individuals with the incidence of HE approaching 1 in 100 in parts of Africa.50, 52 The true incidence of HE is unknown, as many affected patients are asymptomatic.
Hereditary pyropoikilocytosis (HPP) is an uncommon severe hemolytic anemia characterized by erythrocyte morphology reminiscent of that seen in patients after a thermal burn (Figure 4B).53 There is a strong association between HE and HPP, with a third of family members of HPP patients exhibiting typical HE.50, 53, 54 Thus not surprisingly, HPP occurs predominantly in patients of African descent. Many HPP patients suffer from severe hemolytic anemia in infancy that gradually improves, evolving toward typical HE later in life.
Figure 4. Peripheral Blood Smears in Hereditary Elliptocytosis.

A. Hereditary elliptocytosis. Smooth, cigar-shaped elliptocytes are seen. B. Hereditary pyropoikilocytosis. Pronounced microcytosis, poikilocytosis, fragmentation of erythrocytes and elliptocytes are seen.
The principal defect in HE/HPP erythrocytes is mechanical weakness or fragility of the erythrocyte membrane skeleton due to defects in various membrane proteins, 4 including α- and β-spectrin, protein 4.1, and glycophorin C. Most cases of HE and HPP are due to defects in spectrin, the principal structural protein of the erythrocyte membrane skeleton. Spectrin integrity is dependent on the self-association of heterodimers of α- and β-spectrin55, 56 into mature spectrin molecules that are critical for membrane stability and erythrocyte shape and function.57
HE is inherited in an autosomal dominant pattern with rare cases of de novo mutations.58, 59 Mutations in the regions of spectrin where the α- and β-spectrin chains self-associate cause the majority of cases of HE/HPP. Most of these mutations are missense mutations at residues critical for spectrin function.56 In contrast to HS, the HE/HPP syndromes, while also heterogeneous, have been associated with distinct spectrin mutations in persons of similar genetic backgrounds, suggesting a “founder effect” for these mutations.60
Clinical Manifestations
Clinical manifestations of the HE syndromes vary widely, from asymptomatic carriers to patients with severe, transfusion-dependent anemia. The overwhelming majority of HE patients are completely asymptomatic. The diagnosis of HE is typically made incidentally during testing for unrelated conditions. The erythrocyte life span is normal in most patients. Symptomatology may vary between members of the same family, attributed to modifier alleles influencing spectrin expression.
Approximately 10% of HE patients and nearly all HPP patients suffer from compensated hemolysis with mild to moderate anemia. These patients are homozygotes or compound heterozygotes for defects inherited from each of the parents. Hemolytic HE and HPP patients have clinical features similar to HS including pallor, jaundice, anemia, and gallstones. Hemolysis and anemia are exaggerated in infancy. Elliptocytes are rare on peripheral smear before 6 months of age.
Laboratory Findings
The laboratory findings in HE are heterogeneous.
typical HE: varying numbers of elliptocytes on peripheral smear, no anemia or hemolysis
hemolytic HE: elliptocytes and fragmented cells on peripheral smear, normocytic anemia, reticulocytosis, markers of hemolysis, abnormal EMA binding or incubated osmotic fragility
HPP: elliptocytes and fragmented cells on peripheral smear, microcytic anemia, reticulocytosis, markers of hemolysis, abnormal EMA binding or incubated osmotic fragility53, 61
The only laboratory finding in typical HE is the presence of elliptocytes on peripheral blood smear. (Figure 4A.)These normochromic, normocytic elliptocytes number from a few to 100 per cent. In hemolytic HE and HPP, there is anemia, reticulocytosis, hyperbilirubinemia, elevated LDH and other findings of hemolysis. In these conditions, elliptocytes, fragmented cells, and rare ovalocytes, spherocytes, and stomatocytes may be found. In addition to the peripheral blood smear findings found in hemolytic HE, bizarre shaped cells with fragmentation or budding, poikilocytes, pyknocytes, and microspherocytes are seen in HPP (Figure 4B).53 The MCV in HPP is frequently very low, 50–65fL. EMA binding or incubated osmotic fragility testing are abnormal in hemolytic HE and HPP.62
Imaging Studies
In hemolytic HE and HPP, splenomegaly is typically detected on abdominal ultrasound examination or radionuclide scanning. Cholelithiasis may be detected on abdominal ultrasound.
Differential Diagnosis
Conditions with elliptocytes on peripheral smear include the megaloblastic anemias, the hypochromic microcytic anemias (iron deficiency anemia and thalassemia), myleodysplasic syndromes, and myelofibrosis. In these conditions, elliptocytes generally number less than one-third of erythrocytes. History and additional laboratory testing usually clarify the diagnosis of these disorders.
Complications
Complications in hemolytic HE and HPP are similar to those in HS.
Treatment
Treatment is rarely required for patients with typical HE.50 In cases of severe HE and HPP, red blood cell transfusions may be required. Splenectomy has been curative. Many experts apply the same indications for splenectomy in HS to patients with symptomatic HE or HPP. Postsplenectomy, patients with HE or HPP experience resolution of anemia and improvement in clinical symptoms.
Summary
Disorders of the erythrocyte membrane should be considered when evaluating a child with hemolytic anemia. Family history, past medical history, physical examination findings may all provide important clues to the diagnosis. Initial laboratory testing should include a complete blood count, peripheral blood smear, reticulocyte count and either EMA binding or incubated osmotic fragility testing. Patients should be monitored expectantly for complications associated with their disease. Splenectomy is curative in most patients with membrane-linked hemolytic anemia. Once considered routine, careful consideration is given to the risks and benefits of splenectomy before undertaking the procedure.
Key Points.
Disorders of the erythrocyte membrane are an important group of inherited hemolytic anemias.
Red cell membrane disorders are marked by clinical, laboratory, and genetic heterogeneity.
Abnormalities of erythrocyte shape on peripheral blood smear provide clues to the underlying diagnosis.
Hereditary spherocytosis is the most common membrane disorder associated with hemolytic anemia.
Splenectomy is curative in most patients with membrane-associated hemolytic anemia.
Growing recognition of long term risks associated with splenectomy has led to re-evaluation of its use in subsets of patients.
Acknowledgments
Supported in part by grants from the NHLBI, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Da Costa L, Galimand J, Fenneteau O, et al. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013 May 9; doi: 10.1016/j.blre.2013.04.003. doi:pii: S0268-960X(13)00019-2 10.1016/j.blre.2013.04.003. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 2.Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411–26. doi: 10.1016/S0140-6736(08)61588-3. [DOI] [PubMed] [Google Scholar]
- 3.Eber S, Lux SE. Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41:118–41. doi: 10.1053/j.seminhematol.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Mohandas N, Gallagher PG. Red cell membrane: Past, present, and future. Blood. 2008;112:3939–48. doi: 10.1182/blood-2008-07-161166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lusher JM, Barnhart MI. The role of the spleen in the pathoophysiology of hereditary spherocytosis and hereditary elliptocytosis. Am J Pediatr Hematol Oncol. 1980;2:31–39. [Google Scholar]
- 6.Safeukui I, Buffet PA, Deplaine G, et al. Quantitative assessment of sensing and sequestration of spherocytic erythrocytes by the human spleen. Blood. 2012;120:424–30. doi: 10.1182/blood-2012-01-404103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallagher PG. Update on the clinical spectrum and genetics of red blood cell membrane disorders. Curr Hematol Rep. 2004;3:85–91. [PubMed] [Google Scholar]
- 8.Miraglia del Giudice E, Francese M, Nobili B, et al. High frequency of de novo mutations in ankyrin gene (ANK1) in children with hereditary spherocytosis. J Pediatr. 1998;132:117–20. doi: 10.1016/s0022-3476(98)70495-0. [DOI] [PubMed] [Google Scholar]
- 9.Miraglia del Giudice E, Lombardi C, Francese M, et al. Frequent de novo monoallelic expression of beta-spectrin gene (SPTB) in children with hereditary spherocytosis and isolated spectrin deficiency. Br J Haematol. 1998;101:251–4. doi: 10.1046/j.1365-2141.1998.00688.x. [DOI] [PubMed] [Google Scholar]
- 10.Eber SW, Armbrust R, Schroter W. Variable clinical severity of hereditary spherocytosis: relation to erythrocytic spectrin concentration, osmotic fragility, and autohemolysis. J Pediatr. 1990;117:409–16. doi: 10.1016/s0022-3476(05)81081-9. [DOI] [PubMed] [Google Scholar]
- 11.Rocha S, Costa E, Catarino C, et al. Erythropoietin levels in the different clinical forms of hereditary spherocytosis. Br J Haematol. 2005;131:534–42. doi: 10.1111/j.1365-2141.2005.05802.x. [DOI] [PubMed] [Google Scholar]
- 12.Agre P, Asimos A, Casella JF, et al. Inheritance pattern and clinical response to splenectomy as a reflection of erythrocyte spectrin deficiency in hereditary spherocytosis. N Engl J Med. 1986;315:1579–83. doi: 10.1056/NEJM198612183152504. [DOI] [PubMed] [Google Scholar]
- 13.Agre P, Casella JF, Zinkham WH, et al. Partial deficiency of erythrocyte spectrin in hereditary spherocytosis. Nature. 1985;314:380–3. doi: 10.1038/314380a0. [DOI] [PubMed] [Google Scholar]
- 14.Agre P, Orringer EP, Bennett V. Deficient red-cell spectrin in severe, recessively inherited spherocytosis. N Engl J Med. 1982;306:1155–61. doi: 10.1056/NEJM198205133061906. [DOI] [PubMed] [Google Scholar]
- 15.Delhommeau F, Cynober T, Schischmanoff PO, et al. Natural history of hereditary spherocytosis during the first year of life. Blood. 2000;95:393–7. [PubMed] [Google Scholar]
- 16.Christensen RD, Henry E. Hereditary spherocytosis in neonates with hyperbilirubinemia. Pediatrics. 2010;125:120–5. doi: 10.1542/peds.2009-0864. [DOI] [PubMed] [Google Scholar]
- 17.Ribeiro ML, Alloisio N, Almeida H, et al. Severe hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3. Blood. 2000;96:1602–4. [PubMed] [Google Scholar]
- 18.Whitfield CF, Follweiler JB, Lopresti-Morrow L, et al. Deficiency of alpha-spectrin synthesis in burst-forming units-erythroid in lethal hereditary spherocytosis. Blood. 1991;78:3043–51. [PubMed] [Google Scholar]
- 19.Delaunay J, Nouyrigat V, Proust A, et al. Different impacts of alleles alphaLEPRA and alphaLELY as assessed versus a novel, virtually null allele of the SPTA1 gene in trans. Br J Haematol. 2004;127:118–22. doi: 10.1111/j.1365-2141.2004.05160.x. [DOI] [PubMed] [Google Scholar]
- 20.Guarnone R, Centenara E, Zappa M, et al. Erythropoietin production and erythropoiesis in compensated and anaemic states of hereditary spherocytosis. Br J Haematol. 1996;92:150–4. doi: 10.1046/j.1365-2141.1996.00285.x. [DOI] [PubMed] [Google Scholar]
- 21.Michaels LA, Cohen AR, Zhao H, et al. Screening for hereditary spherocytosis by use of automated erythrocyte indexes. J Pediatr. 1997;130:957–60. doi: 10.1016/s0022-3476(97)70283-x. [DOI] [PubMed] [Google Scholar]
- 22.Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes's original 1934 classification to the third millennium. Curr Opin Hematol. 2013;20:222–30. doi: 10.1097/MOH.0b013e32835f5933. [DOI] [PubMed] [Google Scholar]
- 23.Cynober T, Mohandas N, Tchernia G. Red cell abnormalities in hereditary spherocytosis: relevance to diagnosis and understanding of the variable expression of clinical severity. J Lab Clin Med. 1996;128:259–69. doi: 10.1016/s0022-2143(96)90027-x. [DOI] [PubMed] [Google Scholar]
- 24.Hassoun H, Vassiliadis JN, Murray J, et al. Characterization of the underlying molecular defect in hereditary spherocytosis associated with spectrin deficiency. Blood. 1997;90:398–406. [PubMed] [Google Scholar]
- 25.Jarolim P, Murray JL, Rubin HL, et al. Characterization of 13 novel band 3 gene defects in hereditary spherocytosis with band 3 deficiency. Blood. 1996;88:4366–74. [PubMed] [Google Scholar]
- 26.King MJ, Smythe JS, Mushens R. Eosin-5-maleimide binding to band 3 and Rh-related proteins forms the basis of a screening test for hereditary spherocytosis. Br J Haematol. 2004;124:106–13. doi: 10.1046/j.1365-2141.2003.04730.x. [DOI] [PubMed] [Google Scholar]
- 27.Bianchi P, Fermo E, Vercellati C, et al. Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica. 2012;97:516–23. doi: 10.3324/haematol.2011.052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King MJ, Zanella A. Hereditary red cell membrane disorders and laboratory diagnostic testing. International journal of laboratory hematology. 2013;35:237–43. doi: 10.1111/ijlh.12070. [DOI] [PubMed] [Google Scholar]
- 29.D'Alcamo E, Agrigento V, Sclafani S, et al. Reliability of EMA binding test in the diagnosis of hereditary spherocytosis in Italian patients. Acta Haematol. 2011;125:136–40. doi: 10.1159/000322253. [DOI] [PubMed] [Google Scholar]
- 30.Bolton-Maggs PH, Langer JC, Iolascon A, et al. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol. 2012;156:37–49. doi: 10.1111/j.1365-2141.2011.08921.x. [DOI] [PubMed] [Google Scholar]
- 31.King MJ, Telfer P, MacKinnon H, et al. Using the eosin-5-maleimide binding test in the differential diagnosis of hereditary spherocytosis and hereditary pyropoikilocytosis. Cytometry B Clin Cytom. 2008;74:244–50. doi: 10.1002/cyto.b.20413. [DOI] [PubMed] [Google Scholar]
- 32.Smith J, Rahilly M, Davidson K. Extramedullary haematopoiesis secondary to hereditary spherocytosis. Br J Haematol. 2011;154:543. doi: 10.1111/j.1365-2141.2011.08692.x. [DOI] [PubMed] [Google Scholar]
- 33.Rabhi S, Benjelloune H, Meziane M, et al. Hereditary spherocytosis with leg ulcers healing after splenectomy. South Med J. 2011;104:150–2. doi: 10.1097/SMJ.0b013e318200c6ba. [DOI] [PubMed] [Google Scholar]
- 34.Young NS. Hematologic manifestations and diagnosis of parvovirus B19 infections. Clin Adv Hematol Oncol. 2006;4:908–10. [PubMed] [Google Scholar]
- 35.Lefrere JJ, Courouce AM, Girot R, et al. Six cases of hereditary spherocytosis revealed by human parvovirus infection. Br J Haematol. 1986;62:653–8. doi: 10.1111/j.1365-2141.1986.tb04088.x. [DOI] [PubMed] [Google Scholar]
- 36.Delamore IW, Richmond J, Davies SH. Megaloblastic anaemia in congenital spherocytosis. Br Med J. 1961;1:543–5. doi: 10.1136/bmj.1.5225.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baird RN, Macpherson AI, Richmond J. Red-blood-cell survival after splenectomy in congenital spherocytosis. Lancet. 1971;2:1060–1. doi: 10.1016/s0140-6736(71)90380-1. [DOI] [PubMed] [Google Scholar]
- 38.Schilling RF. Risks and benefits of splenectomy versus no splenectomy for hereditary spherocytosis - a personal view. Br J Haematol. 2009;145:728–32. doi: 10.1111/j.1365-2141.2009.07694.x. [DOI] [PubMed] [Google Scholar]
- 39.Crary SE, Ramaciotti C, Buchanan GR. Prevalence of pulmonary hypertension in hereditary spherocytosis. Am J Hematol. 2011;86:E73–6. doi: 10.1002/ajh.22182. [DOI] [PubMed] [Google Scholar]
- 40.Hayag-Barin JE, Smith RE, Tucker FC., Jr Hereditary spherocytosis, thrombocytosis, and chronic pulmonary emboli: a case report and review of the literature. Am J Hematol. 1998;57:82–4. doi: 10.1002/(sici)1096-8652(199801)57:1<82::aid-ajh15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 41.Schilling RF, Gangnon RE, Traver MI. Delayed adverse vascular events after splenectomy in hereditary spherocytosis. J Thromb Haemost. 2008;6:1289–95. doi: 10.1111/j.1538-7836.2008.03024.x. [DOI] [PubMed] [Google Scholar]
- 42.Casale M, Perrotta S. Splenectomy for hereditary spherocytosis: complete, partial or not at all? Expert review of hematology. 2011;4:627–35. doi: 10.1586/ehm.11.51. [DOI] [PubMed] [Google Scholar]
- 43.Schilling RF. Risks and benefits of splenectomy versus no splenectomy for hereditary spherocytosis--a personal view. Br J Haematol. 2009;145:728–32. doi: 10.1111/j.1365-2141.2009.07694.x. [DOI] [PubMed] [Google Scholar]
- 44.Wood JH, Partrick DA, Hays T, et al. Contemporary pediatric splenectomy: continuing controversies. Pediatric surgery international. 2011;27:1165–71. doi: 10.1007/s00383-011-2929-x. [DOI] [PubMed] [Google Scholar]
- 45.Rescorla FJ, Engum SA, West KW, et al. Laparoscopic splenectomy has become the gold standard in children. Am Surg. 2002;68:297–301. discussion 01-2. [PubMed] [Google Scholar]
- 46.Rice HE, Oldham KT, Hillery CA, et al. Clinical and hematologic benefits of partial splenectomy for congenital hemolytic anemias in children. Ann Surg. 2003;237:281–8. doi: 10.1097/01.SLA.0000048453.61168.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buesing KL, Tracy ET, Kiernan C, et al. Partial splenectomy for hereditary spherocytosis: a multiinstitutional review. Journal of pediatric surgery. 2011;46:178–83. doi: 10.1016/j.jpedsurg.2010.09.090. [DOI] [PubMed] [Google Scholar]
- 48.Grace RF, Mednick RE, Neufeld EJ. Compliance with immunizations in splenectomized individuals with hereditary spherocytosis. Pediatr Blood Cancer. 2009;52:865–7. doi: 10.1002/pbc.21961. [DOI] [PubMed] [Google Scholar]
- 49.Dhermy D, Garbarz M, Lecomte MC, et al. Hereditary elliptocytosis: clinical, morphological and biochemical studies of 38 cases. Nouv Rev Fr Hematol. 1986;28:129–40. [PubMed] [Google Scholar]
- 50.Gallagher PG. Hereditary elliptocytosis: spectrin and protein 4.1R. Semin Hematol. 2004;41:142–64. doi: 10.1053/j.seminhematol.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 51.Dhermy D, Schrevel J, Lecomte MC. Spectrin-based skeleton in red blood cells and malaria. Curr Opin Hematol. 2007;14:198–202. doi: 10.1097/MOH.0b013e3280d21afd. [DOI] [PubMed] [Google Scholar]
- 52.Glele-Kakai C, Garbarz M, Lecomte MC, et al. Epidemiological studies of spectrin mutations related to hereditary elliptocytosis and spectrin polymorphisms in Benin. Br J Haematol. 1996;95:57–66. doi: 10.1046/j.1365-2141.1996.d01-1869.x. [DOI] [PubMed] [Google Scholar]
- 53.Zarkowsky HS, Mohandas N, Speaker CB, et al. A congenital haemolytic anaemia with thermal sensitivity of the erythrocyte membrane. Br J Haematol. 1975;29:537–43. doi: 10.1111/j.1365-2141.1975.tb02740.x. [DOI] [PubMed] [Google Scholar]
- 54.Costa DB, Lozovatsky L, Gallagher PG, et al. A novel splicing mutation of the {alpha}-spectrin gene in the original hereditary pyropoikilocytosis kindred. Blood. 2005;106:4367–69. doi: 10.1182/blood-2005-05-1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaetani M, Mootien S, Harper S, et al. Structural and functional effects of hereditary hemolytic anemia-associated point mutations in the alpha spectrin tetramer site. Blood. 2008;111:5712–20. doi: 10.1182/blood-2007-11-122457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ipsaro JJ, Harper SL, Messick TE, et al. Crystal structure and functional interpretation of the erythrocyte spectrin tetramerization domain complex. Blood. 2010;115:4843–52. doi: 10.1182/blood-2010-01-261396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morrow JS, Rimm DL, Kennedy SP, et al. Of membrane stability and mosaics: the spectrin cytoskeleton. In: Hoffman J, Jamieson J, editors. Handbook of Physiology. London: Oxford: 1997. pp. 485–540. [Google Scholar]
- 58.Coetzer T, Lawler J, Prchal JT, et al. Molecular determinants of clinical expression of hereditary elliptocytosis and pyropoikilocytosis. Blood. 1987;70:766–72. [PubMed] [Google Scholar]
- 59.Coetzer T, Palek J, Lawler J, et al. Structural and functional heterogeneity of alpha spectrin mutations involving the spectrin heterodimer self-association site: relationships to hematologic expression of homozygous hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood. 1990;75:2235–44. [PubMed] [Google Scholar]
- 60.Gallagher PG. Red Cell Membrane Disorders. Hematology ASH Education 2005113. 2005;2005:13–18. doi: 10.1182/asheducation-2005.1.13. [DOI] [PubMed] [Google Scholar]
- 61.Coetzer TL, Palek J. Partial spectrin deficiency in hereditary pyropoikilocytosis. Blood. 1986;67:919–24. [PubMed] [Google Scholar]
- 62.King MJ, Jepson MA, Guest A, et al. Detection of hereditary pyropoikilocytosis by the eosin-5-maleimide (EMA)-binding test is attributable to a marked reduction in EMA-reactive transmembrane proteins. Int J Lab Hematol. 2011;33:205–11. doi: 10.1111/j.1751-553X.2010.01270.x. [DOI] [PubMed] [Google Scholar]