

Abstract
Purpose of Review
Maintenance of cellular water and solute homeostasis is critical for survival of the erythrocyte. Inherited or acquired disorders that perturb this homeostasis jeopardize the erythrocyte, leading to its premature destruction. This report reviews recent progress in our understanding the determinants of erythrocyte hydration and its related disorders.
Recent Findings
The molecular and genetic bases of primary disorders of erythrocyte hydration are poorly understood. Recent studies have implicated roles for the anion transporter, SLC4A1, and the Rh-associated glycoprotein, RhAG. The most common secondary disorder associated with perturbed hydration of the erythrocyte is sickle cell disease, where dehydration contributes to disease pathology and clinical complications. Advances in understanding the mechanisms regulating erythrocyte solute and water content, particularly associated with KCl cotransport and Gardos channel activation, have revealed novel signaling mechanisms controlling erythrocyte hydration. These signaling pathways may provide innovative strategies to prevent erythrocyte dehydration in sickle cell disease.
Summary
Clinical, translational and biologic studies all contribute to our knowledge of erythrocyte hydration. Understanding the mechanisms controlling erythrocyte water and solute homeostasis will serve as a paradigm for other cells and may reveal new therapeutic targets for disease prevention and treatment.
Keywords: Erythrocyte, hydration, anion exchanger, RhAG, phosphorylation
Introduction
This is a review of recent advances in our understanding of the mechanisms controlling erythrocyte hydration. For the first time, progress has been made in understanding the genetic bases of primary disorders of erythrocyte hydration. In parallel, application of novel phosphoproteomic techniques to the study of erythrocyte hydration have yielded a better understanding of the posttranslational modifications regulating signaling pathways that influence cellular water and solute content.
Erythrocyte Hydration
Maintenance of water and solute homeostasis is critical to survival of the erythrocyte. Several pathways mediate water and solute homeostasis in normal red blood cells where cellular volume is primarily controlled via regulation of monovalent cation content (Table 1).[1] The Na+ K+ pump maintains the low sodium, high potassium composition of the human erythrocyte by actively transporting sodium out of and potassium into the cell. Several passive cation leak pathways maintain a balance of outward potassium and inward sodium fluxes to establish the steady state volume of the cell. Cation leak pathways have limited capacity to respond to alterations in monovalent cation leak, and, if this capacity is exceeded, cellular volume changes in parallel with the change in total cation content. Erythrocytes swell when the inward sodium leak exceeds the potassium leak out and shrink when the potassium leak out exceeds the inward sodium leak. Alterations in membrane permeability are detected by the analysis of intracellular sodium, potassium, total cation content, and altered indices of erythrocyte hydration, e.g. increased or decreased mean corpuscular hemoglobin concentration (MCHC).[2]
Table 1.
Principal Ion Transport Pathways of the Human Erythrocyte
|
Monovalent anions, predominantly chloride and bicarbonate, are distributed passively across the erythrocyte membrane, with steady state concentrations dictated by the Donnan equilibrium. Since the sum of all cations must equal the sum of all anions, the presence of impermeant anions in the cell, predominately hemoglobin, 2,3-diphosphoglycerate, and nucleotides, dictates that monovalent anion concentration be lower than total cation concentration. Changes in organic phosphate concentration, pH, or hemoglobin charge due to mutation, alter monovalent anion content and therefore water content, however these effects are generally small. Net anion permeability of the erythrocyte membrane is normally at least three orders of magnitude higher than cation permeability and water permeability is even higher. Water is always at osmotic equilibrium across the red cell membrane.
Disorders of Erythrocyte Hydration
Inherited disorders of erythrocyte hydration are a group of rare disorders ranging from dehydrated to overhydrated cells (Table 2). Depending on the degree of perturbation of water and solute homeostasis, hemolytic anemia of varying degree may result. More common are secondary disorders of erythrocyte hydration (Table 2). In these cases, the perturbation in cell hydration is secondary to another condition, for instance the dehydration that commonly accompanies the hemoglobinopathies. In secondary disorders, altered erythrocyte hydration may be a major contributor to disease pathology.
Table 2.
Abnormalities of Erythrocyte Hydration
|
Primary Disorders of Erythrocyte Hydration
The hereditary stomatocytosis (HSt) syndromes are a heterogeneous, rare group of disorders characterized by stomatocytic or mouth-shaped erythrocyte morphology on peripheral blood smear. HSt erythrocyte membranes exhibit abnormal permeability to sodium and potassium, with consequent modification of intracellular water content, ranging from dehydrated to markedly overhydrated erythrocytes.[3] The molecular basis(es) of the hereditary stomatocytosis syndromes is poorly understood. The variable clinical, laboratory, and pathophysiologic findings associated with the stomatocytosis syndromes suggest these are a complex collection of syndromes caused by various molecular defects. Some investigators subtype these disorders by the temperature dependence of the cation leak.[4]
Overhydrated Stomatocytosis (OHSt, Hydrocytosis)
Overhydrated stomatocytosis refers to a heterogeneous group of disorders associated with moderate to severe hemolytic anemia and altered sodium permeability of the erythrocyte membrane.[3] A dramatic increase in sodium permeability (15-40 times normal) leads to increased intracellular sodium, a lesser decrease in intracellular potassium, an increase in total monovalent cation content and an increase in cell water. Although a significant compensatory increase in Na+ K+ ATPase activity occurs, increased pump activity is unable to compensate for the markedly increased inward sodium leak. Erythrocyte membranes from many hydrocytosis patients lack stomatin, but mutations have not been found in the gene of affected patients and erythrocytes from stomatin knock-out mice are normal.
A recent study of hydrocytosis patients identified heterozygous missense mutations, Ile61Arg or Ser65Phe, in predicted transmembrane domain 2 of the Rh-associated glycoprotein (RhAG) gene. When expressed in Xenopus oocytes, mutant RhAG proteins induced abnormal cation fluxes. Interestingly, wild type RhAG also induced cation fluxes, but much less than the mutants. One model places RhAG with band 3, RhCE, RhD, CD47, LW, glycophorin B, and other proteins in a membrane “macrocomplex” which likely participate in erythrocyte gas exchange. RhAG may function as an ammonium transporter or, as recently hypothesized, as a gas channel with the mutations hypothesized to widen the pore, allowing passage of cations.[5]
Cryohydrocytosis
Cryohydrocytosis is a variant of hydrocytosis with abnormal cation transport and overhydrated erythrocytes.[3] Cryohydrocytosis erythrocytes have a mild cation leak at 37°C and develop a profound increase in cation permeability at low temperature, 5°C. Recently, missense mutations in band 3, the anion exchanger (AE1, SLC4A1), have been identified in patients with cryohydrocytosis.[6] These mutations, Ser731Pro and His734Arg, are located in a region between membrane spanning domain 8 and the final two membrane spanning domains (Figure 1). This region is involved in anion translocation. In vitro, the mutations inactivate band 3- mediated anion exchange. While still expressed at the membrane, these mutations may induce the mutant band 3 protein to produce an abnormal cation leak, a hypothesis supported by studies performed in Xenopus oocytes.[6,7]
Figure 1. A partial model of band 3, the erythrocyte membrane anion transporter.
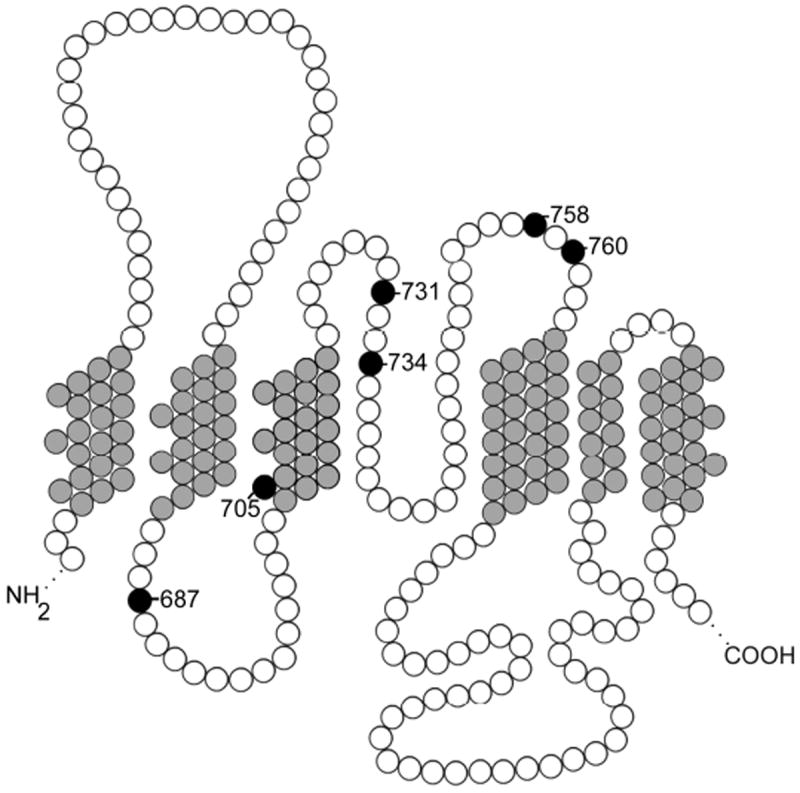
Transmembrane domains seven through twelve, shaded gray, and intracellular and extracellular loops, shaded white, are shown. Missense mutations in this region associated with ablation of anion function and abnormal cation leak, shaded black, are labeled.
Erythrocytes from one hereditary stomatocytosis/cryohydrocytosis variant, HSt Blackburn, do not exhibit the temperature dependence of cryohydrocytosis cells.[8] This variant is associated with a missense mutation, Leu 687Pro, in the same region of band 3 as the other cryohydrocytosis variants. It, too, induces abnormal cation fluxes in Xenopus oocytes.[6] Additional missense mutations have recently been reported in this region of band 3. Band 3 Ceigne, Gly796Arg, was identified in a patient with stomatocytosis and dyserythropoiesis.[9] The temperature dependence of the cation leak was not described.
Band 3 New Haven, Glu758Lys, was discovered in two unrelated patients with wellcompensated hereditary spherostomatocytic anemia. Xenopus oocytes expressing mutant band 3 New Haven, displayed glycophorin A-dependent surface expression and anion transport, as well as glycophorin A-independent pharmacologically distinct rubidium flux and small, glycophorin A-independent currents. These data suggest that the increased cation transport associated with the mutation reflects activation of endogenous cation permeability pathways rather than through mutant band 3.[10] Thus whether this group of missense mutations located in the central region of the band 3 transmembrane domain (Figure 1) cause conductive translocation of cations through the mutant protein itself or lead to activation of endogenous cation leak pathways remains unclear. The majority of HSt patients studied do not have mutations in either the band 3 or RhAG genes.
Hereditary Xerocytosis (HX, Dessicocytosis, Dehydrated Stomatocytosis)
The hereditary xerocytosis syndromes are characterized by minimal to mild anemia, small numbers of stomatocytes on peripheral blood smear, and erythrocyte dehydration due to net potassium loss that is not accompanied by a proportional gain of sodium.[3] Erythrocyte cations demonstrate a slight increase in sodium content, a greater decrease in potassium concentration, and, thus, a decrease in the net intracellular cation content and cell dehydration.
The molecular bases of the hereditary xerocytosis syndromes are unknown. A locus for both hereditary xerocytosis and the related disorder familial pseudohyperkalemia has been mapped to 16q23-q24, but the affected gene has not yet been identified.[11,12]
Rh-Null Disease
Rhnull and Rhmod disease are inherited erythrocyte disorders characterized by mild to moderate anemia with stomatocytes on peripheral blood smear and deficiency of Rh proteins in the erythrocyte membrane. The importance of the Rh locus for integrity of the erythrocyte membrane was recognized through the discovery of rare individuals whose erythrocytes lack (Rhnull) or have markedly decreased (Rhmod) Rh antigen expression. Rh proteins are part of a multiprotein complex that includes 2 Rh proteins and 2 Rh-associated glycoproteins (RhAG).[13] The genetic bases of the Rh deficiency syndromes are heterogeneous, with at least two groups defined.[14] The “amorph type” is due to defects involving the RH30 locus encoding the RhD and RhCE polypeptides. The “regulatory type” of Rhnull and Rhmod phenotypes results from suppressor or “modifier” mutations at the RH50 locus encoding the Rh-associated glycoprotein. When one chain of the Rh-RhAG complex is absent, the entire Rh multiprotein complex is either not transported to or assembled at the membrane.
Secondary Disorders of Erythrocyte Hydration
Erythrocyte dehydration is a secondary effect of several erythrocyte disorders including sickle cell disease, thalassemia, hemoglobin C, and hereditary spherocytosis (Table 2). In sickle cell disease, complex interactions between the polymerization of hemoglobin S and activity of several water and solute transport systems leads to cellular dehydration. At least three pathways mediate cellular dehydration of sickle erythrocytes, a KCl cotransport pathway, a sickling-induced nonselective cation leak pathway, and a calcium-activated potassium channel (the Gardos channel).
KCl cotransport pathways, which normally regulate reticulocyte volume, function pathologically in sickle erythrocytes by exceeding their target hemoglobin concentration, priming the erythrocyte to sickle.[15] The polymerization of hemoglobin S activates an abnormal permeability pathway, Psickle.[2] Upon deoxygenation, this nonselective cation leak pathway whose molecular identity is unknown, is activated, inducing a calcium leak and turning on the Gardos channel. This leads to rapid transport of KCl and water from the cell.
These populations of dehydrated erythrocytes exhibit decreased deformability, increased fragility, and are much more prone to hemoglobin S polymerization and sickling. Dense cells are trapped in the microcirculation and rapidly removed. Because hemoglobin S polymerization is a concentration-dependent process, a number of therapeutic strategies aimed at reducing polymerization by modifying the intracellular concentration of hemoglobin S have been employed or are in development.[16]
KCl Cotransport
KCl cotransport mediates the strictly coupled electroneutral transport of potassium and chloride. Influenced by the oxygenation state of hemoglobin, KCl cotransport is regulated by internal pH, cell volume, and low intracellular magnesium. Its functional state is regulated by transporter phosphorylation/dephosphorylation. Activation of KCl cotransport by cellular swelling occurs following dephosphorylation by protein phosphatases 1 and 2 and alteration of a serine/threonine kinase, and this activation is blocked by several kinase and phosphatase inhibitors,[17] although until very recently the phosphorylation sites that control KCl activity upon changes of cellular tonicity were unknown.
Identification of the phosphorylation sites controlling KCl cotransport, and analysis of their dynamic changes upon osmotic challenge, has been hampered for year by a number of technologic hurdles. Application of novel phosphoproteomic techniques to this fundamental question was performed by Rinehart and colleagues (Figure 2).[18] These studies utilized novel, titanium dioxide-based phosphopeptide enrichment,[19] followed by novel techniques that quantitate dynamic changes in phosphorylation under different experimental conditions, SILAC (stable isotope labeling of amino acids in cell culture) that uncovers relative changes in a “discovery” mode and MRM (multiple reaction monitoring) that measures absolute percentage of phosphorylation of specifically targeted sites.[20-22] A general schema for performing quantitative phosphoproteomics using these techniques or iTRAQ (isobaric tagging technology) for relative and absolute quantitation,[23] are shown in Figure 2. Together, with the use of these techniques, Rinehart and colleagues identified two sites of phosphorylation, T991 and T1048, in the KCC3 transporter that mediate its activation in response to osmotic stress, both in transfected HEK293 cells and in mature human erythrocytes.[18] These two sites were conserved among all known KCl cotransporters from early chordates to humans, implying a broad role in KCl cotransport.
Figure 2. Proteomic workflows for identification and quantitation of phosphorylated proteins and peptides.
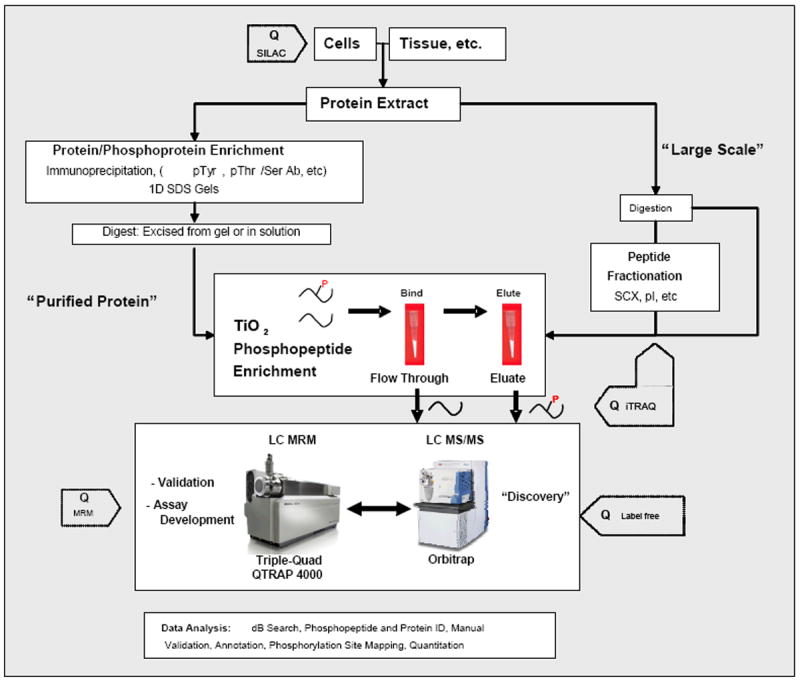
Workflow is divided into two options: one for TiO2 enrichment of phosphopeptides after the protein(s) of interest are purified or immunoprecipitated and another for more complex phosphopeptide mixtures directly extracted from tissue or whole cell extracts. The former is a more targeted approach to discover numerous sites of phosphorylation on proteins of interest, whereas the latter is directed towards more global discovery based approaches. For most of these approaches, three different quantitative methods (SILAC, iTRAQ, and label-free quantitation) can be introduced at steps indicated by “Q” to determine changes of phosphorylation between two or more samples.
The post translational control of KCl cotransport is clearly complex with many unresolved issues. Identification of two phosphorylation sites with non-additive effects on KCl cotransport activity permit layers of regulation, both by differing signaling pathways as well as varying levels of phosphorylation/dephosphorylation. Because the sites have non-additive effects, dephosphorylation may be cooperative or independent and stochastic. This complexity is heightened by observations that some tyrosine kinase inhibitors inactivate cotransport, while others activate it. Another critical goal to our understanding of KCl cotransport is the identification of the kinases and phosphatases that control its activity. Ultimately, unraveling the complexity of KCl cotransport, including the contributions from various conditions such as cellular volume, pH, oxygenation state, magnesium content, etc. will provide us with a clearer understanding of cellular function.
The Gardos Channel (KCNN4, K(Ca)3.1))
The Gardos channel is a calcium-sensitive, intermediate-conductance, potassium selective channel present in many cell types. Described by George Gardos to mediate calcium-activated potassium fluxes in erythrocytes, it has subsequently been found in many cell and tissue types where it plays critical roles in normal and pathologic processes such as immune function and vascular biology.[24,25] In recent studies, Gardos channels were found in numerous cells contributing to atherosclerotic lesions including lymphocytes, macrophages, endothelial cells, and vascular smooth muscle cells.[26] In vivo, Gardos channel blockade suppressed the proliferation and migration of vascular smooth muscle cells, decreased lymphocyte activation, and decreased the size of atherosclerotic lesions, suggesting that such a strategy may be useful in treatment of acquired vascular disease.
The function of the Gardos channel is unknown in normal, mature erythrocytes, although it has been speculated to protect the cell from complement-mediated colloid osmotic lysis or to play a role in erythroid differentiation. Normally, the Gardos channel is inactive, yet when fully activated, Gardos has the capacity to mediate rapid erythrocyte dehydration, far exceeding that of other red cell transporters.[2,27] Erythrocytes from mice lacking the Gardos channel exhibit macrocytosis, increased susceptibility to osmotic lysis, and decreased filterability, suggesting a role for the channel in the establishment of steady state hydration.[28] These mice develop progressive splenomegaly and splenic overload during the course of their lifetime, suggesting a degree of chronic hemolysis.
Similar to KCl cotransporters, signaling cascades that mediate Gardos channel function are critical to cellular function. Gardos channels have been shown to require phosphatidylinositol 3 phosphate (PI[3]P) for activity, an activity inhibited by PI[3]P phosphatase myotubularin-related protein 6 (MTMR6)[29,30] (MTMR6) and localized to a 14 amino acid region in the COOH-terminus.[31] In a remarkable series of experiments, this region was shown to recruit nucleoside diphosphate kinase (NDPK) which activates the Gardos channel by phosphorylating a histidine residue in the 14 amino acid region.[32] Protein histidine phosphatase 1 (PHPT-1) directly binds and dephosphorylates this histidine, inhibiting Gardos channel activity.[33] Study of the Gardos channel, along other mammalian proteins known to undergo reversible histidine phosphorylation such as the beta subunit of heterotrimeric guanosine triphosphate-binding (G) proteins and the enzyme adenosine 5’-triphosphate-citrate lyase, will provide exciting insight into this novel mechanism of cell signaling.[34]
Clinical Implications and Future Directions
In sickle cell disease, because hemoglobin S polymerization is a concentration-dependent process, dehydrated sickle erythrocytes are much more prone to hemolysis, contributing to clinical complications such as pulmonary hypertension, leg ulcers, priapism, and stroke. Thus a number of therapeutic strategies aimed at reducing sickle polymerization by modifying the intracellular concentration of hemoglobin S have been employed or are in development.[35] Preventing cellular dehydration and decreasing the relative amount of sickle hemoglobin by modifying intracellular water and solute content of sickle erythrocytes via blockade of the pathways that lead to potassium loss and cellular dehydration is an appealing strategy in the prevention of complications of sickle cell disease. Trials inhibiting KCl cotransport with magnesium [36-38] or blocking the Gardos channel via imidazole antimycotics,[39] arginine,[40] or senicapoc[41,42] have been reported. In a recent pilot study of senicapoc, a Gardos channel blocker, sickle cell patients demonstrated reduced numbers of dehydrated cells, increased hemoglobin, and decreased hemolysis.[41]
In normal patients, variation in indices of erythrocyte hydration, including volume and hemoglobin concentration, are genetically determined.[43,44] We hypothesize that variants in the genes encoding the proteins involved in erythrocyte volume homeostasis contribute to alterations in erythrocyte hydration in normal controls and patients with sickle cell disease. Based on the presence or absence of genetic variations and their ability to ameliorate or worsen cellular hydration homeostasis, we also hypothesize that there will be variability in the clinical response to pharmacologic interventions aimed at modifying erythrocyte water and solute in sickle erythrocytes. Ultimately, specific patient pharmacogenomic profiles will predict response to therapeutic interventions to modify erythrocyte hydration and thus ameliorate clinical severity in sickle cell disease.
Conclusions
Studies from both the clinic and the laboratory are enhancing our knowledge of normal and abnormal erythrocyte hydration. Ongoing work should include rigorous interrogation of genetic variants discovered in the clinic as well as translation of laboratory advances in regulation of erythrocyte homeostasis into therapeutic targets. Together, these studies will provide tools to prevent, ameliorate, or cure the clinical consequences of perturbation of erythrocyte hydration in patients with these disorders.
Acknowledgments
Supported by NHLBI Proteomics Center N01-HV-28186 with instrumentation support from NCRR UL1 RR024139 and 1S10RR024617-01; Cincinnati Comprehensive Sickle Cell Center U54HL07087; the Leducq Transatlantic Network in Hypertension and the Howard Hughes Medical Institute; and HL65448 and DK62039.
Footnotes
The authors have no conflicts of interest to declare.
References and Recommended Reading
Papers of particular interest, published within the annual period of review, have been highlighted as
-
*
of special interest
-
**
of outstanding interest
- 1.Brugnara C. Erythrocyte membrane transport physiology. Curr Opin Hematol. 1997;4:122–127. doi: 10.1097/00062752-199704020-00008. [DOI] [PubMed] [Google Scholar]
- 2.Lew VL, Bookchin RM. Ion transport pathology in the mechanism of sickle cell dehydration. Physiol Rev. 2005;85:179–200. doi: 10.1152/physrev.00052.2003. [DOI] [PubMed] [Google Scholar]
- 3.Delaunay J. The hereditary stomatocytoses: genetic disorders of the red cell membrane permeability to monovalent cations. Semin Hematol. 2004;41:165–172. doi: 10.1053/j.seminhematol.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Ellory JC, Robinson HC, Browning JA, Stewart GW, Gehl KA, Gibson JS. Abnormal permeability pathways in human red blood cells. Blood Cells Mol Dis. 2007;39:1–6. doi: 10.1016/j.bcmd.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 5**.Bruce LJ, Guizouarn H, Burton NM, Gabillat N, Poole J, Flatt JF, Brady RL, Borgese F, Delaunay J, Stewart GW. The monovalent cation leak in overhydrated stomatocytic red blood cells results from amino acid substitutions in the Rh-associated glycoprotein. Blood. 2009;113:1350–1357. doi: 10.1182/blood-2008-07-171140. This manuscript reports the first genetic defects in overhydrated stomatocytosis. [DOI] [PubMed] [Google Scholar]
- 6.Bruce LJ, Robinson HC, Guizouarn H, Borgese F, Harrison P, King MJ, Goede JS, Coles SE, Gore DM, Lutz HU, et al. Monovalent cation leaks in human red cells caused by single amino-acid substitutions in the transport domain of the band 3 chloride-bicarbonate exchanger, AE1. Nat Genet. 2005;37:1258–1263. doi: 10.1038/ng1656. [DOI] [PubMed] [Google Scholar]
- 7.Guizouarn H, Martial S, Gabillat N, Borgese F. Point mutations involved in red cell stomatocytosis convert the electroneutral anion exchanger 1 to a nonselective cation conductance. Blood. 2007;110:2158–2165. doi: 10.1182/blood-2006-12-063420. [DOI] [PubMed] [Google Scholar]
- 8.Coles SE, Ho MM, Chetty MC, Nicolaou A, Stewart GW. A variant of hereditary stomatocytosis with marked pseudohyperkalaemia. Br J Haematol. 1999;104:275–283. doi: 10.1046/j.1365-2141.1999.01191.x. [DOI] [PubMed] [Google Scholar]
- 9*.Iolascon A, De Falco L, Borgese F, Esposito MR, Avvisati RA, Izzo P, Piscopo C, Guizouarn H, Biondani A, Pantaleo A, et al. A novel erythroid anion exchange variant (Gly796Arg) of hereditary stomatocytosis associated with dyserythropoiesis. Haematologica. 2009;94:1049–1059. doi: 10.3324/haematol.2008.002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10*.Stewart AK, Vandorpe DH, Heneghan JF, Chebib F, Stolpe K, Akhavein A, Edelman EJ, Maksimova Y, Gallagher PG, Alper SL. The glycophorin A (GPA)-dependent, spherostomatocytosis mutant AE1 E758K induces GPA-independent, endogenous cation transport in amphibian oocytes. Am J Physiol Cell Physiol. 2009 doi: 10.1152/ajpcell.00444.2009. References 9 and 10 describe missense mutations in band 3 associated with ablation of anion transport function of band 3 and novel cation leaks in mutant cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carella M, Stewart G, Ajetunmobi JF, Perrotta S, Grootenboer S, Tchernia G, Delaunay J, Totaro A, Zelante L, Gasparini P, et al. Genomewide search for dehydrated hereditary stomatocytosis (hereditary xerocytosis): mapping of locus to chromosome 16 (16q23-qter) Am J Hum Genet. 1998;63:810–816. doi: 10.1086/302024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iolascon A, Stewart GW, Ajetunmobi JF, Perrotta S, Delaunay J, Carella M, Zelante L, Gasparini P. Familial pseudohyperkalemia maps to the same locus as dehydrated hereditary stomatocytosis (hereditary xerocytosis) Blood. 1999;93:3120–3123. [PubMed] [Google Scholar]
- 13.Van Kim CL, Colin Y, Cartron JP. Rh proteins: Key structural and functional components of the red cell membrane. Blood Rev. 2005 doi: 10.1016/j.blre.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Westhoff CM. The structure and function of the Rh antigen complex. Semin Hematol. 2007;44:42–50. doi: 10.1053/j.seminhematol.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joiner CH, Rettig RK, Jiang M, Risinger M, Franco RS. Urea stimulation of KCl cotransport induces abnormal volume reduction in sickle reticulocytes. Blood. 2007;109:1728–1735. doi: 10.1182/blood-2006-04-018630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brugnara C. Sickle cell disease: from membrane pathophysiology to novel therapies for prevention of erythrocyte dehydration. J Pediatr Hematol Oncol. 2003;25:927–933. doi: 10.1097/00043426-200312000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Adragna NC, Di Fulvio M, Lauf PK. Regulation of K-Cl cotransport: from function to genes. J Membr Biol. 2004;201:109–137. doi: 10.1007/s00232-004-0695-6. [DOI] [PubMed] [Google Scholar]
- 18**.Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138:525–536. doi: 10.1016/j.cell.2009.05.031. Using novel phosphoproteomics techniques this report identifies the amino acids responsible for KCl response to osmotic stress, a process widely generalizable to many cell types across species. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods. 2007;4:231–237. doi: 10.1038/nmeth1005. [DOI] [PubMed] [Google Scholar]
- 20.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 21.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104:5860–5865. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leitner A, Lindner W. Chemical tagging strategies for mass spectrometry-based phospho-proteomics. Methods Mol Biol. 2009;527:229–243. doi: 10.1007/978-1-60327-834-8_17. [DOI] [PubMed] [Google Scholar]
- 24.Jensen BS, Strobaek D, Olesen SP, Christophersen P. The Ca2+-activated K+ channel of intermediate conductance: a molecular target for novel treatments? Curr Drug Targets. 2001;2:401–422. doi: 10.2174/1389450013348173. [DOI] [PubMed] [Google Scholar]
- 25.Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C, Alper SL, Melvin JE. Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J Biol Chem. 2004;279:47681–47687. doi: 10.1074/jbc.M409627200. [DOI] [PubMed] [Google Scholar]
- 26*.Toyama K, Wulff H, Chandy KG, Azam P, Raman G, Saito T, Fujiwara Y, Mattson DL, Das S, Melvin JE, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118:3025–3037. doi: 10.1172/JCI30836. This report demonstrates the role of the Gardos channel in the pathobiology of atherosclerosis, making it an attractive target to prevent acquired vascular disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lew VL, Etzion Z, Bookchin RM. Dehydration response of sickle cells to sickling-induced Ca(++) permeabilization. Blood. 2002;99:2578–2585. doi: 10.1182/blood.v99.7.2578. [DOI] [PubMed] [Google Scholar]
- 28.Grgic I, Kaistha BP, Paschen S, Kaistha A, Busch C, Si H, Kohler K, Elsasser HP, Hoyer J, Kohler R. Disruption of the Gardos channel (KCa3.1) in mice causes subtle erythrocyte macrocytosis and progressive splenomegaly. Pflugers Arch. 2009;458:291–302. doi: 10.1007/s00424-008-0619-x. [DOI] [PubMed] [Google Scholar]
- 29.Srivastava S, Ko K, Choudhury P, Li Z, Johnson AK, Nadkarni V, Unutmaz D, Coetzee WA, Skolnik EY. Phosphatidylinositol-3 phosphatase myotubularin-related protein 6 negatively regulates CD4 T cells. Mol Cell Biol. 2006;26:5595–5602. doi: 10.1128/MCB.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srivastava S, Li Z, Lin L, Liu G, Ko K, Coetzee WA, Skolnik EY. The phosphatidylinositol 3-phosphate phosphatase myotubularin- related protein 6 (MTMR6) is a negative regulator of the Ca2+-activated K+ channel KCa3.1. Mol Cell Biol. 2005;25:3630–3638. doi: 10.1128/MCB.25.9.3630-3638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava S, Choudhury P, Li Z, Liu G, Nadkarni V, Ko K, Coetzee WA, Skolnik EY. Phosphatidylinositol 3-phosphate indirectly activates KCa3.1 via 14 amino acids in the carboxy terminus of KCa3.1. Mol Biol Cell. 2006;17:146–154. doi: 10.1091/mbc.E05-08-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava S, Li Z, Ko K, Choudhury P, Albaqumi M, Johnson AK, Yan Y, Backer JM, Unutmaz D, Coetzee WA, et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol Cell. 2006;24:665–675. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 33*.Srivastava S, Zhdanova O, Di L, Li Z, Albaqumi M, Wulff H, Skolnik EY. Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1. Proc Natl Acad Sci U S A. 2008;105:14442–14446. doi: 10.1073/pnas.0803678105. This series of reports, references 29-33, reveals that histidine phosphorylation/dephosphorylation is a critical component of Gardos channel activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klumpp S, Krieglstein J. Reversible phosphorylation of histidine residues in proteins from vertebrates. Sci Signal. 2009;2:pe13. doi: 10.1126/scisignal.261pe13. [DOI] [PubMed] [Google Scholar]
- 35.Raghupathy R, Billett HH. Promising therapies in sickle cell disease. Cardiovasc Hematol Disord Drug Targets. 2009;9:1–8. doi: 10.2174/187152909787581354. [DOI] [PubMed] [Google Scholar]
- 36.De Franceschi L, Bachir D, Galacteros F, Tchernia G, Cynober T, Alper S, Platt O, Beuzard Y, Brugnara C. Oral magnesium supplements reduce erythrocyte dehydration in patients with sickle cell disease. J Clin Invest. 1997;100:1847–1852. doi: 10.1172/JCI119713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Franceschi L, Bachir D, Galacteros F, Tchernia G, Cynober T, Neuberg D, Beuzard Y, Brugnara C. Oral magnesium pidolate: effects of long-term administration in patients with sickle cell disease. Br J Haematol. 2000;108:284–289. doi: 10.1046/j.1365-2141.2000.01861.x. [DOI] [PubMed] [Google Scholar]
- 38*.Hankins JS, Wynn LW, Brugnara C, Hillery CA, Li CS, Wang WC. Phase I study of magnesium pidolate in combination with hydroxycarbamide for children with sickle cell anaemia. Br J Haematol. 2008;140:80–85. doi: 10.1111/j.1365-2141.2007.06884.x. This report describes initial results of hydroxyurea combined with magnesium treatment in modulation of erythrocyte hydration via blockade of KCl cotransport in sickle cell disease. [DOI] [PubMed] [Google Scholar]
- 39.Brugnara C, Gee B, Armsby CC, Kurth S, Sakamoto M, Rifai N, Alper SL, Platt OS. Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J Clin Invest. 1996;97:1227–1234. doi: 10.1172/JCI118537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romero JR, Suzuka SM, Nagel RL, Fabry ME. Arginine supplementation of sickle transgenic mice reduces red cell density and Gardos channel activity. Blood. 2002;99:1103–1108. doi: 10.1182/blood.v99.4.1103. [DOI] [PubMed] [Google Scholar]
- 41*.Ataga KI, Smith WR, De Castro LM, Swerdlow P, Saunthararajah Y, Castro O, Vichinsky E, Kutlar A, Orringer EP, Rigdon GC, et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood. 2008;111:3991–3997. doi: 10.1182/blood-2007-08-110098. This is a report of safety and initial efficacy of senicapoc on preventing erythrocyte dehydration via Garods channel blockade in sickle cell disease. [DOI] [PubMed] [Google Scholar]
- 42.Ataga KI, Stocker J. Senicapoc (ICA-17043): a potential therapy for the prevention and treatment of hemolysis-associated complications in sickle cell anemia. Expert Opin Investig Drugs. 2009;18:231–239. doi: 10.1517/13543780802708011. [DOI] [PubMed] [Google Scholar]
- 43.Soranzo N, Spector TD, Mangino M, Kuhnel B, Rendon A, Teumer A, Willenborg C, Wright B, Chen L, Li M, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41:1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferreira MA, Hottenga JJ, Warrington NM, Medland SE, Willemsen G, Lawrence RW, Gordon S, de Geus EJ, Henders AK, Smit JH, et al. Sequence variants in three loci influence monocyte counts and erythrocyte volume. Am J Hum Genet. 2009;85:745–749. doi: 10.1016/j.ajhg.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]