

Abstract
Background
The phylogeny of the genus Methanobrevibacter was established almost 25 years ago on the basis of the similarities of the 16S rRNA oligonucleotide catalogs. Since then, many 16S rRNA gene sequences of newly isolated strains or clones representing the genus Methanobrevibacter have been deposited. We tried to reorganize the 16S rRNA gene sequences of this genus and revise the taxonomic affiliation of the isolates and clones representing the genus Methanobrevibacter.
Results
The phylogenetic analysis of the genus based on 786 bp aligned region from fifty-four representative sequences of the 120 available sequences for the genus revealed seven multi-member groups namely, Ruminantium, Smithii, Woesei, Curvatus, Arboriphilicus, Filiformis, and the Termite gut symbionts along with three separate lineages represented by Mbr. wolinii, Mbr. acididurans, and termite gut flagellate symbiont LHD12. The cophenetic correlation coefficient, a test for the ultrametric properties of the 16S rRNA gene sequences used for the tree was found to be 0.913 indicating the high degree of goodness of fit of the tree topology. A significant relationship was found between the 16S rRNA sequence similarity (S) and the extent of DNA hybridization (D) for the genus with the correlation coefficient (r) for logD and logS, and for [ln(-lnD) and ln(-lnS)] being 0.73 and 0.796 respectively. Our analysis revealed that for this genus, when S = 0.984, D would be <70% at least 99% of the times, and with 70% D as the species "cutoff", any 16S rRNA gene sequence showing <98% sequence similarity can be considered as a separate species. In addition, we deduced group specific signature positions that have remained conserved in evolution of the genus.
Conclusions
A very significant relationship between D and S was found to exist for the genus Methanobrevibacter, implying that it is possible to predict D from S with a known precision for the genus. We propose to include the termite gut flagellate symbiont LHD12, the methanogenic endosymbionts of the ciliate Nyctotherus ovalis, and rat feces isolate RT reported earlier, as separate species of the genus Methanobrevibacter.
Background
Methanogens are members of the domain Archaea, and fall within the kingdom Euryarchaeota [1]. They are obligate anaerobes and can be unambiguously differentiated from other organisms since they all produce methane as a major catabolic product [2]. A significant source of global atmospheric methane (≤ 1.30 μmol g [fresh weight]-1 h-1) is contributed by termites, which are the terrestrial arthropods, that exist in high biomass densities [3]. This methane production has been attributed to the methanogenic Archaea, which reside in their gut and the symbiotic role of these methanogens in the gut of termites has already been reported [3-7].
Methanogens that reduce CO2 with H2 to form methane are common inhabitants of the gastrointestinal tract ecosystems. Methanobrevibacter is one such major intestinal genus of the Methanobacteriaceae family that can reduce CO2 with H2 to form methane [8]. A majority of the species of the genus Methanobrevibacter were isolated from the gastrointestinal ecosystems. Mbr. ruminantium, the type species, was isolated from bovine rumen whereas Mbr. smithii was isolated from human colon [8,9]. Ferrai et al. isolated Mbr. oralis from the human oral cavity [10]. Three different species were isolated from termite hindguts: Mbr. cuticularis, Mbr. curvatus, and Mbr. filiformis [3,4]. Recently, Miller and Lin proposed the formal nomenclature for five methanogens isolated from animal feces earlier [8,11]. They represent four new species named Mbr. gottschalkii, Mbr. thaueri, Mbr. woesei, and Mbr. wolinii. The eleventh species, Mbr. arboriphilicus, was isolated from decaying cottonwood trees [12]. Recently, one more species, Mbr. acididurans, was isolated from an anaerobic digester [13].
Phenotypic differentiation of species of Methanobrevibacter is unsatisfactory because of the lack of distinguishing morphological, biochemical, and physiological characteristics [3]. The limited number of markers and the lack of information about their distribution among strains and the phenotypic differences between strains mandate the use of more powerful molecular tools for establishing phylogenetic relationships [8]. Two important genotypic markers widely used in recent bacterial taxonomy are the 16S rRNA gene sequence data and DNA-DNA hybridization data. Many researchers reported the correlation between 16S rRNA gene sequence similarity values and genomic DNA relatedness. For the domain Bacteria, Wayne et al. [14] proposed that phenotypically related bacterial strains showing 70% or greater genomic DNA relatedness constitute a single bacterial species. In contrast, those having <70% but >20% similarity are considered to be different species within a genus [15]. 16S rRNA gene sequence similarity value below 97% corresponds to DNA reassociation value not more than 60% whereas 16S rRNA gene similarities over 97%, require genomic DNA reassociation studies to assess relation of two organisms as a single or separate species [16]. In contrast to this generalized view, Boone et al. [17] considered that a sequence similarity of 98% or less as an evidence for separate species within the methanogens. Since the study deals with methanogens, novel species proposals will be based on the latter study by Boone et al. [17].
The statistical implications of this correlation between these two parameters are of great interest in prokaryotic systematics. Devereux et al (1990) proposed that, if the extent of DNA hybridization (D) was considered equivalent to 16S rRNA gene sequence similarity (S), then logS = K logD, where K was a constant [18]. Consistent with this assumption, a very significant correlation was found between logS and logD for many taxa including the family Methanobacteriaceae [18,19]. Thus, 16S rRNA sequence similarity can be a good predictor of extent of DNA hybridization, and being powerful, reliable and convenient, it can be used for the determination of taxonomic affiliation of newly isolated strains or clones to a particular genera [16-19]. This is specifically advantageous in the studies of environmental samples where many organisms are detected only by their rRNA sequence.
The phylogeny of the genus Methanobrevibacter was established almost 25 years ago on the basis of the similarities of the 16S rRNA oligonucleotide catalogs [2]. Since then, many 16S rRNA gene sequences of newly isolated strains or clones representing the genus Methanobrevibacter have been deposited in the GenBank and other public databases. Many reports describing Methanobrevibacter taxonomy based on 16S rRNA gene sequence analysis restricted themselves to Methanobrevibacter species present in the feces of higher animals [8,11,20] and gut of termites [5,6]. A relatively recent report studied the acquisition of methanogenic archaeal symbionts by anaerobic ciliates, both free-living and intestinal, on the basis of 16S rRNA gene sequence similarity [7]. Till date, there is no report describing Methanobrevibacter taxonomy using all the 16S rRNA gene sequences that are available in the database.
It was therefore felt necessary to reorganize the 16S rRNA gene sequences of this genus and revise the taxonomic affiliation of the isolates and clones representing the genus Methanobrevibacter. We studied the correlation between 16S rRNA gene sequence similarity and the extent of DNA hybridization for the genus Methanobrevibacter based on the available DNA hybridization data [8,21,22]. Since majority of available sequences in the database were from clones of environmental samples, we have used 16S rRNA gene sequence as predictor for taxonomy of the genus. In addition, we have deduced group specific nucleotide positions showing specific nucleotide substitutions in the 16S rRNA gene sequences of this genus. Furthermore, we propose to include termite gut flagellate symbiont LHD12, the methanogenic endosymbionts of the ciliate Nyctotherus ovalis, and rat feces isolate RT reported earlier [6-8], as separate species in the genus Methanobrevibacter. Till sufficient information is made available about other markers, our report on Methanobrevibacter taxonomy will prove to be very useful for those who use 16S rRNA gene sequence data for identification of their isolates.
Results
Sequence retrieval and phylogenetic analysis
A total of 120 sequences specific for the 16S rRNA gene for the genus Methanobrevibacter, were available in the public database as of May 10, 2003. A majority of the sequences (86 sequences) were from Methanobrevibacter specific 16S rRNA gene clones whereas only 34 sequences were from isolated strains of the genus Methanobrevibacter. Moreover, all the available sequences were partial with length varying from 287 bp to 1481 bp and only half of these were larger than 1 kb. It was reported earlier that phylogenetic trees based on partial sequence have the same topologies as those based on complete sequence with the established groups being identical but some deep branches differing slightly [23,24]. We therefore included only those sequences (total 82 sequences) that were larger than 600 bp, since this sequence length ensured the inclusion of all those sequences that would otherwise be excluded with larger sequence length criteria. An initial similarity analysis of the sequences showed the presence of sixteen sequences grouping in three different sets of replicate sequences with 100% similarity. Amongst all such sequences, only one representative sequence was used from each set for the further analysis. Moreover, some sequences aligning for regions shorter than 500 bp like, Mbr. oralis, many endosymbionts of the ciliate Nyctotherus spp., and methanogenic clones associated with rumen ciliates [25], were also excluded. Thus, a total of 54 sequences, of which twenty sequences representing Methanobrevibacter strains whereas thirty-four sequences representing Methanobrevibacter spp. specific 16S rRNA gene clones, giving an alignment of 802 nucleotide positions (bases 286 to 1120, E. coli numbering) were used for the final analysis. An initial similarity analysis revealed that the percentage similarity values ranged from 87–100% for all these sequences whereas they showed less then 84% sequence similarity with the representatives of other genera of the family Methanobacteriaceae.
Based on the phylogenetic analysis of the 54 sequences, the genus Methanobrevibacter could be clustered into seven distinct phylogenetic groups comprising of more than one strain and/or clone in each group as shown in Fig. 1. These groups were designated as Ruminantium, Smithii, Woesei, Curvatus, Arboriphilicus, Filiformis, and the Termite gut symbiont group. Mbr. wolinii, Mbr. acididurans and the termite gut flagellate symbiont clone LHD12 formed three separate lineages in the tree and were thus not considered in any of the above mentioned groups. In most cases, the same groupings were obtained regardless of the method used: neighbor-joining, parsimony, maximum likelihood or the UPGMA. The groupings were done taking in view that all the members of a particular group arise from a common node, i.e., monophyletic, and have the same topology regardless of the method that was used. The CCC analysis, the parameter that measures the correlation between similarity values calculated during tree building and the observed similarity, was found to be high (r = 0.913). A branch that showed a separate lineage with a recent evolution in the Arboriphilicus group as seen in the phylogenetic tree (Fig. 1) was represented by a strain that was an endosymbiont of the ciliate Nyctotherus ovalis [7]. This strain with accession number AJ132639 was designated as strain NO by us.
Figure 1.
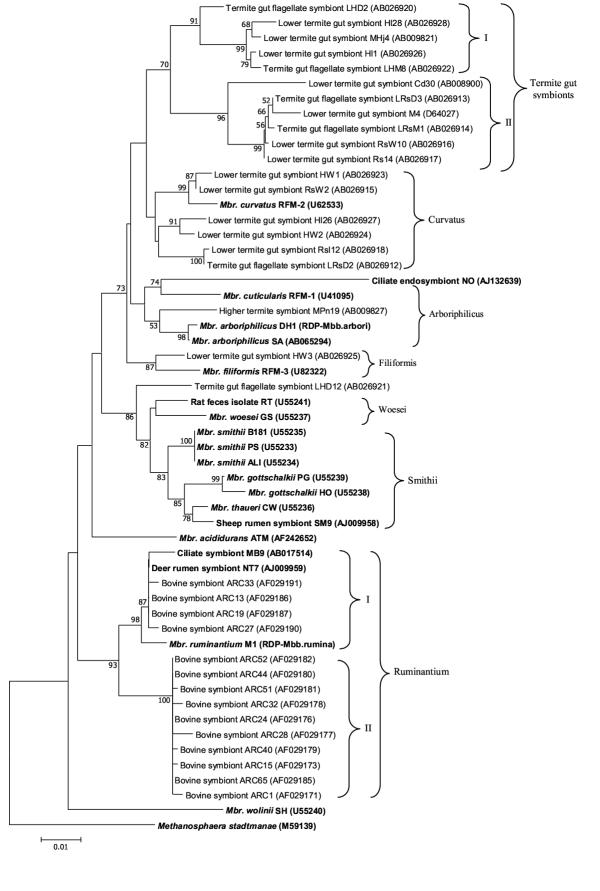
Phylogenetic tree of the members of the genus Methanobrevibacter based on the 16S rRNA gene. The tree was generated using CLUSTAL W program [30] for sequence alignment (786 bp) and by the neighbor joining method using Kimura 2 parameter distances in MEGA 2.1 software. Only positions 286–1120 (E. coli numbering) were considered with Msp. stadtmanae as the outgroup. Numbers at nodes indicate percent bootstrap values above 50 (1000 replicates). Bar indicates Jukes-Cantor evolutionary distance. Bold letters indicate isolated strains whereas normal font indicates clones.
Correlation of D and S
Using the available data, a highly significant relationship was found between the logarithmic transformations of S and D (Fig. 2). The correlation coefficient (r) between logS and logD was 0.73, which was significant at P < 0.0001 (Fig. 2a). Moreover, a high CCC (0.913), that tests the ultrametric properties of the sequences, proved that the biological relationship between D and S is valid for the genus Methanobrevibacter [18,19]. To look for an empirical relationship that might be useful to predict D from S, the complementary log log transformation [ln(-lnD) versus ln(-lnS)] produced a much better correlation coefficient, 0.796, which was significant at P < 0.0001, than found with the normal log transformations of D and S (Fig. 2b). In the absence of a systematic variation in the error, this analysis suggested that D would be less than 70 % at least 99 % of the time when S = 0.984 (Fig. 2c). These results were in good agreement with the proposal of Boone et al. [17], who proposed that a sequence similarity of 98% or less be considered as evidence for a separate species within the methanogens and we base our novel species proposals on this. Estimates of D for a given S were calculated from regression of ln(-lnD) vs ln(-lnS) and standard deviation (SD) of residuals in D, where ln(-lnD) = 0.5077 [ln(-lnS)] + 1.8999 and the SD of the residuals in D was 0.3498.
Figure 2.
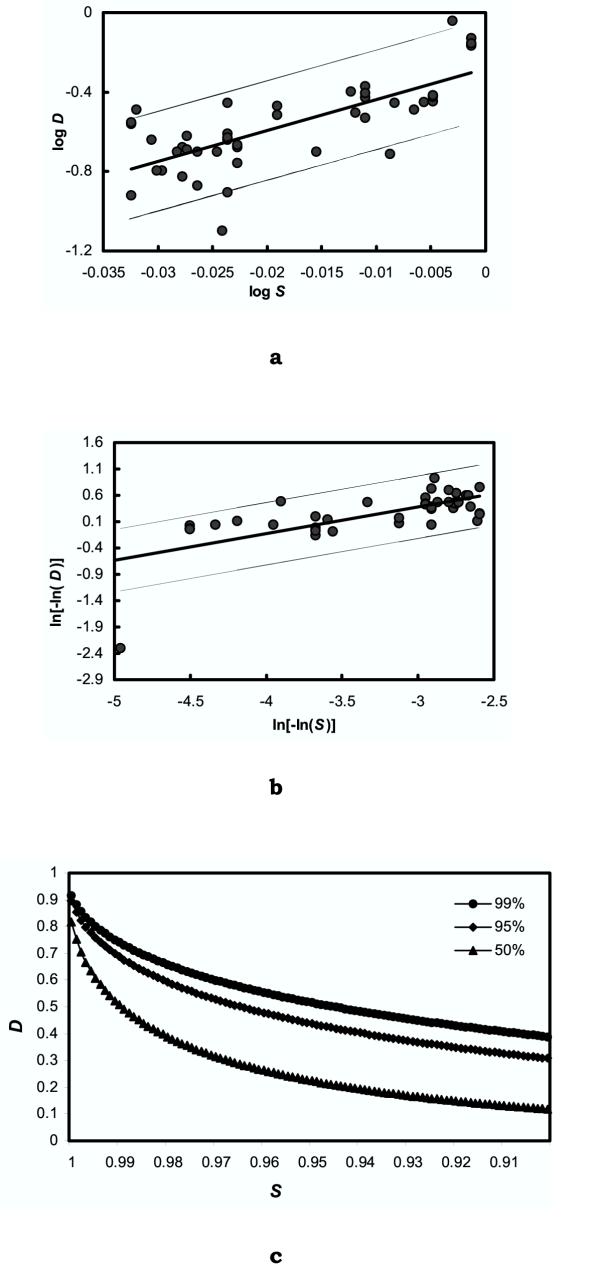
Relationship between the 16S rRNA gene sequence similarity (S) and the extent of DNA hybridization (D). (a) The correlation between logD and logs, (b) Complementary log log, i.e., [ln(-lnD) and ln(-lnS)] plot for the same, and (c) Probability of D for S from 0.90 to 1.00. The line was determined by linear regression of the values. The extent of DNA hybridization was measured by the membrane filter method and the data used for DNA hybridization was as reported earlier [8, 21, 22]. The Distribution of D was calculated from the equation ln(-lnD) = 0.5077 [ln(-lnS)] + 1.8999 and the SD of the residuals in D, which was 0.3498.
Description of the phylogenetic groups
Ruminantium group
The group comprised of seventeen sequences (Fig. 1.) of which three were from isolated strains, represented by Mbr. ruminantium, a ciliate symbiont strain MB9 [26] and a red deer rumen inhabitant (NT7). Similar to the observations made by Whitford et al. [20], the sequences formed two subgroups: Mbr I, with four bovine rumen clones, along with three strains NT7, MB9 and Mbr. ruminantium; and subgroup Mbr II, with ten bovine rumen clones. Since many clone sequences were identical, we chose only few representatives from each group. The percent similarity values for all the sequences of this group used in the analysis here varied from 96.77 to 100%. The sub-clusters originally contained nine sequences in Mbr I (similarity values ranged from 99.4 to 100%) that were 98.5 to 98.8% similar to Mbr. ruminantium, and fifteen in Mbr II (99.3 to 100% similar to each other). Members of Mbr II had sequence similarity ranging from 97.2 to 97.7% with Mbr. ruminantium. On the basis of the study by Boone et al. [17], it had been suggested that Mbr I could be considered as members of species Mbr. ruminantium whereas Mbr II are separate species of this genus since the percentage similarity values for the latter are <98%. Thus, this group contained two distinct species represented by subgroup Mbr I, a mixture of both isolated strains (Mbr. ruminantium, strain NT7 and MB9) and cloned (bovine rumen symbionts) sequences, and subgroup Mbr II, containing only cloned sequences from bovine rumen.
Smithii group
This group was represented by seven isolates (Fig. 1) of which six were methanogenic archaea from feces of higher animals and remaining one was from a different habitat: strain SM9, isolated from sheep rumen (Jarvis GN, Strompl C, Moore ERB, Joblin KN: 1999 Unpublished data). The strains isolated from feces of higher animals like, PG: pig, HO: horse, CW: cow, and humans: B181 and ALI, were reported earlier by Lin and Miller [8]. Recently three members of this group were designated as new species from this group [11], namely Mbr. gottschalkii (HO and PG), and Mbr. thaueri (CW). The overall percentage similarity for this group was 97.46 to 100%. The strain SM9 from sheep rumen forms a part of this group and probably represents another strain of the Smithii group since it was not very distinct from the other members of the group with respect to the similarity values, 97.96 to 99.11%, and these values are in the limits as described earlier [16,17]. Majority of the cloned sequences available in the database demonstrating similarity to Mbr. smithii, could not be included in the study since the sequences of these clones were determined at the 3' end of the 16S rRNA gene and thus showed no overlap with the other sequences. Sequences of methanogens associated with rumen protozoa have been shown to be highly related to Mbr. smithii [20].
Woesei group
The group was represented by only two sequences that represented strains isolated from feces of goose (GS) and rat (RT) reported earlier by Lin and Miller [8]. Of the two, one was recently designated to be a novel species of the genus Methanobrevibacter and named formally as Mbr. woesei GS [11]. It shared 97.4% similarity with the other strain RT of the group. Since this value was less than the cut off as suggested by Boone et al. [17], strain RT can be regarded to be a new species of the genus. Since actual experimental DNA hybridization data was not available for the two strains, quantitative analysis of the data in Fig. 2 clearly indicated that when S = 0.984, D would be <70% at least 99% of the times and with 70% D as the species "cutoff", strain RT almost certainly represents a new species. Strain RT showed percent similarity ranging from 96.1 to 97.5% whereas strain GS showed 95.7 to 97.2% similarity with the other members of the Smithii group members, clearly indicating that even if the site of isolation of the strains is same, they may still be phylogenetically different since most of the members of the Smithii group were isolated from animal feces like the strains GS and RT. Separate species status of both these strains was further supported by the low DNA hybridization values [8] that ranged from 32 to 48 for RT against the three Mbr. smithii strains and 18 to 41 for GS against Mbr. smithii PS, Mbr. gottschalkii (HO and PG), and Mbr. thaueri (CW).
Curvatus group
Seven sequences represented this group of which only one, Mbr. curvatus, was a well-characterized strain isolated from hindgut contents of Reticulitermes flavipes [3]. All the remaining were clones from methanogenic archaea associated either with flagellated protists in termite gut or attached to the gut epithelium of the termites [6]. The percent similarity values for the group range from 97.21 – 99.87. In specific, the group comprised of either clones or isolates from termite gut contents, indicating a habitat specific group and a common evolution of the members.
The termite gut symbionts RsW2 and HW1, both from gut walls of R. speratus and H. sjoestedti [6] respectively, clustered together with Mbr. curvatus with high percent similarity (more than 98.5%) and hence could be regarded as strains of that species. The other cloned gut symbiont sequences formed two distinct lineages (Fig. 1). One contained clones HW2 and HI26, from gut wall and whole gut fractions of H. sjoestedti respectively. The extent of percent similarity between them was 98.98 % and these again represent strains of same species. The third lineage contained LRsD2 from the flagellate Dinenympha parva present in the gut of R. speratus and RsI12 from whole gut fraction of R. speratus, these too represent a homogenous cluster with percent sequence similarity of 99.87 % and represent a single species. Both these lineages of the cloned gut symbionts can be regarded as different species since their intra lineage sequence similarity values were below 98%. No group specific signature positions could be deduced for the Curvatus group.
Arboriphilicus group
This group contained five sequences (Fig. 1) out of which three were well-characterized isolates and one was a newly reported endosymbiont (strain NO) of the ciliate Nyctotherus ovalis [7]. The remaining was a clone, MPn19 from gut contents of the soil feeding higher termite Pericapritermes nitobei [5]. The type strain of Mbr. arboriphilicus was isolated from enrichments of decaying cottonwood tissue, whereas Mbr. cuticularis was isolated from hindgut contents of a termite R. flavipes. The percentage similarity values for the group ranged from 92.59 to 99.74%. No group specific signature positions could be deduced for this group.
The clone MPn19 showed a separate lineage but clustering with the two Mbr. arboriphilicus sequences with more than 97% similarity values thereby indicating that it may warrant a separate species status provided the D values are <70%. The strain NO was the most interesting since it formed a separate lineage. The strain NO sequence showed very low similarity values of 92.59 to 93.5% with the other members of the group. The maximum percentage similarity shared by strain NO was with Mbr. cuticularis (93.5%) and such low sequence similarity value is in support of it being a distinct species. Quantitative estimate from Fig. 2, indicating a high probability of D being <70% at such low S between strain NO and other members of the group, clearly supported the inclusion of strain NO as a separate species of the Arboriphilicus group.
Filiformis group
Similar to the Woesei group, only two members represented the filiformis group, a well characterized strain, Mbr. filiformis, isolated from hindgut contents of a termite R. flavipes, and the lower termite gut symbiont clone HW3 isolated from the gut wall of H. sjoestedti [6]. Both of them being termite gut associated, with 98.23% sequence similarity can be regarded as strains of the same species. Furthermore, both showed very less percentage similarity with the members of the group Arboriphilicus with values ranging from 92.28 to 97% for Mbr. filiformis, and 92.15 to 96.93% for HW3. Quantitative estimate from Fig. 2, indicating a high probability of D being <70% at such low S between the two and the other members of the Arboriphilicus group, clarifies their grouping as a separate group.
Termite gut symbiont group
All the eleven sequences that belonged to the termite gut symbiont group were from Methanobrevibacter specific 16S rRNA gene clones and there are no well-characterized species in this group. The sequences shared a percentage similarity from 94.22 to 99.87. The group showed two subgroups with subgroup I represented by five clones whereas subgroup II by six clones (Fig. 1). Termite gut symbionts HI1, HI28, LHD2, LHM8, and MHj4 formed one lineage, whereas LRsD3, LRsM1, RsI4, RsW10, M4 and Cd30 formed the other. The tree topology for this group was in accordance with the one reported earlier [5], with M4 and Cd30 going together and MHj4 forming a separate branch but all originating from a common node (Fig. 1).
Subgroup I represented methanogenic symbionts isolated from the gut of a wood feeding lower termite, H. sjoestedti [6]. HI1, HI28, and MHj4 were isolated from the whole gut fractions, whereas LHD2 and LHM8 were isolated from the flagellates Dinenympha and Microjoenia, respectively associated with the gut of the termite [6]. The overall sequence similarity for subgroup I was 97.46% to 99.62%. LHD2 formed a separate lineage with similarity values 97.46% to 98.1% against other members of subgroup I, indicating that it may represent a new species distinct from the other four members of the subgroup.
The second subgroup contained methanogenic symbionts isolated from the gut of wood feeding lower termites, R. speratus and Cryptotermes domesticus [5,6]. RsI4 and M4, and Cd30 were isolated from the whole gut fractions of R. speratus and C. domesticus, respectively, whereas RsW10 was found associated with the gut wall of R. speratus. LRsD2 and LRsM1, were isolated from the flagellates Dinenympha and Microjoenia, respectively associated with the gut of the termite R. speratus. The overall sequence similarity for subgroup II was 96.64% to 99.87%. Cd30 formed a separate lineage with similarity values 96.64% to 97.2%, against other members of subgroup II, indicating that it may represent a new species distinct from the other five members of the subgroup.
Single member lineages
The phylogenetic analysis of the genus showed three separate lineages that were represented by single members of which two were well characterized strains, Mbr. wolinii and Mbr. acididurans, whereas one was termite gut flagellate clone LHD12 (Fig. 1). The strain Mbr. wolinii SH was isolated from sheep feces by Lin and Miller [8], was later formally designated as a new species of the genus Methanobrevibacter [11]. The sequence similarity values of the strain with the other members of the Smithii group, that shared a common habitat with the strain SH, were found to be low, 92.83% to 94.64%, whereas it showed percent similarity values ranging from 89.68 to 94.49 with all the other members of the genus Methanobrevibacter. As reported earlier [8], the strain showed no or very low genomic DNA reassociation with Mbr. smithii PS and the other Methanobrevibacter isolates from horse, pig, cow, and goose feces. Based on the 16S rRNA gene sequence and genomic DNA reassociation studies, the strain SH represents a new taxon at the species level [8].
Mbr. acididurans, an anaerobic acid tolerant methanogen (strain ATM), branching separately, shared the least homology with any other sequence (90.78 to 96.04%) and had already been defined as a new species of the genus Methanobrevibacter [13]. The strain showed similarity values ranging from 94.69 to 96.02% with the member of the ruminantium group.
The clone LHD12, isolated from a flagellate, Dinenympha present in the gut of a termite Hodotermopsis sjoestedti [6] and had less than 98 % similarity with any of the sequences used in the analysis with values ranging from 92.46 to 97.83% supporting its separate descent in the tree. It showed maximum sequence similarity of 97.83% with the rat feces strain RT, a member of the Woesei group, whereas with the members of the Smithii group with which it shared a common decent, the percentage similarity was 95.44 to 97.47%. Based on the 16S rRNA gene sequence similarity values, genomic DNA reassociation values estimated by Fig. 2, and the separate lineage shown in the phylogenetic tree, this can be regarded as separate species of this genus.
Group specific signature positions
The group specific signature positions deduced for the Ruminantium, Smithii, the Termite gut symbiont groups and the single member lineages are given in Table 1. The termite gut symbiont group was divided in two subgroups based on the phylogenetic analysis and each of theses subgroups was represented by a number of signature positions as shown. Parsimony and maximum likelihood methods as well as neighbor-joining supported these groupings. The majority of these signature positions corresponded to those suggested by Woese [27] since each position had a different but conserved base in the neighbor sequences. Furthermore, since the sequence positions are conserved, these signature positions are less likely to change in the future when new sequences corresponding to new taxonomic groups are added.
Table 1.
Group specific signature positions for the genus Methanobrevibacter.
| Group No. | Group Name | No. of sequences used | E. coli position | I | II | III | IV | V | VI | VII | VIII | IX | X | XI |
| I | Ruminantium | 5 | 637 | U | A | A | A/G | A | G | A | G | A | A | G |
| II | Smithii | 7 | 987 | G | A | A | G | G | G | G | G | A | C | G |
| 1216 | C | U | U | C | C | C | C | C | U | G | C | |||
| III | Woesei | 2 | None | |||||||||||
| IV | Curvatus | 7 | None | |||||||||||
| V | Arboriphilicus | 5 | None | |||||||||||
| VI | Filiformis | 2 | None | |||||||||||
| VII | Termite gut symbiont subgroup I | 5 | 629 | A | A | A | A | A | A | G | A | A | A | A |
| 820 | G | G | G | G | G | G | A | G | G | G | G | |||
| 838 | G | G | G | G | G | G | A | G | G | G | G | |||
| 844 | A | A | A | A | A | A | U | A | A | A | A | |||
| 1133 | G | G/U | G | G | G | G | A | U | G | U | G | |||
| 1140 | P1 | P/A | P | P | P | P | U | U | P | P | P | |||
| VIII | Termite gut symbiont subgroup II2 | 6 | 537 | G | G | G | G | G | G | G | A | G | G | G |
| 554 | C | C | C | C | C | C | C | U | C | C | C | |||
| 591 | G | A | A | A | A | A | A | U | U | G | A | |||
| 610 | C | C | C | C | C | C | C | U | C | C | C | |||
| 627 | G | G | G | G | G | G | G | A | G | G | G | |||
| IX | Clone LHD12 | 1 | 555 | U | A | A | C | C | C | C | C | U | U | U |
| 611 | C | C | U/C | C | C | C | C | C | U | U | C | |||
| X | Mbr. wolinii | 1 | 571 | A | A | A | A | A | A | A | A | A | G | A |
| XI | Mbr. acididurans | 1 | 600 | C | C | C | U | C | U | U | U | C | U | U |
| 827 | U | U | U | U | U | U | U | U | U | U | G |
1P = gap 2 Excluding the sequence of Clone Cd30
Placement of methanogenic endosymbionts of the ciliate Nyctotherus
Strain NO deserves a special mention as it formed a distinct lineage in the Arboriphilicus group in the phylogenetic tree (Fig. 1). At the time of the study, ten 16S rRNA gene sequences representing methanogenic endosymbionts of Nyctotherus (N. ovalis, N. cordoformis, N. velox) were available from GenBank. These strains are currently placed in the genus Methanobrevibacter [7]. Out of these, only one sequence, representing methanogenic endosymbiont of N. ovalis from Periplaneta americana var. Amsterdam (AJ132639) was greater than 1 kb and hence others were not considered for the construction of the phylogenetic tree. Initial analysis revealed that the sequence AJ132639 was 100% identical to other two sequences, methanogenic endosymbiont of N. ovalis from P. americana var. Dar es Salaam (AJ132641) and methanogenic endosymbiont of N. ovalis from Blaberus var. Amsterdam (AJ132643). Preliminary analysis involving all the ten sequences revealed that majority of the methanogenic endosymbionts of N. ovalis clustered together with the Arboriphilicus group but formed a distinct lineage (data not shown). The strain NO sequence showed the least percent similarity values of 92.15% to 93.5% with the other members of the Arboriphilicus group and such low sequence similarity value indicating a high probability of D being <70% (Fig. 2) is in support of it being considered a distinct species.
We aligned the 16S rRNA gene sequence of the strain NO (AJ132639) along with other members of the Arboriphilicus and Filiformis group (U41095, U82322, AB009827, AB026925, and AB065294), representative species of the genus Methanobrevibacter (U55233, U55240, U62533 and RDP-Mbb.rumina.), Msp. stadtmanae (M59139) and that of E. coli (J01859, Fig. 3). It was clearly seen that there was an insertion of one additional triplet 'AAG' at E. coli position 934. Similarly, there was an insertion of a single base 'A' at E. coli position 1032. Furthermore, there was a deletion of one base each at E. coli positions 970, 995 and 999. As a result of these insertions and deletions, the strain formed a separate branch in the tree. Hence placement of methanogenic endosymbionts of Nyctotherus in the genus Methanobrevibacter needs to be reconsidered and it should be regarded as a separate species of the genus.
Figure 3.
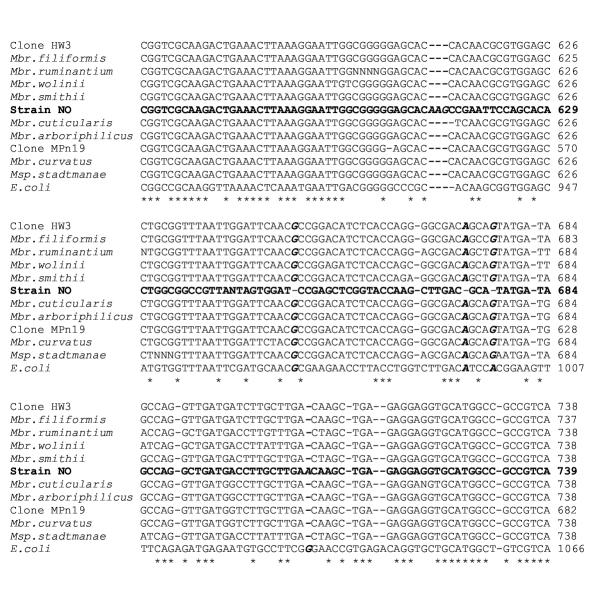
Alignment of sequences showing differences in the 16S rRNA gene sequence of endosymbiont of Nyctotherus ovalis strain NO Sequence for strain NO is shown in bold letters. The positions with differences in the 16S rRNA gene sequence of strain NO with other members are shown in bold-italics. See text for GenBank Accession numbers against the strain names used above. "*" indicates conserved positions amongst the sequences used.
Discussion
The aim of the present investigation was to carry out phylogenetic analysis of the genus Methanobrevibacter based on 16S rRNA gene sequence and propose some guidelines for the delineation of species identity or for the identification of new isolates of the genus Methanobrevibacter. Moreover, we reorganized the sequences in the genus, especially the sequences from the clones representing uncultured bacteria. This information will be very useful when 16S rRNA gene sequence analysis is used alone for this purpose.
We were able to group 54 sequences in seven distinct groups and designate group specific signature positions for three groups, Ruminantium, Smithii and the Termite gut symbionts. The majority of the sequences (86) represented clones from uncultured organisms either from rumen or termite gut. A few of these clones (for e.g. HW1, RsW2, HW3) were closely related to cultured isolates at the 16S rRNA sequence level, and the majority of them formed distinct lineages. The clones from the rumen environment were closely related to Mbr. ruminantium whereas those from termite gut formed separate lineages. Similarly, all the isolates from feces of higher animals were closely related. This indicates a correlation between ecological habitat, physiology and 16S rRNA based phylogeny. The presence of large number of cloned sequences in the termite gut that formed lineages different from any of the cultured isolates emphasizes uniqueness of this environment and also the diversity that is likely to be present in the genus.
16S rRNA gene sequence analysis is undoubtedly an important parameter for species delineation and identification. However, other markers like cell wall composition, bile sensitivity, formate utilization, and requirements for acetate, CoM, 2-methylbutyrate can be useful for species delineation and identification and also to study the activities of Methanobrevibacter strains in native habitats. In addition to 16S rRNA gene sequence similarity, genomic DNA reassociation values are also essential for the correct identification of a strain and together these two markers are the most important aspects of taxonomic affiliation of any strain. A correlation between these two markers exists and it depends on the taxa in study. Since many studies involve the use of 16S rRNA gene sequence analysis as the basis of clonal affiliation, a prediction of the extent of DNA hybridization from sequence similarity will help enormously not only in the environmental studies, but also in the most common studies involving well isolated strains.
Designation of a newly isolated strain as a new species requires critical analysis. The grouping of a new strain within the genus Methanobrevibacter based on 16S rRNA gene sequence, as reported earlier [4] should be supported by: 1) bootstrap values of 99% for the node from which the new strain and the other members of the genus, radiate; 2) the possession of a signature sequence (5'-tgt gag (a/c)aa tcg cg-3', corresponding to E. coli positions 375–388) which is shared only with members of this genus; and 3) a nucleotide bulge (5'-Tn-3', n = 6 or 8; corresponding to a stem-loop structure at E. coli positions 200–218) also shared with other members of the genus except Mbr. curvatus (which instead possess the sequence 5'-ttc tta tgt t-3'). Moreover, all these sequences shared >87% similarity amongst each other whereas they were <84% similar to the sequence of the outgroup and representatives of other generas of the family Methanobacteriaceae, indicating that sequences with more than 87% similarity can be considered to be of the genera Methanobrevibacter. Further support was provided by the estimates of D for a given S, according to which D <20% at an S of <84% at least 95% of the times (Fig. 2).
We observed that very few reports are available on the relationship between 16S rRNA gene sequence similarity and genomic DNA relatedness on methanogenic archaea as compared to that done with bacteria. Therefore it will be difficult to give conclusive statements about these two parameters for the genus Methanobrevibacter and one can predict D given S with an error that is known. It was reported earlier that for the genus Methanobrevibacter, the 16S rRNA gene sequence similarity of >99% corresponds to >70% genomic DNA similarity, whereas the strains of Methanobrevibacter with less than 99% 16S rRNA gene sequence similarity, showed less than 50% DNA reassociation values [8]. Our analysis revealed that for this genus, when S = 0.984, D would be <70% at least 99% of the times, and with 70% D as the species "cutoff", any 16S rRNA gene sequence showing <98% sequence similarity can be considered as a separate species.
Conclusion
Our detailed analysis reveals that any strain showing less than 87% 16S rRNA gene sequence similarity should not be included in the genus. A very significant relationship between D and S was found to exist for the genus Methanobrevibacter, implying that it is possible to predict D from S with a known precision for the genus. The available information allows concluding that newer isolates or clones showing greater than 98% 16S rRNA gene sequence similarity should be considered as strains of that particular species. The exceptions to this statement have been observed only with the genus Methanococcus [28]. The presence of 16S rRNA sequences with nonultrametric properties and the experimental error associated with D and the inherent statistical error in using S to estimate evolutionary distance account for most of the variability of D given S. Given the relative ease in determining S by automated sequencing, the ability to estimate D will be of great utility for systematic studies. For organisms that have never been isolated but have been detected in natural samples by rRNA sequence alone (like the majority of sequences in the present study), the ability to estimate D will provide a clearer understanding of their genetic and phenotypic diversity.
Methods
Sequence retrieval and phylogenetic analysis
The 16S rRNA gene sequences used in this study were either from isolated strains or clones of genus Methanobrevibacter and were retrieved in the fasta format from GenBank except for Mbr. ruminantium (RDP-Mbb.rumina.) and Mbr. arboriphilicus (RDP-Mbb.arbori.), which were obtained from RDP – Ribosomal Database Project [29]. The alignment of all the sequences was done using CLUSTAL W program [30]. The phylogenetic tree was constructed using 786 bp long aligned sequences by the neighbor joining method using Kimura 2 parameter distances in MEGA 2.1 software [31]. The resulting tree was compared with the parsimony method (PHYLIP package) and the maximum-likelihood method using the fastDNAml program [32]. The 16S rRNA gene sequence of Methanosphaera stadtmanae (M59139) was used as the outgroup for the analysis.
Determination of cophenetic correlation coefficient
Cophenetic correlation coefficient (CCC) was calculated to validate the phylogenetic inference [19]. The similarity matrix was prepared using the Dnadist program in the PHYLIP analysis package using the Jukes Cantor corrections. The 16S rRNA similarity matrices were then used to calculate cophenetic matrices using the UPGMA method with the Neighbor program in the PHYLIP package. A cophenetic matrix consisted of the estimated similarity values derived from the calculation of the UPGMA tree. The CCC is the correlation coefficient (r) calculated from the linear regression between the corresponding values of the similarity matrix and cophenetic matrix. A Visual Basic program for Microsoft Excel 2000 was written to do the calculations since even for a small set of data, it becomes nearly impractical to do them by hand. This program can be downloaded at the following address: http://webpages.ull.es/users/jmhernan/CCC.htm.
Correlation between D and S
Using the DNA hybridization data as reported earlier for the eleven strains: Mbr. arboriphilicus (M1), Mbr. ruminantium (DH1), Mbr. smithii (strains PS, B181 and ALI), Mbr. gottschalkii (HO and PG), Mbr. thaueri (CW), Mbr. woesei (GS), Mbr. wolinii (SH) and Methanobrevibacter sp. (RT) [8,31], logD was plotted against logS [18]. The method as described by Keswani and Whitman [19] was also used, in which a [ln(-lnD) versus ln(-lnS)] plot described the relationship between S and D. To help in the calculations and plotting of D against S, an Excel sheet was downloaded from the following address: http://www.arches.uga.edu/~whitman/template_d_s.xls.
Group specific signature positions
Group specific signature positions were identified using the aligned sequence files retrieved from the Ribosomal Database Project website. Each aligned base was assigned a number based on E. coli 16S rRNA gene sequence numbering (Accession Number: J01859). Nucleotide positions that were conserved in all the strains of a given group, but differed in the closest related sequences outside the group, were considered signature positions [27]. To deduce signature positions for a larger sequence length, only those sequences that were greater than 80% of the sequence of E. coli were considered for the analysis. The reference sequences, outside the Methanobrevibacter genus, used to find signature positions for Methanobrevibacter were: Methanobacterium bryantii (M59124), M. congolense (AF233586), M. formicicum (M36508), M. palustre (AF095263), M. subterraneum (X99044), M. uliginosum (AF095265), M. thermoautotrophicum (X15364), M. wolfei (X89406), Methanothermobacter defluvii (X99046) and Methanosphaera stadtmanae (M59139).
Authors' contributions
ASD carried out the sequence retrieval, analysis and participated in drafting the manuscript. KJ continued the sequence retrieval and analysis, determined signature positions, participated in drafting the manuscript, and helped in the statistical analysis. JMG participated in the sequence analysis, determined the signature positions, and carried out the statistical analysis. VJP participated in sequence retrieval and analysis. MSP, DRR and YSS conceived of the study, and participated in its design and coordination. YSS was also involved in the analysis and interpretation of results and drafting the manuscript. All authors read and approved the final manuscript.
Contributor Information
Abhijit S Dighe, Email: abhijitdighe@hotmail.com.
Kamlesh Jangid, Email: jangidk@nccs.res.in.
José M González, Email: jmhernan@ull.es.
Vyankatesh J Pidiyar, Email: vpidiyar@hotmail.com.
Milind S Patole, Email: milindpatole@hotmail.com.
Dilip R Ranade, Email: drranade@vsnl.com.
Yogesh S Shouche, Email: yogesh@nccs.res.in.
References
- Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Fox GE, Magrum LJ, Woese CR, Wolfe RS. Methanogens: reevaluation of a unique biological group. Microbiol Rev. 1979;43:260–296. doi: 10.1128/mr.43.2.260-296.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter JR, Breznak JA. Physiological ecology of Methanobrevibacter cuticularis sp. nov. and Methanobrevibacter curvatus sp. nov., isolated from the hindgut of the termite Reticulitermes flavipes. Appl Environ Microbiol. 1996;62:3620–3631. doi: 10.1128/aem.62.10.3620-3631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter JR, Crosby LD, Breznak JA. Methanobrevibacter filiformis sp. nov, a filamentous methanogen from termite hindguts. Arch Microbiol. 1998;169:287–292. doi: 10.1007/s002030050574. [DOI] [PubMed] [Google Scholar]
- Ohkuma M, Noda S, Kudo T. Phylogenetic relationships of symbiotic methanogens in diverse termites. FEMS Microbiol Lett. 1999;171:147–153. doi: 10.1016/S0378-1097(98)00593-X. [DOI] [PubMed] [Google Scholar]
- Tokura M, Ohkuma M, Kudo T. Molecular phylogeny of methanogens associated with flagellated protists in the gut and with the gut epithelium of termites. FEMS Micribiol Ecol. 2000;33:233–240. doi: 10.1016/S0168-6496(00)00065-9. [DOI] [PubMed] [Google Scholar]
- van Hoek AH, van Alen TA, Sprakel VS, Leunissen JA, Brigge T, Vogels GD, Hackstein JH. Multiple acquisition of methanogenic archaeal symbionts by anaerobic ciliates. Mol Biol Evol. 2000;17:251–258. doi: 10.1093/oxfordjournals.molbev.a026304. [DOI] [PubMed] [Google Scholar]
- Lin C, Miller TL. Phylogenetic analysis of Methanobrevibacter isolated from feces of humans and other animals. Arch Microbiol. 1998;169:397–403. doi: 10.1007/s002030050589. [DOI] [PubMed] [Google Scholar]
- Miller TL, Wolin MJ, Kusel E. Isolation and characterization of methanogens from animal feces. Syst Appl Microbiol. 1986;8:234–238. [Google Scholar]
- Ferrai A, Brusa T, Rutili A, Canzi E, Biavati B. Isolation and characterization of Methanobrevibacter oralis, sp. nov. Curr Microbiol. 1994;29:7–12. [Google Scholar]
- Miller TL, Lin C. Description of Methanobrevibacter gottschalkii sp. nov., Methanobrevibacter thaueri sp. nov, Methanobrevibacter woesei sp nov and Methanobrevibacter wolinii sp nov. Int J Syst Evol Microbiol. 2002;52:819–822. doi: 10.1099/ijs.0.02022-0. [DOI] [PubMed] [Google Scholar]
- Miller TL. Methanobrevibacter. In: Boone DR, Castenholz RW, Garrity GM, editor. In Bergey's manual of systematic bacteriology. 2. I. New York: Springer-Verlag; 2001. pp. 218–226. [Google Scholar]
- Savant DV, Shouche YS, Prakash S, Ranade DR. Methanobrevibacter acididurans sp. nov., a novel methanogen from a sour anaerobic digester. Int J Syst Evol Microbiol. 2002;52:1081–1087. doi: 10.1099/ijs.0.01903-0. [DOI] [PubMed] [Google Scholar]
- Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, Moore WEC, Murray RGE, Stackebrandt E, Starr MP, Truper HG. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol. 1987;37:463–464. [Google Scholar]
- Johnson JL. Nucleic acids in bacterial classification. In: Kreig NR, Holt JG, editor. In Bergey's manual of systematic bacteriology. I. Baltimore: Williams & Wilkins; 1984. pp. 8–11. [Google Scholar]
- Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol. 1994;44:846–849. [Google Scholar]
- Boone DR, Whitman WB, Rouviere P. Diversity and taxonomy of methanogens. In: Ferry JG, editor. In Methanogens. New York: Chapman and Hall, Inc; 1993. pp. 35–80. [Google Scholar]
- Devereux R, He SH, Doyle CL, Orkand S, Stahl DA, LeGall J, Whitman WB. Diversity and origin of Desulfovibrio species: a phylogenetic definition of a family. J Bacteriol. 1990;172:3609–3619. doi: 10.1128/jb.172.7.3609-3619.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keswani J, Whitman WB. Relationship of 16S rRNA sequences similarity to DNA hybridisation in prokaryotes. Int J Syst Evol Microbiol. 2001;51:667–678. doi: 10.1099/00207713-51-2-667. [DOI] [PubMed] [Google Scholar]
- Whitford MF, Teather RM, Forster RJ. Phylogenetic analysis of methanogens from the bovine rumen. BMC Microbiol. 2001;1:5. doi: 10.1186/1471-2180-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa S, Morii H, Akagawa-Matsushita M, Koga Y, Hayano K. Characterization of Methanobrevibacter arboriphilicus SA isolated from a paddy field soil and DNA-DNA hybridization among M. arboriphilicus strains. Int J Syst Bacteriol. 1993;43:683–686. [Google Scholar]
- Miller TL, Wolin MJ. Methanogens in human and animal intestinal tracts. Syst Appl Microbiol. 1986;7:223–229. [Google Scholar]
- Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA. 1985;82:6955–6959. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimdt TM, DeLong EF, Pace NR. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol. 1991;173:4371–4378. doi: 10.1128/jb.173.14.4371-4378.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokura M, Chagan I, Ushida K, Kojima Y. Phylogenetic study of methanogens associated with rumen ciliates. Curr Microbiol. 1999;39:123–128. doi: 10.1007/s002849900432. [DOI] [PubMed] [Google Scholar]
- Tokura M, Tajima K, Ushida K. Isolation of Methanobrevibacter sp. as a Ciliate-associated ruminal methanogen. J Gen Appl Microbiol. 1999;45:43–47. doi: 10.2323/jgam.45.43. [DOI] [PubMed] [Google Scholar]
- Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keswani J, Orkand S, Premaachandran U, Mandelco L, Franklin MJ, Whitman WB. Phylogeny and taxonomy of mesophilic Methanococcus spp. and comparison of rRNA, DNA hybridization, and phenotypic methods. Int J Syst Bacteriol. 1996;46:727–735. doi: 10.1099/00207713-46-3-727. [DOI] [PubMed] [Google Scholar]
- Maidak BL, Larsen N, McCaughey MJ, Overbeek R, Olsen GJ, Fogel K, Blandy J, Woese CR. The ribosomal database project (RDP) Nucleic Acids Res. 1994;22:3485–3487. doi: 10.1093/nar/22.17.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Bioinformatics http://http//:www.ebi.ac.uk/clustalw
- Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA2: Molecular Evolutionary Genetics Analysis software. Bioinformatics. 2001;17:1244–1245. doi: 10.1093/bioinformatics/17.12.1244. [DOI] [PubMed] [Google Scholar]
- Olsen GJ, Matsuda H, Hagstrom R, Overbeek R. fastDNAmL: a tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput Appl Biosci. 1994;10:41–48. doi: 10.1093/bioinformatics/10.1.41. [DOI] [PubMed] [Google Scholar]