

Abstract
A medicinal chemistry exploration of the human phosphodiesterase 4 (hPDE4) inhibitor cilomilast (1) was undertaken in order to identify inhibitors of phosphodiesterase B1 of Trypanosoma brucei (TbrPDEB1). T. brucei is the parasite which causes African sleeping sickness, a neglected tropical disease that affects thousands each year, and TbrPDEB1 has been shown to be an essential target of therapeutic relevance. Noting that 1 is a weak inhibitor of TbrPDEB1, we report the design and synthesis of analogs of this compound, culminating in 12b, a sub-micromolar inhibitor of TbrPDEB1 that shows modest inhibition of T. brucei proliferation.
Keywords: Antiprotozoal agents, Cilomilast, Phosphodiesterase inhibitors, TbrPDEB1 Trypanosoma brucei
Neglected tropical diseases (NTDs) are a group of pathologies that mainly affect the poor in the developing world. The World Health Organization has defined 17 different NTDs, and this list includes human African trypanosomiasis (HAT), or sleeping sickness, which is a parasitic disease caused by the protozoan parasite Trypanosoma brucei. Currently, 50 million people are at risk within rural sub-Saharan Africa and the estimated incidence is around 10,000 cases a year.1 Melarsoprol and eflornithine, the current front-line therapies for advanced HAT, have been the drugs of choice for the treatment of second stage HAT. Though effective, these drugs are limited by serious and sometimes lethal side effects, inconvenient route of administration and increasing incidence of drug resistance. Though new efforts have produced new combination therapies (such as nifurtimox/eflornithine combination therapy (NECT)),2 further work is needed to develop the next generation of therapies that are safe, effective, and inexpensive. With this in mind, our research efforts are focused on repurposing established classes of agents that inhibit druggable human targets.3
Phosphodiesterases (PDEs) are one such target; a family of enzymes that inactivate the cellular signaling agents cAMP or cGMP. In recent years it was shown that cAMP plays a key role in T. brucei biology. T. brucei expresses five cAMP-specific PDEs, two of which (TbrPDEB1 and TbrPDEB2) have been shown to be together essential for parasite proliferation by both RNAi and small molecules.4, 5 Initial screening and early optimization experiments have uncovered a susceptibility of TbrPDEB1 and B2 to human PDE4 (hPDE4) inhibitor chemotypes, and that the inhibition of these enzymes leads to parasite death in vitro. For example, we showed that piclamilast (2, Figure 1), a hPDE4 inhibitor, inhibited TbrPDEB1 and B2 (IC50 4.7 and 11.4 µM, respectively), which translated to inhibition of T. brucei brucei (Tbb) cell growth (EC50= 9.6 µM).5 At the same time, others identified 3 as a potent TbrPDE inhibitor via a high-throughput screening campaign.6 This racemic compound remains the most potent TbrPDE inhibitor described to date, despite several further reports.7–9 A key limitation of all the TbrPDEB inhibitors identified to date is the lack of selectivity over hPDE4, which is likely to lead to various characteristic PDE4 side effects, such as nausea and emesis.
Figure 1.

The structure of 1, highlighting the tail (red) and head region (blue) explored in this work. Also shown are other hPDE4 inhibitors previously studied as inhibitors of TbrPDEB1.5, 6 Human PDE4 data shown is from previous reports.13, 14
We were surprised at the divergence in TbrPDEB1 activity between closely related hPDE4 inhibitors: roflumilast (4) a close analog of 2, was completely inactive.5 Thus, for the purpose of studying a wider variety of hPDE4 inhibitors as starting points for TbrPDEB inhibitors, we investigated cilomiliast, 1, a related hPDE4 inhibitor. Compound 1 (Ariflo, SB-207,499) is an orally active and selective hPDE4 inhibitor developed by GlaxoSmithKline for the treatment of respiratory disorders such as chronic obstructive pulmonary disease (COPD).10, 11 This compound has a reported IC50 of 84 nM against hPDE4B,12 and we observed an IC50 against TbrPDEB1 of 16.4 µM. Given the prior art of repurposing hPDE4 inhibitors for TbrPDEB1, we felt that this result warranted additional medicinal chemistry explorations for trypanosomal PDE inhibitors.
Our investigation into the SAR of 1 as a TbrPDEB1 inhibitor involved first assessing the relative stereochemistry of the headgroup (Figure 1, blue), as well as the importance of the carboxylate functionality. We also wished to determine whether a stereochemically simplified headgroup replacement could be achieved. Secondly, a key structural feature of the TbrPDEB1 binding site, predicted by homology modeling and confirmed by crystallography,5, 15 is a pocket adjacent to the binding site (termed the “parasite-“ or “Ppocket”) that is deeper in comparison to the same region in hPDE4. Thus, guided by the SAR studies of the catechol diethers 2 and 3 reported previously that were intended to explore the “parasite pocket” of the enzyme, we now report exploration of the cyclopentyl ether (Figure 1, red) to longer, chain-extended versions, and we then studied the protein-ligand interactions in silico by carrying out molecular docking using the recently published crystal structure of TbrPDEB1.15
Initial analogues 5–8 (where R1=cyclopentyl) were prepared by the procedure shown in Scheme 1, based on the previously published preparation for 1.11 Analogs where R1=benzyl were synthesized using an analogous route (see Supporting Information). In the interest of exploring simplified headgroup replacements, piperidine analogues were also synthesized (Scheme S2, Supporting Information).
Scheme 1a.

a Reagents and conditions: (a) LiOH, H2O, MeOH, THF, rt, 2 h.
We opted to first test compounds at 10 µM concentrations; those that were above 50% inhibition at this concentration were subjected to dose-response experiments. We have previously noted similarity between compound activity against TbrPDEB1 and B2. Thus, for efficiency, we focused our first round of compound assays on TbrPDEB1, and assumed similar (within 2-3 fold activity) against TbrPDEB2.
While compound 1 is a 16.4 µM inhibitor of TbrPDEB1, the esters 5a and 5b were below the minimum percent inhibition cutoff to obtain an IC50 (Table 1). This is consistent with the SAR for hPDE4 previously reported.11 The benzyl analogue of cilomilast (compound 7) inhibits TbrPDEB1 with activity similar to 1, though it retains some potency against hPDE4 (IC50=0.54 µM). Notably, the compounds with a cis-orientation of the cyano and carboxylate groups are consistently more potent (compare 1 and 7 with 8a and 8b). Replacement of the cyclohexane headgroup with a piperidine ring resulted in significant loss of activity (Table S3, Supporting Information). Others have shown that preparation of analogs with catechol substituents that project into the P-pocket (c.f. 3) can lead to improved potency and selectivity.6, 7 We assumed that our catechol-derived analogs bind in a similar orientation as 2 and 3, and used this hypothesis to design analogs of 7. Thus, we replaced the phenyl group with three pyridine isomers (10a-c). As shown in Scheme 2, ester 5b was debenzylated using catalytic hydrogenation to provide phenol 9. This could be alkylated with the appropriate bromomethylpyridine, providing 10a-c following basic hydrolysis. These compounds were much less active (Table 2), and one (10b) was not soluble enough in the assay conditions to obtain inhibition data.
Table 1.
Cilomilast analogs tested against TbrPDEB1
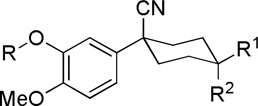 | |||||
|---|---|---|---|---|---|
| Cmpd | R | R1 | R2 | TbrPDEB1 (%)a | TbrPDEB1 IC50b(µM) |
| 1 | 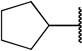 |
COOH | H | 16 ± 6.65 | |
| 5a | 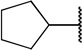 |
COOCH3 | H | 31 ± 8 | |
| 5b |  |
COOCH3 | H | 40 ± 14 | |
| 8a | 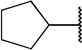 |
H | COOH | 9.3 ± 9.3 | |
| 8b | 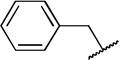 |
H | COOH | 20 ± 4.3 | |
| 6a | 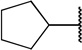 |
H | COOCH3 | 26 ± 3.6 | |
| 6b | 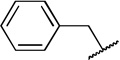 |
H | COOCH3 | 28 ± 8.9 | |
| 7 | 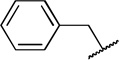 |
COOH | H | 77 ± 6.4 | 12 ± 1.18 |
Inhibitor concentration 10.0 µM, tested in at least two independent replicate experiments all error was within 15%.
Tested in at least two independent replicate experiments.
Scheme 2a.

aReagents and conditions: (a) H2, Pd/C, MeOH, rt, 3 h; (b) Appropriate (bromomethyl)pyridinium bromide, K2CO3, DMF, 60°C, 4 h; (c) LiOH, H2O, MeOH, THF, rt, 2 h; (d) Appropriate dibromoalkane, DMF, 60°C, 4 h; (e) Appropriate phenol, K2CO3, DMF, 60°C, 4 h;
Table 2.
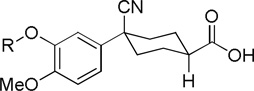
Cilomilast analogs tested against TbrPDEB1 varying catechol substituents.
 | |||
|---|---|---|---|
| Cmpd | R | TbrPDEB1 (%)a | TbrPDEB1 IC50c (µM) |
| 1 | Cyclopentyl | 16.4 ± 6.7 | |
| 10a | 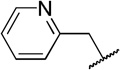 |
33±12 | |
| 10b | 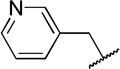 |
ndb | |
| 10c | 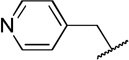 |
11 ± 4.4 | |
| 12a | 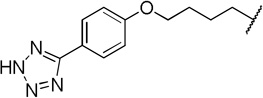 |
69 ± 1.4 | 7.9 ± 4.63 |
| 12b | 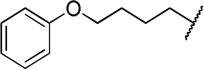 |
96 ± 0.66c | 0.95 ±0.14 |
| 12c | 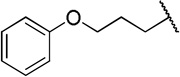 |
66 ± 2.2 | 11.0 ± 0.13 |
| 12d | 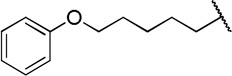 |
54 ± 8 | 9.8 ± 1.27 |
| 12e | 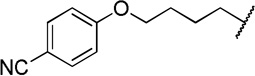 |
87 ± 12 | 3.6 ± 0.3 |
| 12f | 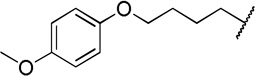 |
79 ± 3.4 | 3.5 ± 0.41 |
| 12g | 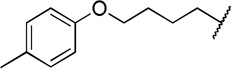 |
74 ± 1.7 | 4.9 ± 2.81 |
| 12h | 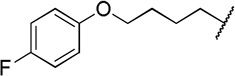 |
79 ± 0.42c | 9.2 ± 5.1 |
Inhibitor concentration 10.0 µM, tested in at least two independent replicate experiments, all error was within 15%;
nd=no data due to insufficient solubility in the assay conditions.
Tested in at least two independent replicate experiments.
We then prepared chain extended analogs of 7 by alkylation of 9 using various dibromoalkanes to provide the bromides 11, which could be utilized to alkylate a range of phenols. This provided analogs 12 after hydrolysis. (The synthesis and characterization of 13, the ester of precursor to 16a is included in the Supporting Information).
The direct analog of 3 (12a) showed an approximate two-fold improvement over 1 (IC50=7.9 µM Table 2). The unadorned phenyl analog (12b), with the same spacer length as 12a, was more than 17-fold more potent than 1. Alteration of the chain length (12c and 12d) led to loss in potency, leading to compounds approximately equipotent to 1. Compound 12b (IC50 of 0.95 µM) is the most active compound in the series to date, being 17 times more active than 1 against TbrPDEB1, though it is 25-fold more potent against hPDE4 (IC50=0.038 µM). This represents a selectivity improvement over 1 (which is 182-fold more potent against hPDE4 than TbrPDEB1).
Exploration of para-substitution of the phenyl group with a range of electron donating, withdrawing, and neutral substituents resulted in loss of potency (12e-h).
To determine whether compounds 1 and 12b had effects in T. brucei growth, we tested these for dose-response using an Alamar blue cell viability assay,16 and found that, while neither compound inhibited growth of mammalian cells (NIH 3T3, TC50 > 50 µM), we observed weak activity for 12b (EC50 = 26 µM) in T. brucei cellular assays. Compound 1 showed no effect upon T. brucei growth (EC50 > 50 µM). This is surprising, given the close concordance we and others observed between TbrPDEB1 enzyme inhibition and cellular growth inhibition with compounds 2 and 35, 6
We considered two possible explanations for the lack of cellular activity. First, the essentiality of both TbrPDEB1 and B2 were demonstrated, as simultaneous knockdown by RNAi was required in order to impact on cellular proliferation.4 We have previously shown that inhibitors have a similar inhibitory profile when tested against both TbrPDEB1 and B2,5 which is perhaps not unexpected given the high sequence similarity in the binding site, and, based on this, we have been screening only against TbrPDEB1. In order to rule out the possible situation where 12b inhibits only TbrPDEB1, allowing a compensatory effect from TbrPDEB2, we tested 12b against TbrPDEB2, and observed anIC50 of 2.1 µM, which is approximately equivalent to the activity against TbrPDEB1.
A second possible explanation for the lack of cellular activity of 1 and 12b, despite the structural similarity between 1–3, and 12b, could be limited trypanosome cell permeability caused by the negatively charged carboxylate anion.17 We anticipated that compound 13, the ester precursor of 12b would likely display improved cellular penetration and act as a prodrug in trypanosome cells. Though 11d is inactive (> 100 µM) against TbrPDEB1 and B2, we observed activity in T. brucei cell cultures (EC50=9.8 µM), which is suggestive that the uncharged ester can permeate the parasite cells and be hydrolyzed to the active species (12b).
We looked towards molecular modeling to help explain our observations. Two crystal structures have been published for 1: (i) a complex with hPDE4B (1XLX) and (ii) a complex with hPDE4D (1XOM).18 The latter crystal structure, with a higher resolution, displays two possible binding modes for the bound ligand where the cyclohexyl showed two alternative conformations (Supporting Information, Figure S1). The structural differences between the human and the parasite phosphodiesterase are important for repositioning the human enzyme inhibitor to a trypanosomal enzyme inhibitor. For this reason we compare the enzyme active sites in the TbrPDEB1 and hPDE4D crystal structures in the top panel of Figure 2.15
Figure 2.

(A). The docked conformation of 1 (green carbon atoms) in TbrPDEB1 (4I15, gold carbon atoms) overlaid with one of the two bound conformations of the same ligand reported in the crystal structure of the human PDE4D-1 complex (1XOM, cyan). Amino acid side chain differences are shown as sticks. TbrPDEB1 residue numbering is used, with the corresponding hPDE4D residues in brackets. The interactions of the ligand with Gln 874 and the Mg2+ ion are displayed in blue dashed lines. (B). Docked conformation of 12b in TbrPDEB1. The surface of the active site and the P-pocket are shown. Mg2+ and Zn2+ are shown as green and grey spheres respectively. Two residues defining the P-pocket (Met868 and Tyr845) are also shown in sticks. In both figures, Mg2+ and Zn2+ ions in the active site are shown as green and grey spheres respectively. Images were generated using PyMol (version 1.5.0.4, Schrodinger).
The predicted conformation of 3 bound to TbrPDEB1, overlaid with 1 as observed in the crystal structure of a hPDE4D (1XOM) is shown in the Supporting Information, Figure S2. This overlay reveals similar interactions for the methoxy group of these two ligands with the glutamine residue in the back pocket (Gln874 in TbrPDEB1) and rationalized our efforts to explore the P-pocket of the enzyme by replacing the cyclopentyl of 1 with different groups able to fill the P-pocket.
Docking (POSIT v.1.0.2., OpenEye Scientific Software, Santa Fe, NM) was used to rationalize the results explained above. Firstly, we observe that the compounds with a cis-orientation of the cyano and carboxylate functionality are more potent than the trans-isomers. This can be explained by the observation that, in the cis-isomer, the cyano group is predicted to occupy an area at the entrance of the active site and the carboxylate group is in close contact with the magnesium ion (Figure 2A). The carboxylate group is in fact predicted to interact with this metal, explaining the loss of potency resulting from the removal or substitution of this group with an ester, leading to loss of a predicted electrostatic interaction. Following docking, the distal phenyl group of compound 12b is optimally placed in the P-pocket of the enzyme, as shown in Figure 2B. The phenyl ring packs against two methionine residues and a tyrosine residue, and, in addition, there is possibly a weak hydrogen bond between the C-alpha hydrogen of Gly873 and the oxygen atom of the phenoxy group of this ligand (Figure S3, Supporting Information).19 Visual inspection of the poses obtained by molecular docking confirms that the ideal linker connecting the two phenyl groups of the ligand contains four methylene groups, explaining the ~10-fold loss in potency observed for 12c and 12d.
Substitutions in the para position of the distal phenyl group of 12b do not increase potency (Table 2). Modeling suggests that a small polar substituent in para position could have been tolerated by interacting with the hydroxyl group of Tyr845, and there was a strong rationale for introducing the tetrazole-containing substituent (12a) given the good potency of compound 3. However the, inconsistencies in the SAR between these two series may be rationalized by slight differences in binding conformation, leading to differences in positioning of the P-pocket substituents (see e.g. Figure S2).
Finally, the 1000-fold higher activity of 3 over the matched analog 12a is clearly due to the bicyclic headgroup; the cycloheptyl N-substituent in this compound is predicted to fill the pocket in the entrance of the active site much better than the cyclohexyl system of cilomilast derivatives (Figure S2, Supporting Information). Further, the substituted bicyclic ring system is predicted to give increased lipophilic interactions with apolar side chains (such as a methionine and two phenylalanine residues). In our docked pose, the carbonyl group of the ligand is predicted to interact with the magnesium ion.
In summary, we have identified a hPDE4 inhibitor chemotype that can be repurposed to discover new inhibitors of TbrPDEB1, one of which shows sub-micromolar activity toward TbrPDEB1 and a modest cellular growth inhibition of T. brucei. As with other repurposed hPDE4 chemotypes, however, we are faced with a formidable barrier of selectivity over hPDE4 which we have yet to surmount. On the other hand, nonetheless, we have shown an ability to improve compound potency within this chemotype, and this knowledge will be useful in informing other PDE inhibitor repurposing programs that are continuing.
Supplementary Material
Acknowledgements
We are grateful to Takeda, Inc. for obtaining the hPDE4 potency data, and to Drs. Geert Jan Sterk and Stefan Ochiana for helpful discussions. Free academic licenses from Open Eye Scientific Software and ChemAxon for their suites of programs are gratefully acknowledged. This work was funded by the National Institutes of Health (R01AI082577).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
Compound syntheses and a tabulation of the compounds with their Northeastern registry numbers and screening data is available in the Supporting Information. The screening data has been made freely available as a shared data set at www.collaborativedrugdiscovery.com. Also included are biological assay and molecular modeling details. This Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.World Health Organization. [Accessed: 7/21/2014]; http://www.who.int/trypanosomiasis_african/en.
- 2.Priotto G, Kasparian S, Ngouama D, Ghorashian S, Arnold U, Ghabri S, Karunakara U. Clin. Infect. Dis. 2007;45:1435. doi: 10.1086/522982. [DOI] [PubMed] [Google Scholar]
- 3.Pollastri MP, Campbell RK. Future Med. Chem. 2011;3:1307. doi: 10.4155/fmc.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oberholzer M, Marti G, Baresic M, Kunz S, Hemphill A, Seebeck T. FASEB J. 2007;21:720. doi: 10.1096/fj.06-6818com. [DOI] [PubMed] [Google Scholar]
- 5.Bland ND, Wang C, Tallman C, Gustafson AE, Wang Z, Ashton TD, Ochiana SO, McAllister G, Cotter K, Fang AP, Gechijian L, Garceau N, Gangurde R, Ortenberg R, Ondrechen MJ, Campbell RK, Pollastri MP. J. Med. Chem. 2011;54:8188. doi: 10.1021/jm201148s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, Luginbuehl E, Seebeck T. J. Infect. Dis. 2012;206:229. doi: 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orrling KM, Jansen C, Vu XL, Balmer V, Bregy P, Shanmugham A, England P, Bailey D, Cos P, Maes L, Adams E, van den Bogaart E, Chatelain E, Ioset JR, van de Stolpe A, Zorg S, Veerman J, Seebeck T, Sterk GJ, de Esch IJ, Leurs R. J. Med. Chem. 2012;55:8745. doi: 10.1021/jm301059b. [DOI] [PubMed] [Google Scholar]
- 8.Ochiana SO, Gustafson A, Bland ND, Wang C, Russo MJ, Campbell RK, Pollastri MP. Bioorg. Med. Chem. Lett. 2012;22:2582. doi: 10.1016/j.bmcl.2012.01.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang C, Ashton TD, Gustafson A, Bland ND, Ochiana SO, Campbell RK, Pollastri MP. Bioorg. Med. Chem. Lett. 2012;22:2579. doi: 10.1016/j.bmcl.2012.01.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spina D. Br. J. Pharmacol. 2008;155:308. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christensen SB, Guider A, Forster CJ, Gleason JG, Bender PE, Karpinski JM, DeWolf WE, Jr, Barnette MS, Underwood DC, Griswold DE, Cieslinski LB, Burman M, Bochnowicz S, Osborn RR, Manning CD, Grous M, Hillegas LM, Bartus JO, Ryan MD, Eggleston DS, Haltiwanger RC, Torphy TJ. J. Med. Chem. 1998;41:821. doi: 10.1021/jm970090r. [DOI] [PubMed] [Google Scholar]
- 12.Duplantier AJ, Bachert EL, Cheng JB, Cohan VL, Jenkinson TH, Kraus KG, McKechney MW, Pillar JD, Watson JW. J. Med. Chem. 2007;50:344. doi: 10.1021/jm060904g. [DOI] [PubMed] [Google Scholar]
- 13.Hatzelmann A, Morcillo EJ, Lungarella G, Adnot S, Sanjar S, Beume R, Schudt C, Tenor H. Pulm. Pharmacol. Therapeut. 2010;23:235. doi: 10.1016/j.pupt.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 14.Ehrman TM, Barlow DJ, Hylands PJ. Bioorg. Med. Chem. 2010;18:2204. doi: 10.1016/j.bmc.2010.01.070. [DOI] [PubMed] [Google Scholar]
- 15.Jansen C, Wang H, Kooistra AJ, de Graaf C, Orrling KM, Tenor H, Seebeck T, Bailey D, de Esch IJ, Ke H, Leurs R. J. Med. Chem. 2013;56:2087. doi: 10.1021/jm3017877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Räz B, Iten M, Grether-Bühler Y, Kaminsky R, Brun R. Acta Tropica. 1997;68:139. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- 17.Gleeson MP. J. Med. Chem. 2008;51:817. doi: 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- 18.Card GL, England BP, Suzuki Y, Fong D, Powell B, Lee B, Luu C, Tabrizizad M, Gillette S, Ibrahim PN, Artis DR, Bollag G, Milburn MV, Kim S-H, Schlessinger J, Zhang KYJ. Structure. 2004;12:2233. doi: 10.1016/j.str.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Scheiner S, Kar T, Gu Y. J. Biol. Chem. 2001;276:9832. doi: 10.1074/jbc.M010770200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.