

Abstract
African Americans (AAs) have a 1.5 times higher risk of colorectal carcinoma (CRC) than Caucasians. Gene silencing through CpG island hypermethylation has been associated with the genesis or progression of microsatellite instability (MSI) largely due to 1 target for hypermethylation being the DNA mismatch repair gene hMLH1; there is anecdotal evidence of an increased incidence of MSI among AAs. P16 and hMLH1 can be inactivated by hypermethylation of their respective promoter regions, abrogating the ability to regulate cell proliferation and repair processes. We studied such methylation, as well as hMHS2 expression in colorectal cancers from AA patients to determine if MSI is associated with epigenetic silencing. Experiments were conducted on matched normal and colon cancer tissues from AA patients (n = 51). A total of 5 microsatellite markers (D2S123, D5S346, D17S250, BAT25 and BAT26) were used to evaluate MSI status. P16 and hMLH1 promoter methylation status was determined following bisulfite modification of DNA and using methylation specific PCR, while immunohistochemistry (IHC) was used to examine expression of hMLH1 and hMSH2. A total of 22 (43%) cancers demonstrated microsatellite instability-high (MSI-H), while 27 were microsatellite stable (MSS) and 2 were microsatellite instability-low (MSH-L). Most of the MSI-H tumors were proximal, well differentiated and highly mucinous. Most patients in the MSI-H group were females (68%). The p16 promoter was methylated in 19 of 47 (40%) tumors. A total of 7 of these CRCs demonstrated MSI-H (33%). The hMLH1 promoter was methylated in 29 of 34 (85%) tumors, of which 13 CRCs demonstrated MSI-H (87%). hMLH1 and hMSH2 staining was observed in 66% and 38% of MSI-H tumors, respectively. Overall, the prevalence of MSI-H colorectal tumor was 2–3-fold higher, while the defect in the percentage expression of mismatch repair (MMR) genes (hMLH1 and hMSH2) was similar in AA patients compared to the U.S. Caucasian population. Similar numbers of AA MSS tumors with p16 and hMLH1 methylation likely indicate hemimethylation of genes that might reflect environmental or genetic influences that might be more common in the AA population.
Colon cancer has been estimated to account for approximately 13% of all cancer-related deaths.1 Epidemiological data show that between 1974 and 1985, 5-year survival for colon cancer has remained unchanged in African Americans, while rates have improved for Caucasians. Furthermore, U.S. mortality rates from colon cancer continue to be greater in AAs than in Caucasians.1,2 These epidemiological observations suggest that AAs constitute a unique population regarding the development and presentation of colon cancer. These include a greater preponderance of neoplasia on the right side of the colon.3 Proximal colon cancers are genetically different from distal (rectosigmoid) lesions. AAs are more likely to have hyperploidy and allelic imbalance. Moreover, the karyotypic characteristics of malignant cells from rectal adenocarcinomas differ from Caucasians.3 Also, epidemiological studies note higher consumption of saturated fats and ethanol in the AA population, as well as higher smoking rates.4 Some of these differences may influence the presentation of colorectal cancer in the AA population.
The progression of some colon cancers is accompanied by MSI and is associated with DNA-repair defects, which occur both in some sporadic and hereditary human colon cancers. DNA methylation is part of the normal regulation of gene products that often silences gene expression. Such alteration in DNA methylation can silence MMR genes like hMLH1, as well as the cell cycle regulating gene p16. Downregulation of the p16 gene, which negatively controls cell cycle progression, represents a possible mechanism for tumorigenesis. One study indicated that a distinct pattern of genetic and epigenetic events, involved in activation of oncogenes like K-ras and inactivation of tumor suppressor genes p16 and p53, occurs in colorectal cancer.5 MSI is a hallmark of DNA MMR deficiency that in turn appears to be primarily due to inherited and/or acquired alterations in the MMR genes, hMLH1 and hMSH2. Thus, the presence of MSI correlates with the absence of either hMLH1 or hMSH2.6–8 Further studies showed that pathogenic missense mutations within hMLH1 and hMSH2 (10–20% of all cases with hereditary nonpolyposis colorectal cancer [HNPCC]) are not necessarily associated with a negative immunohistochemistry result, but the missense mutation may still lead to MSI.8 In sporadic colon carcinomas, loss of hMLH1 expression is frequently the result of hypermethylation of the promoter region of hMLH1, whereas loss of hMSH2 expression seems to occur only through somatic genetic mutation.9,10 We, therefore, examined the tumors of AA CRC patients for MSI, methylation of p16 and hMLH1 and expression of hMSH2.
Materials and methods
Patient selection
A total of 51 AA patients, who underwent surgical resection of colorectal cancers at Howard University Hospital or at the Johns Hopkins Hospital between 1998 and 2001, are included in this study. After approval from both the Howard University and the Johns Hopkins University Institutional Review Boards, formalin-fixed, paraffin-embedded archival tissue was collected. Clinical data obtained on each patient included race, age, site of primary tumor, mucin production, tumor differentiation and family history of cancer. Family data identified those pedigrees that met either the Amsterdam I or Amsterdam II criteria for HNPCC.
Isolation of DNA and MSI testing
To assess microsatellite instability, microdissected tumors were used. Tumor blocks from Howard University Hospital and Johns Hopkins Hospital were cut into 5-μm sections on SuperFrost plus slides (Fisher Scientific, Pittsburgh, PA) for tumor differentiation. Two techniques were used for the MSI studies according to our previous reports.11 We utilized the reference panel of 5 pairs of microsatellite primers (D2S123, D5S346, D17S250, BAT25 and BAT26), recommended to determine the presence of microsatellite instability in colorectal cancer specimens.12
Methylation-specific PCR (MSP)
p16 methylation was analyzed as previously reported.12 The hMLH1 primers used for methylation were; forward 5′ACGTAGACGTTTTATTAGGGTCGC; reverse 5′CCTCATCGTAACTACCCGCG, and for unmethylated state were forward; 5′TTTTGATGTAGATGTTTTATTAGGGTTGT, and reverse; 5′ACCACCTCATCATAACTACCACA.
Histopathological analysis
Independent pathologists who were not aware of the MSI status evaluated specific histopathologic characteristics. Grading of the degree of neoplastic transformation was done by staining with hematoxylin-eosin (H&E). Tumors were classified as proximal (proximal to the splenic flexure) or distal. The tumor nodes metastasis (TNM) system of the International Union Against Cancer was used for tumor staging. Mucin production was evaluated using the modified criteria of Wiggers et al.,13 reported as absent (no extra cellular mucin production), focal (when extra cellular mucin production was present in <50% of the cells) or predominant (when the area of extra cellular mucin production was present in ≥50% of the cells).
Immunohistochemistry
Tissue in paraffin embedded blocks was used. Sections (5 μm) were mounted on charged glass slides. Sections were then deparaffinized with xylene for 2 × 10 min and rehydrated using graded ethanol series. Antigen retrieval was performed using a microwave oven for 20 min with occasional interruption to avoid tissue degradation by excessive heat. The slides were then treated with hydrogen peroxide, followed by protein blocking using primary and secondary antibody, streptavidin-biotin complex, amplification reagent, streptavidin-peroxidase and substrate-chromogen solution and then, counterstained with hematoxylin, rinsed in with ethanol, dried and observed using a light microscope. A negative control was run without the primary antibody. All immunohistochemistry reagents were purchased from DAKO (Carpinteria, CA), and antibodies (hMLH1 clone G168-15 and hMSH2 clone 556349) were purchased from PharMingen (San Diego, CA).
Statistical analysis
MSI was originally identified with 3 levels (MSS, MSI-L, and MSI-H). However, for the statistical analysis, the MSS and MSI-L groups were assigned a “No” response and the MSI-H group was assigned a “Yes” response. Age and the stage of cancer were continuous variables, while gender, location, mucin production, differentiation, p16, hMLH1 and hMSH2 were categorical variables. For statistical analysis mucin production was categorized as <50% compared to ≥50%. Statistical analysis was performed using the LOGISTIC and FREQ procedures of the SAS System (SAS/STAT 2001; Cary, NC). Results are reported based on asymptotic chi-square or the Fisher’s exact test as appropriate.
Results
Demographics and gene alterations
The clinical and pathologic characteristics of the patients are given in Table I. Of 51 tissue samples analyzed, 22 were from males and 29 from females. The mean age was 68 years and all patients self-identified themselves as AAs. These were subdivided into 2 groups by MSI testing: 29 (57%) were MSS+MSI-L and 22 (43%) were MSI-H. Patients in the MSI-H group were slightly older than patients in the MSS+MSI-L groups (mean ages were 69 and 67 years, respectively). Interestingly, the majority of the MSI-H group were females (68%), but there was no statistically significant relationship between MSI status and gender (Table I). One patient was MSI-H and had a strong family history of colon cancer, which met the Amsterdam criteria for HNPCC. Two other patients met the Bethesda criteria,14 based on the age of diagnosis of colorectal cancer. However, tissue samples from these 2 patients were MSS.
TABLE I.
CLINICOPATHOLOGICAL FEATURES OF CRC CASES
| All1 | MSS+MSI-L1 | MSI-H1 | Odds ratio | p | |
|---|---|---|---|---|---|
| Number of patients | 51 | 29(57)2 | 22(43)2 | ||
| Mean age | 68 | 67 | 69 | >.53 | |
| Gender | |||||
| (M) | 22(43) | 15(52) | 7(32) | >.16 | |
| (F) | 29(57) | 14(58) | 15(68) | 2.3 | |
| Site | 49 | 29 | 20 | <.07 | |
| Proximal | 29(59) | 14(48) | 15(75) | 3.2 | |
| Distal | 20(41) | 15(52) | 5(25) | ||
| Mucin production | 51 | 29 | 22 | <.023 | |
| None | 35(69) | 24(82) | 11(50) | ||
| <50% | 5(10) | 1(3) | 4(18) | ||
| >50% | 11(21) | 4(14) | 7(32) | 4.8 | |
| Differentiation | 51 | 29 | 22 | >.74 | |
| Well | 2(4) | 1(3) | 1(5) | ||
| Moderate | 38(74) | 23(80) | 15(68) | ||
| Poor | 11(22) | 5(17) | 6(27) | ||
| Stage | 51 | 29 | 22 | >.65 | |
| I | 1(2) | 1(4) | 0(0) | ||
| II | 20(39) | 10(40) | 10(45.5) | ||
| III | 24(47) | 14(56) | 10(45.5) | ||
| IV | 6(12) | 4(15) | 2(9) | ||
| P16 methylation | 47 | 26 | 21 | >.37 | |
| (MSP) | |||||
| Methylated | 19(40) | 12(46) | 7(33) | 1.7 | |
| Unmethylated | 28(60) | 14(54) | 14(67) | ||
| hMLH1 methylation | 34 | 19 | 15 | >.99 | |
| (MSP) | |||||
| Methylated | 29(85) | 16(84) | 13(87) | .76 | |
| Unmethylated | 5(15) | 3(16) | 2(13) | ||
| hMLH1 expression by IHC | 34 | 19 | 15 | >.51 | |
| Normal | 15(44) | 10(53) | 5(33) | ||
| Partial loss | 16(47) | 8(42) | 8(53) | ||
| Complete loss | 3(9) | 1(5) | 2(13) | ||
| hMSH2 expression by IHC | 31 | 18 | 13 | ||
| Normal | 18(58) | 10(55) | 8(62) | >.99 | |
| Partial loss | 12(39) | 7(39) | 5(38) | ||
| Complete loss | 1(3) | 1(6) | 0(0) |
Parentheses indicate percentages: based on column values.
Indicates percentages based on row values.
Indicates a statistically significant difference in comparison of MSI-H and MSS subjects with respect to mucin production <50%.
Location of the primary lesion
A total of 29 of the lesions were located in the proximal colon (59%). The prevalence of proximal lesions in the MSI-H group (75%) was higher than in the MSS group (48%).
Mucin production
Among 51 samples, 35 (69%) were negative (absence of mucin production), 5 (10%) had focal mucin production, and 11 (21%) were predominantly mucin producing (Table I). The percentage of MSI-H lesions, which were mucin producing (32%) was higher than mucin producing lesions in the MSS+MSI-L group (14%). The odds ratio of patients with mucin production having MSI-H was 4.8 times that of the patients without mucin production (p < 0.02).
Differentiation
Most of the tumors were moderately differentiated (74%). The percentage of moderately differentiated tumors was slightly lower in the MSI-H group (68%) when compared to the MSS group, but the difference was not statistically significant.
TNM staging
The majority of patients in our series were stage III or higher (Table I), with 45.5% of MSI-H lesions at stage II, 45.5% stage III, and the remainder 9%, stage IV. There was no statistically significant differences in the stage of the lesions between MSI and MSS groups (p > 0.65).
p16 and hMLH1 methylation
To investigate if sporadic MSI-H tumors in AA patients were associated with simultaneous methylation of p16 and hMLH1, we determined the methylation status of the p16 and hMLH1 genes (Fig. 1). A total of 19 patients (40%) in our series showed methylation of tumor suppressor gene p16. A total of 68% of these lesions were in females and 32% were in males. We observed a lack of MSI-H lesions in males with p16 promoter methylation, while all females with p16 promoter methylation showed MSI-H lesions. A total of 7 of the 21 MSI-H tumors demonstrated p16 promoter methylation (33%), while 11 of 24 MSS tumors showed methylation of p16 (46%); these differences were not statistically significant (p > 0.37). A total of 85% (29 of 34) of patients showed methylation of hMLH1. A total of 67% of these lesions were in females and 30% were in males. A total of 55% males and 40% females with hMLH1 promoter methylation showed MSI-H patterns. Of the 15 MSI-H tumors, 13 demonstrated hMHL1 promoter methylation (87%), while 16 of 18 MSS (89%) tumors showed methylation of hMLH1 and the differences were not statistically significant (p > 0.99).
Figure 1.
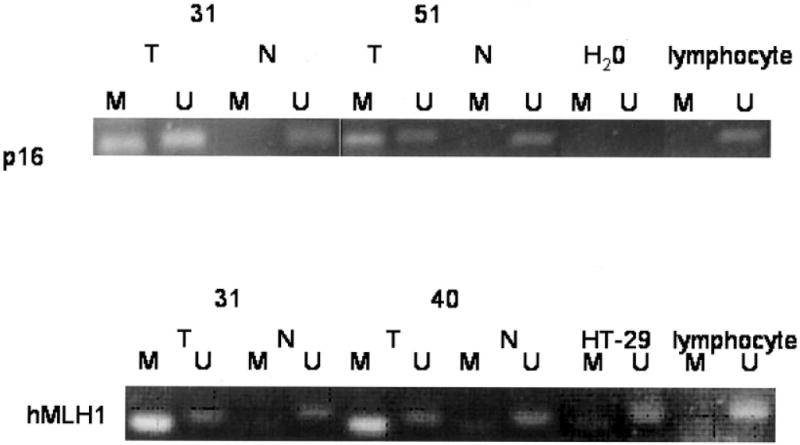
MSP analysis of the promoter region of p16INK4a and hMLH1 in CRC patients. The presence of visible PCR product in lanes “U” indicates the presence of unmethylated genes of p16INK4a or hMLH1, the presence of product in lanes “M” indicates the presence of methylated genes. Water controls for PCR reaction are also shown. MSP of p16INK4a in normal and tumors is shown. Patients 31 and 51 are methylated at p16INK4a. Patients 31 and 40 are methylated at hMLH1. MSP of hMLH1 in normal and tumors is shown. Lymphocyte DNA was used as control for unmethylated at hMLH1. Semimethylated HT-29 DNA is used as positive control for methylation and unmethylation of hMLH1.
Immunohistochemistry
Protein expression for hMLH1 and hMSH2 was examined immunohistochemically in paraffin-embedded tissue sections (Fig. 2). Nonneoplastic cells in all sections demonstrated positive nuclear staining for both hMLH1 and hMSH2. A total of 34 cases were analyzed by IHC for hMLH1 nuclear protein staining. A total of 66% (10/15) of the MSI-H tumors showed partial or complete loss of hMLH1 expression. A total of 15 patients showed complete hMLH1 expression. A total of 5% of MSS+MSI-L tumors had hMLH1 loss. A total of 30 cases showed presence of hMSH2 protein by IHC staining, of which 13 cases were MSI-H. A total of 85% of MSI-H cases showed complete nuclear staining, while 15% showed partial loss of hMSH2 expression. A total of 25 patients showed complete nuclear expression of hMSH2. Most of the MSS tumors showed normal or partial nuclear staining for hMSH2 (89%). For some samples, tissue was not available to perform IHC. A total of 3 cases with unmethylated p16 or hMLH1/hMSH2 are shown in Figure 2 as an example of abundant expression of hMLH1 and hMSH2 nuclear protein.
Figure 2.

Immunohistochemical analysis of hMLH1 and hMSH2 expression in normal epithelial and tumor cells. (a) Normal colonic mucosa showing positive nuclear staining of hMLH1 protein in normal crypt epithelial cells. (b) Tumor showing partial or (c), no hMLH1 protein expression. Normal epithelial cells (c,d) and tumor (e) stained with anti h-hMSH2 antibody showing positive nuclear and negative staining for hMSH2, respectively.
Discussion
Colorectal cancers are characterized by multiple chromosomal and epigenetic abnormalities. Microsatellite instability is one of the molecular mechanisms leading to genomic instability. Recent evidence suggests that patients with CRC manifesting MSI have a different prognosis and response to chemotherapeutic agents.15–19 Several studies report that up to 86% of tumors in patients with HNPCC exhibit microsatellite instability.20 On the other hand, 80–85% of sporadic colorectal cancers lack MSI. In our series of AA patients, the incidence of microsatellite instability was 43%, a rate which is much higher than reported in the literature for other ethnic groups (12–17%).21,22 One of the reasons for the difference in these data could be the large number of right-sided cancers in our sample; MSI is more frequently found in right-sided tumors. This is consistent with a study using the Surveillance, Epidemiology, and End Results (SEER) registry data from the National Cancer Institute, demonstrating that proximal colon carcinoma rates in AAs were considerably higher than in whites.3 These findings may reflect the pathogenesis of colorectal cancer in the AA race, and point to a higher incidence of MSI in this ethnic group compared to the general population.
Clinicopathological features known to be associated with the presence of MSI include location of the primary tumor proximal to the splenic flexure, poorly differentiated cancers, predominance of mucin producing cells in lesions with MSI and peritumoral lymphocytic infiltration.21 In this study of AA patients, CRC location and frequency of mucin production are in accordance with previous publications. However, the degree of differentiation of the cancers was different in our series. Most of the tumors in the MSI-H group were moderately differentiated (68%). The MSS group in our analysis consisted predominantly of moderately differentiated cancers (80%; not significant). This may represent a difference in histopathological expression of MSI-H tumors in AAs, which needs further investigation. Data regarding lymphocytic infiltration into the tumors were not available for this series of patients.
In this study, we examined the frequency and significance of MSI in an AA population with sporadic CRC. However, some individuals enrolled with apparent sporadic disease could harbor germline mutations. Novel germline mutations in mismatch repair genes hMLH1 and hMSH2 have been demonstrated in AA patients within HNPCC families.23 At least one patient in our study fell into this germline mutation category. A total of 3 patients in our series satisfied the Bethesda criteria based on age of cancer diagnosis; one of these patients also met the requirements for HNPCC by the Amsterdam criteria. This patient showed MSI-H in malignant tissue consistent with an inherited mutation causing cancer. But the tissue from the other two such patients were microsatellite stable. This may be explained by the low positive predictive value (27%) of the Bethesda Guidelines for HNPCC.
There are suggestions that MSI-H in the colon may occur as a function of age.24 This may sound counterintuitive in the context of HNPCC in which young patients get colon cancer with MSI. However, at the same time, with age we tend to methylate CpG islands, and this has been proposed to be a mechanism for the epigenetic inactivation of hMLH1 in sporadic, non-HNPCC tumors. It is also interesting that many MSI tumors were from females, although MSI-H CRC has not been readily reported to be a female-predominant condition. Our results suggest that being female might influence the pathway for CRC development. The chance of female patients having MSI-H was 2.3 times higher than males (p = 0.16). Breivik et al.,27 also indicated a relationship between MSI in CRC and gender and age. These investigators found MSI was more frequently present among younger male and older female patients. In addition, epidemiological studies reported gender differences in the site distribution of CRC cancer with proximal cancers most frequently among older women.26 One of the earliest hypotheses addressing the role of anatomical subsets and gender in the development of CRC postulated an influence of estrogen on bile acid secretion.21,25,27 Accordingly, estrogen through its influence on serum cholesterol levels could increase the concentration of bile acids, which have toxic, trophic, and promoting effects on the colonic epithelium. Previous studies have shown a high incidence of methylation of the hMLH1 and p16 promoters in CRC arising in our female patients.27,28 In our series, 68% of patients with p16 methylation were female. Also, the high proportion of MSI-H tumors seen in female patients series may explain why some investigations report that females with CRC have a better prognosis than males. Of note, survival rates are lower in AA patients with colorectal cancer. However, patients with high levels of microsatellite instability as a group have better outcomes. One possible explanation for this discrepancy is that AAs have decreased access to medical care and present late in the course of disease.29,30 A total of 88% of tumors from our study showed some degree of methylation of the hMLH1 promoter. This is consistent with the contribution of a hypermethylated hMLH1 mismatch repair gene being responsible for MSI in these cases of sporadic CRC in this population. A total of 40% of the MSI-H lesions in females were hMLH1 methylated. Thus, hMLH1 methylation and reduced or lost expression reveals that this epigenetic mechanism may be the cause of inactivation of both alleles of hMLH1 genes of these tumors. The association of higher levels of CpG island methylation with more advanced histologic changes suggests that CpG island methylation plays a role in colorectal carcinoma. However, the pathophysiology of hypermethylation (why, when, where) remains unclear. Cancers can be classified according to their degree of methylation, and those cancers with high degrees of methylation (the CpG island methylator phenotype, or CIMP) represent a clinically and etiologically distinct group that is characterized by “epigenetic instability.” It is possible that cancers in our study fall in this category but CpG island methylation involving multiple genes would need to be done in the future. Furthermore, CIMP-associated cancers seem to have a distinct epidemiology, unique histology, different precursor lesions and distinct molecular features. In this study we have 34 hMLH1 negative tumors, of which 10 of 15 were MSI-H (66%). This is close to published results.31 Our hMLH1 and hMSH2 immunohistochemistry results are consistent with the MSI status of the patients. Nuclear expression results showed an inverse relationship between hMLH1 and hMSH2 markers with MSI-H. In addition, the IHC technique proved to be a reliable methodology to evaluate the tumors with DNA MMR defect.
In conclusion, most of the clinicopathologic characteristics of MSI-H lesions in our study of AA patients are similar to those reported previously in Caucasians, including the location of the primary tumor and extent of mucin production. However, most of the MSI-H tumors in our study group were moderately-differentiated, which may indicate a different pathologic expression of MSI-H lesions in AAs. The defect in the MMR genes (hMLH1 and hMSH2) is similar (70–80%) in AA patients with MSI-H colorectal cancers compared to the general population.32 Our results suggest a much higher proportion of CRC tumors with MSI in AAs compared to the general population, 43% vs. <20%, respectively. This may have significant implications in the treatment of AA patients, since MSI-H lesions are often right-sided and may show a different response to chemotherapeutic agents like 5FU.15,33 This investigation confirms the higher percentage of MSI-H seen in our previously reported results.11
Acknowledgments
Grant sponsor: National Cancer Institute, NIH; Grant number: CA102681; Grant sponsor: Marcia Johnson Award, Howard University.
References
- 1.Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin. 2002;52:23–47. doi: 10.3322/canjclin.52.1.23. [DOI] [PubMed] [Google Scholar]
- 2.Carethers JM. Racial and ethnic factors in the genetic pathogenesis of colorectal cancer. J Assoc Acad Minor Phys. 1999;10:59–67. [PubMed] [Google Scholar]
- 3.Troisi RJ, Freedman AN, Devesa SS. Incidence of colorectal carcinoma in the U.S.: an update of trends by gender, race, age, subsite, and stage, 1975–1994. Cancer. 1999;85:1670–6. [PubMed] [Google Scholar]
- 4.Okuyemi KS, Ahluwalia JS, Ebersole-Robinson M, Catley D, Mayo MS, Resnicow K. Does menthol attenuate the effect of bupropion among African American smokers? Addiction. 2003;98:1387–93. doi: 10.1046/j.1360-0443.2003.00443.x. [DOI] [PubMed] [Google Scholar]
- 5.Maekawa M, Sugano K, Ushiama M, Fukayama N, Nomoto K, Kashiwabara H, Fujita S, Kakizoe T. Heterogeneity of DNA methylation status analyzed by bisulfite-PCR-SSCP and correlation with clinicopathological characteristics in colorectal cancer. Clin Chem Lab Med. 2001;39:121–8. doi: 10.1515/CCLM.2001.021. [DOI] [PubMed] [Google Scholar]
- 6.Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 1997;57:4749–56. [PubMed] [Google Scholar]
- 7.Thibodeau SN, French AJ, Cunningham JM, Tester D, Burgart LJ, Roche PC, McDonnell SK, Schaid DJ, Vockley CW, Michels VV, Farr GH, Jr, O’Connell MJ. Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res. 1998;58:1713–8. [PubMed] [Google Scholar]
- 8.Lindor NM, Burgart LJ, Leontovich O, Goldberg RM, Cunningham JM, Sargent DJ, Walsh-Vockley C, Petersen GM, Walsh MD, Leggett BA, Young JP, Barker MA, et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol. 2002;20:1043–8. doi: 10.1200/JCO.2002.20.4.1043. [DOI] [PubMed] [Google Scholar]
- 9.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–11. [PubMed] [Google Scholar]
- 10.Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, Thibodeau SN. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58:3455–60. [PubMed] [Google Scholar]
- 11.Ashktorab H, Smoot DT, Carethers JM, Rahmanian M, Kittles R, Vosganian G, Doura M, Nidhiry E, Naab T, Momen B, Shakhani S, Giardiello FM. High incidence of microsatellite instability in colorectal cancer from African Americans. Clin Cancer Res. 2003;9:1112–7. [PubMed] [Google Scholar]
- 12.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 13.Wiggers T, Arends JW, Schutte B, Volovics L, Bosman FT. A multivariate analysis of pathologic prognostic indicators in large bowel cancer. Cancer. 1988;61:386–95. doi: 10.1002/1097-0142(19880115)61:2<386::aid-cncr2820610231>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 14.Cravo ML, Fidalgo PO, Lage PA, Albuquerque CM, Chaves PP, Claro I, Gomes T, Gaspar C, Soares JO, Nobre-Leitao C. Validation and simplification of Bethesda guidelines for identifying apparently sporadic forms of colorectal carcinoma with microsatellite instability. Cancer. 1999;85:779–85. [PubMed] [Google Scholar]
- 15.Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–31. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145:148–56. [PMC free article] [PubMed] [Google Scholar]
- 17.Lothe RA, Peltomaki P, Meling GI, Aaltonen LA, Nystrom-Lahti M, Pylkkanen L, Heimdal K, Andersen TI, Moller P, Rognum TO, et al. Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res. 1993;53:5849–52. [PubMed] [Google Scholar]
- 18.Risio M, Reato G, di Celle PF, Fizzotti M, Rossini FP, Foa R. Microsatellite instability is associated with the histological features of the tumor in nonfamilial colorectal cancer. Cancer Res. 1996;56:5470–4. [PubMed] [Google Scholar]
- 19.Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K, Boland CR. Use of 5-fluorouracil and survival in patients with microsatellite-unstable col-orectal cancer. Gastroenterology. 2004;126:394–401. doi: 10.1053/j.gastro.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 20.Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–6. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 21.Gryfe R, Gallinger S. Microsatellite instability, mismatch repair deficiency, and colorectal cancer. Surgery. 2001;130:17–20. doi: 10.1067/msy.2001.112738. [DOI] [PubMed] [Google Scholar]
- 22.Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, Chadwick RB, Kaariainen H, Eskelinen M, Jarvinen H, Mecklin JP, de la Chapelle A. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998;338:1481–7. doi: 10.1056/NEJM199805213382101. [DOI] [PubMed] [Google Scholar]
- 23.Weber TK, Chin HM, Rodriguez-Bigas M, Keitz B, Gilligan R, O’Malley L, Urf E, Diba N, Pazik J, Petrelli NJ. Novel hMLH1 and hMSH2 germline mutations in African Americans with colorectal cancer. JAMA. 1999;281:2316–20. doi: 10.1001/jama.281.24.2316. [DOI] [PubMed] [Google Scholar]
- 24.Nakagawa H, Nuovo GJ, Zervos EE, Martin EW, Jr, Salovaara R, Aaltonen LA, de la Chapelle A. Age-related hypermethylation of the 5′ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res. 2001;61:6991–5. [PubMed] [Google Scholar]
- 25.DeCosse JJ, Ngoi SS, Jacobson JS, Cennerazzo WJ. Gender and colorectal cancer. Eur J Cancer Prev. 1993;2:105–15. doi: 10.1097/00008469-199303000-00003. [DOI] [PubMed] [Google Scholar]
- 26.McMichael AJ, Potter JD. Do intrinsic sex differences in lower alimentary tract physiology influence the sex-specific risks of bowel cancer and other biliary and intestinal diseases? Am J Epidemiol. 1983;118:620–7. doi: 10.1093/oxfordjournals.aje.a113672. [DOI] [PubMed] [Google Scholar]
- 27.Breivik J, Lothe RA, Meling GI, Rognum TO, Borresen-Dale AL, Gaudernack G. Different genetic pathways to proximal and distal colorectal cancer influenced by sex-related factors. Int J Cancer. 1997;74:664–9. doi: 10.1002/(sici)1097-0215(19971219)74:6<664::aid-ijc18>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Ward R, Meagher A, Tomlinson I, O’Connor T, Norrie M, Wu R, Hawkins N. Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut. 2001;48:821–9. doi: 10.1136/gut.48.6.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canto M, Piantadosi S, Helzlsouer K, Crum R, P G. Impacts of race and health insurance on colorectal cancer mortality. Gastroenterology. 1994;106:A376. [Google Scholar]
- 30.Marcella S, Miller JE. Racial differences in colorectal cancer mortality. The importance of stage and socioeconomic status. J Clin Epidemiol. 2001;54:359–66. doi: 10.1016/s0895-4356(00)00316-4. [DOI] [PubMed] [Google Scholar]
- 31.Fleisher AS, Esteller M, Wang S, Tamura G, Suzuki H, Yin J, Zou TT, Abraham JM, Kong D, Smolinski KN, Shi YQ, Rhyu MG, et al. Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res. 1999;59:1090–5. [PubMed] [Google Scholar]
- 32.Wright CL, Stewart ID. Histopathology and mismatch repair status of 458 consecutive colorectal carcinomas. Am J Surg Pathol. 2003;27:1393–406. doi: 10.1097/00000478-200311000-00001. [DOI] [PubMed] [Google Scholar]
- 33.Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int J Cancer. 2003;106:66–73. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]