

Abstract
Purpose
An inherited mutation in KRAS (LCS6-variant, rs61764370) results in altered control of the KRAS oncogene. We studied this biomarker’s correlation to anti-EGFR monoclonal Antibody therapy (moAbs) response in metastatic colorectal cancer patients (mCRC) patients.
Experimental Design
LCS6-variant and KRAS/BRAF mutational status was determined in 512 mCRC patients treated with salvage anti-EGFR moAbs therapy, and findings correlated with outcome. Reporters were tested in colon cancer cell lines to evaluate the differential response of the LCS6-variant allele to therapy exposure.
Results
21.2% (109/512) of mCRC patients had the LCS6-variant (TG/GG), which was found twice as frequently in the BRAF mutated versus the wt group (p = 0.03). LCS6-variant patients had significantly longer PFS with anti-EGFR moAbs monotherapy treatment in the whole cohort (16.85 vs 7.85 weeks, p = 0.019) and in the double wt (KRAS and BRAF) patient population (18 vs 10.4 weeks, p = 0.039). Combination therapy (moAbs plus chemotherapy) led to improved PFS and OS for non-variant patients, and brought their outcome to levels comparable to LCS6-variant patients receiving anti-EGFR moAbs monotherapy. Combination therapy did not lead to improved PFS or OS for LCS6-variant patients. Cell line studies confirmed a unique response of the LCS6-variant allele to both anti-EGFR moAbs monotherapy and chemotherapy.
Conclusions
LCS6-variant mCRC patients have an excellent response to anti-EGFR moAbs monotherapy, without any benefit of additional chemotherapy. These findings further confirm the importance of this mutation as a biomarker of anti-EGFR moAbs response in mCRC patients, and warrant further prospective confirmation.
Keywords: KRAS-variant, LCS6, let-7, Cetuximab, Panitumumab, colon cancer, rs61764370
Introduction
In the western world colorectal cancer (CRC) remains a major public health problem, with an estimated 136,830 new cases and 50,310 deaths estimated to occur in 2014 in the United States alone (1). The incorporation of two monoclonal antibodies targeting epidermal growth factor receptor (anti-EGFR moAbs), cetuximab and panitumumab, in mCRC clinical practice, either given as monotherapy or in combination with chemotherapy has proven to provide a modest clinical benefit in pretreated patients (2-5). Nevertheless, their efficacy is restricted to a subset of patients, as non-randomized retrospective studies (6-9), retrospective analysis of prospective randomized trials (10-13), a summary of the above mentioned publications and a grand European consortium study (14) have demonstrated. For example, the presence of tumor acquired KRAS mutations are predictive of resistance to anti-EGFR moAbs therapy and are associated with worse prognosis and shorter survival.
While tumor acquired KRAS mutational status testing is now mandatory for the initiation of anti-EGFR moAb treatment, approximately 50-65% of the mCRC patients with KRAS wt tumors still derive no benefit from these treatments, implying that additional genetic determinants of resistance, or perhaps sensitivity, exist (7, 14-16). Mounting evidence from retrospective studies suggest that the BRAF V600E mutation also confers resistant to anti-EGFR MoAbs (14, 17-20), and, although not entirely clear yet, it also appears that PIK3CA-mutant tumors derive no or little benefit from anti-EGFR moAbs treatment (14, 21-23).
MicroRNAs (miRNAs), discovered almost 20 years ago (24), are an abundant class of highly conserved, endogenous, non-coding, small RNA molecules, 18-25 nucleotides in length, which negatively regulate gene expression by binding to partially complementary sites in the 3′-untranslated region (UTR) of their target mRNAs (25, 26). The binding of miRNAs to their target mRNAs is critical for the regulation of mRNA levels and subsequent protein expression, and this regulation can be affected by single-nucleotide polymorphisms (SNPs) or mutations in miRNA target sites in the 3′UTR of genes. Such 3′UTR variants appear to be capable of playing a role in human diseases like cancer (or conferring an increased risk for certain diseases like cancer) (27, 28). A rapidly accelerating area of research is the systematic genomic evaluation of these sites, which are emerging as potential powerful biomarkers in the growing area of personalized medicine (29, 30). Such variants appear to affect not only gene expression, but also tumor biology, drug response and drug resistance (31-33), likely due to the critical role of miRNAs in managing the response to cytotoxic cancer therapy (33).
The Lethal-7 (let-7) family of miRNAs was among the first discovered, and its differential expression has been found in a number of cancers (34). The KRAS oncogene is a validated direct target of the let-7 miRNA family, as let-7 induces KRAS down-regulation upon binding to the 3′-UTR of the KRAS mRNA (34, 35). Recently, a functional variant was identified in a let-7 complementary site (LCS6) in the KRAS 3′UTR mRNA (36). This variant (rs61764370) consists of a T to G base substitution, and has been found to alter the binding capability of mature let-7 to the KRAS mRNA, resulting in both increased expression of the KRAS oncogenic protein and its downstream pathways (37), as well as altered let-7 miRNA levels in vivo, possibly due to a negative feedback loop. Consistent with the oncogenic nature of KRAS, the variant G-allele has been shown to confer an increased risk of non-small cell lung cancer (NSCLC) in moderate smokers (36), triple negative breast cancer (37) and, in a subset of women, ovarian cancer (38, 39). More recently, significantly worse survival and platinum resistance was found in ovarian cancer patients with the G-allele (40). These findings indicate the functional and clinical significance of the KRAS 3′UTR LCS6-variant.
In the mCRC anti-EGFR moAb therapy setting to date, the KRAS LCS6-variant has been evaluated in four studies with selected populations and with contradicting (conflicting) results (30, 41-43). Graziano et al (41) found within a KRAS and BRAF wt patients’ population treated with salvage irinotecan-cetuximab combination therapy, that LCS6-variant carriers had a statistically significant worse progression free survival (PFS) and overall survival (OS). While in Sebio et al (43), again mCRC patients treated primarily with Irinotecan and cetuximab moAbs were found to be significantly more likely to be nonresponders (p=0.004), in this study the KRAS LCS6-variant did not predict a different outcome in a cohort of people treated with non-moABs based treatment, suggesting that the predictive properties of this mutation were primarily for cetuximab. In contrast to these studies, in a study where patients were given salvage cetuximab monotherapy (30), KRAS LCS6-variant carriers exhibited a longer PFS and OS, and had a better objective response rate (ORR). In addition, in the most recent study (42) of 180 mCRC patients receiving Nordic FLOX (bolus 5-fluorouracil/folinic acid and oxaliplatin) versus 355 patients receiving Nordic FLOX + cetuximab in the NORDIC-VII trial (NCT00145314), there were no significant differences in outcome for KRAS LCS6-variant patients. But, in fact, addition of cetuximab seemed to improve response rate for LCS6-variant carriers more than non-variant carriers (from 35% to 57% versus 44% to 47%), however the difference was not statistically significant (interaction p= 0.16). These conflicting results have led investigators to question if different chemotherapy agents given in addition to cetuximab could impact the response to cetuximab for KRAS LCS6-variant patients (44).
Here our goal was to clarify the role of this mutation as a biomarker of cetuximab response by evaluating the KRAS LCS6-variant along with other molecular markers (KRAS and BRAF) in a series of 512 mCRC patients who underwent either salvage anti-EGFR moAbs monotherapy or moAbs in combination with chemotherapy. We show in our patient cohort as well as in cell lines that the KRAS LCS6-variant allele predicts a positive response to moAbs monotherapy, without any additional benefit of cytotoxic chemotherapy.
Materials and methods
Patients’ characteristics
A total of 559 mCRC patients, 300 treated in the University Hospital of Leuven with anti-EGFR moAb monotherapy or moAb in combination with chemotherapy, as well as 148 patients from the Universite Paris Descartes treated with cetuximab-based salvage combination chemotherapy (14), and 111 previously published mCRC patients (30) treated with cetuximab monotherapy after failing fluoropyrimidine, irinotecan and oxaliplatin containing regimens (30, 45) had tumor tissue available and amenable for analysis of the KRAS LCS6-variant. The mutational status of the KRAS and BRAF genes in the above mentioned patients’ populations has been previously published (14, 30). KRAS LCS6-variant status was successfully determined in 512 of the 559 mCRC patients tested. Molecular characteristics were correlated with ORR, PFS and OS.
Genetic analyses
Formalin-fixed, paraffin-embedded normal tissue from the patients’ specimens was macroscopically dissected using a scalpel blade and DNA was isolated as previously described (14, 30). DNA was amplified using, as previously described (25), a custom-made Taqman genotyping assay (Applied Biosystems,Foster City, CA) designed specifically to identify the T or variant G allele of the KRAS-variant (rs61764370) with the forward primer: 50-GCCAGGCTGGTCTCGAA-30, reverse primer: 50-CTGAATAAATGAGTTCTGCAAAACAGGT T-30, VIC reporter probe: 50-CTCAAGTGATTCACCCA C-30, and FAM reporter probe: 50-CAAGTGATTCACCCAC-30. The KRAS and BRAF mutational status was determined as previously described (14, 30).
Luciferase reporter analysis
KRAS 3′UTR reporter constructs were created by amplifying the entire KRAS 3′UTR from cal27 cells (heterozygous for the LCS6-variant) using the following primers: Forward: CGTATGACTCGAGATACAATTTGTACTTTTTTCTTAAGGCATAC Reverse: ATGAGCGGCCGCTAGGAGTAGTACAGTTCATGACAAAAA
The amplicons were subcloned into the Xho1 and Not1 sites in the 3′UTR of the renilla luciferase gene of the psiCHECK2 dual-luciferase vector (Promega). Reporters with the LCS6-variant (G-allele) and without the variant (T-allele) were confirmed by sequencing.
Dual-luciferase reporters (50ng) containing either the KRAS 3′UTR T-allele or G-allele were transfected into HCT-116 cells (grown at log phase), in a 24-well plate using Lipofectamine 2000 according to the manufacturers protocol. After a 16-hour incubation, the cells were lysed and lysates were analyzed for dual luciferase activities by quantitative titration using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturers protocol. Renilla luciferase was normalized to firefly luciferase. The mean and standard deviation of 4 independent experiments preformed in duplicate were graphed. The p-value was calculated by students t-test.
For drug response analysis, dual-luciferase reporters (500ng) were transfected into HCT-116 cells (grown at log phase) in a 6mm plate using Lipofectamine 2000 according to the manufacturers protocol. Following a 6-hour transfection the cells were trypsonized and re-plated at a density of 5.0×104 cells/well in a 24-well plate along with the indicated amount of each drug. Following a 16-hour incubation the cells were lysed and lysates were analyzed for dual-luciferase activities as described above. The mean and standard deviation of independent experiments preformed in duplicate were graphed.
Statistical analyses
The distribution of genotypes was tested for Hardy-Weinberg Equilibrium and the x2 test was p=0.8. Because of the low frequency of homozygotes for the variant allele, patient samples that were either heterozygous (TG) or homozygous (GG) for the variant allele were considered positive for the KRAS LCS6-variant and entered the analyses as one group. Progression Free Survival (PFS) and Overall Survival (OS) were measured as previously described (14, 30).
The two-tailed Fisher’s exact test was used to compare proportions between non-variant (46) TT patients and LCS-variant (TG and GG) patients. PFS and OS were estimated by Kaplan-Meier method and their association with genotypes was tested with the log-rank test. The association of genotypes with objective response was determined by contingency table and the Fisher’s exact test. To fully explore the possible influence of the KRAS LCS6-variant, analysis were performed in the whole mCRC population, in the patients harboring no tumor acquired mutations in the KRAS and BRAF genes (double wt population) and in the KRAS mutated population. The level of significance was set at a two-sided p value of <0.05. All statistical tests were performed using the statistical package SPSS version 13.
Results
KRAS LCS6-variant in the entire patient cohort
In the 512 mCRC patients 102 (19.9%) patients were heterozygous for the KRAS LCS6-variant and 7 (1.3%) patients homozygous for the variant, thus 109 (21.2%) with the KRAS LCS6-variant overall. KRAS tumor acquired mutations in codons 12, 13 and 61 were found in 184 patients (33%) and the BRAF V600E mutation in 29 patients (5.3%). All patients received anti-EGFR moAbs-based salvage treatment. There were no statistically significant differences found between KRAS LCS6-variant and non-variant carriers for sex or age at diagnosis. The characteristics of the 559 patients have been previously published (14, 30) and are also presented here in Supplemental Table S1.
As shown in Table 1, the distribution of the KRAS LCS6-variant genotypes was different among patients harboring tumor acquired KRAS and BRAF mutations. Specifically, while the percentage of patients with the KRAS LCS6-variant was the same in KRAS wt and mutant groups (20% in each), variant patients were found twice as frequently in the BRAF V600E mutant group vs in the BRAF wt group (20%), resulting in a statistically significant difference (p = 0.030).
Table 1.
Distribution of the KRAS 3′-UTR LCS6 genotypes according to mutation, clinical and demographic data in the mCRC patients’ cohort
| TG | TT or GG | ||||
|---|---|---|---|---|---|
| N | % | N | % | p-value | |
| Age (median, min - max) | 61 | 22 - 89 | 61 | 37 - 80 | 0.654 |
| SEX | |||||
| Male | 229 | 57.4 | 59 | 54.6 | 0.662 |
| Female | 170 | 42.6 | 49 | 45.4 | |
| KRAS status | |||||
| Mutant | 138 | 36.3 | 36 | 34.6 | 0.818 |
| Wild type | 242 | 63.7 | 68 | 65.4 | |
| BRAF status | |||||
| Mutant | 17 | 4.3 | 11 | 10.2 | 0.030 |
| WT | 379 | 95.7 | 97 | 89.8 | |
| TREATMENT | |||||
| Monotherapy | 128 | 32.1 | 32 | 29.6 | 0.726 |
| Combination | 271 | 67.9 | 76 | 70.4 | |
Outcome and Survival analysis in the entire patient cohort
In the cohort as a whole, there were no significant differences in median PFS or OS between the non-variant patients and the LCS6-variant patients (Supplemental Figure S1A and 1B). Similarly, there were no differences in PFS or OS in the double (KRAS and BRAF) wt or in the KRAS mutated patients’ cohorts comparing LCS6-variant and non-variant patients. Finally, there were no significant correlations regarding response (n=483) and skin rash (n=359) with the LCS6-variant and non-variant patients in the whole and in the double wt patients’ cohorts (Supplemental Table S3).
In the cohort as a whole however, in univariate analysis, tumor acquired KRAS mutation (HZ: 1.688, p=0.0001, 95% CI: 1.395 - 2.042), BRAF mutations (HZ: 2.206, p=0.0001, 95%CI: 1.501 - 3.243) and type of treatment (HZ: 1.748, p=0.0001, 95%CI: 1.450 - 2.108), were correlated with PFS and OS. Multivariate analysis revealed that the above factors have an independent association with decreased PFS and OS (Supplemental Table 4), and thus were incorporated into the below analysis.
Progression free survival with monotherapy versus combination treatment
Next, we separately analyzed patients that received moAbs monotherapy versus moAbs combination therapy. From 501 patients with known treatment, 160 (32%) received anti-EGFR moAbs as monotherapy and 341 (68%) were treated with multiple chemotherapy combinations plus EGFR moABs therapy. Of the monotherapy patients, 32 (20%) had the LCS6-variant, and of the combination treatment patients, 75 (22%) had the LCS6-variant (NS). There were also no significant differences in tumor acquired KRAS and BRAF mutations between patients that received anti-EGFR moAbs monotherapy versus combination therapy (Supplemental Table S2).
The median PFS of the whole monotherapy treated patients’ population was 10.43 weeks (95% CI: 7.73-13.12 weeks). There was a statistically significant benefit of monotherapy for the LCS6-variant patients vs non-variant patients, with a PFS of 16.86 weeks (95% CI: 10.2-23.51 weeks) versus 7.85 weeks (95% CI: 3.897-11.817 weeks) (Figure 1A) (p = 0.019, log-rank test). The median PFS of the whole combination therapy patients’ population was 18 weeks (95% CI: 15.87-20.12 weeks), and there was no statistically significant difference observed between the LCS6-variant versus non-variant patients (18 weeks (95% CI: 9.97-26.02 weeks vs 18.43 weeks (95% CI: 16.16-20.69 weeks) vs) (Figure 1B) (p = 0.760, log-rank test). There was strong evidence for an interaction effect for PFS (p=0.051).
Figure 1. LCS6-variant patients have improved PFS vs non-variant patients in response to EGFR moAbs monotherapy for all patients, with no benefit of additional chemotherapy.
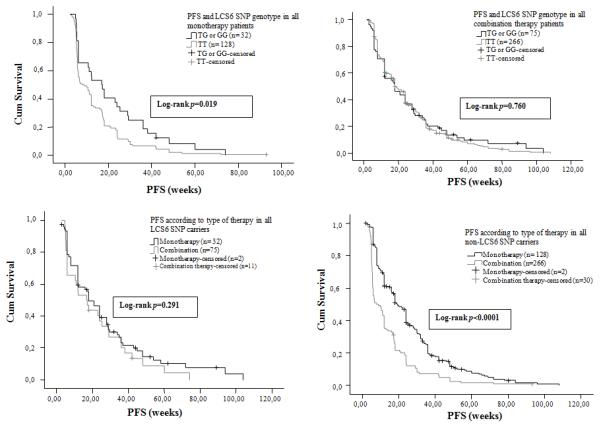
(A) Median progression-free survival (PFS) by KRAS LCS6 genotype in all patients treated with anti-EGFR moAbs monotherapy as salvage treatment. (B) Median PFS according to the KRAS 3′-UTR LCS6 SNP genotype status in all patients treated with anti-EGFR moAbs based combination chemotherapy as salvage treatment. (C) Median PFS according to type of therapy in all KRAS 3′-UTR LCS6 SNP carriers. (D) Median PFS according to type of therapy in all non-KRAS 3′-UTR LCS6 SNP carriers.
Interestingly, there was no improved PFS for LCS6-variant patients that received moAbs therapy [16.86 weeks, (95% CI: 8.55-25.18 weeks)] versus combination therapy [18 weeks, (95% CI: 13.37-22.64 weeks)] (Figure 1C) (p = 0.291, log-rank test). In contrast, there was a significant benefit in PFS with the addition of chemotherapy for non-variant patients [p < 0.0001, log-rank test], 7.86 weeks for monotherapy (95% CI: 3.9-11.82 weeks) versus 19.29 weeks for combination therapy (95% CI: 17-21.58 weeks) (Figure 1D). Of note, there was no significant difference in median PFS for monotherapy treated LCS6-variant patients versus combination treated non-variant patients.
In the double (KRAS and BRAF) wt patients’ population the median PFS of the monotherapy treated patients was 12 weeks (95% CI: 8.38-15.61 weeks), and again a statistically significant difference was observed between non-variant patients and LCS6-variant patients (10.43 weeks (95% CI: 6.74-14.11 weeks) versus 18 weeks (95% CI: 5.16-30.83 weeks)) (Figure 2A) (p = 0.039, log-rank test). The PFS for the combination therapy treated patients was 28.71 weeks (95% CI: 24.98-32.43 weeks), and no statistically significant difference (p = 0.39, log-rank test) was observed between the non-variant patients and LCS6-variant patients (28.3 weeks (95% CI: 24.15-32.45 weeks) versus 28.85 weeks (95% CI: 14.82-42.87 weeks)) (Figure 2B). Here again, there was no significant improvement (p = 0.061, log-rank test) between PFS for LCS6-variant patients that received moAbs monotherapy [18 weeks, (95% CI: 5.1-30.8 weeks)] versus combination therapy [28.85 weeks, (95% CI: 14.83-42.87 weeks)] (Figure 2C), while there was for non-variant patients [p < 0.0001, log-rank test, PFS for moAbs monotherapy 10.43 weeks, (95% CI: 6.75-14.15 weeks) versus combination therapy 28 weeks, (95% CI: 24.1-31.8 weeks) (Figure 2D). Again, there was no significant difference in median PFS between LCS6-variant patients receiving moAbs monotherapy and non-variant patients receiving combination therapy (18 versus 28.8 weeks).
Figure 2. LCS6-variant patients have improved PFS vs non-variant patients in response to EGFR moAbs monotherapy for double wt patients, with no benefit of additional chemotherapy.
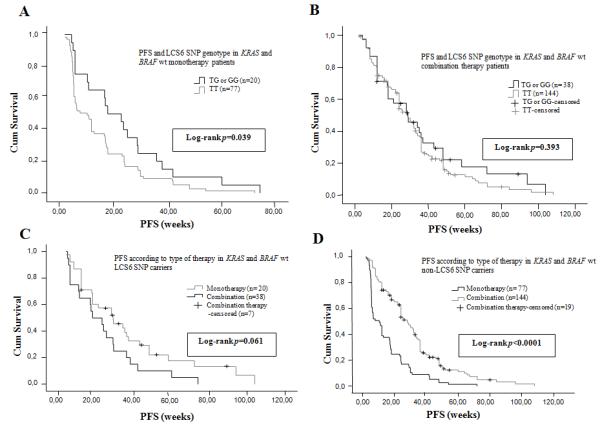
(A) Median PFS according to the KRAS 3′-UTR LCS6 SNP genotype status in the double (KRAS and BRAF) wt patients’ population treated with anti-EGFR moAbs monotherapy as salvage treatment. (B) Median PFS according to the KRAS 3′-UTR LCS6 SNP genotype status in the double (KRAS and BRAF) wt patients’ population treated with anti-EGFR moAbs based combination chemotherapy as salvage treatment. (C) Median PFS according to type of therapy in the double (KRAS and BRAF) wt KRAS 3′-UTR LCS6 SNP carriers. (D) Median PFS according to type of therapy in the double (KRAS and BRAF) wt non-KRAS 3′-UTR LCS6 SNP carriers.
Overall survival analysis correlated with treatment
The median OS of the whole monotherapy patients’ population was 33.14 weeks (95% CI: 26.70-39.57 weeks) and no statistically significant difference (p = 0.139, log-rank test) was observed between the non-variant patients and the LCS6-variant patients (28.85 weeks (95% CI: 22.53-35.18 weeks) versus 45 weeks (95% CI: 35.02-54.97 weeks)) (Figure 3A). The median OS of the whole combination therapy patients’ population was 44 weeks (95% CI: 40.11-47.88 weeks) and no statistically significant difference (p = 0.759, log-rank test) was observed between the non-variant patients and the LCS6-variant patients (44 weeks (95% CI: 40.06-47.93 weeks) versus 43 weeks (95% CI: 29.8-56.2 weeks)) (Figure 3B). For OS the interaction term was clearly non-significant (p=0.248).
Figure 3. LCS6-variant patients do not have improved OS with the addition of chemotherapy for all patients.
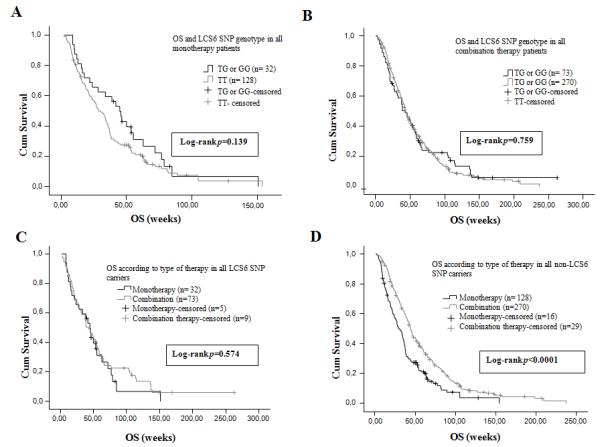
(A) Median overall survival (OS) according to the KRAS 3′-UTR LCS6 SNP genotype status in all patients treated with anti-EGFR moAbs monotherapy as salvage treatment. (B) Median OS according to the KRAS 3′-UTR LCS6 SNP genotype status in all patients treated with anti-EGFR moAbs based combination chemotherapy as salvage treatment. (C) Median OS according to type of therapy in all KRAS 3′-UTR LCS6 SNP carriers. (D) Median OS according to type of therapy in all non-KRAS 3′-UTR LCS6 SNP carriers.
There was no significant difference in OS for LCS6-variant patients that received moAbs monotherapy [45 weeks, (95% CI: 35-55 weeks)] versus combination therapy [43 weeks, (95% CI: 29.8-56.2 weeks)] (Figure 3C) (p = 0.574, log-rank test), yet there was a significant benefit for OS with the addition of chemotherapy for non-variant patients [moAbs monotherapy 28.86 weeks, (95% CI: 22.53-35.18 weeks) versus combination therapy 44 weeks, (95% CI: 40-47.93 weeks)] (p < 0.0001, log-rank test) (Figure 3D). Again, the difference in OS for LCS6-variant patients receiving monotherapy and non-variant patients receiving combination therapy was non-significant.
In the double (KRAS and BRAF) tumor wt patients’ population the median OS of the monotherapy patients was 37 weeks (95% CI: 30.82-43.17 weeks). A trend towards a statistically significant difference was observed between the non-variant patients, 35.71 weeks (95% CI: 32.03-39.4 weeks), and the LCS6-variant patients, 55.43 weeks (95% CI: 36.98-73.87 weeks) (Figure 4A) (p = 0.087, log-rank test). In this population, the median OS of the combination therapy patients was 55 weeks (95% CI: 48.3-61.7 weeks), and there was no statistically significant difference (p = 0.649, log-rank test) between the non-variant patients, 57 weeks (95% CI: 49.4-64.6 weeks), and the LCS6-variant patients, 54 weeks (95% CI: 45.46-62.53 weeks) (Figure 4B). Again, there was no significant improvement (p = 0.705, log-rank test) between OS for LCS6-variant patients that received moAbs monotherapy [55.43 weeks, (95% CI: 37-73.87 weeks)] versus combination therapy [54 weeks, (95% CI: 45.47-62.54 weeks)] (Figure 4C), while there was for non-variant patients [p < 0.0001, log-rank test, OS for moAbs monotherapy 35.71 weeks, (95% CI: 32-39.4 weeks) versus combination therapy 57 weeks, (95% CI: 49.4-64.6 weeks) (Figure 4D). Again, the difference in OS for LCS6 G variant patients receiving monotherapy and KRAS wt patients receiving combination therapy was non-significant.
Figure 4. LCS6-variant patients do not have improved OS with the addition of chemotherapy for double wt patients.
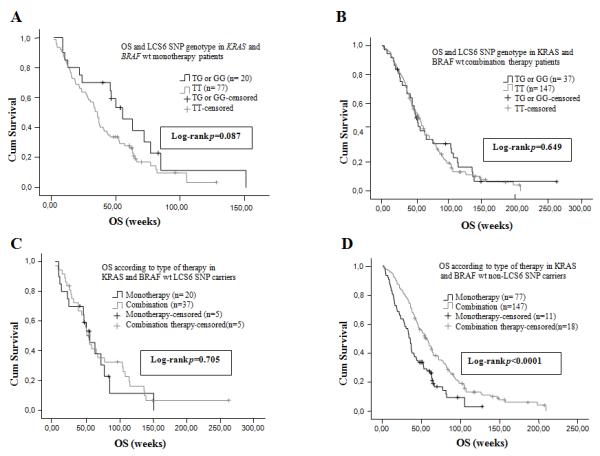
(A) Median OS according to the KRAS 3′-UTR LCS6 SNP genotype status in the double (KRAS and BRAF) wt patients’ population treated with anti-EGFR moAbs monotherapy as salvage treatment. (B) Median OS according to the KRAS 3′-UTR LCS6 SNP genotype status in the double (KRAS and BRAF) wt patients’ population treated with anti-EGFR moAbs based combination chemotherapy as salvage treatment. (C) Median OS according to type of therapy in the double (KRAS and BRAF) wt KRAS 3′-UTR LCS6 SNP carriers. (D) Median OS according to type of therapy in the double (KRAS and BRAF) wt non-KRAS 3′-UTR LCS6 SNP carriers.
The LCS6-variant is not prognostic in KRAS and BRAF mutated patients
In the KRAS and BRAF mutated patients’ population no statistical significant differences regarding PFS and OS were observed in patients treated with either anti-EGFR moAbs monotherapy or with moAbs in combination with chemotherapy (data not shown). Median PFS times were identical between LCS6-variant and non-variant patients, with no significant improvement (p = 0.641, log-rank test) between PFS for LCS6-variant patients that received moAbs monotherapy [6 weeks, (95% CI: 0-13.25 weeks)] versus combination therapy [12 weeks, (95% CI: 6.45-17.56 weeks)] (Supplemental Figure S2A). However, there was a significant improvement in PFS for non-variant patients treated with combination therapy [p < 0.0001, log-rank test, PFS for moAbs monotherapy 6 weeks, (95% CI: 4.46-7.53 weeks) versus combination therapy 12 weeks (95% CI: 9.72-14.28 weeks)] (Supplemental Figure S2B).
Likewise, for OS, there was no significant difference (p = 0.303, log-rank test) between OS for LCS6-variant patients that received moAbs monotherapy [28.43 weeks, (95% CI: 9.47-47.39 weeks)] versus combination therapy [23 weeks, (95% CI: 10.8-35.19 weeks)] (Supplemental Figure S2C), while there was for non-variant patients [p = 0.002, log-rank test, OS for moAbs monotherapy 21.29 weeks, (95% CI: 15-27.55 weeks) versus combination therapy 31 weeks, (95% CI: 25.65-36.34 weeks) (Supplemental Figure S2D).
The LCS6-variant and response to moAbs therapy
From the whole population of 483 patients that were evaluable for both response and LCS6-variant genotyping, 147 (30.4%) had received anti-EGFR moAbs as monotherapy and 336 (69.6%) with multiple chemotherapy combinations. In the monotherapy group, 123 (83.6%) patients were non-responders (stable disease (SD) and progressive disease (PD)), and 24 (16.4%) were responders (partial response (PR) and complete response (CR)). There were significantly more LCS6-variant patients in the monotherapy responders group versus the non-responders group (11/24 versus 19/123)(Fisher’s exact test p=0.002). In the combination with chemotherapy group, 252 (75%) patients were non-responders (SD and PD) and 84 (25%) were responders (PR and CR). There was no statistically significant difference between the proportion of the non-variant and LCS6-variant patients in these groups (Fisher’s exact test p=1).
In the 270 double (KRAS and BRAF) wt populations, 90 (33.3%) received anti-EGFR moAbs as monotherapy and 180 (66.6%) with multiple chemotherapy combinations. In the monotherapy group, 71 (78.8%) patients were non-responders (SD and PD), and 19 (21.2%) were responders (PR and CR). Again there were significantly more LCS6-variant patients in the responders versus the non-responders groups (9/19 versus 11/71)(Fisher’s exact test p=0.010). In the combination with chemotherapy group, 102 (56.6%) patients were nonresponders (SD and PD) and 78 (43.4%) were responders (PR and CR). There was no statistically significant difference between the proportion of non-variant and LCS6-variant patients in the groups (Fisher’s exact test p=1).
The affect of the LCS6-variant on gene expression upon therapy exposure
Because patients with the LCS6-variant in this analysis were sensitive to EGFR moAbs monotherapy, it suggested that they are not EGFR-independent, as tumor-acquired KRAS mutant patients are (30). To better understand how these patients may respond differently to moAbs monotherapy, we created dual-luciferase reporters containing the entire KRAS 3′UTR with the LCS6-variant (G-allele) or without the variant (T-allele). We used this system to test the hypothesis that moAbs therapy and chemotherapy may differentially impact expression of KRAS for those with the LCS6-variant allele versus the non-variant allele.
We found, as has been previously reported in other cell lines (36), that the LCS6-variant allele (G-allele reporter) displayed 1.8-fold higher expression at baseline in HCT-116 colon cancer cells when compared to the non-variant allele (T-allele reporter, Figure 5A). This finding supports previous evidence that this is a functional mutation that permits KRAS overexpression in tumors. Next, we exposed these cells to cetuximab, and found that while there was no impact on KRAS expression in the non-variant allele reporter system, there was a significant increase in the overexpression of KRAS for the LCS6-variant allele reporter system. We found similar increased overexpression of KRAS for the LCS6-variant allele with exposure to 5-FU. In contrast, we saw little to no change in expression of the LCS6-variant allele compared to non-variant allele with exposure to Irinotecan (Figure 5B). These results indicate that the LCS6-variant allele leads to KRAS protein overexpression in response to specific chemotherapy treatments as well as MoAbs therapy, a finding not seen in the presence of the non-variant allele.
Figure 5. The KRAS LCS6-variant causes over-expression of a KRAS reporter.
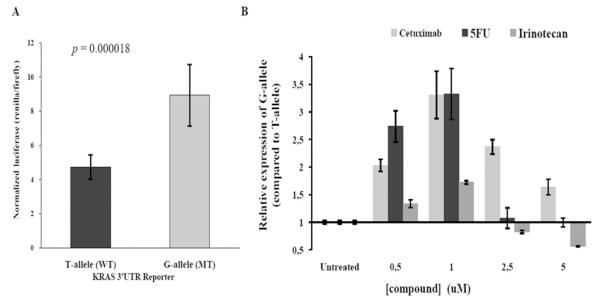
(A) HCT-116 colon cancer cells were transfected with dual-luciferase reporters harboring either the full-length KRAS 3′UTR T-allele or G-allele (LCS6-variant), as indicated. Dual-luciferase activities were measured and renilla was normalized to firefly. Results are graphed as the mean and standard deviation of the mean of four independent experiments preformed in duplicate. The p-value was calculated using the students t-test. (B) The KRAS LCS6-variant exhibits altered gene expression in response to anti-cancer agents. HCT-116 colon cancer cells transfected with either the KRAS LCS6-variant or non-variant 3′UTR reporters were exposed to various concentrations of anti-cancer compounds (as indicated). The results are expressed as the relative expression of the LCS-variant versus the non-variant allele. Graphed is the mean and standard deviation of the mean for two experiments performed in duplicate.
Discussion
Here we have shown a statistically significant improvement in median PFS for all LCS6-variant mCRC patients treated with anti-EGFR moAbs monotherapy. In fact, we find a favorable prognosis for LCS6-variant patients across all cohorts studied in response to anti-EGFR moAbs, including in KRAS or BRAF mutant patients. This improved prognosis is not enhanced by the addition of chemotherapy, and in fact, LCS6-variant patients appeared to experience no benefit from the addition of chemotherapy to anti-EGFR moAbs therapy. This finding is in contrast to non-variant patients, who derived a significant benefit from the addition of chemotherapy to anti-EGFR moAbs across all cohorts, and only then achieved comparable outcomes as those of LCS6-variant patients. This clinical finding was supported by cell line studies indicating that the LCS6-variant allele responds differently than the non-variant allele in response to chemotherapy and anti-EGFR moAbs exposure, with increased expression and likely further dependence on the KRAS pathway (47).
A different distribution of the LCS6 genotypes according to the KRAS and BRAF mutational status was observed in our mCRC patients’ population than that observed in prior reports. LCS6-variant patients were equally likely to have acquired KRAS tumor mutations as not, but, LCS6-variant patients were significantly more likely to be in the BRAF mutated group. In a previously studied mCRC population similar to ours, Graziano et al (41) found an increased prevalence of the LCS6-variant in the KRAS mutant, but not in BRAF mutant patients (41). While one explanation for our different results could be that we used tumor DNA for the majority of testing, this seems unlikely, since, it has previously been well documented that the genotype of normal and tumor tissue is the same in LCS6-variant patients (36). Another hypothesis could be that in the later stages of CRC carcinogenesis, the LCS6-variant allele mediates the selection of less differentiated and more aggressive clones that harbor BRAF mutations, and perhaps our cohort was more advanced. Additionally, there could be a selective pressure to develop KRAS or BRAF mutations in the presence of the LCS6-variant, depending on exposure to specific therapies, and prior therapy likely differed between our two studies.
The finding that this single base pair change in the 3′UTR of KRAS leads to a significant difference in both baseline expression as well as response to chemotherapy in a luciferase reporter construct is intriguing. While tumor acquired KRAS mutations are always turned on, these cell line reporter studies further indicate how fundamentally different this mutation is than a simple tumor acquired KRAS mutation. By their nature, miRNA-binding disrupting mutations, such as the KRAS LCS6-variant, are dependent on trans-activating factors, such as miRNAs, that change in response to stress. It has been know for several years that miRNAs are used to dynamically regulate the response to cytotoxic cancer therapy (33). It is perhaps not surprising therefore that a mutation such as the LCS6-variant would be predictive of cancer treatment response, as cancer treatments will lead to changes in the very factors that regulate the mutation, and subsequent downstream gene and pathway expression. However, further molecular studies of the exact mechanisms by which this mutation alters response to EGFR moAbs treatment are still required in tissue and animal models.
Recently, two large studies of colon cancer patients investigating outcome, found that the LCS6 variant allele predicted a good prognosis, especially when in combination with tumor acquired mutations in KRAS, in both early (48) and late stage (49) patients. These authors hypothesized that at least in early stage colon cancer, the LCS6-variant plus KRAS mutations could lead to too much KRAS, and tumor cell senescence. Based on our cell line data, indicating that anti-EGFR moAbs monotherapy leads to significantly higher KRAS expression, as does 5FU, but not Irinotecan, we hypothesize that this may be a viable explanation of the very favorable anti-EGFR moAbs monotherapy response in advanced KRAS LSC6-variant patients as well. It does further support the hypothesis that the combination of therapy delivered with anti-EGFR moAbs monotherapy is critical, as there appears to be no benefit of additional chemotherapy in our study, and in fact chemotherapy could possibly be a detriment to LCS6-variant mCRC patients.
As also true for other cancers, an important step in the development of CRC, seems to be the deregulation of miRNAs. Over the past few years miRNAs have been brought to the central stage of molecular oncology and have substantially changed the way we view and understand gene regulation (50). The KRAS LCS6-variant was the first mutation in a miRNA binding site to be implicated in cancer risk, and while it certainly will not be the last (36), it appears to also play a significant predictive role that could guide therapy decisions. Our findings here suggest that patients carrying the LCS6-variant are biologically different then non-variant patients, have a higher probability of benefit from anti-EGFR moAbs monotherapy, and deserve prospective clinical studies to determine what, if anything, they should receive in addition to cetuximab treatment in the mCRC setting.
Supplementary Material
Translational relevance.
The KRAS-variant (LCS6-variant or rs61764370) represents a new type of germ-line mutation, which is located in the non-protein coding region (the 3′ untranslated region), of the KRAS oncogene. This mutation disrupts binding of the let-7 microRNA (miRNA) to KRAS, and leads to KRAS and downstream cellular pathway signaling disruption, oncogenesis, and altered tumor biology. The KRAS-variant has been found to be a strong biomarker of treatment response in many cancers, yet appears to act fundamentally differently than tumor acquired KRAS mutations. Here we show that this mutation predicts a positive response to anti-EGFR monoclonal Antibody therapy (moAbs) in a large cohort of metastatic colorectal cancer (mCRC) patients, without any benefit of additional chemotherapy for these patients. Cell line reporter studies also shed light on the functional biology of this mutation. These findings bring this powerful mutation one step closer to helping direct therapy for mCRC patients, allowing improved outcome and personalized treatment.
Acknowledgements
ZS was a recipient of a research fellowship from the Hellenic Society of Medical Oncology (Hesmo), JBW was supported by two R01 Grants, CA131301-04 and CA157749-01A1.
Footnotes
Potential conflicts of interest: JBW is a discoverer and listed as an inventor on the patent by Yale University of the KRAS-variant, and has founded a company that has licensed this patent from Yale University; H-JL has served as an advisor for Merck Serono and BMS, has received honoraria from Merck Serono and clinical trial support from Merck Serono and BMS; ZS has received honoraria from Amgen; ST has received honoraria and research funding from Merck Serono; HP has received honoraria from Merck Serono; PL-P has been a consultant for Merck Serono and Amgen, received grants from Merck Serono and Myriad Genetics, and has travel and accommodations expenses covered or reimbursed by Merck Serono and Amgen. All the other authors declare no conflict of interest.
References
- 1.Siegel R, Desantis C, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:104–17. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 3.Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. N Engl J Med. 2000;343:905–14. doi: 10.1056/NEJM200009283431302. [DOI] [PubMed] [Google Scholar]
- 4.Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 5.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–64. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 6.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 7.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–15. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 8.Lievre A, Bachet J-B, Boige V, Cayre A, Le Corre D, Buc E, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–9. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 9.Sartore-Bianchi A, Moroni M, Veronese S, Carnaghi C, Bajetta E, Luppi G, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007;25:3238–45. doi: 10.1200/JCO.2007.11.5956. [DOI] [PubMed] [Google Scholar]
- 10.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–71. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 11.Douillard J-Y, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 12.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 13.Van Cutsem E, Kohne C-H, Hitre E, Zaluski J, Chang Chien C-R, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 14.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 15.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–6. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 16.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 17.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 18.Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet J-B, Lecomte T, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27:5924–30. doi: 10.1200/JCO.2008.21.6796. [DOI] [PubMed] [Google Scholar]
- 19.Saridaki Z, Tzardi M, Papadaki C, Sfakianaki M, Pega F, Kalikaki A, et al. Impact of KRAS, BRAF, PIK3CA mutations, PTEN, AREG, EREG expression and skin rash in >/= 2 line cetuximab-based therapy of colorectal cancer patients. PLoS One. 2011;6:e15980. doi: 10.1371/journal.pone.0015980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Souglakos J, Philips J, Wang R, Marwah S, Silver M, Tzardi M, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer. 2009;101:465–72. doi: 10.1038/sj.bjc.6605164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol. 2009;27:1477–84. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15:3184–8. doi: 10.1158/1078-0432.CCR-08-2961. [DOI] [PubMed] [Google Scholar]
- 23.Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851–7. doi: 10.1158/0008-5472.CAN-08-2466. [DOI] [PubMed] [Google Scholar]
- 24.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 25.Hollestelle A, Pelletier C, Hooning M, Crepin E, Schutte M, Look M, et al. Prevalence of the variant allele rs61764370 T>G in the 3′UTR of KRAS among Dutch BRCA1, BRCA2 and non-BRCA1/BRCA2 breast cancer families. Breast Cancer Res Treat. 2011;128:79–84. doi: 10.1007/s10549-010-1080-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy SDN, Gajula RP, Pakala SB, Kumar R. MicroRNAs and cancer therapy: the next wave or here to stay? Cancer Biol Ther. 2010;9:479–82. doi: 10.4161/cbt.9.7.11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faber C, Kirchner T, Hlubek F. The impact of microRNAs on colorectal cancer. Virchows Arch. 2009;454:359–67. doi: 10.1007/s00428-009-0751-9. [DOI] [PubMed] [Google Scholar]
- 28.Landi D, Gemignani F, Naccarati A, Pardini B, Vodicka P, Vodickova L, et al. Polymorphisms within micro-RNA-binding sites and risk of sporadic colorectal cancer. Carcinogenesis. 2008;29:579–84. doi: 10.1093/carcin/bgm304. [DOI] [PubMed] [Google Scholar]
- 29.Chen K, Song F, Calin GA, Wei Q, Hao X, Zhang W. Polymorphisms in microRNA targets: a gold mine for molecular epidemiology. Carcinogenesis. 2008;29:1306–11. doi: 10.1093/carcin/bgn116. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Winder T, Ning Y, Pohl A, Yang D, Kahn M, et al. A let-7 microRNA-binding site polymorphism in 3′-untranslated region of KRAS gene predicts response in wild-type KRAS patients with metastatic colorectal cancer treated with cetuximab monotherapy. Ann Oncol. 2011;22:104–9. doi: 10.1093/annonc/mdq315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mishra PJ, Bertino JR. MicroRNA polymorphisms: the future of pharmacogenomics, molecular epidemiology and individualized medicine. Pharmacogenomics. 2009;10:399–416. doi: 10.2217/14622416.10.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sethupathy P, Collins FS. MicroRNA target site polymorphisms and human disease. Trends Genet. 2008;24:489–97. doi: 10.1016/j.tig.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, et al. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111–6. doi: 10.1158/0008-5472.CAN-07-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jerome T, Laurie P, Louis B, Pierre C. Enjoy the Silence: The Story of let-7 MicroRNA and Cancer. Curr Genomics. 2007;8:229–33. doi: 10.2174/138920207781386933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–47. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 36.Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–40. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paranjape T, Heneghan H, Lindner R, Keane FK, Hoffman A, Hollestelle A, et al. A 3′-untranslated region KRAS variant and triple-negative breast cancer: a case-control and genetic analysis. Lancet Oncol. 2011;12:377–86. doi: 10.1016/S1470-2045(11)70044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pharoah PD, Palmieri RT, Ramus SJ, Gayther SA, Andrulis IL, Anton-Culver H, et al. The role of KRAS rs61764370 in invasive epithelial ovarian cancer: implications for clinical testing. Clin Cancer Res. 2011;17:3742–50. doi: 10.1158/1078-0432.CCR-10-3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratner E, Lu L, Boeke M, Barnett R, Nallur S, Chin LJ, et al. A KRAS-variant in ovarian cancer acts as a genetic marker of cancer risk. Cancer Res. 2010;70:6509–15. doi: 10.1158/0008-5472.CAN-10-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ratner ES, Keane FK, Lindner R, Tassi RA, Paranjape T, Glasgow M, et al. A KRAS variant is a biomarker of poor outcome, platinum chemotherapy resistance and a potential target for therapy in ovarian cancer. Oncogene. 2012;31:4559–66. doi: 10.1038/onc.2011.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graziano F, Canestrari E, Loupakis F, Ruzzo A, Galluccio N, Santini D, et al. Genetic modulation of the Let-7 microRNA binding to KRAS 3′-untranslated region and survival of metastatic colorectal cancer patients treated with salvage cetuximab-irinotecan. Pharmacogenomics J. 2010;10:458–64. doi: 10.1038/tpj.2010.9. [DOI] [PubMed] [Google Scholar]
- 42.Kjersem JB, Ikdahl T, Guren T, Skovlund E, Sorbye H, Hamfjord J, et al. Let-7 miRNA-binding site polymorphism in the KRAS 3′UTR; colorectal cancer screening population prevalence and influence on clinical outcome in patients with metastatic colorectal cancer treated with 5-fluorouracil and oxaliplatin +/− cetuximab. BMC Cancer. 2012;12:534. doi: 10.1186/1471-2407-12-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sebio A, Pare L, Paez D, Salazar J, Gonzalez A, Sala N, et al. The LCS6 polymorphism in the binding site of let-7 microRNA to the KRAS 3′-untranslated region: its role in the efficacy of anti-EGFR-based therapy in metastatic colorectal cancer patients. Pharmacogenet Genomics. 2013;23:142–7. doi: 10.1097/FPC.0b013e32835d9b0b. [DOI] [PubMed] [Google Scholar]
- 44.Winder T, Zhang W, Khoueiry AE, Yang D, Pohl A, Lurje G, et al. Association of a germ-line variant in the K-ras 3′ untranslated region with response and progression-free survival in patients with mCRC treated with single-agent cetuximab (IMCL-0144) or in combination with cetuximab (EPIC) independent of K-ras mutation status. ASCO Annual Meeting Proceedings (Post-Meeting Edition).2009. [Google Scholar]
- 45.Lenz HJ, Van Cutsem E, Khambata-Ford S, Mayer RJ, Gold P, Stella P, et al. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J Clin Oncol. 2006;24:4914–21. doi: 10.1200/JCO.2006.06.7595. [DOI] [PubMed] [Google Scholar]
- 46.Thwaites SE, Gurung B, Yao J, Kable K, Robertson P, Ryan BJ, et al. Excellent outcomes of simultaneous pancreas kidney transplantation in patients from rural and urban Australia: a national service experience. Transplantation. 2012;94:1230–5. doi: 10.1097/TP.0b013e3182708e04. [DOI] [PubMed] [Google Scholar]
- 47.Paranjape T, Slack FJ, Weidhaas JB. MicroRNAs: tools for cancer diagnostics. Gut. 2009;58:1546–54. doi: 10.1136/gut.2009.179531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smits KM, Paranjape T, Nallur S, Wouters KAD, Weijenberg MP, Schouten LJ, et al. A let-7 microRNA SNP in the KRAS 3′UTR is prognostic in early-stage colorectal cancer. Clin Cancer Res. 2011;17:7723–31. doi: 10.1158/1078-0432.CCR-11-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan BM, Robles AI, Harris CC. KRAS-LCS6 genotype as a prognostic marker in early-stage CRC--letter. Clin Cancer Res. 2012;18:3487–8. doi: 10.1158/1078-0432.CCR-12-0250. author reply 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slaby O, Svoboda M, Michalek J, Vyzula R. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Mol Cancer. 2009;8:102–107. doi: 10.1186/1476-4598-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.