

1. INTRODUCTION
The amide bond is one of the most important functional groups in chemistry and biology.1 Due to the strong resonance interaction between nN and π*C=O orbitals (Figure 1),2 the vast majority of amides are planar; however, some deviations from planarity have been often observed in peptides3–14 and small molecules alike.15–34 Other notable properties of amides resulting from the amide bond resonance1,2 include (i) relatively short N–C(O) bonds, (ii) high rotational barrier for cis-trans isomerization (typically 15–20 kcal/mol), (iii) resistance of the carbonyl group towards nucleophilic addition and hydrolysis, (iv) minimized ability to engage in coordination at the nitrogen atom, and (v) lower C=O infrared stretching frequencies and more upfield shifts in 13C NMR spectra as compared to other carboxylic acid derivatives. Perhaps because the planar geometry of amides is such an intrinsic part of how we understand amide chemistry, distorted amides that deviate from these norms have attracted the widespread attention of organic chemists.3–59 To do so, a number of methods to modify the spatial arrangement of substituents around the amide bond have been devised in the last two decades (Figure 2). They include (i) steric repulsion,11–34 (ii) conformational effects (ring or allylic strain),35–43 (iii) electronic delocalization (as manifested in amides of XXN–C(O) type where X is an electronegative substituent),44–54 and (iv) steric restriction55–59. Of these four classes, geometrically-restricted amides (bridged, twisted amides)55–59 are particularly interesting because these amides do not necessarily suffer from excessive steric hindrance around the amide bond11–43or the severe electronic influence of neighboring substituents.44–54 Moreover, bridged amides can be more easily modified and diversified when compared to other classes of distorted amides,59 and thus represent one of the most straightforward and wide-ranging methods of constraining an amide bond into a non-planar conformation.
Figure 1.

Resonance descriptions of the amide bond.
Figure 2.

Types of distorted amide bonds: a) steric repulsion; b) conformational effects; c) anomeric amides; d) steric restriction.
In this article, we will provide a comprehensive survey of the synthesis and reactivity of bridged lactams that have been published between 1938 – when Lukeš proposed for the first time60 that incorporating the amide bond nitrogen atom into a bridgehead position in a bicyclic system would result in a distortion of the resulting bond – until 2012. Owing to the fact that the lone pair of electrons of the nitrogen atom is no longer in conjugation with the adjacent π orbitals of the carbonyl group,1,2 bridged amides have properties that differ from those of planar amides. These include enhanced reactivity toward amide bond hydrolysis61–66 and toward nucleophilic attack at the carbonyl atom,67–69 different regiochemistry of amide protonation and alkylation reactions,70–74 different spectroscopic16–18,75 and physical properties.76–83 Besides the insight that such compounds provide into the nature of the amide bond, twisted amides are analogous to the transition state encountered by peptides as they undergo cis-trans isomerisation,84–87 a critical feature of protein folding.
Bridged lactams have been the subject of previous reviews,55–59 however a comprehensive review on this topic has not been published prior to this work. In contrast, other strained molecules88,89 including sterically-hindered amides,90–94 anomeric amides95–97 and anti-Bredt olefins,98–105 have been frequently reviewed in recent years.
The review is arranged by the type of bridged lactams and their method of synthesis. We limited the scope of the review to compounds in which the overall sum of carbon atoms forming the core 1-azabicyclo scaffold is less than or equal to ten. Amides contained in more flexible systems generally have properties analogous to planar amide bonds despite their bridged structures. Some derivatives of bridged lactams (bridged enamines, iminium ions, and sultams) featuring the relevant bond at the bridgehead position in 1-azabicyclo scaffold are also covered. Due to the geometric restriction, these compounds can differ in chemical properties from their planar counterparts in a manner similar to bridged/planar amides.
The general structures of bridged lactams and their derivatives that will be reviewed are presented in Figure 3. Bridged lactams are divided depending on the placement of N–C(O) bond on either one-carbon (3) or larger bridge (2). Such division is justified by different chemical properties of these two classes of lactams and supported by distinct methods of their synthesis. In general, bridged lactams of the type 3 are more strained and more reactive than their analogs 2. Furthermore, quinuclidone and admanatanone-derived amides are presented at the beginning of the review because of their historical importance to the advances in the field of bridged lactams, and also because these two classes of compounds provided some of the most strained amide bonds reported to date. The section focusing on the reactivity of bridged lactams emphasizes that the chemical properties of these compounds differ from those of standard amides. In particular, we discuss the hydrolytic behavior, reactivity of the nitrogen atom, reactivity of the carbonyl group, chemical properties of derivatives of bridged lactams. Finally, the specific application of bridged lactams in natural product synthesis and a selection of natural products containing bridged amide bonds are presented.
Figure 3.

Scope of the review: a) types of bridged lactams; b) heterocycles with 1-aza-bridged scaffold.
We hope that this review will serve as a useful reference for chemists involved in probing the effect of amide bond geometry on chemical and biological properties of amides, and those interested in using twisted amides as tools in physical, organic and biological chemistry.
2. GENERAL PROPERTIES OF BRIDGED LACTAMS
2.1. Distortion Parameters of Bridged Lactams
In 1971, Winkler and Dunitz introduced three independent parameters to quantitatively define distortion of amide bonds: twist angle (τ), pyramidalization at nitrogen (χN), and pyramidalization at carbon (χC) (Figure 4).106 Twist angle describes the magnitude of rotation around the N–C(O) bond, while χN and χC define the tetrahedralcharacter of the nitrogen and carbon atom, respectively. A twist angle of 0° corresponds to a planar amide bond and of 90° corresponds to a fully orthogonal bond; χN and χC are 0° for planar bonds, and 60° for fully pyramidalized amide bonds.
Figure 4.

Winkler-Dunitz distortion parameters of amide bonds.
To date, two bridged lactams with perfectly perpendicular amide bonds have been structurally characterized (see 52,107 Scheme 7 and 78,108 Scheme 16). The remaining bridged lactams display distorted geometries of amide bonds characterized by medium τ and χN values. Importantly, for all reported bridged lactams, the χC values are close to 0° regardless of the degree of distortion. This tendency reflects the predominant contribution of the amino ketone form to the resonance structure of amides (Figure 1, A and B).109,110 In this context it is important to note the work by Wiberg and coworkers on the thermochemical stability of amides arising from the third major resonance contributor (Figure 1, C) in which the resonance overlap takes place primarily between carbon and nitrogen with little π electron transfer to oxygen.76,109,110 Here, we will discuss distortion parameters of specific amides in the section focused on the synthesis of bridged lactams. Moreover, since a large number of bridged lactams with varying Winkler-Dunitz parameters have been characterized, these structurally-defined analogues provide an accurate gauge of the degree of distortion in cases when the X-ray values are not available.
Scheme 7.

Synthesis of 2-Quinuclidonium Tetrafluoroborate by Tani and Stoltz
Scheme 16.

Synthesis of 1-Aza-2-adamantanone (“the Most Twisted Amide”) by Kirby
Besides Winkler-Dunitz parameters, distortion of amide bonds can be quantified by the sum of three bond angles at nitrogen (θ).39 For an ideally sp3 hybridized atom θ = 328.4°, while for an sp2 atom θ = 360.0°. In addition, Yamada has proposed a qualitative description of distorted amides based on twist angle and pyramidalization at nitrogen: type A amides with perpendicularly twisted N–C(O) bonds and non-pyramidalized nitrogen atoms, type B amides with planar N–C(O) bonds and sp3 nitrogen atoms, and type C amides with perpendicular N–C(O) bonds and pyramidalized nitrogen atoms.91,92
2.2. Bond Lengths of Bridged Lactams
Upon increased distortion of amide bonds, the length of N–C(O) bond significantly increases, while the C=O bond only slightly shortens.1,15,111 This tendency reflects significantly larger contribution of the amino ketone resonance form to the resonance structure of non-planar amides (Figure 1), and indicates a gradual pyramidalization of nitrogen occurring with rotation around the N–C(O) bond.109,110 As a specific example, in a perfectly perpendicular 1-aza-2-adamantanone (78)108 the N–C(O) bond of 1.475 Å is 0.15 Å longer than the corresponding bond in the planar N-methyl-δ-valerolactam (1.325 Å),111 while the C=O bond of 1.196 Å is 0.037 Å shorter the same bond in the N-methyl-δ-valerolactam (1.233 Å). In this review, the bond lengths of structurally-characterized bridged amides will be given together with their distortion parameters.
2.3. Spectroscopic Properties of Bridged Lactams
Infrared C=O stretching frequencies of amide bonds are sensitive to changes in the extent of resonance stabilization of the nitrogen lone pair, while carbonyl shifts in 13C NMR spectra of amides respond to changes in the charge density of the carbonyl carbon.112 Due to the limited resonance contribution of the zwitterionic form B (Figure 1), IR and 13C NMR spectra of bridged lactams16–18 are characterized by increased νC=O values and more downfield 13C=O resonances as compared to planar amides. In general, IR and 13C NMR values of amide bonds in bridged lactams appear in the region between those for isolated ketones and planar amides. For example, in the perfectly perpendicular 1-aza-2-adamantanone (78), the carbonyl group resonates at 1732 cm−1 IR, while the 13C NMR signal appears at 200.0 ppm.108 In this review, spectroscopic properties of bridged lactams will be discussed only in specific cases and the reader is suggested to consult the primary literature to obtain values of interest.
2.4. Analogy of Bridged Lactams to Bridgehead Olefins
Due to the partial double bond character, bridged amides have been frequently referred to as anti-Bredt lactams.55–59 In general, bridged lactams are more stable and easier to prepare than the corresponding bridgehead olefins because the amide nitrogen atom can adopt sp3 geometry in the amino ketone resonance form without violating the octet rule. In contrast, the only alternative resonance structures for anti-Bredt olefins do not possess closed-shell octets and represent high-energy species.98–105 The importance of the amino ketone resonance form in bridged lactams is manifested by low values of χC and the progressive shortening of the C=O bond with increased distortion of the amide bond.55–59
To allow prediction of stability and reactivity of bridgehead olefins, in 1981, Schleyer introduced “olefin strain” energy parameter113 (defined as the difference between the strain energy of the olefin and that of its parent hydrocarbon and directly related to the enthalpy of hydrogenation) as a guide to evaluate accessibility of bridgehead olefins under experimental conditions. Bridged olefins with olefin strain energy lower than 17 kcal/mol were suggested to be isolable, those with olefin strain energy between 17 and 21 kcal/mol classified as observable, and those with olefin strain energy higher than 21 kcal/mol were grouped as unstable.113 Despite outlined above differences between bridged amides and bridged olefins,114–116 Schleyer’s olefin strain energy provides a useful predictive tool for evaluating the stability and likelihood of isolation of bridged lactams. Figure 5 presents structures of several of the more common ring systems of bridged lactams, the year of their first synthesis, and the corresponding bridged olefins with their olefin strain energy as calculated by Schleyer.117–127 From comparison of these values, it is evident that even highly strained bridged lactams are stable enough for isolation (for example, compare bridged olefins 10, 11, 17 with bridged lactams 18, 19, 25). Furthermore, in the group of superficially similar bridged lactams 20–24, the structure 23 is predicted to be the least stable on the basis of the Schleyer's olefin strain energy; indeed, a successful synthesis of this type of bridged lactams has not been reported so far. In contrast, lactams represented by 20 and 24 have been extensively studied in recent years, which led to many insights into the properties of distorted amide bonds,55–59 in part, because of the stability of the parent lactam scaffolds.
Figure 5.

Comparison of anti-Bredt olefins and lactams: a) calculated olefin strain energy of bridged olefins by Schleyer; b) year of synthesis of the corresponding bridged lactam. (please use double-column format for Figure 5)
2.5. Chemical and Biological Significance of Distorted Amides
Non-planar amides have been frequently employed to investigate fundamental properties of amide bonds, such as proton exchange,70–72 bonding,128–131 rotational barriers,76–77 chemical reactivity.67–69 The effect of amide distortion has been leveraged to control chemical transformations132–140 including hydrolysis,61–66 acylation,132–134 desymmetrization135 and kinetic resolution136,137 of alcohols. Bridged lactams have been applied as intermediates in the synthesis of bioactive natural products141–192 and are even present in the structures of several alkaloids.193–207 Moreover, bridged lactams have been used as model systems208 for activation of traditionally inert C–N bonds209–213 in transition metal catalyzed processes.
The study of distorted amides also has important biological consequences. Twisted amides have been invoked in variety of enzymatic transformations including peptide hydrolysis,61,214–216 protein splicing217–220 and cis-trans isomerization of peptidyl-proline bonds.84–86 Inhibition of the latter process is of considerable interest in treatment of drug-resistant cancer cells221 and neurodegenerative diseases.222 Furthermore, bridged lactams have been utilized as models for activated peptide units223–225 in acylation of serine,226–228 aspartate229,230 and cysteine proteases,231 and reported as novel lead structures232–236 and as constrained analogues237–246 in medicinal chemistry. Finally, it is worth noting that distorted amide bonds, while not twisted per se, are key structural elements of penicillin and other β-lactam antibiotics.247,248
3.SYNTHESIS OF HISTORICALLY IMPORTANT BRIDGED LACTAMS
3.1.Quinuclidone Derivatives
In 1924, Julius Bredt formulated his famous rule, suggesting that bridgehead carbon-carbon double bonds in the camphene and pinene series would be incapable of existence due to the insufficient overlap between their π orbitals.249 Fourteen years later, in 1938, Rudolf Lukeš applied Bredt’s rule to the amide zwitterionic resonance structure, proposing that bicyclic bridged lactams featuring amide nitrogen atom at the bridgehead position would be “sterically impossible” (Scheme 1).60 Being unsuccessful in preparing bridged amides 27 and 29 by condensation under thermal conditions, Lukeš correctly predicted that if such amides were ever made they would exhibit properties of ketones rather than amides.60 At about the same time, R. B. Woodward attempted synthesis of related bicyclic 2-quinuclidones 31 (Scheme 1).250 Although Woodward was unsuccessful in his studies, he also concluded that such compounds would represent a new type of aminoketone (see his comments to this effect in a footnote included in Doering’s paper,251 discussed below).
Scheme 1.

Early Attempts of Synthesis of Bridged Lactams
In 1946, during studies on the autoxidation of quininone, Doering reported the first synthesis (but not isolation) of a bridged lactam (Scheme 2).251 Treatment of the potassium enolate of quninone 32 with molecular oxygen afforded quinic acid 33 and amino ester 34. The amino ester 34 was proposed to arise from a rapid alcoholysis of the corresponding bridged lactam 32c by tert-butanol, which was used as a solvent for the reaction. The inability to directly isolate 32c and its subsequent in situ transformation forecasted the increased reactivity of twisted amides embedded in unsubstituted quinuclidin-2-one scaffolds.
Scheme 2.

Synthesis of 2-Quinuclidone during Autoxidation of Quininone by Doering and Chanley
In 1957, Yakhontov reported that the intramolecular condensation of amino acyl chloride 38, afforded the parent quinuclidin-2-one (Scheme 3).252 However, the isolation of the bridged lactam 18 as reported by Yakhontov has been questioned in the literature on several occasions253,254,107 because of the propensity of unsubstituted quinuclidin-2-ones to polymerize, vigorous conditions utilized for the synthesis of lactam 18 and the lack of any characterization data save elemental analysis of nitrogen.252
Scheme 3.

Synthesis of Unsubstituted 2-Quinuclidone by Yakhontov
During the same time period, Pracejus found that in contrast to the parent 2-quinuclidone 18, the dimethyl analogue 44 could be reliably prepared using an intramolecular condensation of the corresponding amino acyl chloride (Scheme 4).253 Subsequently, the research groups of Pracejus254,255 and Yakhontov256–258 studied the synthesis of 2-quinuclidones 46, in which bridged amide bonds were protected from nucleophilic opening by the presence of methyl substitutents near the amide moieties (Scheme 5). More recently, Greenberg optimized the conditions for preparation of 6,6,7,7-tetramethyl-2-quinuclidone 46c (Scheme 6).259 It was found that the previously published method for the synthesis of qunuclidone 46c256,257 gives almost exclusively polymeric material. Using high dilution techniques, the highly-strained lactam 46c was obtained in modest yield.259
Scheme 4.

Synthesis of 6,6-Dimethyl-2-Quinuclidone by Pracejus (please use double-column format for Scheme 4)
Scheme 5.

Synthesis of Substituted 2-Quinuclidones by Pracejus and Yakhontov
Scheme 6.

Improved Synthesis of 6,6,7,7-Tetramethyl-2-Quinuclidone by Greenberg
In 2006, Tani and Stoltz achieved an unambiguous synthesis of the iconic twisted amide, 2-quinuclidone, using an intramolecular Schmidt reaction as the key step (Scheme 7).107 This method allowed for the rigorously anhydrous conditions required for isolation of the unstable lactam 52 (in water, its t1/2 was found to be less than 15 s).107 The Schmidt reaction produced the parent 2-quinuclidone protected as its trifluoroborate salt, thus preventing the lactam from extensive polymerization. To our knowledge, this was the first example of an N-protonated amide bond to be characterized by X-ray crystallography. The initial Schmidt reaction afforded a mixture of lactams 52 and 53 resulting from migration of the two possible alkyl groups to nitrogen (Scheme 8), from which the desired compound was isolated by crystallization. Through careful optimization, it was found that the use of HBF4 in ether as a solvent provided the best system for this reaction, while other acids resulted in lower regioselectivity. The X-ray structure of the protonated amide 52 indicated a fully orthogonal amide bond (τ = 90.9°; χN = 59.5°; χC = 0.2°; N–C(O) = 1.526 Å; C=O = 1.192 Å).107
Scheme 8.

Proposed Mechanism for Schmidt Reaction of 3-Azidoalkyl Ketone 51
Subsequently, Stoltz reported a gas-phase synthesis of the protonated 2-quniclidonium 52 (Scheme 9).260 Using kinetic proton affinity measurements, the authors determined that lactam 52 is characterized by a much higher basicity (proton affinity = 965 kJ/mol) than typical amides (proton affinity = 880–900 kJ/mol), which is consistent with a low degree of amide resonance in 52.260 DFT calculation suggested that N-protonation of 52 is favored over O-protonation by approximately 90 kJ/mol.260 These studies are in a good agreement with earlier DFT calculations carried out by Greenberg, which suggested that N-protonation of 52 is favored over O-protonation by 100 kJ/mol.70,71
Scheme 9.

Synthesis of Unsubstituted 2-Quinuclidone in a Gas Phase by Stoltz
In comparison with simple 2-quinuclidones, their benzo-substituted analogues are less prone to hydrolysis and polymerization. In 1980, Blackburn reported the synthesis of lactam 60 by intramolecular amide coupling reaction under standard conditions in high yield (Scheme 10).261 Shortly thereafter, Brown and coworkers reinvestigated the synthesis of this and related benzoquinuclidones.223–225 Under their conditions, DCC was applied as a more efficient coupling reagent (Scheme 11).223 Utilizing a similar protocol, Brown has accomplished the synthesis of related benzo-fused bridged lactams (Scheme 12).224,225 Notably, these lactams did not require special precautions during synthesis and isolation despite highly-strained structures. X-ray structures of amides 64a–c indicated a progressive decrease of amide bond distortion in the series 64a (τ = 30.7°; χN = 57.2°; χC = 9.0°; N–C(O) = 1.401 Å; C=O = 1.216 Å), 64b (τ = 33.2°; χN = 52.8°; χC = 11.0°; N–C(O) = 1.413 Å; C=O = 1.225 Å), and 64c (τ = 15.3°; χN = 38.6°; χC = 4.3°; N–C(O) = 1.370 Å; C=O = 1.233 Å).262 Interestingly, lactams 64a and 64b were characterized by almost completely pyramidal nitrogen atoms with only moderate tortional distortion of p-orbitals. Subsequently, a similar divergence between χN and τ values has also been found in other types of bridged lactams (see Section 4.3). Infrared stretching frequencies of C=O bonds demonstrated that lactam 60 is the most twisted compound in the series 60 (1755 cm−1), 64a (1705 cm−1), 64b (1712 cm−1), and 64c (1677 cm−1).223–225
Scheme 10.

Synthesis of Benzo-2-Quinuclidone by Blackburn
Scheme 11.

Synthesis of Benzo-2-Quinuclidones by Brown
Scheme 12.

Synthesis of Expanded Ring Systems of Benzo-2-Quinuclidones by Brown
Additional examples of synthesis of 2-quinuclidones include oxidation of amines to the corresponding lactams under Gif conditions (Scheme 13)263 and stereoselective rearrangement of modified Cinchona alkaloids to bridged lactams as reported by Hoffmann (Scheme 14).264,265 Both are reminiscent of the attempted reactions reported by Doering (cf. Scheme 2),251 but are low yielding and limited to specific cases.
Scheme 13.

Synthesis of 2-Quinuclidones under Gif Conditions by Jankowski
Scheme 14.

Synthesis of 2-Quinuclidones from Modified Cinchona Alkaloids by Hoffmann
3.2. Adamantanone Derivatives
Due to the enforced proximity in rigid molecular frameworks, derivatives of adamantane are useful templates to study stereoelectronic effects. In 1990, Rebek reported a remarkable rate enhancement in cyclization reactions involving nitrogen atoms of lactam and imide functions prepared from Kemp triacid (Scheme 15).266 The involvement of bridged imide 71a and imidinium 72a was inferred on the basis of racemization of optically active precursors267 and deuterium labeling studies.
Scheme 15.

Synthesis of Bridged Imides from Kemp Triacid by Rebek
In 1998, Kirby, while studying the reverse anomeric effect,268,269 achieved the synthesis of 1-aza-2-adamantanone 78 (Scheme 16).270,108 The amino acid precursor 77 was prepared from the commercially available Kemp triacid 73 in an eight-step sequence.108 A proximity-induced cyclization carried out by sublimation afforded lactam 78 in quantitative yield. The X-ray structure of amide 78 indicated a perfectly perpendicular amide bond (τ = 90.5°; θ = 325.7°; N–C(O) = 1.475 Å; C=O = 1.196 Å).108 Moreover, the chemical271,108 (see Sections 7.1–7.2) and spectroscopic properties108 (see Section 2.3) of 78 are in full agreement with keto-amine-like character of the amide bond in this system. Like the unstabilized 2-qunuclidone 52 prepared by Stoltz,107 lactam 78 also undergoes rapid hydrolysis in water (t1/2< 50s).272
The methyl substituents in 1-aza-2-adamantanone derivatives like 78 help to keep the amino group in close proximity to the carboxylic acid, thus facilitating condensation to the lactam(Figure 6).273,274 Accordingly, computational studies on the stability of 78 revealed that methyl substituents destabilize the amino acid form and contribute to the overall stability of Kirby’s amide.275
Figure 6.

Role of methyl substituents in synthesisof 1-aza-2-adamantanones from the corresponding amino acids.
In 2003, Coe prepared bridged lactam 85, which is a higher homologue of 1-aza-adamantanone, as an intermediate in the synthesis of nicotinic receptor ligands (Scheme 17).232 The key reaction involved base-catalyzed condensation of the open-form amino ester to the bridged lactam 85 under thermal conditions. The final precursor 84 was quickly assembled from cyclopentene 81, using Heck reaction and reductive amination as key transformations. The infrared stretching frequency of the C=O group (1728 cm−1) and instability to the aqueous conditions were consistent with a highly-strained structure of lactam 85.
Scheme 17.

Synthesis of Benzo-1-aza-2-adamantanone by Coe
4. SYNTHESIS OF BRIDGED LACTAMS WITH N–(CO) BOND ON TWO-CARBON OR LARGER BRIDGE, ([m.(≥ 2).n] Type)
The synthesis of bridged lactams with the N–(CO) bond placed on a bridge having two or more carbons is challenging because of the strain associated with distorted amide bonds and general lack of stabilizing features on the lactam skeleton (cf. alkyl-quinuclidones and adamantine-derived lactams). In some cases, the high reactivity of non-planar amide bonds may be incompatible with the reaction conditions used for their synthesis. In this section, bridged lactams are classified based on the reaction type employed for their preparation. Condensation reactions that directly form the amide bond can be used to prepare this type of lactams. However, alternative approaches with the amide bond already present in the precursor usually lead to more strained and diverse examples.
4.1. Condensation Reactions Forming N–C(O) Bond
The first synthesis of a 1-azabicyclo[3.3.1]nonan-2-one derivative was reported by Walker and coworkers in 1949 (Scheme 18).276 During studies on hydrogenation reactions catalyzed by copper chromite, these researches performed an intramolecular condensation of the cyano diester 86 to the bridged lactam 87, which was further reduced under the reaction conditions to 1-azabicyclo[3.3.1]nonane 88 in 39% yield. Concurrently, Albertson reported that the hydrogenation of another cyano ester 89 over Raney nickel catalyst gave two compounds, one of which was originally assigned as bridged lactam 90 (Scheme 19a).277 However, upon reinvestigation of this transformation, Albertson found that the compound originally proposed as lactam 90 was more consistent with the bicyclic enaminone 93 (Scheme 19b).278
Scheme 18.

Synthesis of 1-Azabicyclo[3.3.1]nonan-2-one 87 by Walker
Scheme 19.

a) Condensation to 1-Azabicyclo[3.3.1]nonan-2-one by Albertson; b) Revision of Proposed Structure of 90
In 1980, Hall reported the synthesis of 1-azabicyclo[3.3.1]nonan-2-one 96 applying vacuum pyrolysis conditions (Scheme 20).279 Although the yield was very low, lactam 96 could be separated from the polymer 97 by sublimation from the reaction mixture. The condensation of the corresponding acid chloride using a protocol developed by Yakhontov and Pracejus for the synthesis of 2-quinuclidones (Schemes 3–5)252–258 was unsuccessful in this case. Based on its infrared stretching frequency of 1680 cm−1, stability in water, and moderate tendency to undergo polymerization, Hall proposed lactam 96 to be only slightly distorted from planarity.279 However, subsequent structural characterization of related lactams containing [3.3.1] bridged system280–283 and mechanistic studies by Greenberg284,70,71 clearly demonstrate that 1-azabicyclo[3.3.1]nonan-2-ones are sufficiently non-planar to protonate at nitrogen and display other keto-amine-like characteristics.
Scheme 20.

Synthesis of 1-Azabicyclo[3.3.1]nonan-2-one by Hall
In 1981, Buchanan improved the synthesis of 1-azabicyclo[3.3.1]nonan-2-ones by using a gem-dialkyl effect to facilitate cyclization to the bridged amides (Scheme 21).280,281 With substrate 99 containing a phenyl substitutent at C–3 position (cf. the methyl groups in the Kirby’s 1-aza-2-adamantanone, Figure 6), both thermal cyclization under high vacuum and condensation from the corresponding acyl chloride afforded the desired lactam 87, albeit again in low yield. It is worth noting, that in analogy to bridged lactams featuring [2.2.2] ring system (Section 3.1), the classic intramolecular amine-acid chloride condensation approach provides synthetically more useful results when 1-azabicyclo[3.3.1]nonan-2-one scaffolds are stabilized by additional substitutents (cf. Schemes 26 and 27). On the basis of NMR studies, Buchanan concluded that lactam 87 exists in solution in the chair-boat conformation.280 Related compounds, such as lactam 96 (Scheme 20)279 and the analogous anti-Bredt olefin, bicyclo[3.3.1]non-1-ene,285 also favor the chair–boat conformation, which has been explained on the basis of their preference to place the trans double bond in the larger ring.102 Buchanan reported the X-ray structure of lactam 87 (τ = 20.8°; χN = 48.8°; χC = 5.9°; N–C(O) = 1.374 Å; C=O = 1.201 Å).282 These values together with the infrared stretching frequency of 1695 cm−1 indicate a moderate distortion of the amide bond in 87.
Scheme 21.

Synthesis of 5-Phenyl-1-Azabicyclo[3.3.1]nonan-2-one by Buchanan
Scheme 26.

Synthesis of Quinolino-1-Azabicyclo[3.3.1]nonan-2-ones by Cuny
Scheme 27.

a) Synthesis of Aza-Bridged Lactams by Denzer and Ott; b) Synthesis of Bridged Lactam Precursors via Dearomatization of Quinazolines
The synthesis of 1-azabicyclo[3.3.1]nonan-2-ones was further improved when Steliou introduced Bu2SnO as an efficient promoter for difficult lactamizations.286 Under high dilution (0.005 M), the synthesis of lactam 96 was thus achieved in 77% yield (Scheme 22). However, this reagent was ineffective for synthesis of eight-membered and larger lactams. Subsequently, Sim applied Bu2SnO in the synthesis of the structurally-related 1-azabicyclo[3.3.1]nonane-2,6-dione (Scheme 23).283 The X-ray structure of lactam 103 (τ = 16.3°; χN = 49.1°; χC = 5.8°; N–C(O) = 1.377 Å; C=O = 1.217 Å),283 and the infrared stretching frequency of 1680 cm−1 mirror the properties of 87.282 This study further confirmed that lactams with the N–C(O) bond placed at one of the larger bridges are characterized by large χN values and moderate twist angles (cf. lactams 64a–c prepared by Brown (Scheme 12)).262 A tin-mediated lactamization protocol was also employed by Gerlach in a rare example of the synthesis of an enantiomerically-enriched bridged lactam (Scheme 24).287 Earlier, Pracejus had prepared lactams 44 and 46a–b in enantiomerically enriched form by resolution of the intermediate amino esters with dibenzoyl-d-tartaric acid.253–255
Scheme 22.

Improved Synthesis of 1-Azabicyclo[3.3.1]nonan-2-one by Steliou
Scheme 23.

Application of Bu2SnO in Synthesis of 1-Azabicyclo[3.3.1]nonane-2,6-dione by Sim
Scheme 24.

Application of Bu2SnO in Synthesis of (+)-(R)-1-Azabicyclo[3.3.1]nonan-2-oneby Gerlach
In another study, Najera noticed a significant difference in cyclization rates between diastereoisomeric amino esters to form bridged lactams 107 (Scheme 25).288 The endo lactam was obtained quantitatively during removal of the benzyl group, whereas amino lactam 106 was more resistant to condensation. However, the corresponding lactam 107-exo was generated in a separate step after treatment of the amino ester with LDA. Based on NOE analysis, these authors proposed that lactam 107-endo exists in a twist boat-boat conformation due to steric interactions between the axial methyl group and an axial hydrogen atom, while the lactam 107-exo adopts the usual chair-boat conformation.
Scheme 25.

Synthesis of 4-Methyl-5-tosyl-1-azabicyclo[3.3.1]nonan-2-ones by Najera
As is the case with benzo-2-quinuclidones (see Schemes 10–12),223–225,261,262 appendage of a fused aromatic ring improves the stability of 1-azabicyclo[3.3.1]nonan-2-ones. Cuny reported intramolecular lactamization to yield quinolino-substituted 1-azabicyclo[3.3.1]nonan-2-ones using thionyl chloride (Scheme 26).289 Modification of the reaction conditions resulted in the concomitant introduction of the chloride in the 2-position of quinoline ring. Lactam 110 was further derivatized via nucleophilic aromatic substitution. Denzer and Ott reported the synthesis of two different ring systems of bridged lactams, while studying new 1,5-benzodiazocine derivatives with potential biological activity (Scheme 27).290 The amino acid precursors were readily prepared by dearomatization of the corresponding quinazolines. Cyclization using mixed anhydride protocol offered an advantage over the standard conditions with thionyl chloride/triethylamine in terms of yields of isolated products. The highly twisted nature of amide bonds in these lactams was confirmed by infrared stretching frequencies (112: 1720 cm−1; 114: 1780 cm−1), rapid hydrolysis in water, and chemical reactivity (see Schemes 116 and 137). Compound 114 is one of the very few examples of reported bridged lactams bearing a [3.2.1] scaffold.
Scheme 116.

Hydrolysis of Medium Bridged Twisted Lactams by Aubé
Scheme 137.

Reactions of Medium-Bridged Twisted Lactams with Heteroatom Nucleophiles by Aubé
4.2. Heck Reactions
Grigg has pioneered the use of Heck reaction for the synthesis of bridged lactams featuring the N–(CO) bond on external bridge (Scheme 28).291,292 With a catalyst system consisting of Pd(OAc)2 and PPh3, 6-exo-trig and 7-exo-trig cyclizations of enamide substrates occurred in high yields to generate a variety of bridged amide scaffolds. Isomerization of the double bond in products was rarely observed. An attempt to form the bridged lactam with a [3.2.1] ring system was unsuccessful, reflecting a high ring strain present in the transition state (Scheme 29).292 Subsequently, Grigg extended this method by combining the Heck reaction with ring-closing metathesis (RCM) in a tandem process (Scheme 30).293,294 In a telescoped sequence, bridged lactams 124a and 118a were prepared from simple acyclic starting materials in a one-pot transformation.294 This methodology was also employed for the synthesis of analogous bridged sulfonamides (see Section 6.5).291–294 More recently, Paquette reported the synthesis of [4.3.1], [3.3.2] and [4.3.2] bridged lactams via Heck reaction utilizing similar conditions to those reported by Grigg (Scheme 31).295 The enamide substrates were synthesized by RCM of acyclic dienes, again providing an efficient route to bridged lactams.
Scheme 28.

Synthesis of Bridged Lactams via Heck Reaction by Grigg
Scheme 29.

Unsuccessful Synthesis of 1-Azabicyclo[3.2.1]oct-3-en-7-one via Heck Reaction by Grigg
Scheme 30.

Synthesis of Bridged Lactams via Tandem RCM-Heck Reaction by Grigg
Scheme 31.

Synthesis of Bridged Lactams via Heck Reaction by Paquette (please use double-column format for Scheme 31)
Judd and coworkers developed an exceptional tandem reaction sequence for the synthesis of bridged lactams employing the Heck reaction as a key step (Scheme 32).296 Starting from commercially available reagents, sequential Ugi and RCM reactions rapidly provided advanced intermediates. The subsequent Heck reaction was accomplished under microwave irradiation conditions using Pd(PPh3)2Cl or immobilized palladium catalysts to deliver bridged lactams 133 in high yields and short reaction times. The X-ray structures of lactams 133 indicate significant degrees of pyramidalization at nitrogen (133a: τ = 33.4°; χN = 49.1°; 133b: τ = 7.1°; χN = 35.3°; 133c: τ = 17.6°; χN = 26.2°; 133d: τ = 19.8°; χN = 31.1°; 133e: τ = 19.8°; χN = 15.9°).296 Impressively, this study provided full structural characterization of four different ring systems of bridged lactams. Currently, the Heck reaction is one of the most reliable methods for the synthesis of bridged lactams with the N–(CO) bond on a larger bridge.
Scheme 32.

a) Synthesis of Bridged Lactams via Heck Reactionby Ribelin; b) Synthesis of Bridged Lactam Precursors via Tandem Ugi-RCM Reaction
4.3. Diels-Alder Reactions
Shea reported the synthesis of bridged lactams containing bridgehead olefins via thermal type II intramolecular acyl-imino Diels-Alder reaction (Scheme 33).297,298 Heating the acetoxy amides under high dilution conditions resulted in elimination of acetic acid and [4+2] cycloaddition of the intermediate N-acyl imines. Under the optimized conditions, the reactions were not allowed to reach full conversion because of the thermal instability of the bridged lactams. The synthesis of lactam 142a required a strictly inert atmosphere because the bridgehead olefin present in 142a readily formed the corresponding epoxide upon exposure to air (see Scheme 154).298 On the other hand, the lower yield of lactam 142d was postulated to result from a competing ene reaction of the N-acyl imine intermediate.298 In all of these Diels-Alder reactions, only one regioisomer was observed due to a short tether between the reactive functionalities (Scheme 34) – products resulting from the exo transition state were not detected.
Scheme 33.

Synthesis of Bridged Lactams via Type II Acyl Imino Diels-Alder Reaction by Shea
Scheme 154.

Reactivity of 1-Azabicyclo[3.3.1]non-5-en-2-one Bearing Anti-Bredt Olefin by Shea
Scheme 34.

Proposed Mechanism for Diels-Alder Reaction of Acetoxyamides 141
The X-ray structures of analogues 142a–c were solved.298 As expected, they show a progressive decrease of the amide bond distortion in the series 142a (τ = 16.7°; χN = 54.9°; χC = 1.4°; N–C(O) = 1.399 Å; C=O = 1.215 Å), 142b (τ = 7.5°; χN = 46.4°; χC = 1.2°; N–C(O) = 1.375 Å; C=O = 1.219 Å), and 142c (τ = 0.9°; χN = 38.2°; χC = 0.2°; N–C(O) = 1.376 Å; C=O = 1.224 Å). Similar to other lactams with the N–C(O) bond placed on the larger bridge, lactams 142 were found to have pyramidalized nitrogen atoms and small twist angles. Furthermore, infrared and NMR spectroscopy provide a clear indication of the gradual changes in the electronic properties of the amide bonds in these lactams (νC=O [cm−1] a: 1703; b: 1660; c: 1645; d: 1641; δC=O [ppm] a: 182.6; b: 180.6; c: 177.4; d: 173.7).298 Interestingly, in the 13C NMR spectra, all of the carbonyl carbons were observed closer to the region expected for a typical amide rather than ketone, while the IR and NMR values of the least distorted lactam 142d are in the region expected for a planar amide. Structural properties of non-planar olefins, including those in compounds 142, have been recently reviewed.56,104
4.4. Carbene Insertion Reactions
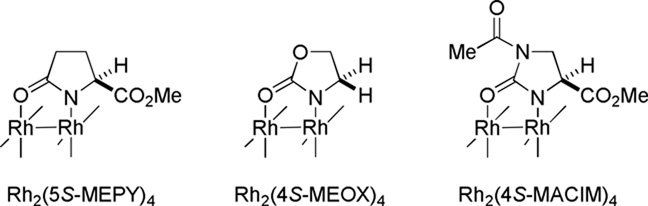
The synthesis of strained non-planar lactams using carbenes is an attractive strategy due to the high reactivity of these intermediates,299–301 which are capable to overcome strain associated with the formation of bridged amide bonds. In 1995, Doyle demonstrated the synthesis of 1-azabicyclo[5.2.1]decan-9-one 145 via Rh-catalyzed C–H insertion reactions with excellent enantioselectivity (Table 1, entries 2–4).302 The less flexible, one-carbon shorter, analogue 144a afforded exclusively the fused lactam 146 (entry 1). Rh2(4S-MACIM)4 catalyst (entry 4) gave the opposite enantiomer of 145 than Rh2(4S-MEOX)4 or Rh2(5S-MEPY)4 (entries 2–3).
Table 1.
Synthesis of 1-Azabicyclo[5.2.1]decan-9-one via Rh-Catalyzed C–H Insertion by Doyle
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | n | 144 | Rh2L4 | ratio of 145:146 |
combined yield (%) |
ee of 145 (%) |
ee of 146 (%) |
| 1 | 0 | 144a | Rh2(5S-MEPY)4 | 1:99 | 67 | n/a | 97 |
| 2 | 1 | 144b | Rh2(4S-MEOX)4 | 74:26 | 95 | 98 | 15 |
| 3 | 1 | 144b | Rh2(5S-MEPY)4 | 33:67 | 67 | 97 | 30 |
| 4 | 1 | 144b | Rh2(4S-MACIM)4 | 61:39 | 81 | 96 | 66 |
 | |||||||
More recently, Aszodi and coworkers reported Rh- and Cu-catalyzed N–H insertion reactions to generate strained lactams containing a [3.2.1] scaffold as a part of their studies on bridged analogues of carbapenem and carbacephem antibiotics (Table 2).233 A remote alkyl substitutent exerted significant influence on the competing C–H insertion process, leading instead to the fused derivative 149 from precursor 147 where R = Et. Williams has applied a similar N–H insertion reaction to prepare some of the most strained bridged lactams isolated to date (see Section 5.1).303,304
Table 2.
Synthesis of 1-Azabicyclo[3.2.1]octan-7-onesvia N–H Insertion by Aszodi
 | |||||
|---|---|---|---|---|---|
| entry | R | 147 | catalyst | yield of 148 (%) |
yield of 149 (%) |
| 1 | H | 147a | Rh2(OAc)4 | 40 | 42 |
| 2 | Et | 147b | Rh2(OAc)4 | 5 | 80 |
| 3 | Et | 147b | Cu(acac)2 | 30 | n/a |
4.5. Reactions via Radical Intermediates
Sundberg developed the synthesis of indole-containing bridged lactams featuring a [5.3.1] ring system using Witkop photocyclization (Scheme 35).305 It was determined that these reactions proceed smoothly in methanol. Sodium carbonate was used as an additive to prevent hydrolysis of the ketal moiety in certain starting materials. An attempt to form a [6.2.2] ring system from the corresponding 4-piperidylmethyl substrate was unsuccessful.305 The authors observed a new type of atropoisomerism in the resulting bridged lactams, with half-lives for the interconversion of atropoisomers 152 and 153 between 2.5 and 8 h at 170 °C (Figure 7).305
Scheme 35.

Synthesis of Indole-1-azabicyclo[5.3.1]undecan-2-onesvia Witkop Photocyclization by Sundberg
Figure 7.

Atropoisomers formed in Witkop photocyclization of (N-chloroacetylpiperidyl)indole 150.
Subsequently, Sundberg reported the photocyclization in the 3-piperidylmethyl series to yield bridged lactams with a [6.3.1] ring system (Scheme 36).306 In this case, yields and diastereoselectivities were slightly improved, while the barrier for interconversion between atropoisomers was lower than in the [5.3.1] ring system (t1/2 in the range of hours at 140 °C). The X-ray structure of lactam 155b306 revealed that lengths of the N–C(O) bond of 1.356 Å and the C=O bond of 1.235 Å are comparable with those encountered in planar amides.
Scheme 36.

Synthesis of Indole-1-azabicyclo[6.3.1]dodecan-2-ones via Witkop Photocyclization by Sundberg
In the course of studying radical cyclization of allenamides, Hsung reported the synthesis of benzo-fused bridged lactam 157 with a [4.2.1] ring system (Scheme 37).307 The product was proposed to arise from 7-endo/5-endo alkene/allene cyclization of an aryl radical intermediate. Beckwith found that the fragmentation of methyl xantates 159, using tributyltin hydride and di-tert-butylperoxide in refluxing toluene, afforded the corresponding ethylidene-substituted bridged lactams containing a [5.3.1] ring system (Scheme 38).308
Scheme 37.

Synthesis of Benzo-1-azabicyclo[4.2.1]nonan-2-one via Radical Cyclization by Hsung
Scheme 38.

Synthesis of 1-Azabicyclo[5.3.1]undecan-10-ones via Radical Fragmentation by Beckwith
4.6. Miscellaneous Examples
During synthetic studies on iso-indolobenzazepine alkaloids, Ruchirawat and coworkers discovered the Friedel-Crafts cyclization of N-benzylbenzazepinones 161 to dibenzo-fused bridged lactams 162 (Scheme 39a).309 In a related work, the same group investigated the use of acyclic amide acetals 163, which participated in a sequential Friedel-Crafts cyclization to afford bridged lactams 164 (Scheme 39b).310 As a part of their research on indole alkaloids, Dolby and Sakai reported a tandem fragmentation–transannular cyclization to give bridged lactam 166 with a [6.2.2] ring system (Scheme 40).311,312 In the context of studies on bis-nor-meptazinols as cholinesterase inhibitors,313 Qiu and co-workers reported the X-ray structure of lactam 167a314 (Figure 8; 167a: τ = 14.4°; χN = 27.8°; χC = 2.5°; N–C(O) = 1.347 Å; C=O = 1.243 Å). Finally, Wärnmark and coworkers reported315,316 the synthesis of bridged lactams 167b and 167c via direct oxidation of the parent amine (Tröger’s base) at the benzylic position with KMnO4 in 25–28% yields (Figure 8). The X-ray structure of the bis-twisted lactam was solved315 and indicates significant distortion of the amide bond in 167c (τ = 43.7°; χN = 57.6°; χC = 4.8°; N–C(O) = 1.437 Å; C=O = 1.209 Å).
Scheme 39.

Synthesis of Bridged Lactams via Friedel-Crafts Reaction by Ruchirawat
Scheme 40.

Synthesis of Indole-1-azabicyclo[6.2.2]dodecan-10-one via Fragmentation/Transannular Condensationby Dolby
Figure 8.

Benzofused bridged lactams: a) Benzo-1-azabicyclo[4.3.1]decan-9-one by Qiu; b) Bridged lactams derived from Tröger’s base by Wärnmark.
5. SYNTHESIS OF BRIDGED LACTAMS WITH N–(CO) BOND ON ONE-CARBON BRIDGE, ([m.1.n] Type)
Bridged lactams featuring the N–(CO) bond on one-carbon bridge are typically more strained than their two-carbon bridged and larger analogues (see Figure 5). Carbene insertion, ring expansion and transannular condensation reactions forming directly the N–(CO) bond are powerful methods for the assembly of this class of compounds. In general, other synthetic methods are less efficient and/or limited to specific structural types. The majority of successful approaches to non-planar one-carbon bridged lactams involve generation of a high-energy intermediate.
5.1. Carbene Insertion Reactions
In 1986, Williams reported the synthesis of highly distorted bridged lactams containing [4.1.1] ring system using Rh-catalyzed carbene N–H insertion chemistry (Scheme 41).303,304 The reaction of diazo β-keto esters 168 proceeded smoothly in the presence of catalytic Rh2(OAc)4 in refluxing benzene to afford bridged lactams in 20–70% yields. Lactams 169 with a sterically-undemanding substituent at the C–8 position (such as 169a) exhibited limited stability at room temperature (t1/2 ~ 1 h in CDCl3, IR = 1795 cm−1). This was proposed to be due to the nucleophilic opening of the strained lactam moiety from the unshielded α face (Figure 9a).304 Higher stability was observed in isopropyl analogues such as 169b–e, in which the i-Pr group hindered the Bürgi-Dunitz trajectory (169b–c: stable in CDCl3, IR = 1795 cm−1; 169d–e: t1/2 ~12 h in CDCl3 at −30 °C, IR = 1805–1810 cm−1). Further improvement was achieved by blocking the bridgehead position to prevent abstraction of the bridgehead methine proton and possible fragmentation (Figure 9b).304 The lactam 169f proved to be significantly more stable than previous analogues, however it decomposed when stored neat at room temperature. The crystalline analogues 169g–h were found to be stable in CDCl3 solution over several days. The X-ray structure of 169h304 (IR = 1785 cm−1) revealed pyramidalized nitrogen atom (θ = 326.8°, which may be compared with θ = 325.7° for Kirby’s amide108 and θ = 324.0° for trimethylamine)304 and a long N–C(O) bond of 1.418 Å. Several 1,3-bridged 2-azetidinones and bridged analogues of β-lactam antibiotics inspired by the William’s work have been reported.242–246
Scheme 41.

Synthesis of 1-Azabicyclo[4.1.1]octan-7-onesvia Rh-Catalyzed N–H Insertion by Williams
Figure 9.

Design of 1-azabicyclo[4.1.1]octan-7-ones with increased stability: a) nucleophilic attack on lactam carbonyl group; b) deprotonation of bridgehead methine proton.
5.2. Schmidt Reactions
The intramolecular Schmidt reaction317–320 has been applied to the synthesis of one-carbon bridged lactams. To date, this method has been used to prepare [4.3.1], [5.3.1], [3.2.1], [4.2.1], and [5.2.1] ring systems of these heterocycles. While two constitutional isomers can, in principle, be formed from the intramolecular Schmidt reactions of 2-azidoalkylketones (Scheme 42),317 the latter pathway (path b) requires particular circumstances to compete with the much more commonly observed path a, which leads to fused bicyclic lactams.318–320 In the last decade, four complementary methods that allow synthesis of bridged lactams via Schmidt reaction have been developed: (1) axial 2-azidoalkyl tethers,208 (2) cation–π interactions involving diazonium cation intermediates,321,322 (3) cation–n interactions involving diazonium cation intermediates,323,324 and (4) two-carbon 2-azidoalkyl tethers.325 In addition, Schmidt reaction of 2-azidoalkylacetals affords bridged orthoamides, which can be converted into bridged lactams.326
Scheme 42.

Regiochemical Options in the Intramolecular Schmidt Reaction of 2-Alkylazido Ketones
In 2005, Aubé reported the synthesis of tricyclic bridged lactams 177 via a tandem Diels-Alder/Schmidt reaction (Table 3).208 This class of bridged lactams was first encountered five years earlier in the context of a total synthesis of stenine.327–329 Mechanistically, an intramolecular Diels-Alder reaction of triene 175 afforded cis-decalone 178, in which the carbon bearing the azidoalkyl side chain is axial relative to the cyclohexanone ring embedded in the bicyclic intermediate (Scheme 43).327 The bridged lactams were obtained by subsequent C→N migration and loss of N2. The intermediate 178-eq containing the leaving N2group in a pseudoequatorial position led to the fused lactam 176, whereas migration of the bond antiperiplanar to the pseudoaxial N2+ afforded the lactam 177. The X-ray structure of lactam 177b indicated that it contains a half-way rotated amide bond (τ = 51.5°; χN = 36.1°; χC = 12.8°; N–C(O) = 1.387 Å; C=O = 1.218 Å;208 see also compound 511b in Figure 15).
Table 3.
Synthesis of Tricyclic Bridged Lactams via Domino Diels-Alder/Schmidt Reaction by Aubé
 | |||||
|---|---|---|---|---|---|
| entry | ketoazide 175 | R1 | R2 | yield of 176 (%) |
yield of 177 (%) |
| 1 | 175a | (CH2)2OBn | H | 43 | 24 |
| 2 | 175b | 4-Br-C6H4 | H | 43 | 23 |
| 3 | 175c | C6H5 | H | 46 | 23 |
| 4 | 175d | H | H | 41 | 22 |
| 5 | 175e | H | i-Pr | 43 | 28 |
| 6 | 175f | H | Ph | 85 | 0 |
Scheme 43.

Proposed Mechanism for Domino Diels-Alder/Schmidt Reaction of Keto-Azidotrienes 175 (please use double-column format for Scheme 43)
Figure 15.

a) Bridged Lactam Bearing Epoxide by Paquette; b) Tricyclic Bridged Lactam by Aubé
Subsequent work led to an improved sequence in which a cation–π directed Schmidt reaction afforded bicyclic bridged lactams as the major products (Scheme 44).321,322 In this case, the combination of an axial azidoalkyl tether and an aromatic group positioned in a 1,3-diaxial relationship with the diazonium cation in the key azidohydrin intermediate 179-ax (Scheme 45) resulted in high selectivity for the rearrangement to bridged lactams. Electron-rich aromatic groups led to high yields, while electron-poor aromatic groups were less effective, providing strong support for the proposed cation–π interactions.322 The X-ray structure of lactam 181b (τ = 43.2°; χN = 33.8°; χC = 16.3°; N–C(O) = 1.363 Å; C=O = 1.234 Å)330 confirmed that the amide bond in the [4.3.1] bicyclic system is significantly distorted from planarity.
Scheme 44.

Synthesis of Bridged Lactams via Cation-π-Directed Schmidt Reaction by Aubé
Scheme 45.

Proposed Mechanism for Schmidt Reaction of 2-Aryl-2-Alkylazido Ketones 179
The Aubé group also reported the synthesis of bicyclic bridged lactams using conformationally-flexible 2-azidoalkylketone substrates containing an α-heteroatomic group (Scheme 46).323 A thiomethyl substitutent in the α-position gave the best results in terms of yields and selectivity. A stabilizing 1,3-diaxial interaction between the diazonium cation and thiomethyl group in the azidohydrin intermediate 182a (Scheme 47) was proposed to explain the regioselectivity of this transformation.323 Recently, cation–π and cation–n directed Schmidt reactions have been investigated using density functional theory, confirming the role of non-bonding interactions involving the diazonium cation on the regioselectivity of the rearrangement.324
Scheme 46.

Synthesis of Bridged Lactams via Cation–n Directed Schmidt Reaction by Aubé
Scheme 47.

Proposed Mechanism for Schmidt Reaction of 2-Alkylazido Ketones 182
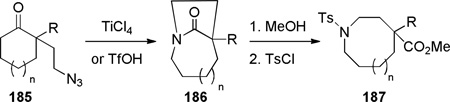
Murphy reported that the intramolecular Schmidt reaction of conformationally-flexible 2-azidoalkylketones containing a two-carbon tether affords predominantly bridged isomers (Table 4).325 Several five-, six- and seven-membered azidoketone substrates produced one-carbon bridged lactams 186, which were converted to the corresponding amino esters 187. The authors observed that the bridged lactam 186a containing a [3.2.1] ring system was unstable to the aqueous work-up conditions due to the high strain associated with the non-planar amide bond.325 Moreover, 185b participated in a competing fragmentation pathway driven by the aryl group.325 The preference for bridged lactams in these examples was explained on the basis of strain developing during the C→N migration to the alternative fused four-membered ring lactams (as also observed in another context331,332).
Table 4.
Synthesis of Bridged Lactams via Schmidt Reaction of 2-(2-Azidoethyl) Ketones by Murphy
 | ||||||
|---|---|---|---|---|---|---|
| entry | n | 185 | R | acid | yield of 186 (%) |
yield of 187 (%) |
| 1 | 0 | 185a | H | TfOH | n/a | 89a |
| 2 | 0 | 185b | 4-MeO-C6H4 | TfOH | n/a | 22a,b |
| 3 | 1 | 185c | H | TfOH | n/a | 94a |
| 4 | 1 | 185d | 4-MeO-C6H4 | TiCl4 | 97 | n/a |
| 5 | 2 | 185e | H | TfOH | 41 | 83 |
Yield from 185.
MsCl instead of TsCl.
Additional examples of bridged lactams prepared by Schmidt reactions have been reported by Aubé208 and Murphy325 (Scheme 48). Although yields were moderate, this methodology offers a concise route to these lactams. The X-ray structure of lactam 190c (τ = 35.9°; χN = 43.7°; χC = 12.4°; N–C(O) = 1.375 Å; C=O = 1.219 Å)333 shows a significantly distorted amide bond and is in good agreement with other bridged lactams featuring a [4.3.1] ring system.
Scheme 48.

Other Examples of Bridged Lactams Synthesized via Intramolecular Schmidt Reaction
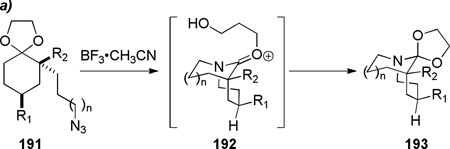
In 2010, Aubé reported that 2-azidoalkylketals undergo Schmidt reaction via iminium ion intermediates to give bridged orthoamides (Table 5a).326 The α-amino ketals 193 could be readily transformed into the parent twisted lactams (Table 5b).326 The synthesis of bridged orthoamides provided the first examples of any intramolecular Schmidt reaction affording exclusively bridged products.331,332,326
Table 5.
a) Synthesis of Bridged Orthoamides via Schmidt Reaction of 2-Azidoalkyl Ketals by Aubé; b) Conversion of Bridged Orthoamides to Bridged Lactams
 | |||||
|---|---|---|---|---|---|
| entry | 191 | n | R1 | R2 | yield of 193 (%) |
| 1 | 191a | 1 | t-Bu | C6H5 | 88 |
| 2 | 191b | 1 | t-Bu | 4-MeO-C6H4 | 92 |
| 3 | 191c | 1 | t-Bu | 4-NO2-C6H4 | 94 |
| 4 | 191d | 1 | H | C6H5 | 59 |
| 5 | 191e | 1 | H | SPh | 78a |
| 6 | 191f | 0 | H | C6H5 | 91 |
| b) |
|---|
 |
TMSOTf.
5.3. Condensation Reactions Forming N–C(O) and C–C or N–C Bonds
Transannular amidation reactions of medium-size ring amino esters have emerged as a versatile method for the synthesis of one-carbon bridged lactams due to the release of transannular strain during the formation of bicyclic systems.
In 1987, using a transannular cyclization under thermal conditions, Schill developed the synthesis of indole-derived bridged lactams 195 as potential precursors to higher analogues of vinca alkaloids (Scheme 49).334–339 The condensation between the amine and pentafluorophenyl esters was applied to the successful synthesis of three different ring systems of bridged lactams.336–338 The presence of an additional substituent (cf. 195b)336 was found to be crucial in obtaining bridged products. In contrast, the cyclization of the indole-derived amino acids was less general, affording only a [4.4.1] ring system in modest yield (Scheme 50).338 Magnus and coworkers reported the synthesis of a related bridged lactam 199 containing a [4.3.1] ring system as a part of their studies towards the synthesis of vinblastine (Scheme 51a).340,341 The transannular cyclization between the methyl ester and amine proceeded smoothly under thermal conditions.340 The nine-membered ring precursor was efficiently prepared from the tetracyclic amine 200 via ring fragmentation and nucleophilic trapping of the resulting iminium ion (Scheme 51b).340More recently, Dennison and coworkers applied a transannular condensation reaction to prepare a complex bridged lactam 205 from the vincristine metabolite 204 to facilitate the structural assignment of 204 (Scheme 52).342
Scheme 49.

Synthesis of Indole-Derived Bridged Lactams via Transannular Amidation by Schill
Scheme 50.

Synthesis of Indole-Derived Bridged Lactams via Transannular Amide Coupling by Schill
Scheme 51.

a) Synthesis of 6-Phenyl-1-azabicyclo[4.3.1]decan-10-one by Magnus; b) Synthesis of Nine-Membered Ring Precursor
Scheme 52.

Synthesis of Bridged Lactam from Vincristine Metabolite by Dennison (please use double-column format for Scheme 52)
In 2009, Aubé reported the synthesis of six different ring systems of one-carbon bridged lactams via a tandem RCM-transannular amidation sequence (Scheme 53).343 The medium-size ring precursors were obtained in high yields from simple dienes using Hoveyda-Grubbs II catalyst. The transannular cyclization took place under thermal conditions with Cs2CO3 or DBU as a base, affording highly strained bridged lactams in the process.
Scheme 53.

Synthesis of One-Carbon Bridged Lactamsvia Tandem RCM/Transannular Amidation by Aubé
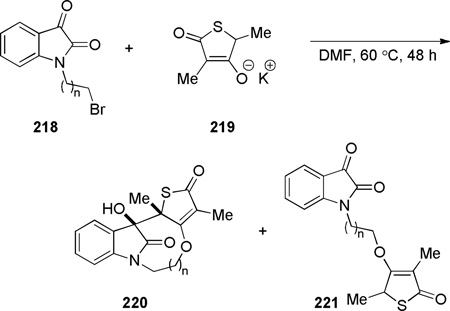
One-carbon bridged lactams have also been prepared by other condensation reactions forming C–C or C–N bonds (Schemes 54–55, Table 6). However, in contrast to the transannular amidation reactions,336–343 these approaches are limited to specific examples. Arata reported the Dieckmann condensation of amido ester 209 to give the corresponding bridged lactam in 30% yield (Scheme 54a).344 Waly found that a sequential treatment of N-acetylaminonitrile 211 with NaH and KOt-Bu afforded the bridged trione 212 (Scheme 54b),345 while Smet reported the synthesis of lactam 214 from 4-bromoisatin (Scheme 54c).346 Nazarenko proposed the intermediacy of bridged lactam 216 in the intramolecular condensation of 1,4-benzothiazin-3-one derivative 215, ultimately affording the more stable 217 from the lactam viaintramolecular transacylation (Scheme 55).347 Chibale reported the synthesis of tetracyclic bridged lactams based on thiolactone-isatin scaffolds (Table 6).235 The X-ray structures of lactams 220c–220e indicated moderate distortions from planarity despite flexible ring systems (220c: τ = 10.4°; χN = 5.1°; χC = 0.7°; N–C(O) = 1.357 Å; C=O = 1.227 Å; 220d: τ = 5.3°; χN = 1.1°; χC = 3.0°; N–C(O) = 1.355 Å; C=O = 1.223 Å; 220e: τ = 0.6°; χN = 8.9°; χC = 4.5°; N–C(O) = 1.361 Å; C=O = 1.223 Å).235
Scheme 54.

Other Examples of Synthesis of One-Carbon Bridged Lactams via Condensation Reactions: a) 1-Azabicyclo[4.2.1]nonane-5,9-dione by Arata; b) 1-Azabicyclo[3.3.1]nonane-4,6,9-trione by Waly; c) 1-Azabicyclo[4.2.1]nonan-9-one by Smet
Table 6.
Synthesis of Tetracyclic Bridged Lactams from Thiolactone-Isatin Hybrids by Chibale
 | |||||
|---|---|---|---|---|---|
| entry | n | 218 | ratio of 220 221 |
combined yield (%) |
ring system of 220 |
| 1 | 1 | 218a | 0:100 | 36 | [5.2.1] |
| 2 | 2 | 218b | 0:100 | 18 | [6.2.1] |
| 3 | 3 | 218c | 78:22 | 45 | [7.2.1] |
| 4 | 4 | 218d | 16:84 | 31 | [8.2.1] |
| 5 | 5 | 218e | 29:71 | 21 | [9.2.1] |
Scheme 55.

Synthesis of Benzo-4-thia-1-azabicyclo[3.2.1]octan-8-one 216 by Nazarenko
5.4. RCM Reactions
Doodeman and Hiemstra carried out an extensive study to probe the synthesis of one-carbon bridged lactams using RCM.348 In a systematic screening of dienes that could afford bridged lactams with eight different ring systems ranging from [4.3.1] to [8.2.1], only one bridged lactam containing a [6.2.1] ring was formed in low yield (Scheme 56). Since polymeric material was obtained in many cases despite high dilution conditions, it is apparent that the starting planar lactam was unable to achieve a suitable conformation in sufficient amounts to permit cyclization.
Scheme 56.

Synthesis of 1-Azabicyclo[6.2.1]undecan-11-one via RCM by Doodeman and Hiemstra
5.5. Aziridinium rearrangement
Aziridinium ions are known to undergo regioselective ring opening with a wide range of nucleophiles and this type of rearrangement has been used extensively to make planar nitrogen-containing heterocycles.349,350 In 1970, Arata reported the synthesis of bridged lactam 225 containing a [4.4.1] ring system via aziridinium rearrangement using a one-pot protocol (Scheme 57).351,352 The proposed mechanism is outlined in Scheme 58,352 and involves the following steps: (1) tautomerization of enamine 224 to iminium ion, (2) addition of the trichloromethyl moiety, (3) formation of the aziridinium 224b, (4) regioselective nucleophilic ring opening to the trichloroderivative 224c, and (5) hydrolysis to give the bridged lactam 225. Subsequently, Miyano and coworkers studied a related rearrangement in the 1-azabicyclo[3.3.1]nonane system (Scheme 59).353,354 In their case, the reaction was carried out step-wise and the trichloromethyl analogue 227 was isolated.353 This intermediate then underwent rearrangement to 1-azabicyclo[3.3.1]nonane 229 upon heating in pyridine. Hydrolysis to the corresponding bridged lactam has not been reported for this example.354
Scheme 57.

Synthesis of Chloro-Substituted Bridged Lactam via Aziridinium Rearrangement by Arata
Scheme 58.

Proposed Mechanism for Rearrangement of 224
Scheme 59.

Synthesis of 5,9,9-Trichloro-1-azabicyclo[3.3.1]nonane by Miyano
5.5. Fragmentation Reactions
Fragmentation reactions have been used to prepare relatively flexible ring systems of one-carbon bridged lactams. In 1994, Bremner reported a photolysis of vinylogous chloroacetamides in water-acetonitrile for the synthesis of bridged lactams containing [6.2.1] and [6.3.1] scaffolds (Scheme 60).355 Improved yields were obtained in acidified aqueous solutions, which could be due to the involvement of iminium 230a in the photoinduced electron transfer step (Scheme 61). The authors proposed that the addition of water could occur at the stage of iminium aromatic cation 230c, which then could undergo C–C bond fragmentation to give bridged lactams. The X-ray structures of 231a and 231b have been solved (231a: θ = 356.4; N–C(O) = 1.322 Å; C=O = 1.236 Å; 231b: θ = 359°; N–C(O) = 1.333 Å; C=O = 1.235 Å).355 These values are comparable to those of planar N-methyl-δ-valerolactam (see Section 2.2).
Scheme 60.

Synthesis of Bridged Lactams viaPhotolysis of Chloroacetamides by Bremner
Scheme 61.

Proposed Mechanism for Cyclization/Fragmentation of 230
Schuman and coworkers reported the photo-oxidation of the tricyclic enamine 232, yielding bridged lactams with a [7.3.1] ring system (Scheme 62).356 The authors proposed that the bridged lactam 233 arose from a singlet oxygen reaction with the electron-rich double bond to give the corresponding dioxetane, which then underwent retro [2+2] (Scheme 63a).356 The trioxygenated lactams 234 and 235 were formed in a sequence starting with the intermolecular redox reaction between the enamine 232 and intermediate 232a, followed by standard steps of the photosensitized oxygen transfer process (Scheme 63b).356
Scheme 62.

Synthesis of 1-Azabicyclo[7.3.1]tridecan-13-onesvia Oxidation of Enamines by Schumann
Scheme 63.

Proposed Mechanism for Oxidation/Fragmentation of Enamines 232 and 232a
During their studies on reactions of arenesulfonylazides with strained indoles, Bailey and coworkers reported an oxidative rearrangement of pyrrolo[1,2–a]indole 236 leading to a benzofused bridged lactam bearing a [6.2.1] ring system (Scheme 64).357 The formation of lactam 237 was not observed in apolar solvents. The reaction involves (1) nucleophilic addition of 236 to the azide, (2) hydration of the imnium 236c, and (3) oxidative cleavage of the C–C bond to give bridged lactam 237 (Scheme 65). The X-ray structure of an oxime derivative of bridged lactam 237 has been solved (θ = 356.6; N–C(O) = 1.372 Å; C=O = 1.215 Å),358 and indicates some differences from the related bridged lactam 231a.
Scheme 64.

Synthesis of 1-Azabicyclo[6.2.1]undecan-11-one by Bailey
Scheme 65.

Proposed Mechanism for Oxidative Fragmentation of 236
5.6. Rearrangements of Nitrogen Ylides
Synthesis of bridged lactams in reactions proceeding via ylide intermediates is a promising approach, however such reactions are not well developed. In 1993, Rudler reported the synthesis of one-carbon bridged lactams containing [5.1.2] and [6.1.2] ring systems using nitrogen ylides, obtained by thermolysis of aminocarbene complexes of chromium (Scheme 66).359 By performing the theromolysis in cyclohexane, the ylides could be isolated in 52–84% yields. Their subsequent thermolysis in toluene afforded bridged lactams 240 in moderate yields. When nitrogen was a part of 6- or 7-membered ring, the rearrangement afforded bridged lactams; however, only fused lactams were obtained from azetidine-and pyrrolidine-containing chromium complexes. The authors proposed that the reaction starts with intramolecular alkyne and carbon monoxide insertion to give 239b (Scheme 67).359 Addition of the tertiary amine to the ketene group in the vinylketene generates intermediate 239c, which then rearranges to the corresponding lactams.
Scheme 66.

Synthesis of Bridged Lactams via Rearrangement of Nitrogen Ylides by Rudler
Scheme 67.

Proposed Mechanism for Rearrangement of 239
Chuche reported the application of spiro pyrazolium ylides to the synthesis of bridged pyrazolin-5-ones (Scheme 68).360 Intermediates 242a could be isolated in good yields by performing flow pyrolysis at 325–350 °C. However, the thermolysis at 400 °C led directly to the bridged pyrazolin-5-ones. The infrared stretching frequencies of 1765 and 1760 cm−1 for 243a and 243c, respectively, indicated significant distortion of amide bonds.
Scheme 68.

Synthesis of 1,4-Bridged Pyrazolin-5-ones via Rearrangement of Spiro Pyrazolium Ylides by Chuche
5.7. Miscellaneous Examples
Kutney reported the synthesis of bis-indole-derived bridged lactams containing [6.3.1] scaffolds via oxidation and Polonovski reaction of the corresponding bridged amines (Scheme 69).361,362 Williams attempted to prepare bridged lactam 251 with the unprecedented [3.2.1] ring system. Despite obtaining promising spectroscopic properties from a neat sample, this material underwent polymerization upon isolation (Scheme 70).304 Toshimitsu reported the use of intramolecular amidoselenation reaction towards the synthesis of bridged lactams,363 however the originally-assigned bridged structure 254 was later found to be incorrect (Scheme 71).364,365 A similar tendency of amides to cyclize via oxygen rather than nitrogen atom was also described by Smissman and coworkers in the context of their work on bridged barbituric acid derivatives (see Section 6.2.2).366–371
Scheme 69.

Synthesis of Vinblastine-Derived Bridged Lactams by Kutney
Scheme 70.

Attempted Synthesis of 1-Azabicyclo[3.1.1]heptan-6-one by Williams
Scheme 71.

Attempted Synthesis of 1-Azabicyclo[3.2.1]octan-8-onesvia Amidoselenation by Toshimitsu
6. SYNTHESIS OF BRIDGED LACTAMS WITH COMPLEX RING SYSTEMS AND RELATED HETEROCYCLES
6.1. Bridged Lactams with Complex Ring Systems
In this section, we discuss the synthesis of bridged lactams embedded in more complex ring systems. It is also possible to classifythese compounds by the type of reaction used for their synthesis (condensation reactions forming N–C(O) bond372–374 or reactions involving radical intermediates).375–377 Bridged monothioimide 263 (Scheme 73)375 and bridged imides 269 (Scheme 74)376,377 can be also classified as heteroatom-containing derivatives of bridged lactams.
Scheme 73.

a) Synthesis of Bridged Monothioimidevia Photochemical Fragmentation/Photocyclization by Sakamoto; b) Rearrangement of One-Carbon Higher Analog
Scheme 74.

Synthesis of Complex Bridged Imides via [2+2] Photocycloaddition by Booker-Milburn
In 1992, during synthetic studies toward the alkaloid stemofoline, Thomas reported the synthesis of bridged lactams containing a rigid tropane-type scaffold (Scheme 72).372–374 Lithium–halogen exchange, followed by intramolecular organolithium addition to carbamates 257 gave tetrahedral intermediates 258, which collapsed to bridged amides upon warming to room temperature. When the reactionswere quenched at −78 °C, transannular ester migration occurred. The X-ray structure of bridged lactam 259c372 (N–C(O) = 1.432 Å) and its infrared stretching frequency of 1746 cm−1indicated non-planar character of the amide bond. However, it should be noted that the synthesis and chemical properties (see Scheme 144)373 of lactams 259 are strongly influenced by the presence of the tropanone ring system.
Scheme 72.

Synthesis of Tropane-Derived Bridged Lactams in Synthetic Studies towards Stemofoline by Thomas
Scheme 144.

Reduction of Tropane-Derived Bridged Lactam by Thomas
In 1990, Sakamoto reported the synthesis of bridged monothioimide via Norrish type I reaction of a thiocarbonyl group (Scheme 73a).375 The authors suggested that the ring strain in the five-membered ring precursor 262 played an important role in the cleavage of C–C(S) bond. In agreement with this hypothesis, the substrate 264 containing a six-membered ring underwent an alternative [2+2] cycloaddition to give the fused enamide 265 (Scheme 73b).
In 2009, Booker-Milburn reported the synthesis of tetracyclic bridged imides 267 via intramolecular [2+2] photocycloaddition (Scheme 74).376 This reaction relies on the use of sensitizers to control [2+2]376 over [5+2]377 mode of cycloaddition of N-alkenyl maleimide substrates (Scheme 75). In the presence of benzophenone sensitizer, [2+2] cycloaddition occurred with high selectivity, while under direct irradiation conditions the synthesis of tricyclic azepines 268 was achieved.376 All bridged imides were isolated as single diastereoisomers. The authors proposed that a sufficiently long lifetime of the triplet excited state allowed the side chain to achieve the lowest energy conformation for the cycloaddition. The X-ray structure of 267f377 indicated that the endocyclic imide carbonyl group (N–C(O) = 1.412 Å; C=O = 1.198 Å) is less planar than the exocyclic carbonyl group (N–C(O) = 1.394 Å; C=O = 1.221 Å).
Scheme 75.

Proposed Mechanism for Photocycloaddition of Imides 266 (please use double-column format for Scheme 75)
6.2. Heteroatom-Containing Derivatives of Bridged Lactams
A sampling of heteroatom-containing derivatives of bridged lactams is depicted in Figure 10a. In general, heterocycles containing heteroatoms adjacent to amide bonds (such as, bridged ureas and bridged urethanes; see Section 6.2.1) are easier to synthesize than the parent bridged lactams because of the stabilizing resonance interaction involving the amide bond and the additional heteroatom (Figure 10b). Another class of heteroatom-containing derivatives of bridged lactams consists of bridged oxazinolactams and bridged diazines (see Section 6.2.2), in which the bridgehead nitrogen atom is accompanied by an α-heteroatom. Amide bonds in these compounds are subjected to similar strain to those in the parent lactams, however transannular interactions in medium-sized rings often contribute to the distortion of these compounds. Finally, bridged imides (such as, barbiturates, oxazolidinediones and hydantoins; see Figure 11 for structures, Section 6.2.3) exhibit a ring-size dependent inhibition of the amide bond resonance and their synthesis can be problematic.
Figure 10.

a) Heteroatom-containing derivatives of bridged lactams; b) Stabilizing resonance interaction in bridged ureas and urethanes.
Figure 11.

Design of conformationally-constrained analogues by Smissman: a) anticonvulsant and anti-steroid drugs containing planar amide bonds; b) potential analogues containing bridged amide bonds (“smissmanones”).
6.2.1. Bridged Ureas and Urethanes
The first successful synthesis of a bridged urea was reported by Hall in 1972 (Scheme 76).378 Treatment of an easily accessible diamine 274 with phosgene at 0 °C resulted in the net acylation of both nitrogen atoms to afford bridged urea 275 in moderate yield. In contrast, the reaction at −75 °C gave the corresponding bis-carbamoyl chloride, which was converted into 275 using Ag2CO3 in refluxing acetonitrile in lower yield.378 The infrared frequency of 275 of 1650 cm−1 was in the region expected for planar tetraalkylureas, indicating that this compound is not particularly strained. In 1980, Hall reported the synthesis of another bridged urea 278 via depolymerization of a polymeric pre-urea under high vacuum (Scheme 77).379
Scheme 76.

Synthesis of 3-Alkyl-1,3-diazabicyclo[3.3.1]nonan-2-ones by Hall
Scheme 77.

Synthesis of 1,3-diazabicyclo[3.3.1]nonan-2-ones by Hall
Concurrently, Hall developed the synthesis of bridged urethanes using phosgene as a carbonyl source (Scheme 78).380,381 The optimum conditions for the synthesis of bridged urethane 281 containing a [3.3.1] ring system proceeded via chloroformate salt 280 (Scheme 78a), while the urethane 284 containing a [3.2.1] ring system was obtained via an alternative route involving an N-carbamoyl chloride (Scheme 78b).380 The first of these reactions proceeds via O→N rearrangement prior to the lactamization step. The infrared frequencies of bridged urethanes (281: 1710 cm−1; 284: 1770 cm−1) were found to be higher than those of the corresponding planar urethanes (1,3-oxazinan-2-one: 1700 cm−1; oxazolidin-2-one: 1730 cm−1). In contrast to these results, treatment of 276 with phosgene resulted in only 5% yield of 278 (Scheme 77), possibly due to competitive formation of a bis phosgene adduct at the free nitrogen atom in the initially formed intermediate.379
Scheme 78.

Synthesis of Bridged Urethanes by Hall: a) 3-Oxa-1-azabicyclo[3.3.1]nonan-2-one; b) 6-oxa-1-azabicyclo[3.2.1]octan-7-one
Scheme 79.

Synthesis of β-Lactamase Inhibitor Containing 1,6-Diazabicyclo[3.2.1]octan-7-one Scaffold by Mangion
Recently, Mangion and coworkers reported the synthesis of β-lactamase inhibitor MK-7655 featuring a bridged urea (Scheme 79).234 A protocol employing triphosgene in the presence of Hünig’s base, followed by treatment with dilute phosphoric acid gave the highest yield. Other carbonyl sources than triphosgene proved ineffective in promoting the cyclization to the bridged urea. The authors proposed that phosphoric acid aids in conversion to 286 by hydrolyzing the intermediate trichloromethyl carbamate. The bridged urea 286 proved sufficiently stable to allow synthesis of MK-7655 in three more steps.
6.2.2. Bridged Oxazinolactams, Oxazinourethanes and Diazines
In 2000, Shea reported the synthesis of bridged oxazinolactams and oxazinourethanes using type II intramolecular nitroso Diels-Alder reaction (Scheme 80).382 This study followed the successful application of acyl-imino Diels-Alder reaction to the synthesis of bridged lactams by the Shea group (see Scheme 33).297,298 The reactive N-acylnitroso dienophiles 290 were generated in situ from the corresponding hydroxamic acids.382 Oxidation using Et4NIO4led to bridged oxazinolactams containing [4.3.1] and [5.3.1] ring systems in high yield. Other oxidants were used for preparation of bridged oxazinourethanes (such as 289c–d).382 The synthesis of [6.3.1] ring system required milder conditions due to decomposition of the acylnitroso dienophile.383 This result was explained on the basis of developing transannular strain in the transition state leading to the 9-membered ring. Ultimately, the desired [6.3.1] oxazinolactam was formed when the N-acylnitroso dienophile was generated thermally from the dimethyl-acetylene cycloadduct (Scheme 81).383 In this case, the more flexible tether led to competitive formation of the regioisomeric bridged oxazinolactam containing [6.2.2] ring system. The X-ray structures of oxazinolactams bearing [4.3.1], [5.3.1] and [6.3.1] ring systems showed that N-oxy amide bonds exhibit comparable degree of strain to the analogous bridged lactams383 (289a: τ = 3.5°; χN = 54.8°; χC = 0.4°; N–C(O) = 1.406 Å; C=O = 1.217 Å; 289b: τ = 10.4°; χN = 52.6°; χC = 1.5°; N–C(O) = 1.398 Å; C=O = 1.219 Å; 293: τ = 16.4°; χN = 49.0°; χC = 4.1°; N–C(O) = 1.388 Å; C=O = 1.219 Å; see Section 4.3 for data on the corresponding bridged lactams298). The authors proposed that the release of transannular interactions in medium-sized rings led to the counterintuitive increase of twist angles with larger ring sizes in this series.
Scheme 80.

Synthesis of Unsubstituted Bridged Oxazinolactams via Type II N-Acylnitroso Diels-Alder Reaction by Shea
Scheme 81.

Synthesis of Unsubstituted [6.2.1] Bridged Oxazinolactamvia Type II N-Acylnitroso Diels-Alder Reaction by Shea
Shea extended this methodology to substituted bridged oxazinolactams (Scheme 82).384 Interestingly, 2-alkyl and 2-oxy-substituted hydroxamic acid precursors resulted in complementary diastereoisomers of the bridged products. This outcome was explained by dipole minimalization in the transition state. High diastereoselectivity was also observed with 3- and 4-substituted hydroxamic acid substrates.
Scheme 82.

Synthesis of Substituted Bridged Oxazinolactams via Type II N-Acylnitroso Diels-Alder Reaction by Shea
Following this account, Shea achieved the synthesis of bridged 1,2-diazines via type II intramolecular N-acylazo Diels-Alder reaction (Scheme 83).385 Initially, the required acylazo dienophiles were generated in situ from the corresponding hydrazides using n-Bu4NIO4 as the oxidant. Subsequently, a two-step protocol involving NBS-promoted synthesis of acylazo precursors and ZnCl2-catalyzed Diels-Alder cycloaddition was developed. This methodology was applied to the synthesis of bridged diazines containing [4.3.1] and [5.3.1] ring systems. The X-ray structures of bridged 1,2-diazines were found to match those of the corresponding bridged oxazinolactams (298a: τ = 0.8°; N–C(O) = 1.394 Å; C=O = 1.216 Å; 298b: τ = 17.6°; N–C(O) = 1.401 Å; C=O = 1.219 Å).385 An unsuccessful attempt to prepare a related bridged diazine with a [3.2.1] ring system was reported by Hoornaert and coworkers.386
Scheme 83.

Synthesis of Bridged 1,2-Diazines via Type II N-Acylazo Diels-Alder Reaction by Shea
In 2005, Shea reported an asymmetric synthesis of bridged oxazinourethane 301 (Scheme 84).387 Treatment of the hydroxamic acid precursor with tert-butyl hydrogenperoxide in the presence of a catalytic amount of ruthenium salen complex 302 at 15 °C under high dilution conditions afforded the desired product with high levels of enantioinduction and in excellent yield. The authors proposed a catalytic cycle, in which the active Ru(IV) catalyst 302b performs a dual role: (1) dehydrogenation of the N-hydroxy formate ester 300 to produce an ion pair 302c, and (2) catalysis of the Diels-Alder reaction to give the bridged oxazinourethane 301 (Scheme 85).
Scheme 84.

Asymmetric Synthesis Bridged Oxazinolactams via Dual Function Catalysis by Shea
Scheme 85.

Proposed Mechanism for Diels-Alder Reaction of Substrate 300
6.2.3. Bridged Barbiturates, Oxazolinediones and Hydantoins (“Smissmanones”)
In a 1964 report, in analogy to known drugs containing planar imide bonds, Smissman proposed bridged bicyclic imides 307–310 as potential anticonvulsant agents (Figure 11).237 However, over the next 20 years, Smissman and coworkers found that synthesis of the proposed bridged imides 307–310 was more difficult than initially expected.366–371 Attempts to prepare bridged barbituric acids, bridged amides or bridged imides via intramolecular alkylation approaches (Scheme 86)366–370 or via intramolecular condensation methods (Scheme 87)371 were unsuccessful due to the propensity of the tested precursors to react with electrophiles at the oxygen rather than nitrogen atom. Moreover, Smissman found that structures of bridged imides reported in literature by other groups were incorrect (Scheme 88).388,389
Scheme 86.

Early Attempts to Synthesize Bridged Lactams from Barbituric Acids by Smissman: a) Alkylation of 5-Iodoalkylbarbituric Acid; b) Electrophilic Activation of N-Allylbarbituric Acid; c) Alkylation of N-Haloalkylbarbituric Acid
Scheme 87.

Early Attempts to Synthesize Bridged Lactams from Amides and Imides by Smissman: a) Intramolecular Condensation; b) Intramolecular N-Alkylation
Scheme 88.

Revision of Initially Reported Structures of Bridged Barbituric Acids: a) Barbituric Acid 322 Reported by Meltzer and Lewis; b) Barbituric Acid 325 Reported by Baumler (please use double-column format for Scheme 88)
Inspired by Smissman’s work,390 in 1984, Brouillette accomplished the synthesis of bridged 2,4-oxazolinediones containing [4.2.1] and [5.2.1] ring systems using acylation of α-hydroxy lactams with aryl chloroformate as a key step (Scheme 89).391 Thus, the reaction of lactams 330 with 4-nitrophenyl chloroformate in refluxing toluene provided bridged oxazolinediones 331b–c in 44–90% yields. Under the same experimental conditions, the corresponding oxazolidinedione containing a [3.2.1] scaffold was not formed, likely because of the higher strain present in this ring system.391 The X-ray structure of oxazolinedione 331b391 indicated that the endocyclic amide bond is more distorted than the exocyclic bond (i.e., the endocyclic N–C(O) bond is 0.038 Å longer and the endocyclic C=O bond 0.014 Å shorter than the corresponding N–C(O) and C=O exocyclic bonds). Later, the X-ray structure of bridged oxazolidinedione 331c was reported by Kubicki (331c: endocyclic N–C(O) = 1.368 Å; C=O = 1.211 Å; exocyclic N–C(O) = 1.383 Å; C=O = 1.193 Å).392
Scheme 89.

Synthesis of Bridged 2,4-Oxazolinediones by Brouillette
Subsequently, Brouillette achieved the first synthesis of bridged hydantoins containing [4.2.1]393 and [5.2.1]240 ring systems (Scheme 90). The exposure of carboxyamides 335 to Pb(OAc)2 resulted in the Hoffman rearrangement to the intermediate isocyanates, which underwent intramolecular attack by the lactam nitrogen atom to afford the desired bridged hydantoins in 48–67% yields. The conditions using 2,6-lutidine in the absence of alcohol proved crucial for the efficient intramolecular N-alkylation of the intermediate isocyanates. Bridged hydantoins were distinguished from the potential O-alkylated products on the basis of 15N NMR experiments (336b: bridgehead nitrogen atom 139.1 ppm; 7-methoxy-3,4,5,6-tetrahydro-2H-azepine: 233.1 ppm).393
Scheme 90.

Synthesis of Bridged Hydantoines by Brouillette
Finally, Brouillette revised394 the structure of bridged barbituric acid 338 as originally reported by Smissman395 (Scheme 91), while Gmünder and Lindenmann reported the attempted synthesis of bridged barbituric acid 341 with an alternative bond connectivity (Scheme 92).396 These examples underline the difficulty of ring closure using N-alkylation and N-acylation via amide nitrogen atom to obtain strained lactams. In recognition of Smissman’s work, bridged amides with the general structures 307–310 are generally referred to as “smissmanones”.241
Scheme 91.

Revision of Structure of Bridged Barbituric Acid Initially Reported by Smissman
Scheme 92.

Attempted Synthesis of Bridged Barbituric Acid by Gmünder and Lindenmann
6.3. Bridged Enamines
Enamines in which nitrogen atom is placed at the bridgehead position in a bicyclic ring system are subject to similar geometrical restrictions as bridged lactams.397 In cases where resonance interaction between the nitrogen lone pair and the π electrons of the double bond is limited, the characteristic nucleophilicity of the carbon atom of enamines decreases with HOMO orbital localized at the nitrogen atom in these systems.
In 1985, Doering published a seminal report on the conjugative interaction of bridged enamines using compound 347 containing a [3.2.2] ring system (Scheme 93).398 The required enamine was prepared from 1-azabicyclo[2.2.2]octan-3-one 344 in a four-step sequence (Scheme 93a). The final step involved equilibration of the allyl amine 346 to the bridged enamine with LiNMe2–HMPA and subsequent fractional crystallization of the corresponding HI salt. Doering demonstrated that the bridged enamine 347 can be equilibrated to 346 using LiNMe2–HMPA or RuH(NO)(PPh3)3 (Scheme 93b).398 The equilibrium constant of 1.0 for both reactions indicates the lack of conjugative interaction between the enamine nitrogen and the olefin double bond in this [3.2.2] ring system. In contrast, planar enamines favor the enamine form by approx. 4 kcal/mol.397
Scheme 93.

a) Synthesis of 1-Azabicyclo[3.2.2]non-2-ene by Doering; b) Equilibrium between Bridged Allylic Amine and Enamine
In 2006, Crawley and Funk reported the synthesis of bridged enamine containing a [3.3.2] ring system during their approach to communesin B (Scheme 94).399 The enamine 349 was formed via a gold-catalyzed 7-exo-dig cyclization of a piperidine nitrogen onto a tethered alkyne in 348. Later, it was found that this cyclization also proceeded spontaneously at room temperature.399 The X-ray structure of enamine 349 has been reported (θ = 337.6°; N–C(CH2) = 1.444 Å; C=CH2 = 1.329 Å).399
Scheme 94.

Synthesis of Bridged Enamine via Au-Catalyzed Hydroamination by Funk
In 2008, Ellman reported the synthesis of endocyclic bridged enamines with a [4.3.1] scaffold via a C–H activation/alkenylation/electrocyclization sequence (Scheme 95).400 A catalyst generated in situ from [Rh(coe)2Cl]2 and the electron-rich monophosphine ligand (4-NMe2)PhPEt2 (used in 1:1 ratio) was identified as optimal for this transformation. The reaction efficiency was highly dependent on the substitution of α,β-unsaturated aldimine moiety and the length of tether connecting the alkyne. The proposed mechanism, shown in Scheme 96,400 involves generation of the hydrido(vinyl)rhodium intermediate 351a, its addition to the alkyne in a 2,1-fashion, reductive elimination to give the cyclic aza-triene 351c, and electrocyclization under the reaction conditions.
Scheme 95.

Synthesis of Bridged Bicyclic Enamines by Tandem Rh-Catalyzed C–H Activation/Alkenylation/Electrocyclization by Ellman
Scheme 96.

Proposed Mechanism for Synthesis of 352
Pearson reported the synthesis of bridged enamines via an intramolecular Schmidt reaction318–320 using azidoalkyl olefins or azidoalkyl alcohols (Scheme 97).401 Azides tethered to five-membered rings afforded mixtures of regioisomeric bridged enamines with [2.2.2] and [3.2.1] scaffolds, while six-membered ring precursors led exclusively to [3.2.2] bridged systems. The use of alkyl-substituted substrates (cf. aryl as in 356) resulted in olefin isomerization in the final products. The mechanism of this rearrangement proceeds via (1) generation of the carbocation, (2) its trapping by the tethered azide, (3) 1,2-carbon migration with expulsion of nitrogen to give bridged iminium ion, and (4) deprotonation to give the twisted enamine product (Scheme 98). The enamine is reprotonated at nitrogen under the reaction conditionsbut the basic work-up gives the final neutral product. Other bridged enamines based on the quinuclidine skeleton402–405 have been reviewed.406,407
Scheme 97.

Synthesis of Bridged Enamines viaIntramolecular Schmidt Reaction by Pearson
Scheme 98.

Proposed Mechanism for Schmidt Reaction of Azidoalkyl Alcohols
Bridged enamines have also been prepared from bridged lactams (see Section 7.2.4).108,408
6.4. Bridged Iminium Ions
Iminium ions containing nitrogen atom at the bridgehead position cannot achieve full resonance stabilization and are typically considered as carbocations.409–412,102 Although several bridgehead imines and iminium ions have been reported,413–419 only a few of these compounds bear nitrogen atom at the bridgehead position in a bicyclic system.420–424,353,354
During studies on solvolysis of modified Cinchona alkaloids containing good leaving groups at the C–9 position, Hoffmann discovered their rearrangement to 1-azabicyclo[3.2.2]nonanes via bridged iminium ions (Scheme 99).420 High yields were obtained in both the quinine (Scheme 99a) and quinidine (Scheme 99b) series.420 In these systems, 1,2-alkyl shift to give strained non-planar iminium ions is favored by stereoelectronic factors. The participation of the aziridinium ions was ruled out on the basis of control experiments probing the ability of activated Cinchona alkaloids to undergo self-quaternization reactions. This rearrangement was subsequently described as “the first Cinchona rearrangement”.421,422
Scheme 102.

Synthesis of Bridged Sultams via Heck Reaction by Paquette
Hoffmann also reported the synthesis of enantiomerically pure 1-azabicyclo[3.2.2]nonanes in quincorine (Table 7) and quincoridine (Table 8) series.423 Treatment of β-amino iodides 366 and 369 with methanol in the presence of AgOBz resulted in stereoselective rearrangement to α-amino ethers via bridged iminium ions. Other silver salts proved less efficient in promoting the reaction or led to partial epimerization of the desired products. The reaction showed good functional group compatibility, tolerating alkynes, ketones, and esters. Notably, the bridged iminium ions were found to be configurationally stable and no equilibration between the quincorine and quincoridine series was observed.
Table 7.
Rearrangement of Quincorine-Derived 2-Iodomethyl-2-Azabicyclo[2.2.2]octanes via Bridged Iminium Ions by Hoffmann
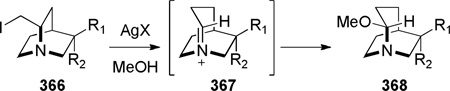 | |||||
|---|---|---|---|---|---|
| entry | 366 | R1 | R2 | AgX | yield of 368 (%) |
| 1 | 366a | CH=CH2 | H | AgOAc | 10 |
| 2 | 366a | CH=CH2 | H | AgOTf | 30 |
| 3 | 366a | CH=CH2 | H | AgOBz | 86 |
| 4 | 366b | Et | H | AgOBz | 66 |
| 5 | 366c | CO2Me | H | AgOBz | 75 |
| 6 | 366d | O= | AgOBz | 76 | |
| 7 | 366e | C≡C-Ph | H | AgOBz | 82 |
| 8 | 366f | C≡CH | H | AgNO3 | 61 |
Table 8.
Rearrangement of Quincoridine-Derived 2-Iodomethyl-2-Azabicyclo[2.2.2]octanes via Bridged Iminium by Hoffmann
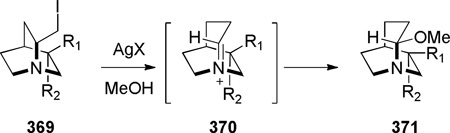 | |||||
|---|---|---|---|---|---|
| entry | 369 | R1 | R2 | AgX | yield of 371 (%) |
| 1 | 369a | CH=CH2 | H | AgOBz | 74 |
| 2 | 369b | Et | H | AgOBz | 62 |
| 3 | 369c | O= | AgOBz | 72 | |
| 4 | 369e | C≡C-Ph | H | AgOBz | 70 |
Applying the same concept, Hoffmann developed the synthesis of functionalized 1-azabicyclo[3.2.2]nonanes from quincorine (Table 9) and quincoridine (Table 10).424 A wide range of carbon, nitrogen and sufur nucleophiles was used to capture bridged iminium ions 373 and 376 with complete diastereoselectivity.
Table 9.
Reactions of Quincorine-Derived Bridged α-Amino Ether 372 with Nucleophiles via Bridged Iminium Ion 373 by Hoffmann
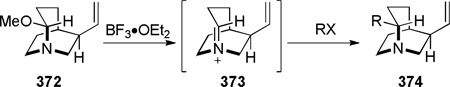 | |||
|---|---|---|---|
| entry | RX | R | yield of 374 (%) |
| 1 | Me3SiCN | -CN | 83 |
| 2 | Me3SiN3 | -N3 | 81 |
| 3 | Bu3SnC≡CH | -C≡CH | 39 |
| 4 | Me2C=C(OMe)OSiMe3 | -C(CO2Me)Me2 | 66 |
Table 10.
Reactions of Quincoridine-Derived Bridged α-Amino Ether 375 with Nucleophiles via Bridged Iminium Ion 376 by Hoffmann
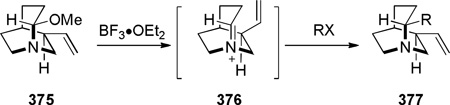 | |||
|---|---|---|---|
| entry | RX | R | yield of 377 (%) |
| 1 | Me3SiCN | -CN | 68 |
| 2 | Me3SiN3 | -N3 | 75 |
| 3 | Bu3SnC≡CH | -C≡CH | 70 |
| 4 | allylMgBr | -CH2CH=CH2 | 48 |
| 5 | Me3SiCH2C≡CH | -CH=C=CH2 | 24 |
| 6 | Me2C=C(OMe)OSiMe3 | -C(CO2Me)Me2 | 70 |
| 7 | Me3SiSC6H5 | -SC6H5 | 59 |
Miyano reported the formation of bridged iminium ions in 1-azabicyclo[3.3.1]nonane system (Scheme 100).353,354 Aziridinium rearrangement of pyrrolizidine 227 provided the bicyclic precursors 229/378 (Scheme 100a; see also Section 5.5). The unexpected stability of these products was proposed to arise from the gauche conformation between the lone pair of electrons at nitrogen and the two C–Cl bonds.353 Both chlorine atoms could be displaced by alkoxides upon heating in alcoholic solvents via bridged cation 379 (Scheme 100b).354 Extensive NMR studies suggested that introduction of two substituents at the C–9 position in 1-azabicyclo[3.3.1]nonane system forces the ring in a double-chair conformation (cf. Section 4.1).
Scheme100.

a) Synthesis of 1-Azabicyclo[3.3.1]nonanes by Miyano; b) Generation of Bridged Iminium Ions from 1-Azabicyclo[3.3.1]nonanes
6.5. Bridged Sultams
In contrast to amide bonds, incorporation of a sulfonamide moiety into a bridged bicylic ring system does not result in its hyperreactivity.425 Bridged sulfonamides (sultams) are the area of considerable current interest due to their potential application in medicinal chemistry426,427 and stereoselective synthesis.428,429 The chemistry of fused sultams has been recently reviewed.430
In the last twenty years, three general approaches to the synthesis of bridged sultams have been reported: i) Heck reactions,292–295,431–436 ii) radical cyclization reactions,437–439 iii) alkylation reactions.440,441
In 1991, Grigg demonstrated that Heck cyclizations of ortho-bromoarylsulfonamides onto tethered alkenes proceed in good yields to give the corresponding bridged sultams (Scheme 101).292 In contrast to the analogous Heck reaction leading to bridged lactams (see Schemes 28 and 29),291–294 sulfonamide-containing substrates allowed for the preparation of sultams bearing [3.2.1] and [3.2.2] ring systems.292 This trend was explained by the lower rotational barrier around the N–S(O)2 bond in sulfonamides as compared to the N–C(O) bond in amides. The regioselectivity of the reaction leading to the [3.2.2] ring system was significantly improved by using a catalyst system comprising of Pd(OAc)2, PPh3, and Tl2CO3.292 Subsequently, Grigg investigated cascade and one-pot RCM-Heck reactions to prepare bridged sultams.293,294 Recently, a similar Heck reaction protocol was utilized by Paquette to construct bridged sultams containing [4.3.1] and [4.2.1] ring systems (Scheme 102),295,431 while Evans reported the synthesis of bridged sultams containing [3.2.1] scaffolds via a tandem Heck reaction/hydrogenation process (Scheme 103).432–436
Scheme 101.

Synthesis of Bridged Sultams via Heck Reaction by Grigg
Scheme 103.

Synthesis of Bridged Sultams via Tandem Heck Reaction/Hydrogenation by Evans
In 1999, Paquette reported a seminal study on the synthesis of bridged sultams via intramolecular radical cyclization reactions of cyclic unsaturated halomethylsulfonamides (Scheme 104).437 This reaction was successfully applied to the synthesis of five different ring systems of unsubstituted bridged sultams, however bridged sultam containing a [2.2.1] scaffold did not form under these reaction conditions. The X-ray structure of 392c indicated O–S–N–lone pair dihedral angles of −90° and 40°. In contrast, the lone pair of electrons at nitrogen in planar sulfonamides is found in a plane bisecting the O–S–O angle. Despite these structural differences, all bridged sultams 392 were found to be hydrolytically stable. Using radical cyclization reaction, Paquette also reported the synthesis of bridged disulfonimide with a [3.2.2] ring system (Scheme 105),438 and complex tricyclic bridged sultams.439 The X-ray structure of 394438 showed a large θ value (394: θ = 345.8°; 392c: θ = 325.2°), indicating a relatively flexible sulfonimide bond in this compound.
Scheme 104.

Synthesis of Bridged Sultams via Radical Cyclization by Paquette
Scheme 105.

Synthesis of Bridged Disulfonimide via Radical Cyclization by Paquette
Subsequently, available ring systems of bridged sulfonamides have been expanded to include bridged sultams bearing sulfur atom at the apex position.440,441 In 2006, Paquette reported the synthesis of a bridged sultam containing a [4.2.1] ring system via an intramolecular alkylation reaction (Scheme 106a).440 Attempts to close the bicyclic system via RCM proved unsuccessful (Scheme 106b).440 More recently, de Meijere and coworkers demonstrated that related bridged sultams can be generated by dialkylation of simple monocyclic precursors (Scheme 107).441 The X-ray structures of several bridged sultams have been reported (400a: θ = 310.2°; 400b: θ = 325.3°; 400d: θ = 340.0°; 400e: θ = 341.6°),441 and indicate progressive decrease of the pyramidalization at nitrogen in this series.
Scheme 106.

a) Synthesis of “Apex” Bridged Sultam by Paquette; b) Unsuccessful Approach via RCM
Scheme 107.

Synthesis of Bridged Sultams via Double Alkylation by de Meijere
An alternative approach to bridged sultams was reported by Hanson and coworkers (Scheme 108).442 As a part of the reaction pairing strategy to access structurally diverse sultams,443–445 these researchers developed a sequential sulfonylation-SNAr protocol starting from ortho-flurobenzenesulfonyl chloride and cyclic amino alcohols to afford bridged sultams with [4.2.1] and [5.2.1] ring systems in good overall yields.442 This protocol was recently employed for the synthesis of a 40-member library of bridged sultams bearing [4.2.1] scaffolds.446
Scheme 108.

Synthesis of Benzofused Bridged Sultams via Sulfonylation/SNAr of Amino Alcohols by Hanson
Finally, Blakey reported the synthesis of heteroatom-derived bridged sultams via rhodium-catalyzed allene sulfonamidation (Scheme 109).447 The reaction showed good substrate scope, allowing for the synthesis of densly functionalized sulfonamides. The mechanism was proposed to involve (1) sulfonamidation to give 2-amidoallylcations, (2) rearrangement to strained N-sulfonylimino-cyclopropanes, and (3) formal [3+3] cycloaddition with acyclic nitrones (Scheme 110).
Scheme 109.

Synthesis of Heteroatom-Derived Bridged Sultams via Rh-Catalyzed Allene Sulfamidation by Blakey
Scheme 110.

Proposed Mechanism for Synthesis of 407
7. REACTIVITY OF BRIDGED LACTAMS AND RELATED HETEROCYCLES
Due to the limited nN→π*C=O conjugation, distorted amides exhibit reactivity dissimilar to traditional amides.57–59 In general, the C=O group is more electrophilic than in planar amides, while the nitrogen atom might behave as a basic amine. Both of these properties depend on the degree of twist of the amide bond; in extreme cases, the amide bond reacts as an isolated amino-ketone rather than an amide, however it is also hydrolytically labile. Conversely, bridged lactams in which the amide bond is close to planarity tend to react as traditional amides and are hydrolytically stable.
For the purpose of this review reactions of bridged lactams are classified depending on the reacting center of the amide bond (that is, carbonyl group or nitrogen atom), and the type of reaction. Because of its historical importance, the hydrolysis of distorted amide bonds is addressed separately. Reactions of heteroatom-containing derivatives of bridged lactams, bridged enamines and bridged sultams are also addressed in this section.
7.1. Hydrolysis of Bridged Lactams
Planar amide bonds are remarkably stable to hydrolysis, with half-life for neutral hydrolysis at room temperature counted in hundreds of years.448 Due to their enhanced electrophilicity, bridged lactams are more susceptible to hydrolysis than planar amides.61 Bridged lactams characterized by large twist angles are hydrolytically unstable, which complicates their synthesis, isolation, and examination of physico-chemical properties, especially in nucleophilic solvents including water.
Between 1958 and 1965, Pracejus studied the hydrolysis of 2-quinuclidones and their hydrochloride salts (Scheme 111).253,254 Near pH = 5, the half-life for methanolysis of the bridged lactam 44 was less than 5 minutes; however, this compound hydrolyzed only very slowly in water at 20 °C (Scheme 111a).254 Under basic conditions, the lactam 44 underwent solvolysis 104 faster than the planar 2-azabicyclo[2.2.2]octan-3-one at 100-times higher alkali concentration.254 The hydrochloride salt 409 was found to be even more labile towards hydrolysis and alcoholysis than the parent amide (Scheme 111b).254 Furthermore, methyl-substituted 2-quinuclidones were found to hydrolyze in the order corresponding to the degree of steric shielding of the amide bond by the neighboring methyl substituents (44 > 46b (endo) > 46a (exo); see Figure 12 for structures of 46a–b).254
Scheme 111.

a) Hydrolysisof 6,6-Dimethylquinuclidin-2-one; b) Hydrolysis of 6,6-Dimethyl-2-quinuclidinium Chloride by Pracejus
Figure 12.

Determination of pKa of 2-quinuclidones by Pracejus (44, 46a–b) and Yakhontov (46c)
Pracejus determined the pKa of methyl-substituted 2-quinuclidones to be 5.33–5.60,254 while Yakhontov established the pKa of a tetramethyl-substituted 2-quinuclidone at 6.37 (Figure 12).256,257 Although these values are consistent with the high basicity of the amide nitrogen in this [2.2.2] ring system, methyl-substituted 2-quinuclidones did not succeed as good models for reactivity of bridged lactams due to steric hindrance around amide bonds.
In the early 1980s, Blackburn261 and Brown262 independently studied the rate of hydrolysis of benzo-fused bridged lactams derived from 2-quinuclidones (Scheme 112). Blackburn observed an increase of seven and nine orders of magnitude in the rate of basic and acidic hydrolysis of the 2-quinuclidone 60, respectively, as compared to the planar 1-phenyl-2-piperidone.261 Brown measured the rate of hydrolysis of several 2-quinuclidone derivatives characterized by different distortion parameters, finding a good relationship between the rate of hydrolysis and twist angles.262 However, for two of these lactams the correlation was better when pyramidalizations at nitrogen were also considered (Table 11).262
Scheme 112.

a) Hydrolysis of Benzo-2-quinuclidone by Blackburn; b) Hydrolysis of Extended Ring Systems of Benzo-2-quinuclidones by Brown
Table 11.
Winkler-Dunitz Distortion Parameters and Hydrolysis Rate Constants of Benzo-2-quinuclidones by Brown
| entry | ring system | τa [deg] |
χNa [deg] |
χCa [deg] |
k3b [M−1s−1] |
k3/K3c [M−1s−1] |
|---|---|---|---|---|---|---|
| 1 | [2.2.2] | 90.0d | 63.4d | 0.0d | 2.6 × 102 | 2.3 × 104 |
| 2 | [3.2.2] | 30.7 | 57.2 | 9.0 | 6.0 × 101 | 5.6 × 101 |
| 3 | [2.3.2] | 33.2 | 52.8 | 11.0 | 1.7 × 101 | 3.0 × 101 |
| 4 | [3.3.2] | 15.3 | 38.6 | 6.7 | 5.1 × 10−4 | 1.2 × 10−4 |
| 5 | planar acetanilidee | 1.5 | 3.7 | −1.5 | 2.2 × 10−5 | 2.2 × 10−7 |
Determined by X-ray crystallography.
Rate constant for base hydrolysis.
Rate constant for acid hydrolysis.
Calculated values.
N-(4-bromo-2-methylphenyl)-N-methylacetamide.
In 1998, Kirby reported the unusual hydrolytic behavior of perpendicularly twisted 1-aza-2-adamantanone (Scheme 113).272,108 This lactam was rapidly hydrolyzed to the ring-opened amino acid when dissolved in water (t1/2 < 50 s). Similarly, the half-life of 0.3 s for acidic hydrolysis at pH = 5 showed that 1-aza-2-adamantanone is significantly more reactive than methyl-substituted 2-quinuclidones (cf. Scheme 111). The pKa value of 5.2 indicated the expected high basicity of the lactam nitrogen atom.108 Interestingly, below pH 4 1-aza-2-adamantanone was quantitatively converted into the protonated orthoamide 412, which at pH = 4.3 existed in equilibrium with the open-form amino acid. Furthermore, the amino acid 411 gave the parent bridged lactam when dissolved in neutral methanol (Scheme 114).108
Scheme 113.

a) Hydrolysis of 1-Aza-2-adamantanone; b) Proposed Mechanismfor Hydrolysis by Kirby
Scheme 114.

Synthesis of 1-Aza-2-adamantanone from the Corresponding Amino Acid by Kirby
Stolz reported that the perpendicularly twisted 2-quinuclidone 52 is rapidly hydrolyzed in water with t1/2 < 15 s (Scheme 115).107 This compound was also unstable in other nucleophilic solvents, including DMSO, pyridine and MeOH, which led to the nucleophilic cleavage of the amide bond.
Scheme 115.

Hydrolysis of 2-Quinuclidonium Tetrafluoroborate by Stoltz
Aubé studied the hydrolysis of one-carbon bridged lactams characterized by medium twist angles (Scheme 116).449 Tricyclic and bicyclic lactams were found to be stable in aqueous solutions under biologically relevant pH conditions (pH = 4–10), however these compounds rapidly hydrolyzed under more acidic or basic pH conditions. Their stability was proposed to result from incorporation of the amide bond into a one-carbon bridge placed across medium-sized heterocycles, where it benefits from the scaffolding effects of medium-sized rings. The spontaneous re-formation of amide 175b from the seco-amide form 416 in water is a notable feature of such systems.
Studies on the hydrolytic stability of bridged lactams bearing the amide bond on the external bridge in a bicyclic system have also been reported. Judd and coworkers found that lactam 133a containing a [3.3.1] ring system was significantly more labile under acidic conditions than the regioisomeric lactams with [4.3.1] ring systems (Figure 13).296 Denzer and Ott discovered that the hydrolytic behavior of bridged lactams containing an additional nitrogen atom at the bridgehead position in [3.3.1] and [3.2.1] ring systems corresponds to the degree of strain associated with the bridged amide bond (Scheme 117).290 Hall investigated the hydrolytic stability of a family of unsubstituted bridged lactams,279 bridged ureas378,379 and bridged urethanes380,381 (Table 12 and Figure 14). Derivatives containing a [3.3.1] ring system were relatively stable under the tested reaction conditions, while the bridged urethane containing a [3.2.1] scaffold rapidly hydrolyzed in water.380
Figure 13.

Hydrolysis of Bridged Homologues of1-Azabicyclo[3.3.1]nonan-2-oneby Judd
Scheme 117.

Hydrolysis of Aza-Bridged Lactams by Denzer and Ott
Table 12.
Hydrolysis of Bridged Lactams and Ureas by Hall
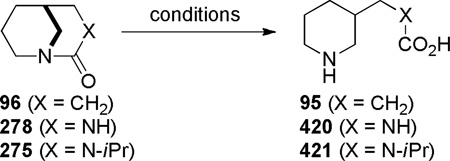 | ||||
|---|---|---|---|---|
| entry | X | lactam | conditions | result |
| 1 | CH2 | 96 | D2O, 28 °C, 7 d | 96 |
| 2 | CH2 | 96 | D2O, 100 °C, 6 h | 96 |
| 3 | CH2 | 96 | D2O, HCl (g), 28 °C | 95 HCl (quant) |
| 4 | N-H | 278 | D2O, 70 °C, 2 h | 278 |
| 5 | N-H | 278 | D2O, HCl (g), 28 °C | 420 HCl (quant) |
| 6 | N-H | 278 | D2O, KOH, 70 °C, 2 h | 278 |
| 7 | N-iPr | 275 | D2O, 100 °C, 20 h | 275 |
| 8 | N-iPr | 275 | 3 M NaOH, 100 °C, 6 h | 420 (quant) |
Figure 14.

Hydrolysis of bridged urethanes by Hall.
Wärnmark investigated acid-catalyzed hydrolysis of twisted bis-amide 167c derived from Tröger’s base (see Figure 8 for structure of 167c).315 A complete hydrolysis was observed in a 0.58 N solution of HCl within 400 min, while kinetic and 18O labeling studies suggested irreversible collapse of the protonated tetrahedral intermediate.315
Arata reported an interesting rearrangement of the one-carbon bridged lactam 225 bearing a leaving group at the bridgehead position on treatment with NaOH (Scheme 118).352 Finally, several theoretical studies addressing the hydrolysis of non-planar lactams have been published.62–66
Scheme 118.

Hydrolysis of 6-Chloro-1-Azabicyclo[4.4.1]undecan-11-one by Arata
7.2. Reactivity of Nitrogen Atom of Bridged Lactams
Two types of reactions of bridged lactams involving the nitrogen atom have been reported: (1) reactions with electrophiles leading to derivatives with intact bicyclic skeleton, and (2) reactions proceeding with scission of the C–NC(O) bond, following electrophilic activation of the lactam nitrogen atom.
7.2.1. N-Protonation and N-Methylation
Although oxygen is the protonation site of planar amide bonds, the limited nN→π*C=O donation in bridged lactams leads to the enhanced basicity of nitrogen.70,71 In general, even moderately distorted bridged lactams favor N-protonation over O-protonation.70,71,284
Pracejus and Yakhontov were the first to show that bridged lactams undergo protonation and methylation reactions at the nitrogen atom (Scheme 119).253–257 The potential products resulting from the cleavage of C–NC(O) bonds were not observed in these systems.
Scheme 119.

N-Protonation and N-Methylation of 2-Quinuclidones by Pracejus and Yakhontov
Kirby demonstrated that 1-aza-2-adamantanone readily forms quaternary ammonium salts upon treatment with Meerwein’s reagent or HCl (Scheme 120).108 The latter product, isolated as the N-protonated hydrate 427, may be considered as a model for a cationic tetrahedral intermediate in acid-catalyzed acyl transfer reactions.450 The X-ray structure of 427108 was characterized by a long C–N bond of 1.552 Å and significantly shortened C–O bonds of 1.382 Å. The stability of 427 resulted from the torsional restriction imposed by the adamantane scaffold.
Scheme 120.

N-Methylation and N-Protonation of 1-Aza-2-adamantanone by Kirby
In an important study, Brown found that methylation of the bridged lactam containing a [3.2.2] ring system takes place at nitrogen, while the less distorted analogue with a [3.3.2] ring system reacts at the oxygen atom under the same experimental conditions (Scheme 121).451 Using DFT calculations, Greenberg predicted that related bridged lactams display higher electron density at nitrogen as compared to planar amides.70,71 Recently, Greenberg reported that protonation of the relatively undistorted 1-azabicyclo[3.3.1]nonan-2-one also proceeds at nitrogen (Scheme 122).284 This finding is important in the light of the hydrolytic stability of 96 (see Table 12)279 and its unstrained structure (see lactam 87 for the amide bond distortion parameters in the [3.3.1] ring system: τ = 20.8°, χN = 48.8°, χC = 5.9°).282,283
Scheme 121.

N-Methylation vs. O-Methylation of Benzo-2-quinuclidones by Brown
Scheme 122.
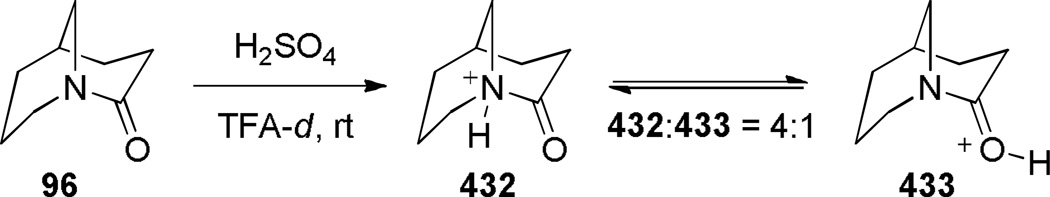
N-Protonation vs. O-Protonation of 1-Azabicyclo[3.3.1]nonan-2-one by Greenberg
Aubé reported several examples of N-protonated and N-methylated lactams bearing a [4.3.1] scaffold (Scheme 123).330 The structures of three of these analogues were examined by X-ray crystallography and provided the first direct crystallographic comparison between any twisted lactam and its corresponding salt. The distortion parameters and bond lengths of the N-protonated lactam 434b330 indicated a significant increase of pyramidalization around the C–N amide bond compared to its neutral analogue (434b: τ = 81.9°; χN = 52.1°; χC = 1.4°; N–C(O) = 1.502 Å; C=O = 1.192 Å; the corresponding bridged lactam 177b: τ = 51.5°; χN = 36.1°; χC = 12.8°; N–C(O) = 1.387 Å; C=O = 1.218 Å).330
Scheme 123.

N-Protonation and N-Methylation of Medium Bridged Twisted Lactams by Aubé
7.2.2. Cleavage of C–NC(O) Bond
Electrophilic activation of the nitrogen atom in bridged lactams may result in the scission of the adjacent C–N bond. Thus, Yakhontov reported cleavage reactions of the C–NC(O) bond in the tetramethyl-substituted 2-quinuclidone using PhLi, PCl5 and acetone cyanohydrin (Scheme 124).256,257 The nucleophile had a pronounced effect on the reaction outcome (cf. Table 14). This reaction was limited to systems capable of forming tertiary carbocations (Scheme 125).256,257
Scheme 124.

Reactions of 6,6,7,7-Tetramethylquinuclidin-2-one with Cleavage of C–NC(O) Bond by Yakhontov
Table 14.
Reactions of 6,6,7,7-Tetramethylquinuclidin-2-one with Cleavage of Amide Bond by Yakhontov
 | ||||
|---|---|---|---|---|
| entry | RH | conditions | 447 | yield (%) |
| 1 | H2O | 100 °C, 0.5 h | 447a | 94 |
| 2 | MeOH | 65°C, 5 h | 447b | 76 |
| 3 | morpholine | Δ, 12 h | 447c | 97 |
| 4 | NH2OH | EtOH, 78 °C, 2 h | 447d | 62 |
| 5 | NH2NH2 | xylenes, Δ Dean-Stark, 5 h |
447e | 63 |
| 6 | NH2NHPh | xylenes, Δ Dean-Stark, 12 h |
447f | 64 |
Scheme 125.

Proposed Mechanism for Synthesis of 436
Aubé reported that one-carbon bridged lactams undergo C–NC(O) bond cleavage reactions under very mild conditions (Scheme 126).208 The mechanism of the MeI-mediated scission was proposed to proceed via the amidinium ion, while the cleavage mediated by DDQ to involve the initial single-electron transfer from the amide bond.208 In all cases, the σ C–N bond furthest away from co-planarity with the carbonyl group in the neutral lactam was selectively cleaved. Tricyclic and bicyclic bridged lactams were also found to participate in regioselective hydrogenolysis reactions of the C–NC(O) bond using Pd(OH)2 in alcoholic solvents (Scheme 127).208 The activation of bridged lactams by hydrogen bonding to nitrogen was proposed to play an important role in facilitating these reactions. In another study, medium-bridged bicyclic lactams containing an internal double bond showed increased reactivity towards the C–NC(O) bond hydrogenolysis (Table 13).343 Recently, Aubé reported the cleavage of a C–NC(S) bond in bridged thioamide containing a [4.3.1] ring system (Scheme 128).452
Scheme 126.

Cleavage of C–NC(O) Bond in Medium-Bridged Twisted Lactams by Aubé: a) Amidinium Salts; b) Oxidative Cleavage
Scheme 127.

Hydrogenolysis of C–NC(O) Bond in Medium-Bridged Twisted Lactams by Aubé
Table 13.
Hydrogenolysis of C–NC(O) Bond in Medium-Bridged Twisted Lactams by Aubé
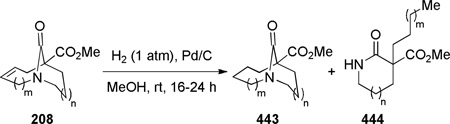 | ||||||
|---|---|---|---|---|---|---|
| entry | 208 | m | n | ring system |
ratio of 443:444 |
combined yield (%) |
| 1 | 208b | 1 | 0 | [4.2.1] | 100:0 | 74 |
| 2 | 208a | 1 | 1 | [4.3.1] | 25:75 | 95 |
| 3 | 208f | 1 | 2 | [4.4.1] | 50:50 | 54 |
| 4 | 208c | 2 | 0 | [5.2.1] | 100:0 | 72 |
| 5 | 208d | 2 | 1 | [5.3.1] | 100:0 | 79 |
| 6 | 208e | 3 | 0 | [6.2.1] | 100:0 | 89 |
Scheme 128.

a) Synthesis of Bridged Thioamide by Aubé; b) Proposed Mechanism Involving Cleavage of C–NC(S) Bond
7.3. Reactivity of Carbonyl Group of Bridged Lactams
The geometric constraints of non-planar amides modify the chemistry of the affected carbonyl group and its derivatives. Typically, amide bonds in bridged lactams are more electrophilic than those in planar amides because of the limited amide bond resonance.67–69 Moreover, torsional restriction imposed by bicyclic scaffolds of bridged lactams often permit isolation of stable tetrahedral intermediates of the amide bond addition reactions.450
For the purpose of this review, the reactivity of carbonyl group of bridged lactams has been classified into the following classes: (1) reactions with heteroatom nucleophiles proceeding with cleavage of amide bonds, (2) reactions with heteroatom nucleophiles proceeding without cleavage of amide bonds, (3) reduction reactions, (4) reactions with organometallic and related reagents, and (5) miscellaneous reactions.
7.3.1. Reactions with Heteroatom Nucleophiles with Cleavage of Amide Bonds
Yakhontov was first to demonstrate the nucleophilic ring opening of bridged lactams with a variety of nucleophiles, such as water, alcohols, amines and hydrazines (Table 14).256,257 The resulting piperidines were isolated in good yields after the basic workup. Yakhontov also observed an unexpected transacylation of the twisted amide bond during reduction of the sterically-hindered 2-quinuclidone 46c with LiAlH4 (Scheme 129). The parent bridged lactam reacted in situ with the amino alcohol product to give amino ester product 449. Concurrently, Pracejus reported a rapid alcoholysis of another 2-quinuclidone under acidic conditions (Scheme 130).253,254 These reactions confirm that amide bonds in bicyclic 2-quinuclidines exhibit properties of isolated amino-ketones.
Scheme 129.

Reduction of 6,6,7,7-Tetramethylquinuclidin-2-one by Yakhontov
Scheme 130.

Alcoholysis of 6,6-Dimethylquinuclidin-2-one by Pracejus
The susceptibility of non-planar amides to nucleophiles has been reported to complicate the synthesis and isolation of some bridged lactams.250 Aszodi and coworkers reported a facile aminolysis of the amide bond in a [3.2.1] bridged lactam 451 during the attempted removal of its p-nitrobenzyl group (Scheme 131).233 Later, this undesired side reaction was minimized by performing the hydrogenolysis in acetone to in situ convert the reactive 4-toluidine into the less nuclophilic N-isopropylaniline.233 Murphy subjected hydrolytically-unstable bridged lactams, prepared by an intramolecular Schmidt reaction, to methanolysis under mild conditions with TfOH and MeOH to facilitate identification of these compounds (Scheme 132).325
Scheme 131.

Aminolysis of [3.2.1] Bridged Lactam by Aszodi
Scheme 132.

Alcoholysis of One-Carbon Bridged Lactams by Murphy
Hall developed a series of polymerization reactions453–455,55 of bridged lactams relying on the inherent strain of amide bonds (Table 15a).279,378–381 Lactam 96 containing a [3.3.1] scaffold279 was more reactive than the corresponding bridged ureas378,379 and urethanes.380,381 The analogous planar lactams, ureas and urethanes did not polymerize under the same reaction conditions (Table 15b).
Table 15.
a) Polymerization of 1-Azabicyclo[3.3.1]nonan-2-one and Related Compounds by Hall; b) Planar Derivatives Inert to Polymerization
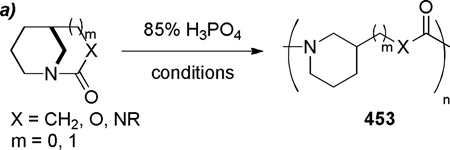 | ||||||
|---|---|---|---|---|---|---|
| entry | X | m | lactam | T [°C] | t (h) | polymerization |
| 1 | CH2 | 1 | 96 | 100 | 0.3 | + |
| 2 | O | 0 | 281 | 105 | 20 | + |
| 3 | O | 1 | 284 | 105 | 27 | + |
| 4 | N-H | 1 | 278 | 98 | 2 | + |
| 5 | N-iPr | 1 | 275 | 200 | 17 | − |
| b) | ||||||
|---|---|---|---|---|---|---|
 |
7.3.2. Reactions with Heteroatom Nucleophiles without Cleavage of Amide Bonds
Diols,108,408 hydrazines232,408 and amines408 have been used as successful nucleophiles to give bridged derivatives of twisted lactams without cleavage of the amide bond. In all reported examples to date, scaffolding effects of adamantane-type or medium-sized rings stabilize the final products.
Kirby and coworkers reported the synthesis of a bridged hemiaminal from 1-aza-2-adamantanone using standard conditions for ketal formation (Scheme 133).108 The same research group prepared a bridged tosylhydrazone as a precursor to amino carbene 459 (Scheme 134),271 which subsequently was shown to react as a singlet carbene.271
Scheme 133.

Synthesis of Bridged Amino-Ketal from 1-Aza-2-adamantanone by Kirby
Scheme 134.

Synthesis and Reactions of Aminocarbene from 1-Aza-2-adamantanone by Kirby
In their search for novel ligands of nicotinic receptor, Coe and coworkers reported the synthesis and Wolff-Kishner reduction of a bridged amino hydrazone to give the tricyclic amine 463 (Scheme 135).232 Impressively, the synthesis of 463 could be conducted on 10–95 mmol scale directly from the ring-opened amino ester 84 (see Scheme 17) without isolating the intermediate bridged lactam and amino hydrazone in excellent overall yields (64–85%). During the course of this study, mixed ethanol-hydrazine intermediates were detected by APCI MS analysis (Scheme 136).232 The bridged hydrazone 462 was obtained upon removal of solvent.
Scheme 135.

Wolff-Kishner Reduction of Bridged Lactam by Coe
Scheme 136.

Proposed Mechanism for Synthesis of Bridged Amidine 462
Finally, Aubé reported the synthesis of bridged amino ketals and amidines from moderately distorted tricylic lactams containing [4.3.1] ring system (Scheme 137).408 These reactions demonstrate that the amide bond does not need to be fully perpendicular to exhibit ketone-like properties.
7.3.3. Reduction of Carbonyl Group
The reduction of bridged lactams occurs under mild conditions to give hemiaminals, which are frequently isolated because of the geometrical constraints prohibiting their further reduction via bridged iminium ions.450
Denzer and Ott were among the first to demonstrate the reduction of bridged lactams with NaBH4, which is considered unreactive towards planar amides (Scheme 138).290 The resulting hemiaminal collapsed to the aldehyde, which underwent further reduction. Interestingly, hydrogenation of 469 over PtO2 resulted in the cleavage of methylene bridge to afford 1,5-benzodiazocinone 471. Subsequently, Coe and coworkers used NaBH4 or LiAlH4 to obtain isolable hemiaminal product from an adamantane-derived bridged lactam (Scheme 139).232 It was proposed that the bis-axially bridged piperidine ring system provided an additional degree of stabilization in this system. Aubé reported that moderately distorted medium-bridged lactams efficiently react with NaBH4, and that the resulting hemiaminals are stable to reaction and isolation conditions (Scheme 140).408 In some cases, mixtures of hemiaminals and primary alcohols were obtained (Scheme 141).408 When an electron-withdrawing group was α to the twisted amide bond, an unusual C–C bond cleavage reaction was observed to occur completely exclusive to breakage of the alternative C–N bond (Scheme 141).408
Scheme 138.

Reduction of 1,5-Diazabicyclo[3.3.1]nonan-2-one by Denzer and Ott
Scheme 139.

Reduction of Adamantane-Derived Bridged Lactam by Coe
Scheme 140.

Reduction of Medium-Bridged Twisted Lactams with Formation of Isolable Hemiaminals by Aubé
Scheme 141.

Reduction of Medium-Bridged Twisted Lactams with Collapse of Hemiaminals by Aubé
Arata studied the reduction of one-carbon bridged lactam containing a leaving group α to the twisted amide bond (Scheme 142a).352 Thus, lactam 225 was converted to hemiaminal 479 by electron transfer reduction using Na/NH3,or by the two-stage hydrogenolysis of the C–Cl bond, followed by amide bond reduction with LiAlH4. Notably, the product of this reaction was the hemiaminal instead of fully reduced amine, as would be expected for planar amides under LAH reduction conditions. However, bridged hemiaminal 479 could be readily converted into the parent 1-azabicyclo[4.4.1]undecane (Scheme 142b). In contrast to the above conversions, the direct reduction of lactam 225 with LiAlH4 afforded quinazoline 478. The proposed mechanism for this reaction is shown in Scheme 143.
Scheme 142.

a) Reduction of Chloro-Substituted Bridged Lactam by Arata; b) Synthesis of 1-Azabicyclo[4.4.1]undecane
Scheme 143.

Proposed Mechanism for Rearrangement of 225
Several examples documenting the reduction of bridged lactams in total synthesis of natural products have been reported. The Thomas group utilized hemiaminal 481, prepared by LiAlH4 reduction of the bridged lactam 480, as a precursor to the corresponding tricyclic amine in their approach towards the tropane alkaloid stemofoline (Scheme 144).372,373 Attempts to deoxygenate the bridged hemiaminal by employing S-methyl xanthate or halides under free-radical conditions were unsuccessful.373 Ultimately, the desired tricyclic amine was prepared in high yield via an intermediate acetate. Several alkaloids from Amaryllidaceae family contain bridged hemiaminals in a [3.2.1] benzofused ring system (Scheme 145).183,188 The Wildman group investigated the reduction of oxohaemanthidine 484 to the corresponding hemiaminal using NaBH4 in methanol,183 while Hendrickson and coworkers synthesized 6,11-dihydroxycrinene 487 under similar conditions.188 Dolby and Sakai studied the reduction of a bridged lactam bearing a relatively flexible [6.2.2] ring system (Scheme 146).311,312 In this case, standard treatment with LiAlH4 achieved exhaustive reduction to the corresponding amine. This suggests that the properties of the amide bond in the [6.2.2] ring system embodied in 488 are similar to those of planar amides. Finally, Harley-Mason determined that a bridged lactam containing a [5.2.2] ring system gives a stable hemiaminal on treatment with triethyloxonium tetrafluoroborate, followed by reduction with sodium borohydride (Scheme 147).155 Attempts to prepare the corresponding amine were unsuccessful, leading only to the reduction of the ester group in the starting material. The bridged hemiaminal 490 was subsequently used as a key intermediate in the synthesis of indole alkaloid condylocarpine (see Scheme 178).155
Scheme 145.

Reduction of Bridged Lactams Derived from Amaryllidaceae Alkaloids by: a) Wildman; b) Hendrickson
Scheme 146.

Reduction of Indole-1-azabicyclo[6.2.2]dodecan-10-one by Dolby
Scheme 147.

Synthesis of Bridged Hemiaminal from Bridged Lactam by Harley-Mason
Scheme 178.

Synthesis of Indole Alkaloids by Harley-Mason: a) Tubifoline and Condyfoline; b) Geissoschizoline; c) Fluorocurarine; d) Condylocarpine (please use double-column format for Scheme 178)
7.3.4. Reactions with Organometallic and Related Reagents
Besides hydride, addition of more sterically-demanding organometallic reagents to bridged lactams has been reported to yield stable hemiaminals.450
By subjecting bicyclic and tricyclic bridged lactams containing a [4.3.1] ring system to organometallic reagents of increasing steric demand, Aubé has shown that the resulting adducts can range from products having stable tetrahedral carbon to the collapsed amino ketone isomers (Tables 16–17).408 Bicyclic lactams afforded the corresponding amino ketones (Table 16), but in contrast the parent tricyclic lactams were found to be more effective in stabilizing the hemiaminal products (Table 17). The observed difference in reactivity was explained on the basis of the scaffolding effect provided by the additional six-membered ring in the latter system. In some cases, intermediary products featuring intramolecular N…C=O interactions involving nN→π*C=O transitions in nine-membered amino ketones were observed.408
Table 16.
Addition of Organometallic Reagents to Medium-Bridged Bicyclic Lactams by Aubé
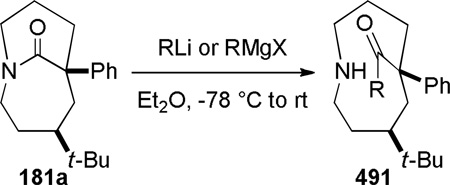 | ||||
|---|---|---|---|---|
| entry | RLi/RMgX | R | 491 | yield (%) |
| 1 | MeLi | Me | 491a | 89 |
| 2 | MeMgI | Me | 491a | 73 |
| 3 | n-BuLi | n-Bu | 491b | 83 |
| 4 | sec-BuLi | sec-Bu | 491c | 93 |
| 5 | tert-BuLi | tert-Bu | 491d | 80 |
Table 17.
Addition of Organometallic Reagent to Medium-Bridged Tricyclic Lactams by Aubé
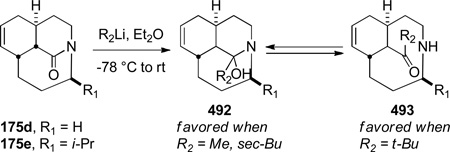 | ||||||
|---|---|---|---|---|---|---|
| entry | 177 | RLi | R | 487/488 | yield (%) | |
| 1 | 177d | MeLi | Me | 487a | 95 | |
| 2 | 177d | sec-BuLi | sec-Bu | 487b | 80 | |
| 3 | 177d | tert-BuLi | tert-Bu | 488a | 90 | |
| 4 | 177e | MeLi | Me | 487c | 92 | |
| 5 | 177e | sec-BuLi | sec-Bu | 487d | 88 | |
| 6 | 177e | tert-BuLi | tert-Bu | 488b | 90 | |
The same group established that bridged lactams can function as effective substrates for the synthesis of bridged α-aminoepoxides using dimethylsulfonium methylide under Corey-Chaykovsky epoxidation conditions (Scheme 148).456 The success of this reaction relied on both the increased electrophilicity of distorted amide bonds and the stability of these bridged aminoepoxides to the reaction and isolation conditions due to the limited nN→σ*C-O delocalization.
Scheme 148.

Corey-Chaykovsky Epoxidation of Bridged Lactams by Aubé
Olefination of bridged lactams provides facile access to bridged enamines featuring decreased overlap between the lone pair of electrons of nitrogen and the olefin π systems (see Section 6.3).397–398 Kirby demonstrated that 1-aza-2-adamantanone undergoes the Wittig reaction to give a bridged enamine under standard conditions employed for olefination of ketones (Scheme 149a),108 Aubé utilized a Petasis olefination to afford exo-cyclic enamine with a [4.3.1] ring system (Scheme 149b),408 while Wärnmark functionalized Tröger’s base-derived bridged lactams using the less reactive carbethoxymethylene-triphenylphosphorane under thermal conditions (Scheme 149c).316
Scheme 149.

a) Wittig Olefination of 1-Aza-2-adamantanone by Kirby; b) Petasis Olefination of Medium Bridged Twisted Lactams by Aubé; c) Wittig Olefination of Tröger’s Base-Derived Twisted Lactams by Wärnmark
During synthetic studies on Aspidosperma alkaloids, Ban demonstrated that under certain conditions cyanide adds to bridged lactam containing a [6.3.1] ring system (Scheme 150).457 Structure of 499 was confirmed by X-ray crystallography. The proposed mechanism for this rearrangement involves cleavage of the bridged amide bond to give the reactive amino acyl cyanide, which undergoes further transformations (Scheme 151).
Scheme 150.

Cyanide-Induced Rearrangement of Indole-1-azabicyclo[6.3.1]dodecan-12-one by Ban
Scheme 151.

Proposed Mechanism for Cyanide-Induced Rearrangement of Lactam 498
7.3.5. Other Reactions Involving Carbonyl Group
Ban has studied164 the alkylation of bridged lactams with a [6.3.1] ring system at the bridgehead position458 (Table 18). In this system, the bridgehead enolate was readily formed in the presence of LDA at −78 °C, but only a few electrophiles could be used to install C-electrophiles.164 For example, when the bridgehead enolate was treated with methyl bromoacetate, the corresponding α-bromo lactam was formed as the major product.
Table 18.
Alkylation of Indole-1-azabicyclo[6.3.1]dodecan-12-one at Bridgehead Position by Ban
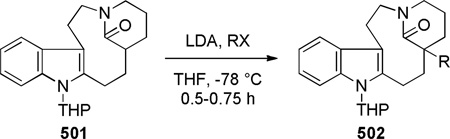 | ||||
|---|---|---|---|---|
| entry | RX | R | 502 | yield (%) |
| 1 | EtI | Et | 502a | 84 |
| 2 | allylBr | allyl | 502b | 71 |
| 3 | ICH2CO2Me | I | 502c | 42 |
| 4 | BrCH2CO2Me | Br | 502d | 34 |
| 5 | ClCH2CO2Me | COCH2Cl | 502e | 66 |
| 6 | (CO2Me)2 | COCO2Me | 502f | 90 |
Schill reported a Friedel-Crafts-type α-arylation of bridged lactam featuring [5.4.1] ring system (Scheme 152).337Although these authors also accomplished a related α-arylation of [4.4.1] ring system,339 the reaction of a bridged lactam with [4.3.1] scaffold was unsuccessful due to its instability to the reaction conditions.336
Scheme 152.

α-Arylation of Indole-1-azabicyclo[5.4.1]dodecan-12-one by Schill
Finally, Judd demonstrated that a complex bridged lactam containing an α,β-unsaturated amide bond on the external bridge in a [4.3.1] ring system could be further functionalized by rhodium-catalyzed conjugate addition of boronic acid with complete diastereoselectivity (Scheme 153).296
Scheme 153.

Rh-Catalyzed Conjugate Addition of Boronic Acid to 1-Azabicyclo[4.3.1]dec-7-en-9-one by Judd
7.4. Miscellaneous Reactions of Bridged Lactams
Shea reported oxidation and hydrolysis reactions of a bridged lactam containing an additional bridgehead olefin in a [3.3.1] ring system (Scheme 154).298 The authors proposed a complex mechanism involving two molecules of the bridged lactam. The moderate yield of the hydrolysis was proposed to be a result of decomposition of the bridged lactam under the reaction conditions.
Paquette investigated photocyclization reactions of bridged lactams containing diene motifs (Schemes 155–156).295 The required substrates were prepared from the monounsaturated bridged lactams via a bromination/elimination sequence (Scheme 155).295 The best results in the elimination step were achieved using TBAF in DMSO as a solvent at high temperature.459 Upon irradiation with 300 nm light, bridged lactam 508, containing a [4.3.1] ring system, underwent disrotatory ring closure to cyclobutene, while bridged lactam 509 with a [4.3.2] scaffold was unreactive under the same conditions (Scheme 156).295 On the basis of computational studies, the authors proposed that a non-planar arrangement of the diene moiety in lactam 509 contributed to this divergence in reactivity. It is worth noting that under similar irradiation conditions bridged sultams react with a preferential cleavage of the N–SO2 bond (see Section 7.7.2).295,431
Scheme 155.

Synthesis of Bridged Lactams with Endocyclic Diene Motifs by Paquette
Scheme 156.

Photochemical Reactions of Bridged Lactams Bearing Endocyclic Dienes by Paquette
The X-ray structures of oxidation products derived from two other bridged lactams have been reported (Figure 15). Lactam 511a was prepared by epoxidation of the corresponding olefin by Paquette (511a: τ = 9.6°; χN = 28.3°; χC = 3.3°; N–C(O) = 1.363 Å; C=O = 1.231 Å).460 The observed diastereoselectivity is similar to that observed in the epoxidation of related bridged sultams (see Scheme 166).434 Aubé reported the X-ray structure of lactam 511b prepared by Sharpless dihydroxylation/acylation of the corresponding olefin.208 As indicated by its Winkler-Dunitz parameters (τ = 72.4°; χN = 49.8°; χC = 7.1°; N–C(O) = 1.418 Å; C=O = 1.212 Å), this lactam contains one of the most distorted amide bonds isolated to date.
Scheme 157.

Reactivity of Bridged Oxazinolactamsby Shea: a) Reduction; b) Alkylation
7.4. Reactivity of Heteroatom-Containing Derivatives of Bridged Lactams
In contrast to bridged lactams, the reactivity of their heteroatom-containing derivatives has been sparsely studied. Relevant studies on these heterocycles are limited to transformations of bridged oxazinolactams and 1,2-diazines developed by Shea.383–385
Bridged oxazinolactams, prepared in the intramolecular N-acylnitroso Diels-Alder reaction (see Section 6.2.1),382–384 have been shown to undergo hydrogenation of the bridgehead olefin over Pd/C, while Na(Hg) chemoselectively cleaved the N–O bond (Scheme 157a).383,384 Shea has also achieved highly diastereoselective alkylation of bridged oxazinolactams using reactive electrophiles (Scheme 157b).384 The alkylation occurs from the more accessible exo face, providing complementary stereochemical outcome to the isomers obtained directly from the N-acylnitroso Diels-Alder reaction (Scheme 82).384 This methodology has been applied to the stereocontrolled synthesis of seven- and eight-membered lactams (Scheme 158),384 and later expanded to include bridged 1,2-diazines (Scheme 159).385
Scheme 158.

Stereoselective Synthesis of Medium Ring Lactams from Oxazinolactams by Shea
Scheme 159.

Reactivity of Bridged 1,2-Diazines by Shea
In 2007, Shea and coworkers reported the use of bridged oxazinolactams in their approach towards synthesis of stenine (Scheme 160).461 The intramolecular N-acylnitroso Diels-Alder reaction of diene 520 gave the bridged tricyclic lactam 521 in good yield and diastereoselectivity. Reduction with Na(Hg) afforded the azepane 522, which was elaborated to the key intermediate 523 in four steps.
Scheme 160.

Application of Bridged Oxazinolactams in Synthetic Studies towards Stenine by Shea (please use double-column format for Scheme 160)
7.5. Reactivity of Bridged Enamines
Although planar enamines are nucleophilic at carbon, bridged enamines are expected to exhibit properties of isolated amino-olefins as a result of the limited nN→π*C=C overlap.397,398 (Doering memorably commented “Of the conventional chemistry of enamines not a vestige remains.”398) However, reports on the reactivity of bridged enamines are relatively rare compared to bridged lactams.
Kirby showed that the bridged enamine derived from 1-aza-2-adamantane is alkylated at nitrogen using MeI in high yield (Scheme 161a).108 This finding is in good agreement with the reactivity of another bridged enamine containing nitrogen at the apex position in a [3.3.1] ring system reported by Kresge (Scheme 161b).462 The ammonium salt 525 was characterized by the X-ray crystallography (θ = 326.4°; N–CH3 = 1.503 Å; N–C(=CH2) = 1.474 Å; C=CH2 = 1.301 Å).108
Scheme 161.

a) N-Methylation of 3,5,7-Trimethyl-2-methylene-1-azaadamantane by Kirby; b) N-Protonation of 9-Methyl-9-Azabicyclo[3.3.1]non-1-ene by Kresge
Ellman examined the reactivity of endocyclic bridged enamines containing a bridgehead double bond (Scheme 162).400 The alkylation with Me2SO4 also proceeded at the nitrogen atom, while the reduction with NaBH4 afforded 529 as the only isolated product. Mechanistic investigation suggested that this reduction proceeds via the corresponding iminium ion. Finally, hydrogenation over Pd/C afforded the fully saturated bicyclic amine as a single diastereoisomer. The X-ray structure of ammonium salt 528 has been reported (θ = 333.6°; N–CH3 = 1.511 Å; N–CH(=C) = 1.490 Å; CH=C = 1.330 Å).400
Scheme 162.

Reactivity of Endocyclic [4.3.1] Bridged Enamines by Ellman
Pearson studied the reactivity of bridged enamines prepared by the intramolecular Schmidt reaction (Scheme 163).401 It was found that hydrogenation and bromination reactions of these enamines proceed in high yields to give bicyclic amines bearing a close resemblance to core common to several Cinchona alkaloids.
Scheme 163.

Reactivity of Bridged Enamines Prepared by Schmidt Reaction by Pearson: a) [3.2.2] Scaffold; b) [2.2.2] Scaffold
7.6. Reactivity of Bridged Sultams
While bridged sultams contain non-planar sulfonamide bonds, these compounds do not show any signs of hyperreactivity.437 It means that their properties can be examined under the conditions which would result in rapid decomposition of the analogous bridged lactams.
7.6.1. General Reactivity
The first reactions of bridged sultams were described in 2001 by Paquette. It was shown that a sultam containing [4.2.1] ring system undergoes smooth deprotonation with tert-butyllithium to give, after quenching with allyl or benzyl bromide, the substitution products in good yields (Scheme 164).438 The same ring system was found to be stable to highly oxidizing conditions with chromyl acetate, providing the α-amino acetate and the bridged keto-sultam in good selectivity (Scheme 164).438,431 Paquette also reported the synthesis of bridged sultams containing diene motifs via bromination/olefination of the corresponding olefins under relatively harsh conditions (Scheme 165).295,440,431,459 The diene products were later used in the photocyclization studies.295,440,431
Scheme 164.

Alkylation and Oxidation of Bridged Sultams by Paquette
Scheme 165.

Bromination/Elimination of Bridged Sultams by Paquette
More recently, Evans studied reactions of unsaturated benzofused bridged sultams with bromine and m-CPBA (Scheme 166).434 In all cases, complete diastereoselectivity was observed, which was ascribed to the steric effect of the aryl group. Interestingly, the bridged sultam bearing an electron-rich aromatic ring participated in a cationic rearrangement upon treatment with bromine in CHCl3.434 Proposed mechanism for this transformation is outlined in Scheme 167.
Scheme 166.

Bromination and Epoxidation of Benzo-Bridged Sultams by Evans
Scheme 167.

Proposed Mechanism for Synthesis of 544
The groups of Paquette463 and Evans434 further extended the ability to functionalize bridged sultams by developing a set of cross-coupling reactions (Table 19). Halogen-lithium exchange followed by quenching with diverse electrophiles or palladium-catalyzed cross-coupling with aryl boronic acids proceeded efficiently, delivering a variety of functionalized bridged sultams.463 Evans also demonstrated that hydrogenation of unsaturated bridged sultams bearing [3.2.1] ring system proceeds with high diastereoselectivity (Scheme 168).434 In contrast to hydrogenolysis of bridged lactams (see Section 7.2.2),208 the cleavage of N–SO2 bond was not observed.
Table 19.
Cross-coupling Reactions of Bridged Sultams by Paquette and Evans
 | ||||
|---|---|---|---|---|
| entry | conditions | R | 548 | yield (%) |
| 1 | MeLi/Me2SO4 | -CH3 | 548a | 70 |
| 2 | MeLi/BrCH2Ph | -CH2Ph | 548b | 58 |
| 3 | tBuLi/ClCOtBu | -C(O)tBu | 548c | 44 |
| 4 | tBuLi/ClCOOBn | -C(O)OBn | 548d | 33 |
| 5 | MeLi/TMSCl | -SiMe3 | 548e | 40 |
| 6 | tBuLi/Bu3SnCl | -SnBu3 | 548f | 57 |
| 7 | tBuLi/PhSeCl | -SePh | 548g | 55 |
| 8 | Pd(PPh3)4, CuI, HC≡CC4H9 | -C≡CC4H9 | 548h | 10 |
| 9 | PPh3, Pd(OAc)2 Cs2CO3, ArB(OH)2 |
C6H5 | 548i | 63 |
| 10 | as above | 2-MeO-C6H4 | 548j | 76 |
| 11 | as above | 4-MeO-C6H4 | 548k | 84 |
| 12 | as above | 2-Naphthyl | 548l | 84 |
Scheme 168.

Hydrogenation of Bridged Sultams by Evans
A single example of an unusual desulfonylative indolization of a bridged sultam was reported by Paquette.464
7.6.2. Photochemical Reactions
Photochemical reactions of bridged sultams have been used to probe the effect of non-planar geometry of sulfonamide bonds on their reactivity.431,295
In 2004, Paquette reported the rearrangement of a bridged sultam with a [4.2.1] bis-unsaturated scaffold to a complex heterocycle containing cyclobutene, thietane dioxide and pyrrolidine rings (Scheme 169).431 The proposed mechanism involves (1) homolytic cleavage of the N–SO2 bond, (2) rebonding to give the bicyclic isomer, (3) thermally-induced [1,5]-hydrogen shift, and (4) photo-induced disrotatory closure to the cyclobutene ring. This reaction constituted the first example of a σ N–SO2 bond cleavage in a bridged sulfonamide.
Scheme 169.

Photoinduced Cleavage of SO2–N Bond of Bridged Sultams by Paquette
In line with the previous report, Paquette observed the photochemical rearrangement of a related benzofused-bridged sultam to a dihydroazepine heterocycle via N–SO2 bond cleavage and [1,5]-sulfonyl migration (Scheme 170a).295 In this case, minor quantities of the alternative cyclobutene product were formed via a photo-induced disrotatory closure. Later, Paquette attempted photochemical reactions of a bridged sultam featuring the sulfur atom at the apex position (Scheme 170b).440 However, under a variety of conditions, polymerization was found to be the predominant reaction pathway. In one case only, a cyclobutene product was isolated in low yield.440
Scheme 170.

Photoisomerization and Cycloaddition of Bridged Sultams by Paquette: a) Benzo[4.3.1] Ring System; b) [4.2.1] Ring System
Paquette also reported photoinduced di-π-methane rearrangement of benzofused bridged sultams bearing a [3.2.1] ring system (Table 20).463 Upon irradiation with 300 nm light, a series of functionalized substrates gave formal contraction products 554. Despite a potential to give two regioisomeric outcomes, in all examples the rebonding step occurred distal to the sulfonamide bond.
Table 20.
Di-π-methane Rearrangement of Bridged Sultams by Paquette
 | ||||
|---|---|---|---|---|
| entry | 382/548 | R | 554 | yield (%) |
| 1 | 382a | H | 554a | 48 |
| 2 | 548a | CH3 | 554b | 40 |
| 3 | 548b | CH2Ph | 554c | 50 |
| 4 | 548c | C(O)tBu | 554d | 40 |
| 5 | 548d | C(O)OBn | 554e | 55 |
| 6 | 548e | SiMe3 | 554f | 64 |
| 7 | 548f | SnBu3 | 554g | 49 |
| 8 | 548g | SePh | 554h | 23 |
| 9 | 548h | C≡CC4H9 | 554i | 50 |
7.6.3. Double Reduction
Bridged sultams have been shown to simultaneously undergo the reduction of both N–SO2 and SO2–C bonds to give planar nitrogen-containing heterocycles.432–435
Evans applied the double reduction of aromatic bridged sultams to the synthesis of pyrrolidines bearing various functional groups (Scheme 171).432–434 The reaction can be conducted by treatment of the bridged sultams with lithium or sodium metal in ammonia.432 The use of lithium/ethylenediamine (Benkeser reduction) afforded partially dearomatized products, while sodium naphthalenide led to extensive decomposition.432
Scheme 171.

Synthesis of Pyrrolidines via Double Reduction of Bridged Sultams by Evans
Evans utilized the double reduction methodology to establish all-carbon quaternary stereocenter in the total synthesis of mesembrane (Scheme 172).434 The required bridged sultam was prepared in high yield via Heck reaction; however, the double reduction was achieved in modest yield due to the competing removal of one of the methoxy groups. The reaction was sensitive to the conditions used; for example sodium and lithium naphthalenide afforded the final product with higher selectivity, however in lower yields.
Scheme 172.

Total Synthesis of Mesembrane via Double Reduction of Bridged Sultams by Evans
8. APPLICATION OF BRIDGED LACTAMS IN TOTAL SYNTHESIS
Bridged lactams have been used on several occasions in total synthesis of natural products.141–192 The ability to provide a rigid molecular framework and to attenuate the basicity of a nitrogen atom are among strategic advantages of applying bridged lactams in target-oriented synthesis. Moreover, in recent years a number of natural products containing bridged amide bonds have been isolated,193–207 suggesting that bridged lactams may be more prevalent in nature than had been previously considered. In the final section of this review, we will cover representative examples of bridged lactams used in total synthesis, with discussion focused on the role of bridged amide bonds.
8.1. Lycopodium Alkaloids
In 2008, Dake reported the total synthesis of Lycopodium alkaloid, (+)-fawcettidine (Scheme 173).141 The congested tetracyclic framework of this alkaloid was introduced by conjugate addition of the thiolate anion to give a complex bridged lactam 564 in 76% yield. Sequential functional group manipulations transformed 564 into the sulfone 565, which was subjected to the Ramberg-Bäcklung ring contraction to afford another bridged lactam containing a [4.3.1] ring system. This intermediate was rapidly converted into (+)-fawcettidine in three steps, including an LAH reduction that suggests that this particular lactam, while in a bridged framework, is not unusually twisted.
Scheme 173.

Total Synthesis of Fawcettidine by Dake (please use double-column format for Scheme 173)
8.2. Communesins
Communesins are indole alkaloids containing a 1-azabicyclo[3.2.2]nonane moiety.142 In 2010, Ma reported the first total synthesis of the potently cytotoxic (−)-communesin F using bridged lactam 569 to construct the 1-azabicyclo[3.2.2]nonane core of this molecule (Scheme 174a).143 Treatment of the allylic alcohol 568 with MsCl and Et3N provided the required lactam 569 in 63% yield. After mesylate displacement with sodium azide, the intermediate 570 was subjected to an unprecedented Staudinger reaction with the bridged amide, using PPh3 at 80 °C, to give the amidine 571 in 83% yield. Subsequent reduction and acetylation afforded the natural product. In this case, the inherent strain of the amide bond in the bridged lactam 570 enabled the rapid synthesis of (−)-communesin F.
Scheme 174.

a) Total Synthesis of Communesin F by Ma; a) Total Synthesis of Communesins A and B by Ma (please use double-column format for Scheme 174)
In 2011, Ma reported another elegant use of bridged lactams during the total synthesis of (−)-communesins A and B (Scheme 174b).144 Oxidative cross-coupling of the dianion derived from 573 afforded the bridged lactam 574 as a single diastereoisomer in a respectable 73% yield. The obtained stereochemical outcome was proposed to arise from a favorable π-stacking interaction between nitrophenyl and indole rings. The lactam 574 was advanced to the cyanide 575, which was subjected to the sequential reduction/reductive amination to afford the aminal 576 in 92% yield. This reaction most likely proceeds via the intermediate amino aldehyde, however the mechanism involving a bridged iminium ion cannot not be excluded. The aminal 576 required only four more steps to complete the synthesis of (−)-communesins A and B.
Biogenetically, communesins have been proposed to originate from two oxidized molecules of tryptamine.142 In 2003, Stoltz put forward a biomimetic hypothesis for the origin of communesins A and B, in which the key step was a Diels-Alder reaction between the quinine methide imine 579 and aurantioclavine derivative 578 to give the highly twisted bridged lactam 580 (Scheme 175a).145,146 It was postulated that the amide bond in 580 would then undergo transamination by the tethered primary amine to afford 581, another key intermediate in the biosynthesis.
Scheme 175.

a) Biomimetic Proposal for Synthesis of Communesins A and B by Stoltz; b) Biomimetic Proposal for Synthesis of Communesin B by Funk; c) Total Synthesis of Perophoramidine by Funk (please use double-column format for Scheme 175)
In 2004, Funk proposed a similar biomimetic pathway for the synthesis of communesin B (Scheme 175b).147 Funk validated this biomimetic proposal during the total synthesis of a related natural product perophoramidine (Scheme 175c).147 The coupling of indole 586 and 3-bromo-3-alkylindolin-2-one 587 afforded the intermediate 589 in 89% yield and ≥20:1 diastereoselectivity. The mechanism for this transformation may involve hetero Diels-Alder reaction to give bridged lactam 588, as proposed in the biomimetic hypothesis, or a conjugate addition pathway. The subsequent transamidation of 589 provided the pentacycle 590, which required only 9 steps to furnish the natural product. Recently, the Funk group synthesized communesin F utilizing a similar indol-2-one cycloaddition to rapidly generate the tetracyclic core of this natural product.148 These studies also featured a bridged lactam (analogous to 569, Scheme 174a) and prepared via trimethylaluminum-mediated condensation of the corresponding amino ester.
8.3. Cripowellins
The aglycon of cripowellins contains an intriguing 1-azabicyclo[5.3.2]dodecan-2-one ring system, in which the amide bond is placed on the larger external bridge. In 2005, Moon and coworkers reported a rapid synthesis of the skeleton of cripowellins by employing ring expansion reaction under reductive conditions (Scheme 176a).149 The precursor 594 was efficiently prepared via oxidative cyclization of the acylic amine 592. The key step proceeded in 51% yield using sodium naphthalenide to afford the bridged keto amide 595 in the following sequence of events: (i) nitrogen deprotection, (ii) nucleophilic addition to the tetrahedral intermediate 595, and (iii) C–C bond fragmentation.
Scheme 176.

a) Approach to Cripowellin Aglycon by Moon; b) Synthesis of 1-epi-Aglycon of Cripowellins A nd B by Enders (please use double-column format for Scheme 176)
In the same year, Enders reported asymmetric synthesis of the 1-epi aglycon of cripowellins (Scheme 176b).150,151 In this approach, the bridged ring system was constructed via the intramolecular Heck reaction of the 9-membered precursor 601, which in turn was easily prepared by the ring-closing metathesis of the appropriately rigidified diene. The asymmetry was introduced by a rare Sharpless asymmetric dihydroxylation of a skipped diene (Scheme 176b, box). The conditions used for the Heck reactions had a significant effect on the outcome of the pivotal cyclization; the neutral conditions employing PPh3 and Et3N in DMF led to the disibstituted Z-olefin 602, while under cationic conditions (dppp, Ag2CO3, toluene) the anti-Bredt olefin 603 was formed as a mixture of E and Z isomers. From intermediate 603, the completion of the synthesis required only four additional steps.
8.4. Quinine Derivatives
The 1946 oxidative degradation of quininone (via 5-vinylquinuclidin-2-one) by Doering and Chanley marks the first synthesis and the first reaction of any bridged lactam (see Scheme 2).251 Subsequently, this process has been applied to the synthesis of piperidines152 and isoquinolinones153 from Cinchona alkaloids.
Recently, Doris and coworkers reported the total synthesis of (+)-mequitazine using oxidative degradation of quinine via an unisolated bridged lactam, 5-vinylquinuclidin-2-one, as the key step (Scheme 177).154 Oxidation of quinine under Swern conditions provided the required quininone. This intermediate was reacted with potassium tert-butoxide in the presence of oxygen to deliver, after alcoholysis of the transient bridged amide bond, the vinyl piperidine in 60% yield. Transannular closure and coupling with phenothiazine completed the synthesis of (+)-mequitazine.
Scheme 177.

Synthesis of Mequitazine by Doris (please use double-column format for Scheme 177)
8.5. Aspidosperma and Strychnos Alkaloids
In classic studies on Strychnos-type alkaloids, Harley-Mason investigated the use of bridged lactams (Scheme 178).155 In particular, his group developed efficient synthesis of bridged lactam 611 containing a [5.2.2] ring system,155 which then served as a key intermediate in the synthesis of tubifoline,156 condyfoline156 and geissoschizoline157 (Schemes 178a and b). A similar strategy relying on bridged lactams bearing the same bicyclic scaffold158 was utilized in the rapid syntheses of several other alkaloids, including fluorocurarine159 (Scheme 178c), condylocarpine155 (Scheme 178d), tubotaiwine,160 dihydronorfluorocurarine,161 and tubifolidine.156 Interestingly, as early as in 1975, Harley-Mason explicitly proposed that lactams in which the amide bond is non-planar might be useful in the synthesis of complex alkaloids.155
In 1981, Ban and coworkers reported the total synthesis of quebrachamine using the bridged lactam 621 containing a [6.3.1] ring system (Scheme 179).162 The requisite lactam was prepared via an intramolecular N-alkylation of 620 using NaH in the presence of KI/18-crown-6, followed by alkylation at the bridgehead position in very good overall yield (see also Table 18). Reduction of 621 followed divergent pathways depending on the conditions used for this step: with LiAlH4 in refluxing dioxane, quebrachamine was formed, whereas in THF, 1,2-dehydroaspidospermidine was obtained via the bridged hemiaminal 623. Using similar strategy, Ban prepared other aspidosperma alkaloids,162–164 including 1-acetylaspidoalbidine,164 deoxyaspidodispermine164 and 1-acetylaspidospermidine.163 Recently, Movassaghi165 reported the use of structurally-related bridged lactams featuring a [6.3.1] ring system166 in the synthesis of complex dimeric aspidosperma alkaloids.
Scheme 179.

Synthesis of Quebrachamine and 1,2-Dehydroaspidospermidine by Ban (please use double-column format for Scheme 179)
The groups of Ziegler167,168 and Bosch169 utilized bridged lactam 626 bearing the amide bond on the larger external bridge in a [6.3.1] scaffold in an alternative approach to quebrachamine (Scheme 180a). The lactam 626 was prepared from the carboxylic acid 625 by treatment with PPA at 90 °C. This reaction is thought to proceed via the highly reactive acylated intermediate 627.168 Subsequent reduction of the bridged lactam with LiAlH4 provided the final alkaloid. Similar synthetic approaches have been reported in the total synthesis of dihydrocleavamine,170–172 tabersonine173 and cleavamine174 by the research groups of Bosch,170,171 Lesma,172 Ziegler,173 and Hanaoka174 (Scheme 180b–d). In general, only modest yields have been obtained in the reduction of these bridged lactams.
Scheme 180.

Synthesis of Indole Alkaloids via Friedel-Crafts Acylation of Bridged Amides: a) Quebrachamine by Ziegler and Bosch; b) Dihydrocleavamine by Bosch and Lesma; c) Tabersonine by Ziegler; d) Cleavamine by Hanaoka (please use double-column format for Scheme 180)
Another elegant example of the use of bridged lactams was reported by Bosch in the total synthesis of (−)-tubifoline (Scheme 181a).175,176 Witkop photocyclization of chloroacetamide 637 gave the late stage intermediate 638 containing bridged amide bond in a [5.2.2] ring system as an inconsequential mixture of the double bond isomers. Tandem lactam/olefin reduction and oxidative cyclization using PtO2/O2 transformed the lactam 638 into the natural product. Similar Witkop photocyclizations to give bridged lactams in the synthesis of Strychnos alkaloids have been reported independently by Snieckus177 and Ishikura.178
Scheme 181.

a) Application of Bridged Amide in Total Synthesis of Tubifoline by Bosch; b) Application in Bridged Amide in Total Synthesis of Strychnine by Magnus
Magnus utilized the bridged lactam 641 featuring the amide bond on the shorter external bridge in a [5.2.2] ring system as an early stage intermediate in the total synthesis of strychnine (Scheme 181b).179 Conjugate addition of the enolate 640 afforded the bridged lactam 641 in 65–98% yield, forming the F ring of strychnine. It was found that the yield of the conjugate addition was significantly improved when the sulfoxide precursor was used. The next step involved oxidation to the α-keto amide via Pummerer rearrangement.
Büchi180,181 and Narisada182 independently reported the total synthesis of velbamine, a degradation product of oncolytic vinblastine and vincristine alkaloids, using bridged lactams as key intermediates (Scheme 182). The approach by Büchi relied on retroaldol cleavage of the β-hydroxyketone 644 to reveal the bridged lactam 645 embedded in a [6.3.1] ring system (Scheme 182a).180,181 This intermediate was converted into velbamine in five more steps. In contrast, Narisada showed that the analogous bridged lactam 650 could be obtained via fragmentation of 649 under oxidative conditions using Pb(OAc)4 in comparable yield (Scheme 182b).182 The methoxy group was eliminated under acidic conditions to give bridged enamide 651, which was converted into velbamine using standard functional group manipulations.
Scheme 182.

Total Synthesis of Velbanamine: a) Büchi; b) Narisada (please use double-column format for Scheme 182)
8.6. Amaryllidaceae Alkaloids
Wildman reported the synthesis of bridged lactams from several Amaryllidaceae alkaloids, such as haemanthidine, oxohaemanthidine and apohaemanthidine, using oxidation with MnO2 (Scheme 183a).183 When oxohaemanthidine was treated with NaOAc/HOAc, the conjugated lactone was formed via hydrolysis of the bridged amide bond and lactonization.184,185 After methylation to 652, this product was used to correlate two common Amaryllidaceae alkaloids, heamanthidine and pretazettine (Scheme 183a).184,185
Scheme 183.

Bridged Lactams in Total Synthesis of Amaryllidaceae Alkaloids: a) Oxohaemanthidine, Oxodihydrohaemanthidine, Oxoapohaemanthidine and Pretazzetine; b) Dihydrocrinine; c) 6,11-Dioxocrinene; d) Haemanthidine (please use double-column format for Scheme 183)
Irie and coworkers reported the use of a related bridged lactam 659 to install the tricyclic core of dihydrocrinine (Scheme 183b).186,187 Transannular conjugate addition of the precursor 658 under ketalization conditions stereoselectively afforded the required lactam. Lower yields of lactam 659 were observed with NaH, NaOtBu and NaNH2.187 Interestingly, the minor product isolated in this reaction was the bridged ethylene orthoamide resulting from the reaction of the twisted amide bond with ethylene glycol (not shown; cf. Table 5 and Scheme 133).187 Similarly, the formation of stable hemiaminals on treatment with LiAlH4 (659→660) that required additional steps to effect full reduction to the amine indicates that bridged amide bonds in these systems possess a significant ketonic character.
Finally, Hendrickson reported the total synthesis of dioxocrinene 486 featuring the bridged amide bond (Scheme 183c)188 and haemanthidine using the bridged lactam 664 as the key intermediate (Scheme 183d).189 Both lactams were constructed from the corresponding esters 662–663 via a sequence involving hydrolysis, homologation with diazomethane, and treatment with dry HCl to effect the cyclization (Scheme 183c–d).188,189 The ester precursors 662–663 were easily prepared via Curtuis rearrangement followed by Friedel–Crafts cyclization, highlighting the efficiency of this strategy. Hendrickson studied the quasi-amide bond properties of some of the prepared bridged lactams, finding these compounds to be sensitive towards hydrolysis/methanolysis and able to be reduced with the mild reducing agent NaBH4.188
8.7. Kopsia Alkaloids
In 2001, Magnus reported the total synthesis of kopsijasminilam, a complex Kopsia alkaloid containing a bridged amide in a pentacyclic ring system (Scheme 184a).190,191 The transannular cyclization of eleven-membered precursor 665 set the stage for the key Diels-Alder reaction. The Diels-Alder adduct was obtained on treatment of the cyanide 666 with acryloyl chloride. Substituents other than cyanide at the hemiaminal carbon, including OH, OMe or SPh, resulted in decomposition during the Diels-Alder step. Polonovski fragmentation of the N-oxide 668 gave the spirocyclic bridged lactam 670 in 90% yield. The synthesis was completed by conjugate reduction/oxidation using catalytic Mn(dpm)3/PhSiH3 under oxygen atmosphere. Magnus utilized a similar concept based on the peroxyaminal fragmentation of the intermediate 672 in the total synthesis of pauciflorine B (Scheme 184b).190,191
Scheme 184.

Total Synthesis of Kopsijasminilam by Magnus; b) Total Synthesis of and Pauciflorine B by Magnus; c) Total Synthesis of Kopsijasminilam by Kuehne (please use double-column format for Scheme 184)
At about the same time, Kuehne applied an alternative fragmentation strategy in the total synthesis of kopsijasminilam (Scheme 184c).192 The key precursor 676 was prepared from alkaloid minovincine. Treatment of 676 with NaCN in refluxing ethanol-water resulted in the nitrogen-assisted fragmentation to give the pentacyclic cyanide 677 in 89% yield. The bridged lactam 678 was obtained after oxidation with potassium tert-butoxide in the presence of oxygen. This intermediate was converted to kopsijasminilam in five more steps.
8.8. Natural Products Containing Bridged Amide Bonds
Several natural products containing bridged amide bonds have been isolated from Stemona,193–196 Daphnum,197–202 Kopsia,203 Strychnos,204 Iboga,205 Amaryllidaceae465 and Lycopodium species206,207 (Schemes 185–189).
Scheme 185.

Stemona Alkaloids Containing Bridged Amide Bonds (please use double-column format for Scheme 185)
Representative bridged lactams in the the structural class of Stemona alkaloids are tuberostemoninols (680–682),193 neotuberostemoninol (683),194 sessilifoliamide I (684),195 and maireistemoninol (685),196 in which the amide bond is placed on a one-carbon bridge in a [4.3.1] ring system, and the biogenetically-related neotuberostemonone (686)196 and epoxytuberostemonone (687)196 containing [6.4.1] bridged systems (Scheme 185). The X-ray structures and spectroscopic properties of neotuberostemoninol (683)194 and sessilifoliamide I (684)195 indicate that amide bonds in these compounds aresignificantly distorted from planarity (683: τ = 49.6°; χN = 30.8°; χC = 12.7°; N–C(O) = 1.402 Å; C=O = 1.215 Å; νC=O 1682 cm−1; 13CC=O 184.7 ppm; 684: τ = 43.9°; χN = 35.3°; χC = 11.0°; N–C(O) = 1.390 Å; C=O = 1.232 Å; νC=O 1656 cm−1; 13CC=O 182.4 ppm).
Daphnum alkaloids containing bridged amide bonds include daphmanidins B–D (688–690),197,198 daphnezomines F, G, and U (691–693),199,200 daphhimalenine A (694),201 and daphnicyclidine J (695),202 which feature 1-azabicyclo[5.2.2]undecane, 1-azabicyclo[5.2.1]decane and 1-azabicyclo[6.2.2]dodecane ring systems (Scheme 186). Infrared stretching frequencies and 13C NMR shifts of daphhimalenine A (694)201 confirm that the amide bond in this alkaloid is non-planar (νC=O 1687 cm−1; 13CC=O 183.8 ppm).
Scheme 186.

Daphnum Alkaloids Containing Bridged Amide Bonds (please use double-column format for Scheme 186)
Pericidine (696)203 and henningsamides (697–699)204 are Kopsia and Strychnos alkaloids, respectively, which contain the amide bond in a [5.2.2] ring system, while 3-oxocoronaridine (700)205 and 3-oxovoacangine (701)205 are oxygenated Iboga alkaloids with bridged amide bonds embedded in complex caged tricyclic structures (Scheme 187). Kopsijasminilam (671) and pauciflorine B (674) (Scheme 187) have been mentioned in the previous section in the context of their synthesis (see Section 8.7);190–192 two other Kopsia alkaloids, deoxykopsijasminilam (702) and pauciflorine A (703)190–192 share their unprecedented skeleton (Scheme 187). The Amaryllidaceae alkaloids containing bridged amide bonds, cripowellins A and B (598–599) (Scheme 188),465 have been already discussed in this review (see Section 8.3).149–151
Scheme 187.

Indole Alkaloids Containing Bridged Amide Bonds (please use double-column format for Scheme 187)
Scheme 188.

Cripowellins Containing Bridged Amide Bonds (please use double-column format for Scheme 188)
Finally, several Lycopodium alkaloids, such as phlegmariurines A–C (704–706)206 and lycoposerramine E (707)206 feature bridged amide bonds placed in a [5.3.4] ring system. These compounds belong to the fawcettimine class of Lycopodium alkaloids207 and can be regarded as degradation products of fawcettidine (see Scheme 173).
9. CONCLUSIONS AND OUTLOOK
In the course of 75 years of inquiry, the chemistry of bridged lactams has provided numerous examples of fascinating molecules. Due to the restricted conformation, these lactams cannot benefit from the full resonance stabilization typical to planar amides. The initial interest in bridged lactams was fuelled by the imagination of organic chemists to test the stability of anti-Bredt amides. Over the years, it has been demonstrated that non-planar amides are highly susceptible to hydrolysis, and in general difficult to prepare or isolate. In the last decade, improved mechanistic understanding and the emergence of new synthetic methods have allowed the synthesis of bridged lactams containing perfectly orthogonal amide bonds. Remarkably, many of these perpendicularly twisted amides have been structurally characterized. Furthermore, new structural classes of bridged lactams have been invented and applied to probe the effect of amide bond distortion on its reactivity. The N-activation of amide bonds, detailed studies on amide bond hydrolysis, structural characterization of N-protonated lactams, and the synthesis of hydrolytically-robust bridged lactams have been described. A large number of bridged lactams have been characterized by X-ray crystallography, which in combination with IR, and 13C NMR spectroscopic data, makes it possible to estimate the relationship between twist angle and chemical reactivity of these compounds. The importance of bridged lactams is further underlined by their successful application to access complex building blocks in target-oriented synthesis and their tantalizing potential as lead structures in medicinal chemistry.
While progress has been considerable, many areas remain to be explored. Expanding the family of highly-strained bridged lactams, systematic examination of their follow-up chemistry, additional insight into the quantitative evaluation of amide bonds, and application of the new reactivity manifolds of non-planar lactams to simple amides are among the challenges that need to be addressed. Finally, as mechanical distortion plays a key role in the enzymatic activation of amide bonds, non-planar lactams will continue to give us a glimpse into the molecular level of enzymatic catalysis.
Scheme 99.

Generation of Bridged 1-Aza Iminium Ions by Hoffmann: a) Quinine Series; b) Quinidine Seires; c) Structures of Quinine and Quinidine
Scheme 189.

Lycopodium Alkaloids Containing Bridged Amide Bonds (please use double-column format for Scheme 189)
Acknowledgement
We thank the National Institute of General Medical Sciences for the support (GM-49093) of our efforts summarized in this review.
References
- 1.Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley; 2000. [Google Scholar]
- 2.Pauling L. The Nature of the Chemical Bond. London: Oxford University Press; 1940. [Google Scholar]
- 3.Chalupsky J, Vondrasek J, Spirko V. J. Phys. Chem. A. 2008;112:693. doi: 10.1021/jp0750699. [DOI] [PubMed] [Google Scholar]
- 4.Mannfors BE, Mirkin NG, Palmo K, Krimm S. J. Phys. Chem. A. 2003;107:1825. [Google Scholar]
- 5.Shin SBY, Yoo B, Todaro LJ, Kirshenbaum K. J. Am. Chem. Soc. 2007;129:3218. doi: 10.1021/ja066960o. [DOI] [PubMed] [Google Scholar]
- 6.Poteau R, Trinquier G. J. Am. Chem. Soc. 2005;127:13875. doi: 10.1021/ja052342g. [DOI] [PubMed] [Google Scholar]
- 7.Ramachandran GN. Biopolymers. 1968;6:1494. doi: 10.1002/bip.1968.360061013. [DOI] [PubMed] [Google Scholar]
- 8.MacArthur MW, Thornton JM. J. Mol. Biol. 1996;264:1180. doi: 10.1006/jmbi.1996.0705. [DOI] [PubMed] [Google Scholar]
- 9.Bednarova L, Malon P, Bour P. Chirality. 2007;19:775. doi: 10.1002/chir.20462. [DOI] [PubMed] [Google Scholar]
- 10.Edison AS. Nat. Struct. Biol. 2001;8:201. doi: 10.1038/84921. [DOI] [PubMed] [Google Scholar]
- 11.Beausoleil E, Lubell WD. J. Am. Chem. Soc. 1996;118:12902. [Google Scholar]
- 12.Halab L, Lubell WD. J. Org. Chem. 1999;64:3312. doi: 10.1021/jo990294a. [DOI] [PubMed] [Google Scholar]
- 13.Halab L, Lubell WD. J. Am. Chem. Soc. 2002;124:2474. doi: 10.1021/ja012442w. [DOI] [PubMed] [Google Scholar]
- 14.Wipf P, Miller CP, Grant CM. Tetrahedron. 2000;56:9143. [Google Scholar]
- 15.Yamada S. Angew. Chem., Int. Ed. 1993;32:1083. [Google Scholar]
- 16.Yamada S. Angew. Chem., Int. Ed. 1995;34:1113. [Google Scholar]
- 17.Yamada S. J. Org. Chem. 1996;61:941. [Google Scholar]
- 18.Yamada S, Nakamura M, Kawauchi I. Chem. Commun. 1997:885. [Google Scholar]
- 19.Yamada S, Matsuda K. Chem. Lett. 2001:750. [Google Scholar]
- 20.Pedone C, Benedetti E, Immirzi A, Allegra G. J. Am. Chem. Soc. 1970;92:3549. [Google Scholar]
- 21.Bennet AJ, Somayaji V, Brown RS, Santarsiero BD. J. Am. Chem. Soc. 1991;113:7563. [Google Scholar]
- 22.Yamada S, Nunami N, Hori K. Chem. Lett. 1998:451. [Google Scholar]
- 23.Evans PA, Holmes AB, Collins I, Raithby PR, Russell K. J. Chem. Soc., Chem. Commun. 1995:2325. [Google Scholar]
- 24.Yamada S, Homma A. Chem. Commun. 2002:2656. doi: 10.1039/b207925a. [DOI] [PubMed] [Google Scholar]
- 25.Cow CN, Britten JF, Harrison PHM. Chem. Commun. 1998:1147. [Google Scholar]
- 26.Matta CF, Cow CC, Sun S, Britten JF, Harrison PHM. J. Mol. Struc. 2000;523:241. [Google Scholar]
- 27.Duspara PA, Matta CF, Jenkins SI, Harrison PHM. Org. Lett. 2001;3:495. doi: 10.1021/ol000349r. [DOI] [PubMed] [Google Scholar]
- 28.Matta CF, Cow CN, Harrison PHM. J. Mol. Struc. 2003;660:81. [Google Scholar]
- 29.Bain AD, Chen H, Harrison PHM. Can. J. Chem. 2006;84:421. [Google Scholar]
- 30.Yamamoto G, Murakami H, Tsubai N, Mazaki Y. Chem. Lett. 1997:605. [Google Scholar]
- 31.Yamamoto G, Tsubai N, Murakami H, Mazaki Y. Chem. Lett. 1997:1295. [Google Scholar]
- 32.Yamamoto G, Nakajo F, Tsubai N, Murakami H, Mazaki Y. Bull. Chem. Soc. Jpn. 1999;72:2315. [Google Scholar]
- 33.Saeed A, Erben MF, Bolte M. J. Org. Chem. 2012;77:4688. doi: 10.1021/jo300360b. [DOI] [PubMed] [Google Scholar]
- 34.Hutchby M, Houlden CE, Haddow MF, Tyler SN, Lloyd-Jones GC, Booker-Milburn KI. Angew. Chem., Int. Ed. 2012;51:548. doi: 10.1002/anie.201107117. [DOI] [PubMed] [Google Scholar]
- 35.Alcorta I, Cativiela C, Elguero J, Gil AM, Jiménez AI. New. J. Chem. 2005;29:1450. [Google Scholar]
- 36.Rauk A, Allen LC, Mislow K. Angew. Chem., Int. Ed. 1970;9:400. [Google Scholar]
- 37.Korn A, Rudolph-Böhner S, Moroder L. Tetrahedron. 1994;50:1717. [Google Scholar]
- 38.Shao H, Jiang X, Gantzel P, Goodman M. Chem. Biol. 1994;1:231. doi: 10.1016/1074-5521(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 39.Otani Y, Nagae O, Naruse Y, Inagaki S, Ohno M, Yamaguchi K, Yamamoto G, Uchiyama M, Ohwada T. J. Am. Chem. Soc. 2003;125:15191. doi: 10.1021/ja036644z. [DOI] [PubMed] [Google Scholar]
- 40.Ohwada T, Achiwa T, Okamoto I, Shudo K, Yamaguchi K. Tetrahedron Lett. 1998;39:865. [Google Scholar]
- 41.Hori T, Otani Y, Kawahata M, Yamaguchi K, Ohwada T. J. Org. Chem. 2008;73:9102. doi: 10.1021/jo801996b. [DOI] [PubMed] [Google Scholar]
- 42.Hosoya M, Otani Y, Kawahata M, Yamaguchi K, Ohwada T. J. Am. Chem. Soc. 2010;132:14780. doi: 10.1021/ja1017877. [DOI] [PubMed] [Google Scholar]
- 43.Aleman C, Jiménez AI, Cativiela C, Perez JJ, Casanovas J. Phys. Chem. B. 2005;109:11836. doi: 10.1021/jp050141t. [DOI] [PubMed] [Google Scholar]
- 44.Gerdes RG, Glover SA, ten Have JF, Rowbottom CA. Tetrahedron Lett. 1989;30:2649. [Google Scholar]
- 45.Rauk A, Glover SA. J. Org. Chem. 1996;61:2337. [Google Scholar]
- 46.Glover SA, Rauk A. J. Org. Chem. 1999;64:2340. [Google Scholar]
- 47.Glover SA, Mo G, Rauk A, Tucker DJ, Turner P. J. Chem. Soc., Perkin Trans. 1999;2:2053. [Google Scholar]
- 48.Glover SA, Mo GN. J. Chem. Soc. Perkin Trans. 2002;2:1728. [Google Scholar]
- 49.Glover SA, Rauk A. J. Chem. Soc., Perkin Trans. 2002;2:1740. [Google Scholar]
- 50.Gillson AME, Glover SA, Tucker DJ, Turner P. Org. Biomol. Chem. 2003;1:3430. doi: 10.1039/b306098p. [DOI] [PubMed] [Google Scholar]
- 51.Banks TM, Bonin AM, Glover SA, Prakash AS. Org. Biomol. Chem. 2003;1:2238. doi: 10.1039/b301618h. [DOI] [PubMed] [Google Scholar]
- 52.Andrews LE, Banks TM, Bonin AM, Clay SF, Gillson AME, Glover SA. Aust. J. Chem. 2004;57:377. [Google Scholar]
- 53.Glover SA, White JM, Rosser AA, Digianantonio KM. J. Org. Chem. 2011;76:9757. doi: 10.1021/jo201856u. [DOI] [PubMed] [Google Scholar]
- 54.Digianantonio KM, Glover SA, Johns JP, Rosser AA. Org. Biomol. Chem. 2011;9:4116. doi: 10.1039/c1ob00008j. [DOI] [PubMed] [Google Scholar]
- 55.Hall HK, Jr, El-Shekeil A. Chem. Rev. 1983;83:549. [Google Scholar]
- 56.Lease TG, Shea KJA. Advances in Theoretically Interesting Molecules. Vol. 2. JAI Press Inc.; 1992. Compilation and Analysis of Structural Data of Distorted Bridgehead Olefins and Amides. [Google Scholar]
- 57.Greenberg A. The Amide Linkage as a Ligand-Its Properties and the Role of Distortion. In: Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley; 2000. [Google Scholar]
- 58.Clayden J, Moran WJ. Angew. Chem., Int. Ed. 2006;45:7118. doi: 10.1002/anie.200603016. [DOI] [PubMed] [Google Scholar]
- 59.Szostak M, Aubé J. Org. Biomol. Chem. 2011;9:27. doi: 10.1039/c0ob00215a. [DOI] [PubMed] [Google Scholar]
- 60.Lukeš R. Collect. Czech., Chem. Commun. 1938;10:148. [Google Scholar]
- 61.Brown R. Studies in Amide Hydrolysis: The Acid, Base, and Water Reactions. In: Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley; 2000. [Google Scholar]
- 62.Lopez X, Mujika JI, Blackburn GM, Karplus M. J. Phys. Chem. A. 2003;107:2304. [Google Scholar]
- 63.Mujika JI, Mercero JM, Lopez X. J. Phys. Chem. A. 2003;107:6099. [Google Scholar]
- 64.Mujika JI, Mercero JM, Lopez X. J. Am. Chem. Soc. 2005;127:4445. doi: 10.1021/ja044873v. [DOI] [PubMed] [Google Scholar]
- 65.Mujika JI, Formoso E, Mercero JM, Lopez X. J. Phys. Chem. B. 2006;110:15000. doi: 10.1021/jp0604037. [DOI] [PubMed] [Google Scholar]
- 66.Wang B, Cao Z. Chem. Eur. J. 2011;17:11919. doi: 10.1002/chem.201101274. [DOI] [PubMed] [Google Scholar]
- 67. Mucsi Z, Tsai A, Szori M, Chass GA, Viskolcz B, Csizmadia IG. J. Phys. Chem. A. 2007;111:13245. doi: 10.1021/jp0759325. For studies on determination of energy changes during the rotation of the amino group in amides, see: Glover SA, Rosser AA. J. Org. Chem. 2012;77:5492. doi: 10.1021/jo300347k. Wiberg KB, Breneman CM. J. Am. Chem. Soc. 1992;114:831.
- 68.Mucsi Z, Chass GA, Viskolcz B, Csizmadia IG. J. Phys. Chem. A. 2008;112:9153. doi: 10.1021/jp8048586. [DOI] [PubMed] [Google Scholar]
- 69.Mucsi Z, Chass GA, Csizmadia IG. J. Phys. Chem. B. 2008;112:7885. doi: 10.1021/jp8023292. [DOI] [PubMed] [Google Scholar]
- 70.Greenberg A, Venanzi CA. J. Am. Chem. Soc. 1993;115:6951. [Google Scholar]
- 71.Greenberg A, Moore DT, DuBois TD. J. Am. Chem. Soc. 1996;118:8658. [Google Scholar]
- 72.Cho SJ, Cui C, Lee JY, Park JK, Suh SB, Park J, Kim BH, Kim KS. J. Org. Chem. 1997;62:4068. [Google Scholar]
- 73.Cox C, Wack H, Lectka T. Angew. Chem., Int. Ed. 1999;38:798. doi: 10.1002/(SICI)1521-3773(19990315)38:6<798::AID-ANIE798>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 74. Morgan J, Greenberg A, Liebman JF. Struct. Chem. 2012;23:9851. Morgan J, Greenberg A. J. Phys. Org. Chem. 2012;25:1422. Morgan J, Greenberg A, Liebman JF. Struct. Chem. 2012;23:197.
- 75.Bednarova L, Malon P, Bour P. Chirality. 2007;19:775. doi: 10.1002/chir.20462. [DOI] [PubMed] [Google Scholar]
- 76.Kemnitz CR, Loewen MJ. J. Am. Chem. Soc. 2007;129:2521. doi: 10.1021/ja0663024. [DOI] [PubMed] [Google Scholar]
- 77.Mujika JI, Matxain JM, Eriksson LA, Lopez X. Chem. Eur. J. 2006;12:7215. doi: 10.1002/chem.200600052. [DOI] [PubMed] [Google Scholar]
- 78.Jean Y, Demachy I, Lledos A, Maseras F. J. Mol. Struc. (Theochem) 2003;632:131. [Google Scholar]
- 79.Greenberg A, Moore DT. J. Mol. Struc. 1997;413:477. [Google Scholar]
- 80.Greenberg A, Thomas TD, Bevilacqua CR, Coville M, Ji D, Tsai JC, Wu G. J. Org. Chem. 1992;57:7093. [Google Scholar]
- 81.Greenberg A, Hsing HJ, Liebman JF. J. Mol. Struc. (Theochem) 1995;338:83. [Google Scholar]
- 82.Greenberg A, DuBois TD. J. Mol. Struc. 2001;567:303. [Google Scholar]
- 83.Kesharwani MK, Ganguly B. J. Comput. Chem. 2011;32:2170. doi: 10.1002/jcc.21800. [DOI] [PubMed] [Google Scholar]
- 84.Liu J, Albers MW, Chen CM, Schreiber SL, Walsh CT. Proc. Natl. Acad. Sci. U. S. A. 1990;87:2304. doi: 10.1073/pnas.87.6.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fischer G. Chem. Soc. Rev. 2000;29:119. [Google Scholar]
- 86.Fischer G, Schmid FX. Peptidyl-prolyl cis/trans isomerases. In: Bakau B, editor. Molecular Chaperones and Folding Catalysts: Regulation, Cellular Functions and Mechanisms. CRC; 1999. [Google Scholar]
- 87.Cox C, Lectka T. Acc. Chem. Res. 2000;33:849. doi: 10.1021/ar990165g. [DOI] [PubMed] [Google Scholar]
- 88.Liebman JF, Greenberg A. Chem. Rev. 1976;76:311. [Google Scholar]
- 89.Hoffmann R, Hopf H. Angew. Chem., Int. Ed. 2008;47:4474. doi: 10.1002/anie.200705775. [DOI] [PubMed] [Google Scholar]
- 90.Yamada S. J. Synth. Org. Chem. Jpn. 1998;56:303. [Google Scholar]
- 91.Yamada S. Rev. Heteroat. Chem. 1999;19:203. [Google Scholar]
- 92.Yamada S. Sterically Hindered Twisted Amides. In: Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley; 2000. [Google Scholar]
- 93.Aubé J. Angew. Chem., Int. Ed. 2012;51:3063. doi: 10.1002/anie.201108173. [DOI] [PubMed] [Google Scholar]
- 94.Clayden J. Nature. 2012;481:274. doi: 10.1038/481274a. [DOI] [PubMed] [Google Scholar]
- 95.Glover SA. Tetrahedron. 1998;54:7229. [Google Scholar]
- 96.Glover SA, Rauk A, Buccigross JM, Campbell JJ, Hammond GP, Mo GN, Andrews LE, Gillson AME. Can. J. Chem. 2005;83:1492. [Google Scholar]
- 97.Glover SA. Adv. Phys. Org. Chem. 2007;42:35. [Google Scholar]
- 98.Fawcett FS. Chem. Rev. 1950;47:219. doi: 10.1021/cr60147a003. [DOI] [PubMed] [Google Scholar]
- 99.Köbrich G. Angew. Chem., Int. Ed. 1973;12:464. [Google Scholar]
- 100.Buchanan GL. Chem. Soc. Rev. 1974;3:41. [Google Scholar]
- 101.Keese R. Angew. Chem., Int. Ed. 1975;14:528. [Google Scholar]
- 102.Warner PM. Chem. Rev. 1989;89:1067. [Google Scholar]
- 103.Shea KJ. Tetrahedron. 1980;36:1683. [Google Scholar]
- 104.Bear BR, Sparks SM, Shea KJ. Angew. Chem., Int. Ed. 2001;40:821. [PubMed] [Google Scholar]
- 105.Vazquez S, Camps P. Tetrahedron. 2005;61:5147. [Google Scholar]
- 106.Winkler FK, Dunitz JD. J. Mol. Biol. 1971;59:169. doi: 10.1016/0022-2836(71)90419-0. [DOI] [PubMed] [Google Scholar]
- 107.Tani K, Stoltz BM. Nature. 2006;441:731. doi: 10.1038/nature04842. [DOI] [PubMed] [Google Scholar]
- 108.Kirby AJ, Komarov IV, Feeder N. J. Chem. Soc., Perkin Trans. 2001;2:522. [Google Scholar]
- 109.Wiberg K. Origin of the Amide Rotational Barrier. In: Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley; 2000. [Google Scholar]
- 110.Wiberg KB. Acc. Chem. Res. 1999;32:922. [Google Scholar]
- 111.Kirby AJ, Komarov IV, Kowski K, Rademacher P. J. Chem. Soc., Perkin Trans. 1999;2:1313. [Google Scholar]
- 112.Silverstein RM, Bassler GC, Morrill TC. Spectroscopic Identification of Organic Compounds. New York: John Wiley and Sons; 1991. [Google Scholar]
- 113.Maier WF, Schleyer PV. J. Am. Chem. Soc. 1981;103:1891. [Google Scholar]
- 114.Kim JS. Bull. Korean Chem. Soc. 1997;18:488. [Google Scholar]
- 115.van Eis MJ, Wijsman GW, de Wolf WH, Bickelhaupt F, Rogers DW, Kooijman H, Spek AL. Chem. Eur. J. 2000;6:1537. doi: 10.1002/(sici)1521-3765(20000502)6:9<1537::aid-chem1537>3.3.co;2-5. [DOI] [PubMed] [Google Scholar]
- 116.Novak I. J. Chem. Inf. Model. 2005;45:334. doi: 10.1021/ci0497354. [DOI] [PubMed] [Google Scholar]
- 117.Wiseman JR. J. Am. Chem. Soc. 1967;89:5966. [Google Scholar]
- 118.Wiseman JR, Chan HF, Ahola CJ. J. Am. Chem. Soc. 1969;91:2812. [Google Scholar]
- 119.Wiseman JR, Chong JA. J. Am. Chem. Soc. 1969;91:7775. [Google Scholar]
- 120.Wiseman RJ, Pletcher WA. J. Am. Chem. Soc. 1970;92:956. [Google Scholar]
- 121.Marshall JA, Faubl H. J. Am. Chem. Soc. 1970;92:948. [Google Scholar]
- 122.Chong JA, Wiseman JR. J. Am. Chem. Soc. 1972;94:8627. [Google Scholar]
- 123.Kim MG, White JD. J. Am. Chem. Soc. 1977;99:1172. [Google Scholar]
- 124.Gudipati MS, Radziszewski JG, Kaszynski P, Michl J. J. Org. Chem. 1993;58:3668. [Google Scholar]
- 125.Ohno M, Itoh M, Umeda M, Furuta R, Kondo K, Eguchi S. J. Am. Chem. Soc. 1996;118:7075. [Google Scholar]
- 126.Tae EL, Zhu Z, Platz M. J. Phys. Chem. A. 2001;105:3803. [Google Scholar]
- 127.Roach P, Warmuth R. Angew. Chem., Int. Ed. 2003;42:3039. doi: 10.1002/anie.200351120. [DOI] [PubMed] [Google Scholar]
- 128.Hudnall TW, Bielawski CW. J. Am. Chem. Soc. 2009;131:16039. doi: 10.1021/ja907481w. [DOI] [PubMed] [Google Scholar]
- 129.Neilson BM, Lynch VM, Bielawski CW. Angew. Chem., Int. Ed. 2011;50:10326. doi: 10.1002/anie.201105032. [DOI] [PubMed] [Google Scholar]
- 130.Borowiak T, Dutkiewicz G, Sosnicki JG, Jagodzinski TS, Hansen PE. J. Mol. Struct. 2008;892:438. [Google Scholar]
- 131.Nanubolu JB, Sridhar B, Ravikumar K. CrystEngComm. 2012;14:2571. [Google Scholar]
- 132.Yamada S. J. Org. Chem. 1992;57:1591. [Google Scholar]
- 133.Yamada S, Sugaki T, Matsuzaki K. J. Org. Chem. 1996;61:5932. [Google Scholar]
- 134.Yamada S, Misono T, Iwai Y, Masumizu A, Akiyama Y. J. Org. Chem. 2006;71:6872. doi: 10.1021/jo060989t. [DOI] [PubMed] [Google Scholar]
- 135.Yamada S, Katsumata H. Chem. Lett. 1998:995. [Google Scholar]
- 136.Yamada S, Ohe T. Tetrahedron Lett. 1996;37:6777. [Google Scholar]
- 137.Yamada S, Katsumata H. J. Org. Chem. 1999;64:9365. [Google Scholar]
- 138.Yamada S, Ichikawa M. Tetrahedron Lett. 1999;40:4231. [Google Scholar]
- 139.Yamada S, Noguchi E. Tetrahedron Lett. 2001;42:3621. [Google Scholar]
- 140.Yamada S, Misono T, Ichikawa M, Morita C. Tetrahedron. 2001;57:8939. [Google Scholar]
- 141.Kozak JA, Dake GR. Angew. Chem., Int. Ed. 2008;47:4221. doi: 10.1002/anie.200800522. [DOI] [PubMed] [Google Scholar]
- 142.Siengalewicz P, Gaich T, Mulzer J. Angew. Chem., Int. Ed. 2008;47:8170. doi: 10.1002/anie.200801735. [DOI] [PubMed] [Google Scholar]
- 143.Zuo Z, Xie W, Ma D. J. Am. Chem. Soc. 2010;132:13226. doi: 10.1021/ja106739g. [DOI] [PubMed] [Google Scholar]
- 144.Zuo Z, Ma D. Angew. Chem., Int. Ed. 2011;50:12008. doi: 10.1002/anie.201106205. [DOI] [PubMed] [Google Scholar]
- 145.May JA, Zeidan RK, Stoltz BM. Tetrahedron Lett. 2003;44:1203. [Google Scholar]
- 146.May JA, Stoltz BM. Tetrahedron. 2006;62:5262. [Google Scholar]
- 147.Fuchs JR, Funk RL. J. Am. Chem. Soc. 2004;126:5068. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]
- 148.Belmar J, Funk RL. J. Am. Chem. Soc. 2012;134:16941. doi: 10.1021/ja307277w. [DOI] [PubMed] [Google Scholar]
- 149.Moon B, Han S, Yoon Y, Kwon H. Org. Lett. 2005;7:1031. doi: 10.1021/ol047520+. [DOI] [PubMed] [Google Scholar]
- 150.Enders D, Lenzen A, Raabe G. Angew. Chem., Int. Ed. 2005;44:3766. doi: 10.1002/anie.200500556. [DOI] [PubMed] [Google Scholar]
- 151.Enders D, Lenzen A, Backes M, Janeck C, Catlin K, Lannou MI, Runsink J, Raabe G. J. Org. Chem. 2005;70:10538. doi: 10.1021/jo0518093. [DOI] [PubMed] [Google Scholar]
- 152.Martinelli MJ, Peterson BC, Khau VV, Hutchison DR, Sullivan KA. Tetrahedron Lett. 1993;34:5413. [Google Scholar]
- 153.Hutchison DR, Khau VV, Martinelli MJ, Nayyar NK, Peterson BC, Sullivan KA. Org. Synth. 1998;Vol. 75:223. Org. Synth. 2004, Coll. Vol. 10, 35. [Google Scholar]
- 154.Leroux S, Larquetoux L, Nicolas M, Doris E. Org. Lett. 2011;13:3549. doi: 10.1021/ol2012567. [DOI] [PubMed] [Google Scholar]
- 155.Harley-Mason J. Pure Appl. Chem. 1975;41:167. [Google Scholar]
- 156.Dadson BA, Harley-Mason J, Foster GH. Chem. Commun. 1968:1233. [Google Scholar]
- 157.Dadson BA, Harley-Mason J. Chem. Commun. 1969:665. (Communication 583). [Google Scholar]
- 158.Harley-Mason J, Taylor CG. Chem. Commun. 1969:1384. [Google Scholar]
- 159.Crawley GC, Harley-Mason J. Chem. Commun. 1971:685. [Google Scholar]
- 160.Dadson BA, Harley-Mason J. Chem. Commun. 1969:665. (Communication 584). [Google Scholar]
- 161.Harley-Mason J, Taylor CG. Chem. Commun. 1970:812. [Google Scholar]
- 162.Ban Y, Yoshida K, Goto J, Oishi T. J. Am. Chem. Soc. 1981;103:6990. [Google Scholar]
- 163.Ban Y, Yoshida K, Goto J, Oishi T, Takeda E. Tetrahedron. 1983;39:3657. [Google Scholar]
- 164.Yoshida K, Sakuma Y, Ban Y. Heterocycles. 1987;25:47. [Google Scholar]
- 165.Medley JW, Movassaghi M. Angew. Chem., Int. Ed. 2012;51:4572. doi: 10.1002/anie.201200387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yates P, MacLachlan FN, Rae ID. Can. J. Chem. 1978;56:1052. [Google Scholar]
- 167.Ziegler FE, Zoretic PA. Tetrahedron Lett. 1968;22:2639. [Google Scholar]
- 168.Ziegler FE, Kloek JA, Zoretic PA. J. Am. Chem. Soc. 1969;91:2342. [Google Scholar]
- 169.Amat M, Lozano O, Escolano C, Molins E, Bosch J. J. Org. Chem. 2007;72:4431. doi: 10.1021/jo070397q. [DOI] [PubMed] [Google Scholar]
- 170.Amat M, Escolano C, Lozano O, Llor N, Bosch J. Org. Lett. 2003;5:3139. doi: 10.1021/ol035199+. [DOI] [PubMed] [Google Scholar]
- 171.Amat M, Escolano C, Lozano O, Gómez-Esqué A, Griera R, Molins E, Bosch J. J. Org. Chem. 2006;71:3804. doi: 10.1021/jo060157v. [DOI] [PubMed] [Google Scholar]
- 172.Danieli B, Lesma G, Passarella D, Silvani A. Tetrahedron Lett. 2000;41:3489. [Google Scholar]
- 173.Ziegler FE, Bennett GB. J. Am. Chem. Soc. 1973;95:7458. doi: 10.1021/ja00803a041. [DOI] [PubMed] [Google Scholar]
- 174.Imanishi T, Nakai A, Yagi N, Hanaoka M. Chem. Pharm. Bull. 1981;29:901. [Google Scholar]
- 175.Amat M, Coll MD, Passarella D, Bosch J. Tetrahedron: Asymmetry. 1996;7:2775. [Google Scholar]
- 176.Amat M, Coll MD, Bosch J, Espinosa E, Molins E. Tetrahedron: Asymmetry. 1997;8:935. [Google Scholar]
- 177.Wu A, Snieckus V. Tetrahedron Lett. 1975;25:2057. [Google Scholar]
- 178.Ishikura M, Takahashi N, Yamada K, Abe T. Heterocycles. 2008;75:107. [Google Scholar]
- 179.Magnus P, Giles M, Bonnert R, Johnson G, McQuire L, Deluca M, Merritt A, Kim CS, Vicker N. J. Am. Chem. Soc. 1993;115:8116. [Google Scholar]
- 180.Büchi G, Kulsa P, Rosati RL. J. Am. Chem. Soc. 1968;90:2448. doi: 10.1021/ja01011a059. [DOI] [PubMed] [Google Scholar]
- 181.Büchi G, Kulsa P, Ogasawara K, Rosati RL. J. Am. Chem. Soc. 1970;92:999. doi: 10.1021/ja00707a043. [DOI] [PubMed] [Google Scholar]
- 182.Narisada M, Watanabe F, Nagata W. Tetrahedron Lett. 1971;39:3681. [Google Scholar]
- 183.Uyeo S, Fales HM, Highet RJ, Wildman WC. J. Am. Chem. Soc. 1958;80:2590. [Google Scholar]
- 184.Wildman WC, Bailey DT. J. Am. Chem. Soc. 1967;89:5514. doi: 10.1021/ja00996a032. [DOI] [PubMed] [Google Scholar]
- 185.Wildman WC, Bailey DT. J. Org. Chem. 1968;33:3749. [Google Scholar]
- 186.Irie H, Uyeo S, Yoshitake A. Chem. Commun. 1966:635. [Google Scholar]
- 187.Irie H, Uyeo S, Yoshitake A. J. Chem. Soc. (C) 1968:1802. doi: 10.1039/j39680001802. [DOI] [PubMed] [Google Scholar]
- 188.Hendrickson JB, Foote C, Yoshimura N. Chem. Commun. 1965:165. [Google Scholar]
- 189.Hendrickson JB, Bogard TL, Fisch ME, Grossert S, Yoshimura N. J. Am. Chem. Soc. 1974;96:7781. [Google Scholar]
- 190.Magnus P, Hobson LA, Westlund N, Lynch V. Tetrahedron Lett. 2001;42:993. [Google Scholar]
- 191.Magnus P, Gazzard L, Hobson L, Payne AH, Rainey TJ, Westlund N, Lynch V. Tetrahedron. 2002;58:3423. [Google Scholar]
- 192.Kuehne ME, Li YL, Wei CQ. J. Org. Chem. 2000;65:6434. doi: 10.1021/jo000398h. [DOI] [PubMed] [Google Scholar]
- 193.Zhong Y, Gao Y, Guo QP, Li WM. Helv. Chim. Acta. 2010;93:133. [Google Scholar]
- 194.Jiang RW, Hon PM, But PPH, Chung HS, Lin G, Ye WC, Mak TCW. Tetrahedron. 2002;58:6705. [Google Scholar]
- 195.Hitotsuyanagi Y, Hikita M, Nakada K, Fukuya H. Heterocycles. 2007;71:2035. [Google Scholar]
- 196.Cai XG, Luo XD. Planta Med. 2007;73:170. doi: 10.1055/s-2006-957064. [DOI] [PubMed] [Google Scholar]
- 197.Kobayashi J, Ueno S, Morita H. J. Org. Chem. 2002;67:6546. doi: 10.1021/jo0258204. [DOI] [PubMed] [Google Scholar]
- 198.Morita H, Ishioka N, Takatsu H, Shinzato T, Obara Y, Nakahata N, Kobayashi J. Org. Lett. 2005;7:459. doi: 10.1021/ol047641+. [DOI] [PubMed] [Google Scholar]
- 199.Morita H, Yoshida N, Kobayashi J. J. Org. Chem. 2000;65:3558. doi: 10.1021/jo000004m. [DOI] [PubMed] [Google Scholar]
- 200.Kubota T, Suzuki T, Ishiuchi K, Kuhara T, Kobayashi J. Chem. Pharm. Bull. 2009;57:504. doi: 10.1248/cpb.57.504. [DOI] [PubMed] [Google Scholar]
- 201.Zhang Y, Di YT, Zhang Q, Mu SZ, Tan CJ, Fang X, He HP, Li SL, Hao XJ. Org. Lett. 2009;11:5414. doi: 10.1021/ol902262g. [DOI] [PubMed] [Google Scholar]
- 202.Morita H, Yoshida N, Kobayashi J. J. Org. Chem. 2002;67:2278. doi: 10.1021/jo0163288. [DOI] [PubMed] [Google Scholar]
- 203.Lim KH, Kam TS. Helv. Chim. Acta. 2007;90:31. [Google Scholar]
- 204.Massiot G, Thepenier P, Jacquier MJ, Henin J, Men-Olivier L, Delaude C. Phytochem. 1991;30:3449. [Google Scholar]
- 205.Nielsen HB, Hazell A, Hazell R, Ghia F, Torssell KBG. Phytochem. 1994;37:1729. [Google Scholar]
- 206.Takayama H, Katakawa K, Kitajima M, Yamaguchi K, Aimi N. Tetrahedron Lett. 2002;43:8307. [Google Scholar]
- 207.Ma X, Gang DR. Nat. Prod. Rep. 2004;21:752. doi: 10.1039/b409720n. [DOI] [PubMed] [Google Scholar]
- 208.Lei Y, Wrobleski AD, Golden JE, Powell DR, Aubé J. J. Am. Chem. Soc. 2005;127:4552. doi: 10.1021/ja050214m. [DOI] [PubMed] [Google Scholar]
- 209.Ito M, Sakaguchi A, Kobayashi C, Ikariya T. J. Am. Chem. Soc. 2007;129:290. doi: 10.1021/ja067777y. [DOI] [PubMed] [Google Scholar]
- 210.Kajita Y, Matsubara S, Kurahashi T. J. Am. Chem. Soc. 2008;130:6058–6059. doi: 10.1021/ja7114426. [DOI] [PubMed] [Google Scholar]
- 211.Yoshino Y, Kurahashi T, Matsubara S. J. Am. Chem. Soc. 2009;131:7494. doi: 10.1021/ja900805y. [DOI] [PubMed] [Google Scholar]
- 212.Miura T, Yarnauchi M, Murakami M. Org. Lett. 2008;10:3085. doi: 10.1021/ol8010826. [DOI] [PubMed] [Google Scholar]
- 213.Ueno S, Chatani N, Kakiuchi F. J. Am. Chem. Soc. 2007;129:6098. doi: 10.1021/ja0713431. [DOI] [PubMed] [Google Scholar]
- 214.Williams A. J. Am. Chem. Soc. 1976;98:5645. [Google Scholar]
- 215.Perrin CL. Acc. Chem. Res. 1989;22:268. [Google Scholar]
- 216.Brown RS, Bennet AJ, Slebocka-Tilk H. Acc. Chem. Res. 1992;25:481. [Google Scholar]
- 217.Poland BW, Xu MQ, Quiocho FA. J. Biol. Chem. 2000;275:16408. doi: 10.1074/jbc.275.22.16408. [DOI] [PubMed] [Google Scholar]
- 218.Romanelli A, Shekhtman A, Cowburn D, Muir TW. Proc. Natl. Acad. Sci. U.S.A. 2004;101:6397. doi: 10.1073/pnas.0306616101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Shemella P, Pereira B, Zhang YM, van Roey P, Belfort G, Garde S, Nayak SK. Biophys. J. 2007;92:847. doi: 10.1529/biophysj.106.092049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Johansson DGA, Wallin G, Sandberg A, Macao B, Aqvist J, Hard T. J. Am. Chem. Soc. 2009;131:9475. doi: 10.1021/ja9010817. [DOI] [PubMed] [Google Scholar]
- 221.Schiene-Fischer C, Aumüller T, Fischer G. Top. Curr. Chem. 2013;328:35. doi: 10.1007/128_2011_151. [DOI] [PubMed] [Google Scholar]
- 222.Rudrabhatla P, Pant HC. J. Alzheimers Dis. 2010;19:389. doi: 10.3233/JAD-2010-1243. [DOI] [PubMed] [Google Scholar]
- 223.Somayaji V, Brown RS. J. Org. Chem. 1986;51:2676. [Google Scholar]
- 224.Wang QP, Bennet AJ, Brown RS, Santarsiero BD. Can. J. Chem. 1990;68:1732. [Google Scholar]
- 225.Wang QP, Bennet AJ, Brown RS, Santarsiero BD. J. Am. Chem. Soc. 1991;113:5757. [Google Scholar]
- 226.Somayaji V, Skorey KI, Brown RS, Ball RG. J. Org. Chem. 1986;51:4866. [Google Scholar]
- 227.Skorey KI, Somayaji V, Brown RS. J. Am. Chem. Soc. 1988;110:5205. [Google Scholar]
- 228.Skorey KI, Somayaji V, Brown RS. J. Am. Chem. Soc. 1989;111:1445. [Google Scholar]
- 229.Somayaji V, Brown RS. J. Am. Chem. Soc. 1987;109:4738. [Google Scholar]
- 230.Somayaji V, Keillor J, Brown RS. J. Am. Chem. Soc. 1988;110:2625. [Google Scholar]
- 231.Keillor JW, Brown RS. J. Am. Chem. Soc. 1991;113:5114. [Google Scholar]
- 232.Bashore CG, Samardjiev IJ, Bordner J, Coe JW. J. Am. Chem. Soc. 2003;125:3268. doi: 10.1021/ja028152c. [DOI] [PubMed] [Google Scholar]
- 233.Aszodi J, Rowlands DA, Mauvais P, Collette P, Bonnefoy A, Lampilas M. Bioorg. Med. Chem. Lett. 2004;14:2489. doi: 10.1016/j.bmcl.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 234.Mangion IK, Ruck RT, Rivera N, Huffman MA, Shevlin M. Org. Lett. 2011;13:5480. doi: 10.1021/ol202195n. [DOI] [PubMed] [Google Scholar]
- 235.Hans RH, Su H, Chibale K. Beilstein J. Org. Chem. 2010;6 doi: 10.3762/bjoc.6.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hans RH, Wiid IJF, van Helden PD, Wan B, Franzblau SG, Gut J, Rosenthal PJ, Chibale K. Bioorg. Med. Chem. Lett. 2011;21:2055. doi: 10.1016/j.bmcl.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 237.Smissman EE, Matuszak AJ, Corder CN. J. Pharm. Sci. 1964;53:1541. doi: 10.1002/jps.2600531229. [DOI] [PubMed] [Google Scholar]
- 238.Brouillette WJ, Brown GB, DeLorey TM, Shirali SS, Grunewald GL. J. Med. Chem. 1988;31:2218. doi: 10.1021/jm00119a025. [DOI] [PubMed] [Google Scholar]
- 239.Brouillette WJ, Grunewald GL, Brown GB, DeLorey TM, Akhtar MS, Liang G. J. Med. Chem. 1989;32:1577. doi: 10.1021/jm00127a029. [DOI] [PubMed] [Google Scholar]
- 240.Brouillette WJ, Jestkov VP, Brown ML, Akhtar MS, DeLorey TM, Brown GB. J. Med. Chem. 1994;37:3289. doi: 10.1021/jm00046a013. [DOI] [PubMed] [Google Scholar]
- 241.Grimm JB, Stables JP, Brown ML. Bioorg. Med. Chem. 2003;11:4133. doi: 10.1016/s0968-0896(03)00400-0. [DOI] [PubMed] [Google Scholar]
- 242.Murakami M, Aoki T, Nagata W. Synlett. 1990:684. [Google Scholar]
- 243.Buynak JD, Rao AS, Adam G, Nidamarthy SD, Zhang H. J. Am. Chem. Soc. 1998;120:6846. [Google Scholar]
- 244.Sierra MA, Rodriguez-Fernandez M, Mancheno MJ, Casarrubios L, Gomez-Gallego M. Tetrahedron. 2008;64:9592. [Google Scholar]
- 245.Urbach A, Dive G, Marchand-Brynaert J. Eur. J. Org. Chem. 2009:1757. doi: 10.1016/j.ejmech.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 246.Urbach A, Dive G, Tinant B, Duval V, Marchand-Brynaert J. Eur. J. Med. Chem. 2009;44:2071. doi: 10.1016/j.ejmech.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 247.Georg GI. The Organic Chemistry of Beta-Lactams. Wiley-VCH; 1992. [Google Scholar]
- 248.Bose AK, Manhas MS, Banik BK, Srirajan V. β-Lactams: Cyclic Amides of Distinction. In: Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley; 2000. [Google Scholar]
- 249.Bredt J, Thouet H, Schmitz J. Liebigs Ann. Chem. 1924;437:1. [Google Scholar]
- 250.Wasserman HH. Nature. 2006;441:699. doi: 10.1038/441699a. [DOI] [PubMed] [Google Scholar]
- 251.Doering WE, Chanley JD. J. Am. Chem. Soc. 1946;68:586. doi: 10.1021/ja01208a017. [DOI] [PubMed] [Google Scholar]
- 252.Yakhontov LN, Rubtsov MV. J. Gen. Chem. USSR. 1957;27:83. [Google Scholar]
- 253.Pracejus H. Ber. 1959;92:988. [Google Scholar]
- 254.Pracejus H, Kehlen M, Kehlen H, Matschiner H. Tetrahedron. 1965;21:2257. [Google Scholar]
- 255.Pracejus H. Ber. 1965;98:2897. [Google Scholar]
- 256.Levkoeva EI, Nikitskaya ES, Yakhontov LN. Dokl. Akad. Nauk. 1970;192:342. [Google Scholar]
- 257.Levkoeva EI, Nikitskaya ES, Yakhontov LN. Khim. Geterotsikl. Soedin. 1971:378. [Google Scholar]
- 258.Kostyanovsky RG, Mikhlina EE, Levkoeva EI, Yakhontov LN. Org. Mass Spectrom. 1970;3:1023. [Google Scholar]
- 259.Greenberg A, Wu GL, Tsai JC, Chiu YY. Struct. Chem. 1993;4:127. [Google Scholar]
- 260.Ly T, Krout M, Pham DK, Tani K, Stoltz BM, Julian RR. J. Am. Chem. Soc. 2007;129:1864. doi: 10.1021/ja067703m. [DOI] [PubMed] [Google Scholar]
- 261.Blackburn GM, Skaife CJ, Kay IT. J. Chem. Res. Synop. 1980:294. [Google Scholar]
- 262.Bennet AJ, Wang QP, Slebocka-Tilk H, Somayaji V, Brown RS, Santarsiero BD. J. Am. Chem. Soc. 1990;112:6383. [Google Scholar]
- 263.Boivin J, Gaudin D, Labrecque D, Jankowski K. Tetrahedron Lett. 1990;31:2281. [Google Scholar]
- 264.Frackenpohl J, Langer P, Hoffmann HMR. Helv. Chim. Acta. 1998;81:1429. [Google Scholar]
- 265.Hoffmann HMR, Frackenpohl J. Organic Chemistry of Cinchona Alkaloids. In: Song CE, editor. Cinchona Alkaloids in Synthesis and Catalysis. Wiley; 2009. [Google Scholar]
- 266.Ballester P, Tadayoni BM, Branda N, Rebek J., Jr J. Am. Chem. Soc. 1990;112:3685. [Google Scholar]
- 267.Jeong KS, Parris K, Ballester P, Rebek J., Jr Angew. Chem., Int. Ed. 1990;29:555. [Google Scholar]
- 268.Jones PG, Kirby AJ, Komarov IV, Wothers PD. Chem. Commun. 1998:1695. [Google Scholar]
- 269.Kirby AJ, Komarov IV, Wothers PD, Feeder N, Jones PG. Pure Appl. Chem. 1999;71:385. [Google Scholar]
- 270.Kirby AJ, Komarov IV, Wothers PD, Feeder N. Angew. Chem., Int. Ed. 1998;37:785. doi: 10.1002/(SICI)1521-3773(19980403)37:6<785::AID-ANIE785>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 271.Ye Q, Komarov IV, Kirby AJ, Jones M., Jr J. Org. Chem. 2002;67:9288. doi: 10.1021/jo0206014. [DOI] [PubMed] [Google Scholar]
- 272.Kirby AJ, Komarov IV, Feeder N. J. Am. Chem. Soc. 1998;120:7101. [Google Scholar]
- 273.Davies JE, Kirby AJ, Komarov IV. Helv. Chim. Acta. 2003;86:1222. [Google Scholar]
- 274.Kirby AJ, Komarov IV, Bilenko VA, Davies JE, Rawson JM. Chem. Commun. 2002:2106. doi: 10.1039/b206639d. [DOI] [PubMed] [Google Scholar]
- 275.Morgan KM, Rawlins ML, Montgomery MN. J. Phys. Org. Chem. 2005;18:310. [Google Scholar]
- 276.Badger GM, Cook JW, Walker T. Chem. Commun. 1949:1141. [Google Scholar]
- 277.Albertson NF. J. Am. Chem. Soc. 1950;72:2594. [Google Scholar]
- 278.Albertson NF. J. Am. Chem. Soc. 1952;74:249. [Google Scholar]
- 279.Hall HK, Jr, Shaw RG, Deutschmann A. J. Org. Chem. 1980;45:3722. [Google Scholar]
- 280.Buchanan GL. J. Chem. Soc. Chem. Commun. 1981:814. [Google Scholar]
- 281.Buchanan GL. J. Chem. Soc. Perkin Trans. 1984;1:2669. [Google Scholar]
- 282.Buchanan GL, Kitson DH, Mallinson PR, Sim GA, White DNJ, Cox PJ. J. Chem. Soc. Perkin Trans. 1983;2:1709. [Google Scholar]
- 283.McCabe PH, Milne NJ, Sim GA. J. Chem. Soc. Perkin Trans. 1989;2:1459. [Google Scholar]
- 284.Sliter B, Morgan J, Greenberg A. J. Org. Chem. 2011;76:2770. doi: 10.1021/jo200195a. [DOI] [PubMed] [Google Scholar]
- 285.Wiseman JR, Kipp JE. J. Am. Chem. Soc. 1982;104:4688. [Google Scholar]
- 286.Steliou K, Poupart MA. J. Am. Chem. Soc. 1983;105:7130. [Google Scholar]
- 287.Brehm R, Ohnhäuser D, Gerlach H. Helv. Chim. Acta. 1987;70:1981. [Google Scholar]
- 288.Alonso DA, Costa A, Mancheno B, Najera C. Tetrahedron. 1997;53:4791. [Google Scholar]
- 289.Hellal M, Cuny GD. J. Org. Chem. 2010;75:3465. doi: 10.1021/jo1003339. [DOI] [PubMed] [Google Scholar]
- 290.Denzer M, Ott H. J. Org. Chem. 1969;34:183. [Google Scholar]
- 291.Grigg R, Sridharan V, Stevenson P, Worakun T. J. Chem. Soc. Chem. Commun. 1986:1697. [Google Scholar]
- 292.Grigg R, Santhakumar V, Sridharan V, Stevenson P, Teasdale A, Thorntonpett M, Worakun T. Tetrahedron. 1991;47:9703. [Google Scholar]
- 293.Grigg R, Sridharan V, York M. Tetrahedron Lett. 1998;39:4139. [Google Scholar]
- 294.Grigg R, York M. Tetrahedron Lett. 2000;41:7255. [Google Scholar]
- 295.Paquette LA, Dura RD, Fosnaugh N, Stepanian M. J. Org. Chem. 2006;71:8438. doi: 10.1021/jo061404y. [DOI] [PubMed] [Google Scholar]
- 296.Ribelin TP, Judd AS, Akritopoulou-Zanze I, Henry RF, Cross JL, Whittern DN, Djuric SW. Org. Lett. 2007;9:5119. doi: 10.1021/ol7023373. [DOI] [PubMed] [Google Scholar]
- 297.Shea KJ, Lease TG, Ziller JW. J. Am. Chem. Soc. 1990;112:8627. [Google Scholar]
- 298.Lease TG, Shea KJ. J. Am. Chem. Soc. 1993;115:2248. [Google Scholar]
- 299.Doyle MP. Chem. Rev. 1986;86:919. [Google Scholar]
- 300.Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- 301.Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem. Rev. 2010;110:70. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- 302.Doyle MP, Kalinin AV. Synlett. 1995:1075. [Google Scholar]
- 303.Williams RM, Lee BH. J. Am. Chem. Soc. 1986;108:6431. [Google Scholar]
- 304.Williams RM, Lee BH, Miller MM, Anderson OP. J. Am. Chem. Soc. 1989;111:1073. [Google Scholar]
- 305.Sundberg RJ, Smith FX. J. Org. Chem. 1975;40:2613. [Google Scholar]
- 306.Sundberg RJ, Luis JG, Parton RL, Schreiber S, Srinivasan PC, Lamb P, Forcier P, Bryan RF. J. Org. Chem. 1978;43:4859. [Google Scholar]
- 307.Shen L, Hsung RP. Org. Lett. 2005;7:775. doi: 10.1021/ol047572z. [DOI] [PubMed] [Google Scholar]
- 308.Adamson G, Beckwith ALJ, Kaufmann M, Willis AC. J. Chem. Soc. Chem. Commun. 1995:1783. [Google Scholar]
- 309.Phakhodee W, Ploypradith P, Sahakitpichan P, Ruchirawat S. Tetrahedron. 2009;65:351. [Google Scholar]
- 310.Phakhodee W, Sahakitpichan P, Deechongkit S, Ruchirawat S. Heterocycles. 2008;75:1963. [Google Scholar]
- 311.Dolby LJ, Sakai SI. J. Am. Chem. Soc. 1964;86:1890. [Google Scholar]
- 312.Dolby LJ, Sakai SI. Tetrahedron. 1967;23:1. doi: 10.1016/s0040-4020(01)83280-4. [DOI] [PubMed] [Google Scholar]
- 313.Xie Q, Wang H, Xia Z, Lu M, Zhang W, Wang X, Fu W, Tang Y, Sheng W, Li W, Zhou W, Zhu X, Qiu Z, Chen H. J. Med. Chem. 2008;51:2027. doi: 10.1021/jm070154q. [DOI] [PubMed] [Google Scholar]
- 314.Zheng W, Xie Q, Li F, Qui ZB. Acta Cryst. 2009;E65:3008. [Google Scholar]
- 315.Artacho J, Ascic E, Rantanen T, Karlsson J, Wallentin CJ, Wang R, Wendt OF, Harmata M, Snieckus V, Wärnmark K. Chem. Eur. J. 2012;18:1038. doi: 10.1002/chem.201103228. [DOI] [PubMed] [Google Scholar]
- 316.Artacho J, Ascic E, Rantanen T, Wallentin CJ, Dawaigher S, Bergquist KE, Harmata M, Snieckus V, Wärnmark K. Org. Lett. 2012;14:4706. doi: 10.1021/ol302022y. [DOI] [PubMed] [Google Scholar]
- 317.Milligan GL, Mossman CJ, Aubé J. J. Am. Chem. Soc. 1995;117:10449. [Google Scholar]
- 318.Lang S, Murphy JA. Chem. Soc. Rev. 2006;35:146. doi: 10.1039/b505080d. [DOI] [PubMed] [Google Scholar]
- 319.Nyfeler E, Renaud P. Chimia. 2006;60:276. [Google Scholar]
- 320.Grecian S, Aubé J. Schmidt Rearrangement Reactions with Alkyl Azides. In: Bräse S, Banert K, editors. Organic Azides: Synthesis and Applications. Chichester, U.K: Wiley and Sons; 2009. [Google Scholar]
- 321.Yao L, Aubé J. J. Am. Chem. Soc. 2007;129:2766. doi: 10.1021/ja068919r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Szostak M, Yao L, Aubé J. J. Org. Chem. 2010;75:1235. doi: 10.1021/jo902574m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Szostak M, Yao L, Aubé J. Org. Lett. 2009;11:4386. doi: 10.1021/ol901771b. [DOI] [PubMed] [Google Scholar]
- 324.Gutierrrez O, Aubé J, Tantillo DJ. J. Org. Chem. 2012;77:640. doi: 10.1021/jo202338m. [DOI] [PubMed] [Google Scholar]
- 325.Macleod F, Lang S, Murphy JA. Synlett. 2010:529. [Google Scholar]
- 326.Szostak M, Aubé J. J. Am. Chem. Soc. 2010;132:2530. doi: 10.1021/ja910654t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Golden JE, Aubé J. Angew. Chem., Int. Ed. 2002;41:4316. doi: 10.1002/1521-3773(20021115)41:22<4316::AID-ANIE4316>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 328.Zeng YB, Aubé J. J. Am. Chem. Soc. 2005;125:15712. doi: 10.1021/ja055629m. [DOI] [PubMed] [Google Scholar]
- 329.Frankowski KJ, Golden JE, Zeng YB, Lei Y, Aubé J. J. Am. Chem. Soc. 2008;130:6018. doi: 10.1021/ja800574m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Szostak M, Yao L, Day VW, Powell DR, Aubé J. J. Am. Chem. Soc. 2010;132:8336. doi: 10.1021/ja101690u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Iyengar R, Schildknegt K, Aubé J. Org. Lett. 2000;2:1625. doi: 10.1021/ol005913c. [DOI] [PubMed] [Google Scholar]
- 332.Iyengar R, Schildknegt K, Morton M, Aubé J. J. Org. Chem. 2005;70:10645. doi: 10.1021/jo051212n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Wrobleski AD. Ph.D. Thesis. University of Kansas; 2003. [Google Scholar]
- 334.Schill G, Priester CU, Windhövel UF, Fritz H. Helv. Chim. Acta. 1986;69:438. [Google Scholar]
- 335.Schill G, Löwer H, Priester CU, Windhövel UF, Fritz H. Tetrahedron. 1987;43:3729. [Google Scholar]
- 336.Schill G, Priester CU, Windhövel UF, Fritz H. Tetrahedron. 1987;43:3747. [Google Scholar]
- 337.Schill G, Priester CU, Windhövel UF, Fritz H. Tetrahedron. 1987;43:3765. [Google Scholar]
- 338.Schill G, Priester CU, Windhövel UF, Fritz H. Tetrahedron. 1990;46:1211. [Google Scholar]
- 339.Schill G, Priester CU, Windhovel UF, Fritz H. Tetrahedron. 1990;46:1221. [Google Scholar]
- 340.Magnus P, Ladlow M, Elliott J, Kim CS. J. Chem. Soc. Chem. Commun. 1989:518. [Google Scholar]
- 341.Magnus P, Mendoza JS, Stamford A, Ladlow M, Willis P. J. Am. Chem. Soc. 1992;114:10232. [Google Scholar]
- 342.Dennison JB, Kulanthaivel P, Barbuch RJ, Renbarger JL, Ehlhardt WJ, Hall SD. Drug Metab. Dispos. 2006;34:1317. doi: 10.1124/dmd.106.009902. [DOI] [PubMed] [Google Scholar]
- 343.Szostak M, Aubé J. Org. Lett. 2009;11:3878. doi: 10.1021/ol901449y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Arata Y, Tanaka KI, Yoshifuji S, Kanatomo S. Chem. Pharm. Bull. 1979;27:981. [Google Scholar]
- 345.Waly MA, Kodier MN, Hossini MS. Chin. Pharm. J. 1994;46:135. [Google Scholar]
- 346.Smet M, Van Oosterwijck C, Van Hecke K, Van Meervelt L, Vandendriessche A, Dehaen W. Synlett. 2004:2388. [Google Scholar]
- 347.Nazarenko KG, Shtil NA, Buth SA, Chernega AN, Lozinskii MO, Tolmachev AA. Tetrahedron. 2008;64:4478. [Google Scholar]
- 348.Doodeman R. Ph.D. Thesis. University of Amsterdam; 2002. [Google Scholar]
- 349.Cossy J, Pardo DG. Chemtracts. 2002;15:579. [Google Scholar]
- 350.Metro TX, Duthion B, Pardo DG, Cossy J. Chem. Soc. Rev. 2010;39:89. doi: 10.1039/b806985a. [DOI] [PubMed] [Google Scholar]
- 351.Arata Y, Kobayashi T. Chem. Pharm. Bull. 1970;18:2361. [Google Scholar]
- 352.Arata Y, Kobayashi T. Chem. Pharm. Bull. 1972;20:325. [Google Scholar]
- 353.Miyano S, Mibu N, Irie M, Fujii S, Fujisaki F, Abe N, Sumoto K. J. Chem. Soc. Perkin Trans. 1987;1:313. [Google Scholar]
- 354.Miyano S, Irie M, Mibu N, Miyamoto Y, Nagata K, Sumoto K. J. Chem. Soc. Perkin Trans. 1988;1:1057. [Google Scholar]
- 355.Bremner JB, Eschler BM, Skelton BW, White AH. Aust. J. Chem. 1994;47:1935. [Google Scholar]
- 356.Schumann D, Geirsson J, Naumann A. Chem. Ber. 1985;118:1927. [Google Scholar]
- 357.Bahadur GA, Bailey AS, Scott PW, Vandrevala MH. J. Chem. Soc. Perkin Trans. 1980;1:2870. [Google Scholar]
- 358.Prout K, Sims M, Watkin D, Couldwell C, Vandrevala M, Bailey AS. Acta Cryst. 1980;B36:1846. [Google Scholar]
- 359.Chelain E, Parlier A, Audouin M, Rudler H, Daran JC, Vaissermann J. J. Am. Chem. Soc. 1993;115:10568. [Google Scholar]
- 360.Coqueret X, Bourelle-Wargnier F, Chuche J. J. Org. Chem. 1985;50:909. [Google Scholar]
- 361.Kutney JP, Balsevich J, Bokelman GH. Heterocycles. 1976;4:1377. [Google Scholar]
- 362.Kutney JP, Balsevich J, Bokelman GH, Hibino T, Honda T, Itoh I, Ratcliffe AH, Worth BR. Can. J. Chem. 1978;56:62. [Google Scholar]
- 363.Toshimitsu A, Terao K, Uemura S. Tetrahedron Lett. 1984;25:5917. [Google Scholar]
- 364.Toshimitsu A, Terao K, Uemura S. J. Chem. Soc. Chem. Commun. 1986:530. [Google Scholar]
- 365.Toshimitsu A, Terao K, Uemura S. J. Org. Chem. 1987;52:2018. [Google Scholar]
- 366.Smissman EE, Robinson RA. J. Org. Chem. 1970;35:3532. doi: 10.1021/jo00836a052. [DOI] [PubMed] [Google Scholar]
- 367.Smissman EE, Robinson RA, Matuszak AJ. J. Org. Chem. 1970;35:3823. doi: 10.1021/jo00836a054. [DOI] [PubMed] [Google Scholar]
- 368.Smissman EE, Ayres JW. J. Org. Chem. 1971;36:2407. doi: 10.1021/jo00816a005. [DOI] [PubMed] [Google Scholar]
- 369.Smissman EE, Ayres JW, Wirth PJ, Abernethy DR. J. Org. Chem. 1972;37:3486. doi: 10.1021/jo00795a020. [DOI] [PubMed] [Google Scholar]
- 370.Smissman EE, Wirth PJ. J. Org. Chem. 1975;40:1576. doi: 10.1021/jo00899a012. [DOI] [PubMed] [Google Scholar]
- 371.Smissman EE, Wirth PJ, Glynn DR. J. Org. Chem. 1975;40:281. doi: 10.1021/jo00899a012. [DOI] [PubMed] [Google Scholar]
- 372.Beddoes RL, Davies MPH, Thomas EJ. J. Chem. Soc. Chem. Commun. 1992:538. [Google Scholar]
- 373.Baylis AM, Davies MPH, Thomas EJ. Org. Biomol. Chem. 2007;5:3139. doi: 10.1039/b708910d. [DOI] [PubMed] [Google Scholar]
- 374.Baylis AM, Thomas EJ. Tetrahedron. 2007;63:11666. [Google Scholar]
- 375.Sakamoto M, Watanabe S, Fujita T, Yanase T. J. Org. Chem. 1990;55:2986. [Google Scholar]
- 376.Roscini C, Cubbage KL, Berry M, Orr-Ewing AJ, Booker-Milburn KI. Angew. Chem., Int. Ed. 2009;48:8716. doi: 10.1002/anie.200904059. [DOI] [PubMed] [Google Scholar]
- 377.Booker-Milburn KI, Anson CE, Clissold C, Costin NJ, Dainty RF, Murray M, Patel D, Sharpe A. Eur. J. Org. Chem. 2001:1473. [Google Scholar]
- 378.Hall HK, Jr, Johnson RC. J. Org. Chem. 1972;37:697. [Google Scholar]
- 379.Hall HK, Jr, Ekechukwu OE, Deutschmann A, Rose C. Polym. Bull. 1980;3:375. [Google Scholar]
- 380.Hall HK, Jr, El-Shekeil A. J. Org. Chem. 1980;45:5325. [Google Scholar]
- 381.Hall HK, Jr, El-Shekeil A. Polym. Bull. 1980;2:829. [Google Scholar]
- 382.Sparks SM, Vargas JD, Shea KJ. Org. Lett. 2000;2:1473. doi: 10.1021/ol005811m. [DOI] [PubMed] [Google Scholar]
- 383.Sparks SM, Chow CP, Zhu L, Shea KJ. J. Org. Chem. 2004;69:3025. doi: 10.1021/jo049897z. [DOI] [PubMed] [Google Scholar]
- 384.Chow CP, Shea KJ, Sparks SM. Org. Lett. 2002;4:2637. doi: 10.1021/ol026075k. [DOI] [PubMed] [Google Scholar]
- 385.Molina CL, Chow CP, Shea KJ. J. Org. Chem. 2007;72:6816. doi: 10.1021/jo070978f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Lahousse H, Martens HJ, Toppet S, Hoornaert GJ. J. Org. Chem. 1982;47:2001. [Google Scholar]
- 387.Chow CP, Shea KJ. J. Am. Chem. Soc. 2005;127:3678. doi: 10.1021/ja050059b. [DOI] [PubMed] [Google Scholar]
- 388.Meltzer RI, Lewis AD. J. Org. Chem. 1956;21:256. [Google Scholar]
- 389.Baumler J, Sorkin E, Erlenmeyer H. Helv. Chim. Acta. 1951;34:459. [Google Scholar]
- 390.Brouillette WJ, Smissman EE, Grunewald GL. J. Org. Chem. 1979;44:839. [Google Scholar]
- 391.Brouillette WJ, Einspahr HM. J. Org. Chem. 1984;49:5113. [Google Scholar]
- 392.Kubicki M, Codding PW. Acta Cryst. 2001;E57:o406. doi: 10.1107/s0108270101003171. [DOI] [PubMed] [Google Scholar]
- 393.Akhtar MS, Brouillette WJ, Waterhous DV. J. Org. Chem. 1990;55:5222. [Google Scholar]
- 394.Brouillette WJ, Friedrich JD, Muccio DD. J. Org. Chem. 1984;49:3227. [Google Scholar]
- 395.Smissman EE, Robinson RA, Carr JB, Matuszak AJ. J. Org. Chem. 1970;35:3821. doi: 10.1021/jo00836a053. [DOI] [PubMed] [Google Scholar]
- 396.Gmünder J, Lindenmann A. Helv. Chim. Acta. 1964;47:66. [Google Scholar]
- 397.Rappoport Z. The Chemistry of Enamines. John Wiley & Sons; 1994. [Google Scholar]
- 398.Doering WV, Birladeanu L, Andrews DW, Pagnotta M. J. Am. Chem. Soc. 1985;107:428. [Google Scholar]
- 399.Crawley SL, Funk RL. Org. Lett. 2006;8:3995. doi: 10.1021/ol061461d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Yotphan S, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2008;130:2452. doi: 10.1021/ja710981b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Pearson WH, Walavalkar R, Schkeryantz JM, Fang WK, Blickensdorf JD. J. Am. Chem. Soc. 1993;115:10183. [Google Scholar]
- 402.Nielsen AT. J. Org. Chem. 1966;31:1053. [Google Scholar]
- 403.Bender DR, Coffen DL. J. Org. Chem. 1968;33:2504. [Google Scholar]
- 404.Warawa EJ, Campbell JR. J. Org. Chem. 1974;39:3511. [Google Scholar]
- 405.O’Neil IA, Wynn D, Lai JYQ. Tetrahedron Lett. 2000;41:271. [Google Scholar]
- 406.Yakhontov L. Heterocycles. 1977;7:1033. [Google Scholar]
- 407.Hamama WS, El-Magid OMA, Zoorob HH. J. Heterocycl. Chem. 2006;43:1397. [Google Scholar]
- 408.Szostak M, Yao L, Aubé J. J. Am. Chem. Soc. 2010;132:2078. doi: 10.1021/ja909792h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Sheridan RS, Ganzer GA. J. Am. Chem. Soc. 1983;105:6158. [Google Scholar]
- 410.Radziszewski JG, Downing JW, Wentrup C, Kaszynski P, Jawdosiuk M, Kovacic P, Michl J. J. Am. Chem. Soc. 1985;107:2799. [Google Scholar]
- 411.Wayne GS, Snyder GJ. J. Am. Chem. Soc. 1993;115:9860. [Google Scholar]
- 412.Eguchi S, Okano T, Takeuchi H. Heterocycles. 1987;26:3265. [Google Scholar]
- 413.Brummond KM, Lu JL. Org. Lett. 2001;3:1347. doi: 10.1021/ol010029n. [DOI] [PubMed] [Google Scholar]
- 414.Brummond KM, Hong SP. J. Org. Chem. 2005;70:907. doi: 10.1021/jo0483567. [DOI] [PubMed] [Google Scholar]
- 415.Yamazaki N, Suzuki H, Kibayashi C. J. Org. Chem. 1997;62:8280. doi: 10.1021/jo9715579. [DOI] [PubMed] [Google Scholar]
- 416.Suzuki H, Yamazaki N, Kibayashi C. Tetrahedron Lett. 2001;42:3013. [Google Scholar]
- 417.Yoshimura Y, Kusanagi T, Kibayashi C, Yamazaki N, Aoyagi S. Heterocycles. 2007;75:1329. [Google Scholar]
- 418.Rodriguez-Escrich S, Sola L, Jimeno C, Rodriguez-Escrich C, Pericas MA. Adv. Synth. Catal. 2008;350:2250. [Google Scholar]
- 419.Wang YF, Toh KK, Ng EPJ, Chiba S. J. Am. Chem. Soc. 2011;133:6411. doi: 10.1021/ja200879w. [DOI] [PubMed] [Google Scholar]
- 420.Braje W, Wartchow R, Hoffmann HMR. Angew. Chem., Int. Ed. 1999;38:2540. doi: 10.1002/(sici)1521-3773(19990903)38:17<2539::aid-anie2539>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 421.Franz MH, Röper S, Wartchow R, Hoffmann HMR. J. Org. Chem. 2004;69:2983. doi: 10.1021/jo030363s. [DOI] [PubMed] [Google Scholar]
- 422.Hoffmann HMR, Frackenpohl J. Eur. J. Org. Chem. 2004:4293. [Google Scholar]
- 423.Röper S, Frackenpohl J, Schrake O, Wartchow R, Hoffmann HMR. Org. Lett. 2000;2:1661. doi: 10.1021/ol0057378. [DOI] [PubMed] [Google Scholar]
- 424.Röper S, Wartchow R, Hoffmann HMR. Org. Lett. 2002;4:3179. doi: 10.1021/ol026342m. [DOI] [PubMed] [Google Scholar]
- 425.Katritzky AR, Rees CW, Scriven EFC. Comprehensive Heterocyclic Chemistry II. Vol. 3. Elsevier; 1996. p. 4. [Google Scholar]
- 426.Drews J. Science. 2000;287:1960. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 427.Sneader W. Drug Discovery: A History. Wiley; 2005. [Google Scholar]
- 428.Davies FA, Chen BC. Chem. Rev. 1992;92:919. [Google Scholar]
- 429.Oppolzer W. Pure Appl. Chem. 1990;62:1241. [Google Scholar]
- 430.Majumdar KC, Mondal S. Chem. Rev. 2011;111:7749. doi: 10.1021/cr1003776. [DOI] [PubMed] [Google Scholar]
- 431.Paquette LA, Barton WRS, Gallucci JC. Org. Lett. 2004;6:1313. doi: 10.1021/ol049679s. [DOI] [PubMed] [Google Scholar]
- 432.Evans P, McCabe T, Morgan BS, Reau S. Org. Lett. 2005;7:43. doi: 10.1021/ol0480123. [DOI] [PubMed] [Google Scholar]
- 433.Evans P. J. Org. Chem. 2007;72:1830. doi: 10.1021/jo062189o. [DOI] [PubMed] [Google Scholar]
- 434.Kelleher S, Quesne PY, Evans P. Beilstein J. Org. Chem. 2009;5 doi: 10.3762/bjoc.5.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Klein JEMN, Geoghegan K, Méral N, Evans P. Chem. Commun. 2010;46:937. doi: 10.1039/b923175g. [DOI] [PubMed] [Google Scholar]
- 436. Geoghegan K, Kelleher S, Evans P. J. Org. Chem. 2011;76:2187. doi: 10.1021/jo200023r. For a recent study on regioselectivity in the Heck reaction forming bridged sultams, see: Geoghegan K, Evans P, Rozas I, Alkorta I. Chem. Eur. J. 2012;18:13379. doi: 10.1002/chem.201201359.
- 437.Paquette LA, Leit SM. J. Am. Chem. Soc. 1999;121:8126. [Google Scholar]
- 438.Paquette LA, Ra CS, Schloss JD, Leit SM, Gallucci JC. J. Org. Chem. 2001;66:3564. doi: 10.1021/jo010216z. [DOI] [PubMed] [Google Scholar]
- 439.Schloss JD, Leit SM, Paquette LA. J. Org. Chem. 2000;65:7119. doi: 10.1021/jo000873b. [DOI] [PubMed] [Google Scholar]
- 440.Preston AJ, Gallucci JC, Paquette LA. J. Org. Chem. 2006;71:6573. doi: 10.1021/jo0611162. [DOI] [PubMed] [Google Scholar]
- 441.Rassadin VA, Grosheva DS, Tomashevskiy AA, Sokolov VV, Yufit DS, Kozhushkov SI, de Meijere A. Eur. J. Org. Chem. 2010:3481. [Google Scholar]
- 442.Samarakoon TB, Loh JK, Rolfe A, Le LS, Yoon SY, Lushington GH, Hanson PR. Org. Lett. 2011;13:5148. doi: 10.1021/ol201962n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Zhou A, Rayabarapu D, Hanson PR. Org. Lett. 2009;11:531. doi: 10.1021/ol802467f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Rolfe A, Samarakoon TB, Hanson PR. Org. Lett. 2010;12:1216. doi: 10.1021/ol100035e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Samarakoon TB, Hur MY, Kurtz RD, Hanson PR. Org. Lett. 2010;12:2182. doi: 10.1021/ol100495w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Loh JK, Yoon SY, Samarakoon TB, Rolfe A, Porubsky P, Neuenswander B, Lushington GH, Hanson PR. Beilstein J. Org. Chem. 2012;8:1293. doi: 10.3762/bjoc.8.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Stoll AH, Blakey SB. Chem. Sci. 2011;2:112. doi: 10.1039/C0SC00577K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Radziecka A, Wolfenden R. J. Am. Chem. Soc. 1996;118:6105. [Google Scholar]
- 449.Szostak M, Yao L, Aubé J. J. Org. Chem. 2009;74:1869. doi: 10.1021/jo802192v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Adler M, Alder S, Boche G. J. Phys. Org. Chem. 2005;18:193. [Google Scholar]
- 451.Werstiuk NH, Brown RS, Wang Q. Can. J. Chem. 1996;74:524. [Google Scholar]
- 452.Szostak M, Aubé J. Chem. Commun. 2009:7122. doi: 10.1039/b917508c. [DOI] [PubMed] [Google Scholar]
- 453.Hall HK, Jr, Schneider AK. J. Am. Chem. Soc. 1958;80:6409. [Google Scholar]
- 454.Hall HK., Jr J. Am. Chem. Soc. 1958;80:6412. [Google Scholar]
- 455.Hall HK., Jr J. Am. Chem. Soc. 1960;82:1209. [Google Scholar]
- 456.Szostak M, Aubé J. J. Am. Chem. Soc. 2009;131:13246. doi: 10.1021/ja906471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Yoshida K, Nomura S, Nishibata Y, Ban Y. Heterocycles. 1986;24:2239. [Google Scholar]
- 458.Hayes CJ, Simpkins NS, Kirk DT, Mitchell L, Baudoux J, Blake AJ, Wilson C. J. Am. Chem. Soc. 2009;131:8196. doi: 10.1021/ja9009875. [DOI] [PubMed] [Google Scholar]
- 459.Dura RD, Paquette LA. Synthesis. 2006:2837. [Google Scholar]
- 460.Gallucci JC, Dura RD, Paquette LA. Acta Cryst. 2007;E63:3417. [Google Scholar]
- 461.Zhu L, Lauchli R, Loo M, Shea KJ. Org. Lett. 2007;9:2269. doi: 10.1021/ol070397c. [DOI] [PubMed] [Google Scholar]
- 462.Chiang Y, Kresge AJ, Walsh PA. J. Org. Chem. 1990;55:1309. [Google Scholar]
- 463.Dura RD, Paquette LA. J. Org. Chem. 2006;71:2456. doi: 10.1021/jo0526587. [DOI] [PubMed] [Google Scholar]
- 464.Preston AJ, Paquette LA. Heterocycles. 2006;70:41. [Google Scholar]
- 465.Velten R, Erdelen C, Gehling M, Göhrt A, Gondol D, Lenz J, Lockhoff O, Wachendorff U, Wendisch D. Tetrahedron Lett. 1998;39:1737. [Google Scholar]