

Abstract
Background
Several aspects of microarray data analysis are dependent on identification of genes expressed at or near the limits of detection. For example, regression-based normalization methods rely on the premise that most genes in compared samples are expressed at similar levels and therefore require accurate identification of nonexpressed genes (additive noise) so that they can be excluded from the normalization procedure. Moreover, key regulatory genes can maintain stringent control of a given response at low expression levels. If arbitrary cutoffs are used for distinguishing expressed from nonexpressed genes, some of these key regulatory genes may be unnecessarily excluded from the analysis. Unfortunately, no accurate method for differentiating additive noise from genes expressed at low levels is currently available.
Results
We developed a multistep procedure for analysis of mRNA expression data that robustly identifies the additive noise in a microarray experiment. This analysis is predicated on the fact that additive noise signals can be accurately identified by both distribution and statistical analysis.
Conclusions
Identification of additive noise in this manner allows exclusion of noncorrelated weak signals from regression-based normalization of compared profiles thus maximizing the accuracy of these methods. Moreover, genes expressed at very low levels can be clearly identified due to the fact that their expression distribution is stable and distinguishable from the random pattern of additive noise.
Background
Microarrays are powerful and cost-effective tools for large-scale analysis of gene expression. While the utility of this technology has been established [1,2], analytical methods are evolving and a matter of contention. Key among the more controversial aspects is the treatment of data from weak spots, which significantly influences outcomes. For example, ratio analysis is commonly employed to determine expression differences between two samples. However any procedure that uses raw intensities to infer relative expression is limited due to the fact that accuracy is signal level dependent, with variation increasing dramatically for low intensity signals [1,3]. Several methods have been developed to diminish the influence of additive noise. One solution is to ignore any genes whose transcripts are present at a low total abundance, to exclude weak spots – arbitrarily (in Kooperberg etal., [3] an intensity cutoff was used such that the relative error in ratios was less than 25%) or with some statistical procedures [4,5]. Other methods proposed for discriminating expressed genes from those not expressed, such as the method of Greller and Tobin [6], are suitable only for bimodal distributions in which the distribution of intensities for these two subsets are non-overlapping, unlike many empirical data sets. However even procedures for flagging and exclusion of weak spots based on solid statistical background [4] remains problematic as these methods discard potentially valuable data. This issue is compounded by the fact that in biological systems several key regulators may be expressed at low levels presumably so that modulation of these regulators can be tightly controlled [7].
The two main sources of heterogeneity in gene expression variations are indicated in Rocke and Durbin [7] as, the "additive component", prominent at low expression levels, and the "multiplicative component", prominent at high expression levels. The intensity measurement yi, j for gene i ∈ I = {i1, ..., in} in sample j ∈ J = {j1, ..., jm} is modeled by the equation: yi, j = αi, j + μi, j × eη + ει, j, where α – is the normal background (and independent of expression level), μ – the expression level in arbitrary units, ε – is first within spot error term (additive), and η – is the second error term, which represents the proportional error (multiplicative) [8,9]. Gene expression data obtained with standard procedure of the local background subtraction will include noisy spots – spots at which expression level is ignorably low and whose intensity ει, j presents additive noise.
We have previously demonstrated the presence of normally distributed noise spots in radioactive labeled Clontech macroarrays and proposed an iterative algorithm for obtaining the parameters of this distribution [2,10]. Herein we have extended the utility of this approach by demonstrating the noncorrelative nature of these spots in both internal and external comparisons. We also present new algorithm modifications for locating the additive noise in gene expression histograms and for estimation of its distribution parameters. Quantization of additive noise variation can therefore be used as a statistically robust criterion to identify measurable but low-level gene expression. It becomes possible to select even genes that are stably expressed at the additive noise level that can be discriminated from additive noise due to their stability.
Results
Data among experiments was first normalized to additive noise. A histogram of all intensity values demonstrates the presence of the normally distributed spots corresponding to cDNA targets that do not hybridize to a detectable extent with the labeled test probes (Fig. 1). These signals were due to noise and therefore fit a random distribution whose properties (mean and SD) were proportional to the total amount of signal on the array and hence can be used for data normalization among array experiments. The mean and variance of the intensity levels of non-expressed genes must be estimated using the same general principles as described in materials and methods. Modifications of this method are required for analysis of different types of arrays.
Figure 1.
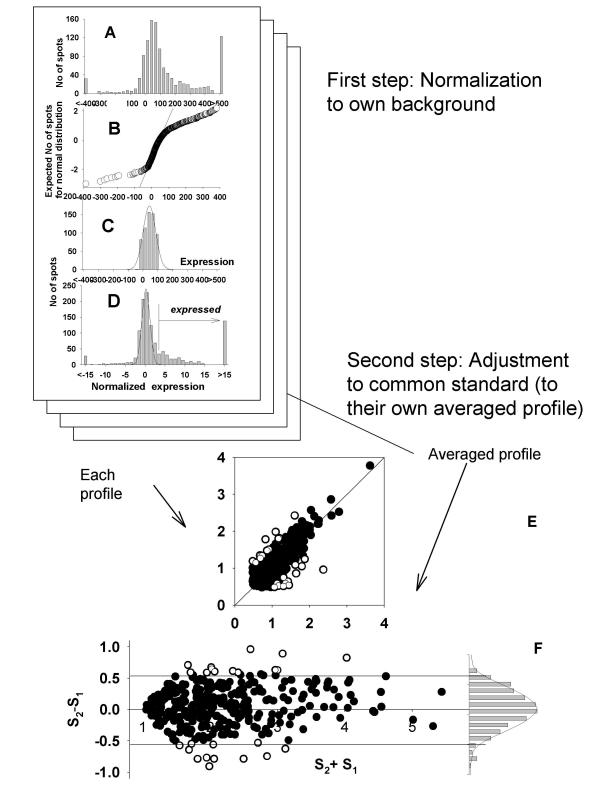
Normalization procedure for array data as shown on Atlas Clontech membranes. A. Histogram of averaged expression data from duplicated spots. B. Normality plot of A. C. Histogram of the trimmed data +/- 2SD about the mean. D. Histogram of the data after z transformation. E. Scatter plot showing the regression line for duplicated expressions (log transformed) after normalization. F. Scatter plot of background genes exhibiting the normality of their profile with average of 0 and SD of 1.
High quality membrane arrays
Atlas arrays (Clontech) are a good example of high quality membrane-based arrays exemplifying high specificity and low levels of additive noise. Additive noise spots consist of up to 50% of all spots on the array. The nearly normal distribution of this noise can be seen in a histogram of all intensity values (Fig. 1A). Parameters of this distribution were estimated by excluding expressed genes one by one as their values exceeded the mean +/- 2 SD of the core of non-discarded values. This process was repeated in an iterative manner until no additional spots were excluded and the resulting non-discarded points (typically between 500 and 600 of the initial set of 1176) represent the set of non-expressed genes. After these exclusions, parameters of additive noise are estimated by non-linear fitting of a normal distribution function to the core of non-excluded values.
The knowledge of the parameters of the additive noise distribution enables selection of expressed genes for the final profile adjustment. The threshold, 3 SDs above the mean of additive noise, is used for selecting genes expressed above additive noise from which the final adjustment is made. The necessity for this exclusion is illustrated in Fig. 1. Figure 1 data was collected experimentally from an early Clontech Atlas array with 600 genes spotted in duplicate and was subsequently normalized to additive noise as described in Materials and Methods. Duplicated spots give two sets of expressions for identical genes on the same membrane. Their ratios are expected to be distributed around 1 with small random variations. This was not true for expressions below some threshold that correlated with our determination of additive noise. These results support the idea about the necessity to exclude noncorrelated weak expressions from comparison. They also demonstrate the utility of excluding values expressed "3SD above mean" to eliminate noncorrelated noise from regression analysis and ratio calculations.
Moderate quality fluorescently labeled microarrays
In moderate quality microarrays with low signal to noise ratios the right portion of the additive noise distribution is distorted by the presence of expressed genes (Fig. 2). This was observed by comparing two expression profiles from a homogenous group. Proposing that the majority of genes are equally expressed in these samples, it is possible to compare ratio variations of the same gene across two arrays. In agreement with Figure 1, we can expect significant variance increase for genes in a noisy area compared to a high expression area. The border between noise and correlated expressions was obtained by using an F-test as follows. The "sliding window" procedure compared the ratios of identical genes across two arrays. The two arrays under comparison were sorted from low to high expression on one and the second's gene list was sorted to match, maintaining parity. An F-test was carried out for the ratios in the window selected gene expressions (10 lowest in sample one) compared with the ratios of the remaining genes. When the window is sliding from low to high expressions we obtain comparative characteristics of ratio variability dependent upon expression level. There is a definite border, when the null hypotheses can't be rejected (Fig 2B), starting with some level of gene expression (equal to 1 in Fig. 2B) when we can see parameter variation providing statistical evidence supporting the appearance of the correlated gene sets. This relatively sharp border in the p-value distribution (Fig. 2B) divides the area of pure noncorrelated noise from noise "contaminated" with highly correlated gene expressions.
Figure 2.
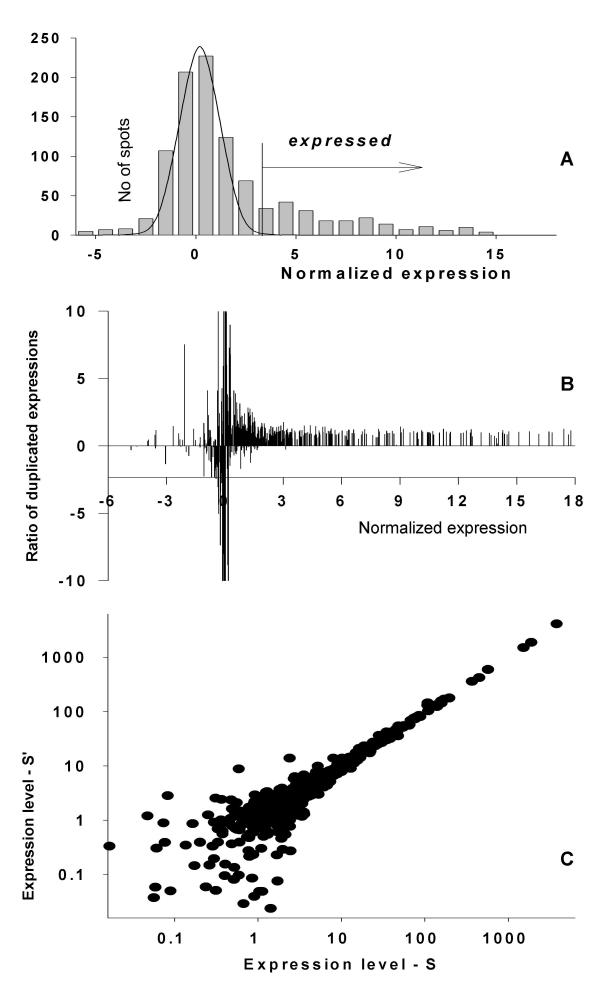
"Contamination" of the normally distributed additive noise with weak expressions on Micromax cDNA arrays. A. Histogram of gene expression distribution. B. The variability of the gene expressions ratios on two arrays ordered by expression levels. C. Scatter plot of gene expressions used in the calculation of ratios in B with gene order preserved.
For this case, a new histogram is created by substituting the right portion of the additive noise distribution with the mirror reflection of the left portion. Curve fitting is then applied to the new histogram, which is minimally altered by the presence of weak expressions (Solid line in Fig. 2A).
High quality fluorescently labeled microarrays
High quality arrays produce another type of problem for localization of the additive noise distribution. The spots at the noise floor are represented by a relatively small portion of all spots (less then 10%) in these arrays, consequently, their distribution is not as prominent as in the previous examples when viewed in a histogram of all spots (Fig. 3A). The automated iterative procedure for selection of additive noise described Materials and Methods will not locate the additive noise distribution in this case. It is necessary to perform a special preliminary step intended to magnify the area of the additive noise distribution and focus the iteration procedure to this area. In these arrays, as in the previous described arrays, variability in expression measurements will increase at or near additive noise levels. The F-test is able to select an area where variation of gene expression ratios is statistically higher when compared with the remaining gene expressions (Fig. 3B). The results of the F-test demonstrate the existence of a sharp border between non-correlated noise and weak gene expressions. This border is used to guide the identification of the additive noise distribution in the area where additive noise is presented by a very clear normal distribution minimally contaminated with the weak gene expressions (Fig. 3C). Application of the iterative procedure to this area leads to an unambiguous determination of the additive noise parameters.
Figure 3.
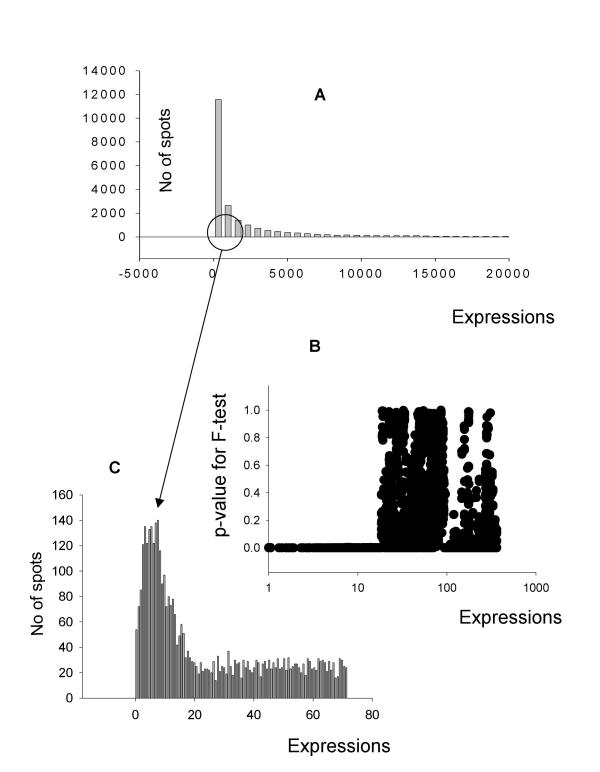
Localization of additive noise through an F-test. A. Histogram of gene expressions from a microarray experiment. B. P value distribution showing correlation of additive noise as determined by F test. A window of 20 genes was created to calculate the groups SD and further windows were created by shifting the index of the ordered SD's by one gene. For example: widow 1 contains genes 1–20 and window 2 contains genes 2–21. As the expression level in these windows increases the SDs become correlated and a p-value threshold becomes apparent. C. Close up histogram of low intensity spots used as background. The Gaussian distribution can clearly be seen; furthermore, the right tail cutoff of the Gaussian distribution is at the expression level corresponds to the p value threshold for the ratio calculation in B.
"Signal from noise" elicitation
With a clear delineation of additive noise parameters it is possible to identify genes expressed distinctly from additive noise using recognized statistical criteria. Genes expressed significantly higher than additive noise are easily identified by paired analysis. Genes with low level signals within additive noise can also be identified as distinctive from additive noise due to their higher stability (lower SD in replicated measurements). Discrimination of expressed vs. nonexpressed genes is not based on an arbitrary cutoff, but a Student-T test is performed to measure the probability that the gene expression belongs to the normally distributed additive noise.
As shown in a representative sample of results, some expressed genes have very low expression levels but high stability (lower SD and a resulting low p value). Conversely, some highly expressed genes are not statistically distinct from additive noise because their high variability (higher SD and a resulting high p value).
Spatial stability of the additive noise distribution
To investigate the noise parameters stability across different regions of the array, we carried out the usual procedures for noise localization and normal distribution fitting utilizing data from the total slide and its different regions. The results that are presented in Fig. 4 demonstrate that in spite of some variations in the noise distributions and its variable "contamination" with weak signals, the estimated parameters demonstrate relative stability of the additive noise in different regions within the slide.
Figure 4.
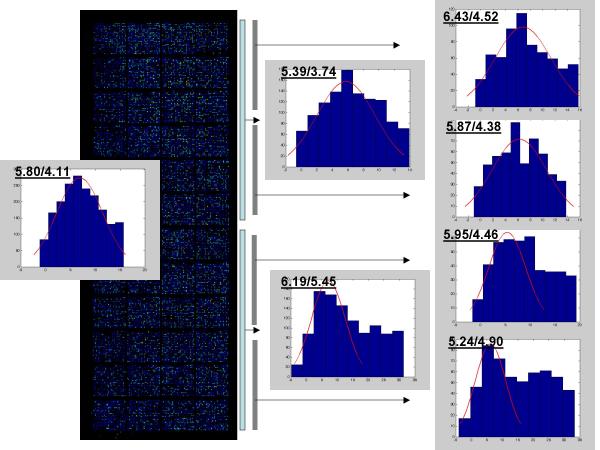
Within slide stability of the additive noise. Localization of the additive noise distribution and estimation of the normal distribution parameters were carried out as described in Materials and Methods. Parameters for additive noise distribution (Mean/SD) were estimated from total data set (left histogram) and from each half or quarters (intermediate and right histograms) of the slide.
Discussion
Normalization of cDNA microarray data is an obligatory step during microarray experiments due to the relatively frequent non-specific errors. Generally, normalization of microarray data is based on the null hypothesis and variance model. In the gene expression model [4,8] at least two types of noises are included. One is additive noise and the other is multiplicative noise. Usually, background is considered as additive noise and the variation between the signal pixels is the representative of multiplicative noise. However, as we demonstrated previously [10,11] and in this article, the additive noise is present even after subtraction of local background from signal (background correction) and is product of hybridization below technological specificity. This additive noise can be observed as isolated or partially overlapping normally distributed signals of low intensity spots. We would like to emphasize the difference between discussing additive noise from around spot local background intensities and additive noise that can vary at different locations within the individual chip [12]. The influence of these variations is removed by the local background correction procedure, subsequently; resulting in additive noise that is relative stable over different localizations on the slide (Fig. 4).
There are versatile distribution assumptions for the background and additive error term in the literature [13]. Rocke and Durbin [8] were the first to suggest using an iterative procedure, similar to what is presented here, for estimation of background parameters. The transformation introduced independently by several research groups has been called the Generalized Logarithm. More recently, Rocke and Durbin demonstrated [14] that there are some alternative log-transformations that produce approximate variance stabilizing transformations for microarray data that are nearly as good as the Generalized Logarithm. Several authors searched variance-stabilizing transformations for gene-expression microarray data [15,16]. It was demonstrated that for data obtained after background extraction these transformations converge to a log-transformation for relatively large expression levels. Our results go further and demonstrate that the apparent deviation of the additive noise distribution from normality is produced by the presence of the weak signals overlapping with the noise. These results enable the skewed distribution presented in Fig. 2 to be treated as a normally distributed additive noise distorted on its right side by the presence of low but distinctive (reproducible) gene expressions.
In this study, the influences of additive noise on the ratio estimates and normalization procedures were investigated. As was clearly demonstrated, it is necessary to exclude from comparisons the non-correlated weak expressions with duplicated spots (two sets of expressions for each gene) on the Clontech Atlas membrane. The ratio variation was dramatically increased even for these identical expressions below some threshold corresponding to the previously described determination of additive noise [10]. This phenomenon represents a main obstacle for data normalization and ratio estimates in microarrays [1]. We presented various methods for localization of additive noise spots. The knowledge of these parameters enables the exclusion of noncorrelated noise from the biased adjustment of the compared profiles and also from ratio estimations – an additional and very helpful characteristic of comparative gene expression analysis.
In fact it is an ad hoc practice to eliminate weak spots on microarrays before subsequent data analyses [3-5]. We have demonstrated that additive noise can be used as an important inner standard for data normalization and for selection of weak signals statistically distinctive from nonspecific noise. In contrast to all previous attempts to exclude the influence of additive noise by cutting off low expressions, there are no meaningless low expressions in our method, only expressions statistically distinctive or not from additive noise: an important discrimination in light of the special importance of regulatory genes which are consistently expressed at low levels [7].
Methods
Microarray data
To demonstrate the applicability of the analysis presented herein data has been analyzed from several distinct array technologies. The first data set is from a previous publication in which liver tissue from Ames dwarf mice was screened using BD Atlas™ arrays (Clontech, CA) [10]. These macroarrays are nylon membrane-based with 600 hundred mouse genes represented by cDNA clones spotted in duplicate. The second data set was obtained from human peripheral blood mononuclear cells from healthy donors screened on Perkin-Elmer Micromax cDNA arrays. These arrays are glass slide-based with 2,600 human genes represented as cDNA clones. The third set of data was obtained from human peripheral blood mononuclear cells from healthy donors screened on in-house printed genome-scale oligo-microarrays. These oligos are synthesized by Qiagen/Operon and printed on Corning UltraGAPS slides using a GeneMachines OmniGrid 100 microarray printer.
Outline of normalization and analysis procedures
Normalization for differences among experiments was conducted using the procedure described in detail elsewhere [10]. In brief, the procedure assumes that intensities corresponding to mRNA not expressed by the tissue will be normally distributed and computes the mean and SD of these nonexpressed genes using an iterative nonlinear curve fitting procedure.
The next step is normalization of each expression profile to its own additive noise, with selection of the genes expressed above additive noise for subsequent adjustment and comparison. For further analysis, data obtained after normalization of each profile to own additive noise are log-transformed with substitution of negative values by the minimal logarithmic value obtained within positive values.
Normalized profiles are adjusted to each other by means of a robust regression analysis of genes expressed above additive noise. All expression profiles of both control and experimental groups are re-scaled to a common standard – the averaged profile of the control group. This analysis is based on the fact that majority of genes are presumably equally expressed in compared samples. In the scatter plot, genes with similar expression levels should be randomly distributed around a line of equity with a small portion of differentially expressed "outliers". Their contribution in the regression analysis is down-weighted in an iterative manner. Our procedure for exclusion of outliers [10,11] is based on the selection of equally expressed genes as a homogenous family of genes with normally distributed residuals (for log-transformed data with exclusion weak expressions) measured as deviations from the regression line calculated against the averaged profile. Outliers are thereafter determined as having deviations not associated with this normal distribution represented by several hundred members.
The next step is identifying a set of similarly expressed genes from the control samples, denoted "reference group", composed of genes expressed above additive noise with low variability of expression as determined by an F-test, and whose residuals approximate a normal distribution, based on the Kolmogorov-Smirnov criterion.
Identification of genes differentially expressed in patients vs. control group. These analyses include:
– Selection with a Student T-test for replicates using the commonly accepted significance threshold of p < 0.05. It employs the commonly accepted sensitivity level used in biologic experiments, however a significant proportion of these genes identified as differentially expressed will be false positive determinations at this threshold level;
– An associative T-test in which the replicated residuals for each gene of the experimental group are compared with the entire set of residuals from the reference group defined above. Null hypotheses are checked to see if gene expressions in the experimental group presented as replicated residuals (deviations from averaged control group profile) are associated with the reference group. The significance threshold is corrected to make improbable the appearance of false positive determinations.
– Genes expressed distinctively from additive noise were determined analytically by association of each replicated gene expression with a normal distribution of additive noise having average equal 0 and standard deviation equal 1. Genes expressed distinctively from additive noise in one group and not distinctive from additive noise in another are selected as another example of differential gene expression.
Authors' contributions
ID invented the method and drafted the paper. NK realized the method in Matlab® and provided critical comments. YT was involved in method testing and applications. MC participated in design of this method and provided data vital for project completion. All authors prepared, read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported by grants NIH NCRR IDeA, and NIH 1 P20 RR1557 from the National Institutes of Health, and BRIN grant #P20RR16478.
Contributor Information
Igor Dozmorov, Email: dozmorovi@omrf.ouhsc.edu.
Nicholas Knowlton, Email: knowltonn@omrf.ouhsc.edu.
Yuhong Tang, Email: tangy@omrf.ouhsc.edu.
Michael Centola, Email: centolam@omrf.ouhsc.edu.
References
- Geiss GK, Bumgarner RE, An MC, Agy MB, van't Wout AB, Hammersmark E, Carter VS, Upchurch D, Mullins JI, Katze MG. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology. 2000;266:8–16. doi: 10.1006/viro.1999.0044. [DOI] [PubMed] [Google Scholar]
- Dozmorov I, Saban MR, Gerard NP, Lu B, Nguyen NB, Centola M, Saban R. Neurokinin 1 receptors and neprilysin modulation of mouse bladder generegulation. Physiol Genmics. 2003;12:239–250. doi: 10.1152/physiolgenomics.00141.2002. [DOI] [PubMed] [Google Scholar]
- Kooperberg C, Fazzio TG, Delrow JJ, Tsukiyama T. Improved background correction for spotted DNA microarrays. J Comput Biol. 2002;9:55–66. doi: 10.1089/10665270252833190. [DOI] [PubMed] [Google Scholar]
- Yang MC, Ruan QG, Yang JJ, Eckenrode S, Wu S, McIndoe RA, She JX. A statistical method for flagging weak spots improves normalization and ratio estimates in microarrays. Physiol Genomics. 2001;7:45–53. doi: 10.1152/physiolgenomics.00020.2001. [DOI] [PubMed] [Google Scholar]
- Chen Y, Kamat V, Dougherty ER, Bittner ML, Meltzer PS, Trent JM. Ratio statistics of gene expression levels and applications to microarray data analysis. Bioinformatics. 2002;18:1207–15. doi: 10.1093/bioinformatics/18.9.1207. [DOI] [PubMed] [Google Scholar]
- Greller LD, Tobin FL. Detecting selective expression of genes and proteins. Genome Res. 1999;9:282–296. [PMC free article] [PubMed] [Google Scholar]
- Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Geneti. 2001;29:389–395. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- Rocke DM, Durbin BA. A model for measurement error for gene expression arrays. J Comput Biol. 2001;8:557–569. doi: 10.1089/106652701753307485. [DOI] [PubMed] [Google Scholar]
- Zien A, Aigner T, Zimmer R, Lengauer T. Centralization: a new method for the normalization of gene expression data. Bioinformatics. 2001;17:S323–31. doi: 10.1093/bioinformatics/17.suppl_1.s323. [DOI] [PubMed] [Google Scholar]
- Dozmorov IM, Centola M. An associative analysis of gene expression array data. Bioinformatics. 2003;19:204–11. doi: 10.1093/bioinformatics/19.2.204. [DOI] [PubMed] [Google Scholar]
- Dozmorov I, Bartke A, Miller RA. Array-based expression analysis of mouse liver genes: Effect of age and of the longevity mutant Prop1df. J Gerontol A Biol Sci Med Sci. 2001;56:B72–B80. doi: 10.1093/gerona/56.2.b72. [DOI] [PubMed] [Google Scholar]
- Kim JH, Shin DM, Lee YS. Effect of local background intensities in the normalization of cDNA microarray data with a skewed expression profiles. Exp Mol Med. 2002;34:224–32. doi: 10.1038/emm.2002.31. [DOI] [PubMed] [Google Scholar]
- Strimmer K. Modeling gene expression measurement error: a quasi-likelihood approach. BMC Bioinformatics. 2003;4:10. doi: 10.1186/1471-2105-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocke DM, Durbin B. Approximate variance-stabilizing transformations for gene-expression microarray data. Bioinformatics. 2003;19:966–72. doi: 10.1093/bioinformatics/btg107. [DOI] [PubMed] [Google Scholar]
- Durbin BP, Hardin JS, Hawkins DM, Rocke DM. A variance-stabilizing transformation for gene-expression microarray data. Bioinformatics. 2002;18:S105–10. doi: 10.1093/bioinformatics/18.suppl_1.s105. [DOI] [PubMed] [Google Scholar]
- Huber W, Von Heydebreck A, Sultmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18:S96–104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]