

Abstract
Close to two decades of research has established that astrocytes in situ and in vivo express numerous G protein-coupled receptors (GPCRs) that can be stimulated by neuronally-released transmitter. However, the ability of astrocytic receptors to exhibit plasticity in response to changes in neuronal activity has received little attention. Here we describe a model system that can be used to globally scale up or down astrocytic group I metabotropic glutamate receptors (mGluRs) in acute brain slices. Included are methods on how to prepare parasagittal hippocampal slices, construct chambers suitable for long-term slice incubation, bidirectionally manipulate neuronal action potential frequency, load astrocytes and astrocyte processes with fluorescent Ca2+ indicator, and measure changes in astrocytic Gq GPCR activity by recording spontaneous and evoked astrocyte Ca2+ events using confocal microscopy. In essence, a “calcium roadmap” is provided for how to measure plasticity of astrocytic Gq GPCRs. Applications of the technique for study of astrocytes are discussed. Having an understanding of how astrocytic receptor signaling is affected by changes in neuronal activity has important implications for both normal synaptic function as well as processes underlying neurological disorders and neurodegenerative disease.
Keywords: Neuroscience, Issue 85, astrocyte, plasticity, mGluRs, neuronal Firing, electrophysiology, Gq GPCRs, Bolus-loading, calcium, microdomains, acute slices, Hippocampus, mouse
Introduction
Astrocytes respond within seconds to stimulation of neurons or neuronal axons with increases in cytoplasmic Ca2+ resulting almost exclusively from activation of astrocytic Gq GPCRs. For example, muscarinic acetylcholine receptors1, cannabinoid receptors2, α1A adrenergic receptors3,4, and group I mGluRs (see below) are all astrocytic Gq GPCR subtypes that acutely respond to neuronal activity. Activation of astrocytic group I mGluRs has been demonstrated most extensively, following stimulation of neuronal glutamatergic afferents in situ (such as acute hippocampal slices)5-7, as well as in adult mouse cortex in vivo following sensory stimulation8. The outcome of activation of astrocytic Gq GPCR signaling on the biology and physiology of astrocytes, neurons, or astrocyte-neuron interactions has been a matter of debate9-12. It will be some time before the function of neuron-to-astrocyte receptor signaling is fully appreciated.
While it is clear that neurons can activate astrocytic receptors using experimental protocols, there are aspects of neuron-to-astrocyte receptor communication that remain poorly understood. First, the actual amount of neuronal activity required to activate astrocytic Gq GPCRs is not well-defined, and second, the ability of astrocytic receptors to exhibit use-dependent plasticity has received little attention. To begin to address these questions, we recently developed a protocol to induce bidirectional scaling of astrocytic group I mGluRs in acute juvenile hippocampal slices in response to long-term changes in neuronal action potential (AP)-dependent synaptic activity. Similar to what has been discovered for bidirectional homeostatic plasticity of neuronal ionotropic glutamate receptors13,14, astrocytic group I mGluRs scale up following blockade of neuronal action potentials and scale down when neuronal action potential frequency is increased15. These compensatory changes in astrocytic receptors can be measured by recording spontaneous and evoked astrocyte Ca2+ transients and comparing the properties of these events to those from astrocytes in control conditions. In this manuscript, we describe the complete methodology for use of this protocol, including preparation of acute hippocampal slices, incubation conditions to induce astrocyte receptor scaling, astrocyte Ca2+ indicator dye loading, Ca2+ imaging techniques using confocal microscopy, and expected effects on astrocyte Gq GPCR activity. Predictable effects on astrocyte Ca2+ signaling properties - which match those recorded previously in cultured cells transfected with different expression levels of Gq GPCRs - provide a “roadmap” that can be used in future studies to assay for changes in astrocytic GPCR expression. The ramifications and potential applications for use of this technique will contribute to our understanding of astrocyte-neuronal interactions in the healthy and diseased brain.
Protocol
The procedures that follow have been approved by the Institutional Animal Care and Use Committee of the University of California, Riverside.
1. Construction of Incubation Chamber and Slice Holder
- Constructing the incubation chamber: Construct the incubation chamber from material that is nontoxic. Make sure that the chamber has controlled air circulation, holds a sufficient amount of ACSF for a slice holder to float, and is large enough so that the slice holder can float at one end of the chamber while the gas dispersion from oxygen lines occurs at the other end. The drawer portion of a pipette storage container with its air-tight lid (Figure 1A) fits these criteria nicely.
- Drill a small hole on the side of the container approximately 1¼ in from the top and ¼ in from the side. Fit a piece of flexible tubing through the hole (Figure 1B). Be careful to drill low enough so that the tubing will not be compressed when the lid is closed, yet high enough so it is above the solution.
- Apply silicone seam sealant (“aquarium seam sealer”) to create a water-tight seal between the oxygen line and the chamber.
- Connect the “outside” end of the tubing to a gas tank (95% oxygen, 5% carbon dioxide) using a male Luer fitting. For a custom fit, cut-to-fit a natural beveled 200 µl pipette tip (Figure 1C).
- Connect the “inside” end of the tubing to a one-to-six line plastic manifold. Cut-to-fit six 20 µl Eppendorf microloader pipette tips to each manifold inlet (Figure 1D). The fine opening of the microloaders is ideal for producing a steady stream of small bubbles.
- To allow for ventilation, drill two tiny holes on the lid of the container (Figure 1E).
- Constructing the slice holder: The slice holder is made from a Floating Bubble Rack (Figure 1F).
- Remove the bottom “legs” of the bubble rack, and cut the top to approximately 1¾ in.
- Glue a piece of Nylon mesh material to the bottom of the round rack using cyanoacrylate glue (such as standard Krazy Glue) to create an eight-well slice holder (Figure 1G). Each well can fit a single mouse hippocampal slice.
- Fit the slice holder at one end of the incubation chamber and the microloader-manifold apparatus at the other end. Allow the microloader tips to rest on the chamber floor (Figure 1H). This setup is designed to ensure sufficient oxygenation to both the top and the bottom of the acute hippocampal slices, and to avoid bubbles coming out directly below the slice holder in order to minimize slice agitation and prevent direct contact of bubbles with the hippocampal slices.
- Rinse the incubation chambers and slice holders thoroughly with ddH2O after each experiment, and decontaminate with 70% EtOH weekly. In the case of incubating slices in different solutions, multiple chambers/slice holders will be needed.
2. Solutions and Drugs
Artificial Cerebrospinal Fluid (ACSF): Prepare 4 L of standard ACSF in ddH2O using the following (in mM): 125 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1.25 NaH2PO4, 26.0 NaHCO3, 15 glucose, and 0.1 Trolox. Measure the osmolarity of the solution using an osmometer; it should come to ~310 mOsm. ACSF compositions for experimental conditions are described below (see Protocol 3). Filter all solutions using a 0.22 µm bottle-top filter into autoclaved glass bottles. Solutions are stable in 4 °C for up to a month.
Slicing Buffer: Prepare slices using a modified ACSF containing (in mM): 125 NaCl, 2.5 KCl, 3.8 MgCl2, 1.25 NaH2PO4, 26.0 NaHCO3, 15 glucose, and 1.3 ascorbic acid. Measure the osmolarity of the solution using an osmometer; it should come to ~310 mOsm. Replacement of CaCl2 with MgCl2 in the slicing buffer improves slice health.
Sulforhodamine 101 (SR-101, 1 µM) is used to identify astrocytes in acute hippocampal slices. Make a 1 mM stock of SR-101 by diluting 60.67 mg SR-101 in 100 ml of a modified low calcium ACSF prepared as follows (in mM): 125 NaCl, 2.5 KCl, 0.5 CaCl2, 6 MgCl2, 1.25 NaH2PO4, 26.0 NaHCO3, 15 glucose, and 1.3 ascorbic acid. Verify that the osmolarity of the low calcium ACSF is ~310 mOsm. Dilute the 1 mM SR-101 stock 1,000 times in modified low Ca2+ ACSF for loading. Store the final SR-101 solution in 4 °C and protected from light until needed for the experiment.
3. Manipulation of Long Term Neuronal Firing Rates in Acute Hippocampal Slices
Use one of two incubation protocols in separate experiments to manipulate long-term neuronal firing rates:
Inhibit neuronal firing: Incubate in tetrodotoxin (TTX, 1 µM): CAUTION: Handle TTX with care as it can be fatal if ingested in sufficient quantities. Gloves and goggles are recommended. TTX completely abolishes AP-driven neuronal firing in acute slices. Incubate slices in 3.5 mM K+ ACSF, plus 1 µM TTX in the experimental condition. In the control condition, incubate slices in 3.5 mM K+ ACSF without TTX. The comparison between the two conditions reveals the up-scaling effect in astrocytic Gq GPCR activity. 3.5 mM K+ ACSF serves as the control condition (as opposed to say, 2.5 mM K+) in order to maximize the effect of TTX treatment.
-- OR --
Increase neuronal firing above basal rates: Incubate in high potassium: Increasing extracellular K+ concentration depolarizes neurons and increases their basal firing rate. Incubation in 5.0 mM K+ ACSF significantly elevates neuronal action potential frequency compared to 2.5 mM K+ ACSF15. Incubate slices in 5.0 mM K+ ACSF for the experimental condition, and for the control condition, incubate slices in standard 2.5 mM K+ ACSF. The comparison between the two conditions reveals the down-scaling effect in astrocytic Gq GPCR activity.
4. Acute Hippocampal Slice Preparation
- Setting up the warm recovery chamber:
- Warm the water bath to 35 °C, then place the prepared incubation chambers inside. Fill the water bath with water up to the height of the ACSF within the incubation chambers (Figure 1G).
- Oxygenate the experimental and control ACSF with 95% O2, 5% CO2. The bubbles emitted from the microloader-manifold apparatus should be small and plentiful but also gentle; there should not be any visible movement of ACSF within the chamber.
- Setting up the ice-cold dissection chamber:
- Take two buckets of ice. Place a bottle of approximately 300 ml slicing buffer into one bucket of ice and keep it oxygenated with 95% O2, 5% CO2 for 20 min. Submerge a 100 mm Petri dish into the other ice bucket, just below the surface of ice. Ensure the side of the Petri dish is in direct contact with the ice. Pour some slicing buffer into the Petri dish and keep it oxygenated as well.
- Chill the cutting edge of the single-edge razor blade by submerging it into the ice cold slicing buffer in the Petri dish for more than 1 min.
- Vibratome setup:
- Turn on the vibratome and make sure the drainage is closed.
- Secure the cutting chamber in the vibratome and pack ice around the cutting chamber. Precool it to 0-4 °C.
- Remove factory greases from the double edge razor blade by soaking it into 70% EtOH for 5 min and then rinsing with ddH2O. Cut it into halves carefully (do not bend the blade) and mount one half blade onto the cutting block.
- Removing the mouse brain:
- Anesthetize a twelve to eighteen-day-old C57BL/6J mouse in a small chamber preloaded with 0.5 ml isoflurane soaked into a Kimwipe or cotton ball. Gently pinch the animal’s toes to make sure there is no pain reflex.
- Decapitate the mouse using a pair of sharp scissors, then remove the scalp using small forceps. Use small bone scissors to cut the skull from the cerebellum to the olfactory bulbs along the longitudinal fissure. Remove the cranial flaps using the small forceps. Gently remove the brain with a spatula, and submerge it into the ice-cold oxygenated slicing buffer in the Petri dish.
- Bisect the mouse brain with the chilled razor blade in the Petri dish to allow more surface area for cooling and oxygenation. Let the bisected hemispheres sit in the ice cold slicing buffer for 2-3 min. The brain should become completely cool and more solid.
Apply a thin layer of cyanoacrylate glue on the platform of the vibratome. Glue both hemispheres to the platform cut side-down and lateral sides up, with the olfactory bulb facing forward. Secure the platform in the cutting chamber, then fill the cutting chamber with ice cold, well-oxygenated slicing buffer.
Continue oxygenating the cutting chamber while preparing 300 µm thick parasagittal slices using the vibratome. Cut the slices at a frequency of 85 Hz, a forward speed of 0.20 mm/sec, and an amplitude of 1.40 mm. NOTE: We have found that the single most important variable in preparing healthy acute hippocampal slices is the quality of the vibratome. Our laboratory uses the Leica VT 1200 S magnetic drive vibratome with vibrocheck to reduce “z” vibration.
After slicing, dissect the hippocampus and the adjacent entorhinal cortex out of each parasagittal slice using sharp forceps. Perform this procedure in the ice-cold, well oxygenated slicing buffer in the cutting chamber of the vibratome. The sharpness of the forceps is very important to the slice health as it minimizes handling of the slices.
Make a transfer pipette by breaking off the long tip of a glass Pasteur pipette and topping the broken part with a pipette bulb. This enables use of the large end of the pipette to transfer slices. Make sure to order pipettes without the cotton plug in the large end (see Materials table). As slices are prepared in nonsterile conditions, it is not necessary to autoclave the pipette prior to use, although a new pipette should be prepared and used for each experiment.
Transfer each hippocampal slice to the incubation chambers in the 35 °C water bath, by dipping the transfer pipette into the ACSF inside each well of the slice holder and aspirating the slice. Minimize movement of the slice during this process. Direct transfer of the slices to the warm incubation bath results in better quality slices than “ramping” the temperature up gradually.
Allow the hippocampal slices to recover in the warm water bath for a total of 45 min, broken down as follows: Incubate slices for 20 min in 1 µM SR-101 diluted in low calcium ACSF, then transfer them to the low calcium ACSF (without SR-101) for 10 min. Subsequently, transfer the slices to control or experimental ACSF for the remaining 15 min of the warm incubation.
After the 45 min warm recovery, carefully move the incubation chambers from the 35 °C water bath to the bench top, and then allow the slices to continue to incubate at room temperature for a total incubation time of 3 hr before beginning the bolus-loading protocol (see below).
5. Bolus Loading of Astrocytes with Ca2+ Indicator
- Preparing Ca2+ indicator bolus-loading dye:
- For each vial of dye (50 µg), add 3.87 µl of fresh dimethyl sulfoxide (DMSO) and vortex thoroughly. The freshness of the DMSO is important for good loading and therefore, crack open a fresh ampule each time.
- Mix in 9 µl of 20% pluronic acid and vortex thoroughly. Mix the dye with 100 µl of the appropriate experimental or control ACSF and vortex well, for a final dye concentration of 10 µM.
- Filter the solution using a centrifuge filter tube. This step prevents the loading pipette from clogging during ejection of the dye.
- Prepare a pipette from a borosilicate glass capillary pulled to a resistance of approximately 1.3 MΩ when filled with dye solution.
Place a hippocampal slice into a recording chamber designed for use with the microscope and continuously perfuse with oxygenated ACSF of the same composition that it was incubated in (1.5 ml/min). Alternate between control and experimental conditions when selecting a hippocampal slice.
- Discard unhealthy looking hippocampal slices. The quality of slices will vary even within a given experiment. There are no set criteria for quantifying slice health; therefore, determination of slice health is subjective and based mostly on experience.
- Generally speaking, keep slices that have a smooth appearing surface and a high percentage of healthy CA1 pyramidal cells (Figure 2A). CA1 pyramidal cells are particularly sensitive and vulnerable to CNS insults, so having a large percentage (≥75%) of healthy CA1 pyramidal neurons may be a useful criteria for accepting or rejecting a slice.
- Discard slices that have approximately >25% dead neurons. Dead neurons have a fried egg-like appearance (Figure 2B). NOTE: We have observed that the angle of the cut plays a major role in whether neurons appear healthy or not. If neuronal dendrites are projecting upwards (out of the slice), then the neurons will be mostly dead. This is presumably because such a large proportion of the neuron volume is contained within the dendritic tree, that severing the dendrites is lethal to the cells. On the other hand, if the dendrites are projecting parallel to or at a downward angle from the slice surface, there will be a high percentage of healthy neurons. Sometimes, therefore, neurons will be mostly dead on one side of the slice and healthy looking on the other side.
Locate a suitable field of s.r. astrocytes 40-70 μm below the slice surface based on size, morphology, and location using differential interference contrast (DIC) optics.
Load the glass pipette with dye solution and lower it to the surface of the slice above this field using a standard patch-clamp microelectrode holder. With the pipette at the surface of the slice, apply back pressure to the pipette to begin ejecting dye. Ejection of dye will be visible under both DIC optics and an appropriate laser such as a 488 line for a green dye.
Slowly lower the pipette to approximately 40 μm below the slice surface using a micromanipulator and allow the dye to eject for approximately 45-60 sec. Then, lower the pipette an additional 35 μm (75 μm below slice surface) and eject dye for approximately 45-60 sec. Slowly retract the pipette tip from the slice. Shorter injection time is likely to result in insufficient dye loading, whereas longer injection tends to increase background loading, which decreases signal-to-noise ratio of the recording.
To ensure that a large number of astrocytes take up the dye, it is usually helpful to inject a second dye bolus a short distance away. Raise the pipette back to the surface of the slice, make sure that the pipette is not clogged, then move the pipette approximately 80-100 μm away from the first injection site, along the stratum radiatum. Repeat bolus injection at this site.
Allow 30-45 min before imaging for the astrocytes to take up the dye and for the background signal to diminish. Leave the slice in the perfusion chamber during this time. Make sure this incubation time is built into the total 4 hr treatment of the experiment. Obtain the next slice and repeat steps 5.2-5.8.
6. Recording Spontaneous and Gq GPCR Agonist-evoked Astrocytic Ca2+ Activity in Hippocampal Slices
- Setting up the confocal microscope for imaging:
- Limiting slice exposure to laser light is of utmost importance, as high exposure can lead to dye bleaching and/or phototoxicity. Using higher optical magnification or an increased zoom setting increases light exposure to the imaged field. Therefore, set the default values for each laser to a high photomultiplier setting, 1x gain and 0.5% laser output power.
- Apply a 1.5x zoom for better visualization of astrocyte processes.
- Set the field resolution to 512 x 512 pixels.
- Set the scan speed to the fastest possible, which is ~1.2 sec per scan using the one way scan mode.
- Collect emission spectra using bandpass filters of 503-548 nm for the 488 nm laser, and 624-724 nm for the 559 nm laser. These settings allow imaging of a field of ~5-8 astrocytes at a relatively rapid speed at a resolution sufficient to observe astrocyte cell bodies and main processes. Ideally, astrocytes in the field will be bright enough to be seen clearly, but without any pixel saturation.
- Confirm the identity of cells loaded with Ca2+ dye as astrocytes by visualizing the SR-101 colabeling using the 559 nm laser.
- Recording astrocyte Ca2+ activity:
- Draw boxes using the image acquisition software over regions of interest (ROIs) within the cell, in this case over astrocyte cell bodies. The boxes should not include background pixels, to achieve the best signal-to-noise ratio possible. Draw one box over background as reference.
- Switch the perfusion to experimental ACSF plus 1 µM TTX (Abcam) through the remainder of the experiment. This eliminates any possible neuronal AP-driven astrocyte calcium responses. Remaining increases in astrocytic Ca2+ concentration will then be due to quantal vesicular release, constitutive (basal) GPCR activity, or some combination of both mechanisms.
- Record fluorescence over time from all ROIs. Any increases in fluorescence over baseline indicate increases in cytoplasmic Ca2+ concentration16, and therefore GPCR activity in astrocytes10,17,18. To avoid any possible early scaling effects on astrocytic receptors by TTX, complete experiments within 40 min from the time when the 1 µM TTX perfusion began.
- After obtaining 10 min of baseline recording of spontaneous Ca2+ activity, apply an agonist of interest (such as DHPG) at sequentially increasing concentrations. Leave a minimum of 5 min between applications to reduce possible receptor desensitization.
- At the end of the recording, apply a cocktail of agonists for other astrocytic Gq GPCRs as a positive control for intact astrocytic Gq GPCR signaling pathways. Components of the agonist cocktail will depend on the receptor of interest. 10 µM of each of the Gq GPCR agonists histamine, carbachol, and 2Na-ATP to stimulate histamine H1 receptors [H1R], muscarinic acetylcholine receptors [mAchR], and purinergic receptors [P2YR], respectively, is a commonly used agonist cocktail.
- Post-experiment image acquisition:
- At the completion of the Ca2+ recording, take still images with the 488 nm and 559 nm lasers, for later confirmation of astrocyte identity and ROI placement. Laser power and HV settings may be altered freely at this point to obtain an optimal image for colocalization, as there is no longer a concern about laser light intensity affecting the data (Figure 2A).
Repeat steps 6.1-6.3 for a total of approximately 8 slices and 40 astrocytes/group. Slices should come from a minimum of 3 different mice.
7. Analysis of Astrocyte Ca2+ Activity
- Defining a Ca2+ elevation: Standardization in defining Ca2+ transients has not been firmly established within the scientific community. What follows is a typical protocol that maximizes sensitivity while limiting detection of false positive events out of baseline noise.
- Have another lab member assign each slice a numeric code in order to analyze them blindly. At the conclusion of analysis, decode each slice.
- Analyze spontaneous and evoked astrocyte Ca2+ elevations offline using imaging analysis software. Redraw and/or adjust the size, shape and the location of the ROIs as desired.
- Score increases in fluorescence intensity over baseline as a Ca2+ elevation if the peak amplitude is greater than two standard deviations (S.D.) above the mean of 30 sec of average baseline fluorescence for at least two consecutive sample points. In particularly noisy recordings (low signal-to-noise), this criteria may need to be adjusted to 3 S.D. above the mean baseline fluorescence. Define the onset of each Ca2+ elevation as the last data point before the fluorescence intensity exceeds one standard deviation above the mean.
- Differentiate between multipeak vs. successive single-peak events. Score an event as “multipeak” when the fluorescence intensity does not return to baseline (below average baseline value +2 SD) for ≤9 consecutive data points (10.8 sec) between peaks. Thus, single peak events will have 10 or more consecutive baseline data points in-between them.
- Classify events as “plateau”-type responses when fluorescence intensity maintains peak amplitude (±10% of peak value) for at least 3.6 sec.
- Analyze the amplitude, frequency, and kinetics of spontaneous and agonist-evoked calcium transients.
- Define peak amplitude of the Ca2+ elevation as the data point with the highest intensity value (in cases of “multipeak” responses use the first peak; see Figure 2B).
- Calculate rise time as the difference between the response onset and the time corresponding to the peak amplitude. NOTE: 0 to 100% rise time may need to be used in order to have a sufficient number of data points to obtain a time value; scan speed is an important variable here.
- Calculate latency as the time between the initiation of agonist perfusion to the response onset. Rise time may be a more useful measure in brain slices, as long wash-in times create a confound in calculating response latencies.
Determine if there are statistically significant differences between the two groups for each parameter using Student’s independent t-test. Use number of astrocytes as the ‘n’. Use Pearson’s chi-square test to compare the Ca2+ activity patterns between control and treatment groups. Use Fisher’s exact 2-tail test to compare percentages of specific Ca2+ activity patterns between control and treatment groups. Express differences as *p < 0.05, **p < 0.01, and ***p < 0.001.
Representative Results
Representative results in Figure 3 show the effect of incubation of acute mouse hippocampal slices in TTX for 4-6 hr on s.r. astrocyte Ca2+ activity. Data include both spontaneous Ca2+ transients and DHPG-evoked group I mGluR Ca2+ responses, from slices incubated in control ACSF vs. ACSF plus TTX. Other than basic characteristic morphological features, stellate process assembly and small soma size (~10 µm), astrocytes are identified in the s.r. by overlay of the Ca2+ indicator OGB-1 AM with the selective astrocyte marker SR-10119,20 (Figure 3A). The numbered boxes over astrocyte cell bodies correspond to the numbered fluorescence over time traces shown in Figure 3B. The group 1 mGluR agonist (RS)-3.5-DHPG is applied to determine the specific scaling effects on group 1 mGluRs in astrocytes. To discriminate between physiology in the specificity of the scaling effect to group 1 mGluRs versus other Gq GPCRs, a cocktail of agonists is applied at the end of each experiment. Here we used 10 µM each of the Gq GPCR agonists histamine, carbamylcholine chloride (carbachol) and adenosine 5’-ATP disodium (Na-ATP). The agonist cocktail also serves as a positive control to identify viable, responsive astrocytes in cases where cells do not respond to DHPG, presumably because those particular astrocytes do not express sufficient amounts of the receptor to elicit a response to DHPG.
We used different concentrations of DHPG, including 5 µM and 15 µM (Figure 3B) as well as 30 µM and 50 µM (Figure 4A) to assist in revealing changes in astrocytic group I mGluRs. The relationship between cellular Ca2+ responses and Gq GPCR expression levels has been examined previously in vitro21-24. First, the threshold to respond to a particular agonist concentration depends on the density of receptors expressed by each cell. In a population of cells, more cells respond with a Ca2+ elevation to a given concentration of agonist when the cells are transfected with higher densities of receptors. After incubating slices in TTX, the percentage of astrocytes in the population responding to a fixed concentration of agonist increases (Figures 3B and 3C). We have found that 5 µM and 15 µM DHPG reveal obvious differences in the percentage of responsive astrocytes between the control and TTX treated cells, whereas 30 and 50 µM DHPG are required to compare group I mGluR responses in 5.0 mM K+ treated vs. control cells (Figure 4A).
The relationship between astrocytic Ca2+ response patterns and agonist concentration has also been examined in situ. Increasing the agonist concentration shifts the pattern of the Ca2+ response in astrocytes from single peak Ca2+ elevations to multipeak and plateau Ca2+ elevations25-27. Based on these previous findings, we predicted that the response pattern to a single concentration of agonist will shift if there are changes in the level of receptor expression. Thus, depending on which of the two scaling methods is being utilized (inhibiting neuronal firing or increasing it), the concentration of agonist necessary to produce a particular response pattern will increase or decrease. For example, astrocytes incubated in TTX shift their DHPG-evoked Ca2+ response pattern to more plateau-type Ca2+ elevations and respond to lower agonist concentrations compared to control astrocytes (Figure 3C). Taking into account the earlier studies, these observations suggest that the group I mGluR receptor expression levels in astrocytes has increased.
Rise time and onset (latency) of the Ca2+ elevations have also been shown to directly correlate to changes in Gq GPCR expression levels in cultured cells22-24. Higher receptor expression levels result in shorter response latencies and faster rise times while reduction in receptor density produces the opposite effect. For astrocytes incubated in TTX, Ca2+ transients evoked by application of DHPG have significantly faster rise times compared to astrocytes incubated in control ACSF (Figure 3D). As previously mentioned, amplitudes of agonist-evoked Ca2+ responses remain unchanged regardless of the agonist concentration or the scaling model15 (Figure 3D).
In addition to the changes observed by directly activating group I mGluRs with DHPG, spontaneous astrocyte Ca2+ activity is also significantly affected by this manipulation. We observed a 2.26 fold increase in the percentage of spontaneously active astrocytes incubated in TTX versus control. This is an increase from only 12.9% of control astrocytes exhibiting spontaneous activity in the soma to 42.1% in the TTX incubated astrocytes (Figure 3E). Because it is known that GPCRs exhibit “intrinsic” or constitutive activity in the absence of agonist21,26,28, and that the level of this intrinsic activity increases with increasing receptor expression levels, these data suggest that the density of astrocytic Gq GPCRs increases following long-term reduction in neuronal action potential firing. Similar to agonist-evoked responses, the rise times of the spontaneous Ca2+ elevations are also increased (Figure 3E).
Representative data using the second protocol, incubation in elevated extracellular potassium (5.0 mM), is depicted in Figure 4. An increase in extracellular K+ from 2.5-5.0 mM results in a significant increase in basal CA3 neuron action potential frequency15. Higher concentrations of DHPG (30 µM and 50 µM) are necessary in order to evoke group I mGluR Ca2+ responses from the astrocytes incubated in high potassium (Figures 4A and 4B). This is consistent with a reduced level of group I mGluR responsiveness in astrocytes following a long-term increase in neuronal action potentials. In addition, the evoked response pattern to a fixed concentration of DHPG shifts from plateau-like responses to weaker single-peak responses (Figure 4B). Examining the percentage of spontaneously active astrocytes in the two potassium conditions reveals that fewer astrocytes incubated in high K+ are spontaneously active compared to the control condition (Figure 4C). This effect is in the opposite direction of the TTX condition in which the percentage of astrocytes exhibiting spontaneous Ca2+ elevations is increased. Last, both evoked and spontaneous Ca2+ elevations have slower rise times in astrocytes incubated in high K+ versus the control condition (Figures 4C and 4D). Overall these data suggest that astrocytic Gq GPCR expression levels scale bidirectionally depending upon the level of neuronal action potential activity over an extended time period.
 Figure 1. Slice incubation chamber fabrication and set up. (A) The drawer portion of a Brinkmann pipette storage container along with its air-tight lid is used to construct the slice incubation chamber. (B) Drill a hole in the side of the container approximately 1¼ in from the top and ¼ in from the side. Fit in a piece of flexible tubing. (C) Connect the microloader-manifold apparatus to the inside end of the flexible tubing. Note cut-to-fit natural 200 µl pipette tip (white arrow). (D) Six 20 µl Eppendorf microloaders are cut-to-fit to a one-to-six line manifold to create a microloader-manifold apparatus. (E) Drill two small holes on the lid. (F) A Floating Bubble Rack for making the slice holder. (G) The bottom “legs” of the floating bubble rack are removed so that a piece of Nylon mesh material can be glued to the bottom. (H) Fill the incubation chamber with sufficient amount of ACSF so that the slice holder floats. Adjust the tubing length so the tips of the microloaders can rest at one corner of the chamber floor. When placing the incubation chamber in the water bath, the water level in the bath should be at the same level as the ACSF in the chamber. Click here to view larger image.
Figure 1. Slice incubation chamber fabrication and set up. (A) The drawer portion of a Brinkmann pipette storage container along with its air-tight lid is used to construct the slice incubation chamber. (B) Drill a hole in the side of the container approximately 1¼ in from the top and ¼ in from the side. Fit in a piece of flexible tubing. (C) Connect the microloader-manifold apparatus to the inside end of the flexible tubing. Note cut-to-fit natural 200 µl pipette tip (white arrow). (D) Six 20 µl Eppendorf microloaders are cut-to-fit to a one-to-six line manifold to create a microloader-manifold apparatus. (E) Drill two small holes on the lid. (F) A Floating Bubble Rack for making the slice holder. (G) The bottom “legs” of the floating bubble rack are removed so that a piece of Nylon mesh material can be glued to the bottom. (H) Fill the incubation chamber with sufficient amount of ACSF so that the slice holder floats. Adjust the tubing length so the tips of the microloaders can rest at one corner of the chamber floor. When placing the incubation chamber in the water bath, the water level in the bath should be at the same level as the ACSF in the chamber. Click here to view larger image.
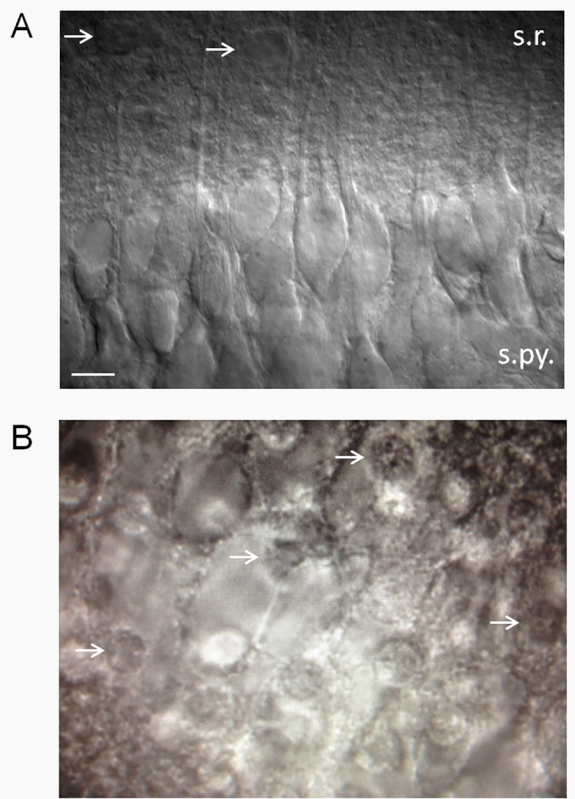 Figure 2. Estimation of slice health. (A) A healthy-looking hippocampal slice using differential interference contrast (DIC) optics. Healthy slices have a smooth, velvety appearance and a high percentage of healthy CA1 pyramidal neurons. Note the apical dendrites projecting into stratum radiatum. Patch-clamp of neurons that look like those shown here reveal a low resting membrane potential (-61 to -62 mV) in standard 2.5 mM K+ ACSF with few spontaneous action potentials. Membrane potential and firing rates will vary as a function of extracellular K+ (Xie et al.15). Arrows point to putative astrocytes. Abbreviations: s.r., stratum radiatum; s.py., stratum pyramidale. Scale bar, 10 µm. (B) Unhealthy slices will have a high percentage of dead CA1 pyramidal neurons, which have the appearance of fried eggs (white arrows point to nuclei of dead neurons - the “yolk” of the fried egg). Click here to view larger image.
Figure 2. Estimation of slice health. (A) A healthy-looking hippocampal slice using differential interference contrast (DIC) optics. Healthy slices have a smooth, velvety appearance and a high percentage of healthy CA1 pyramidal neurons. Note the apical dendrites projecting into stratum radiatum. Patch-clamp of neurons that look like those shown here reveal a low resting membrane potential (-61 to -62 mV) in standard 2.5 mM K+ ACSF with few spontaneous action potentials. Membrane potential and firing rates will vary as a function of extracellular K+ (Xie et al.15). Arrows point to putative astrocytes. Abbreviations: s.r., stratum radiatum; s.py., stratum pyramidale. Scale bar, 10 µm. (B) Unhealthy slices will have a high percentage of dead CA1 pyramidal neurons, which have the appearance of fried eggs (white arrows point to nuclei of dead neurons - the “yolk” of the fried egg). Click here to view larger image.
 Figure 3. Recording amplified Gq GPCR activity and evoked group I mGluR Ca2+ responses after long-term inhibition of neuronal APs by incubation in TTX. (A) Representative images of cells in the recording field incubated in control conditions (upper panels) or in TTX (lower panels) that have taken up Oregon Green BAPTA-1 AM Ca2+ indicator dye (left panels) and SR-101 (middle panels). Scale bar is 10 µm. Overlay of both signals (“merge”) indicates that astrocytes load with Ca2+ indicator. Boxes are drawn over individual astrocyte soma to record fluorescence intensity over time in the green channel to monitor Ca2+ activity in astrocytes. (B) Sample traces from the recording boxes in A) of Ca2+ activity in the astrocytes. Astrocytes incubated in TTX show increased spontaneous activity and more robust evoked group I mGluR Ca2+ responses as evidenced by changes in the pattern of response. Examples of single peak (circle), multipeak (rectangle), and plateau (dotted rectangle) Ca2+ transients are shown. (C) Changes in response patterns are especially evident using different concentrations of the group I mGluR agonist DHPG. More multipeak and plateau responses are evident after incubation in TTX compared to control at a given agonist concentration. (D) Rise times of DHPG-evoked Ca2+ responses are faster in astrocytes incubated in TTX compared to control (upper panel), while amplitudes do not change (lower panel), indicative of “all-or-none” response amplitudes once the threshold to respond has been reached. (E) Rise times of spontaneous astrocyte Ca2+ transients are also faster in TTX incubated vs. control incubated astrocytes (upper panel), while the percentage of astrocytes in the population exhibiting spontaneous Gq GPCR Ca2+ activity increases (lower panel). Click here to view larger image.
Figure 3. Recording amplified Gq GPCR activity and evoked group I mGluR Ca2+ responses after long-term inhibition of neuronal APs by incubation in TTX. (A) Representative images of cells in the recording field incubated in control conditions (upper panels) or in TTX (lower panels) that have taken up Oregon Green BAPTA-1 AM Ca2+ indicator dye (left panels) and SR-101 (middle panels). Scale bar is 10 µm. Overlay of both signals (“merge”) indicates that astrocytes load with Ca2+ indicator. Boxes are drawn over individual astrocyte soma to record fluorescence intensity over time in the green channel to monitor Ca2+ activity in astrocytes. (B) Sample traces from the recording boxes in A) of Ca2+ activity in the astrocytes. Astrocytes incubated in TTX show increased spontaneous activity and more robust evoked group I mGluR Ca2+ responses as evidenced by changes in the pattern of response. Examples of single peak (circle), multipeak (rectangle), and plateau (dotted rectangle) Ca2+ transients are shown. (C) Changes in response patterns are especially evident using different concentrations of the group I mGluR agonist DHPG. More multipeak and plateau responses are evident after incubation in TTX compared to control at a given agonist concentration. (D) Rise times of DHPG-evoked Ca2+ responses are faster in astrocytes incubated in TTX compared to control (upper panel), while amplitudes do not change (lower panel), indicative of “all-or-none” response amplitudes once the threshold to respond has been reached. (E) Rise times of spontaneous astrocyte Ca2+ transients are also faster in TTX incubated vs. control incubated astrocytes (upper panel), while the percentage of astrocytes in the population exhibiting spontaneous Gq GPCR Ca2+ activity increases (lower panel). Click here to view larger image.
 Figure 4. Recording diminished astrocytic Gq GPCR activity and evoked group I mGluR Ca2+ responses following long-term increase in neuronal APs by incubation in elevated extracellular potassium. (A) Representative traces of astrocyte Ca2+ recordings from slices incubated in 5.0 mM K+ ACSF to depolarize neurons and increase their basal firing rate compared to control ACSF (2.5 mM K+ ACSF). Astrocytes incubated with 5.0 mM K+ ACSF exhibit fewer spontaneous somatic Ca2+ transients and weaker DHPG evoked responses compared to astrocytes incubated in control ACSF. (B) A comparison of patterns of evoked responses to multiple concentrations of DHPG reveals weaker response types after long-term increase in neuronal APs. (C) A reduction in the percentage of astrocytes in the population exhibiting spontaneous Ca2+ activity is observed after long-term incubation in elevated K+ compared to control ACSF (upper panel), while rise times of the spontaneous activity become slower (lower panel). (D) Rise times of evoked astrocyte Ca2+ responses to different concentrations of DHPG become slower following 5.0 mM K+ treatment compared to astrocytes incubated in control ACSF. Click here to view larger image.
Figure 4. Recording diminished astrocytic Gq GPCR activity and evoked group I mGluR Ca2+ responses following long-term increase in neuronal APs by incubation in elevated extracellular potassium. (A) Representative traces of astrocyte Ca2+ recordings from slices incubated in 5.0 mM K+ ACSF to depolarize neurons and increase their basal firing rate compared to control ACSF (2.5 mM K+ ACSF). Astrocytes incubated with 5.0 mM K+ ACSF exhibit fewer spontaneous somatic Ca2+ transients and weaker DHPG evoked responses compared to astrocytes incubated in control ACSF. (B) A comparison of patterns of evoked responses to multiple concentrations of DHPG reveals weaker response types after long-term increase in neuronal APs. (C) A reduction in the percentage of astrocytes in the population exhibiting spontaneous Ca2+ activity is observed after long-term incubation in elevated K+ compared to control ACSF (upper panel), while rise times of the spontaneous activity become slower (lower panel). (D) Rise times of evoked astrocyte Ca2+ responses to different concentrations of DHPG become slower following 5.0 mM K+ treatment compared to astrocytes incubated in control ACSF. Click here to view larger image.
Discussion
The described scaling models represent practical methods for researching long-term plasticity of astrocytic group I mGluRs. Imaging spontaneous and evoked Ca2+ events provides a sensitive assay for measuring changes in astrocytic Gq GPCR activity, as firm evidence has been established that astrocyte Ca2+ elevations occur following release from IP3R-sensitive stores downstream of Gq GPCR activation10,12,17,18. The percentage of astrocytes in the population responding to group I mGluR agonist and the pattern of such Ca2+ responses report changes in group I mGluRs by astrocytes.
The specific technique used to load astrocytes with Ca2+ indicator is an important consideration in the design of experiments to look for changes in astrocytic Gq GPCR activity. Bolus-loading or bulk-loading multiple astrocytes, or patch-clamp loading of individual astrocytes can be used to image Ca2+ transients in astrocytes. Each approach offers certain advantages and disadvantages. Directly filling astrocytes with Ca2+ indicator via patch clamp allows unequivocal identification of the cell as an astrocyte without need for a secondary marker such as SR-101. Patch-clamp delivery of indicator also enables recording of Ca2+ activity from small astrocytic compartments including the bushy fine processes, potentially deeper in the slice where cells are healthier and with more intact interactions with synapses (depending on the laser power available). However, patch-clamp loading suffers from low throughput as data are collected one cell at a time. Bulk-loading, by contrast, allows a large number of astrocytes to be loaded with Ca2+ indicator and imaged simultaneously. However, only astrocytes near the surface (<20 µm) of the slice are loaded, with associated concerns about cell health and intact synapses.
The backpressure bolus-loading protocol presented here offers a middle ground, with relatively high throughput and the ability to monitor Ca2+ activity deeper within the slice (40-75 µm). A significant increase in the percentage of spontaneously active astrocytes using the bolus-loading technique is observed compared to bulk-loading, suggesting that the connections among neuronal synapses and the astrocytic processes are more complete15. With good loading, one can often monitor Ca2+ activity in main processes of astrocytes (data not shown) or potentially even smaller compartments using 2-photon microscopy. However, care would need to be exercised in assigning the smaller processes to a particular astrocyte, as the boundaries blend into the nonspecific background staining. An additional concern with the use of bulk-loading or bolus-loading procedures is the need for a secondary marker for astrocyte identification. While it has been known for many years that astrocytes preferentially take up AM ester Ca2+ indicators, the secondary marker SR-101 is often used to verify the loaded cells as astrocytes. SR-101 may in itself alter the intrinsic excitability of neurons29. Use of SR-101 corroborates the need to perform all astrocyte Ca2+ measurements in TTX to limit possible SR-101 effects on neuronal excitability. Assuming that both control and experimental groups include SR-101, the marker in itself should not account for the effects observed in astrocyte Ca2+ signaling following long-term manipulation of neuronal action potentials. SR-101 may be more of a concern in high K+ experiments, however, as it may reduce the difference between 2.5 mM K+ vs. 5.0 mM K+ if the basal firing rate is not altered proportionally.
A very promising approach to deliver Ca2+ indicator to astrocytes has emerged recently which offers an attractive alternative to the more traditional approaches using Ca2+ dyes. Significant advances have been made over the past few years with genetically encoded calcium indicators (GECIs) targeted to astrocytes30-32. GECIs can be delivered to astrocytes by in vivo microinjection of adeno-associated viral vectors into a brain region of interest such as the hippocampus. Expression of GECIs is achieved after approximately two weeks following viral infection32. There are numerous advantages presented by the use of GECIs in astrocytes. First, the vectors are targeted to astrocytes using an astrocyte-specific promoter, so the labeled cells are astrocytes32. Second, the signal-to-noise now seems comparable to what can be obtained using patch-clamp delivery of dye, but without the invasiveness of having had a patch pipette on the cell32. Third, the indicators can be delivered and expressed in adult tissue, which is problematic using bulk-loading delivery methods. Furthermore, the expression is mosaic, offering the ability to differentiate among multiple astrocytes. Thus, several astrocytes can potentially be imaged simultaneously, while also recording in the soma and fine branchlets at the same time. Therefore, potentially one single technique could be used in place of three separate techniques (bulk-loading, bolus-loading, and patch-clamp loading) to record scaling activity of astrocytic Gq GPCRs, greatly increasing efficiency.
One potential drawback of using viral-mediated delivery of Ca2+ indicators to astrocytes is the possible effect on slice health32. The adeno-associated viral vectors used to deliver the GECIs have been shown previously to cause reactive gliosis of astrocytes33. Preparation of brain slices in general likely initiates early stages of pathology including release of inflammatory molecules10. Therefore, combined with the long incubation times required to induce scaling of astrocytic receptors, use of GECIs delivered using viral vectors would need to receive additional consideration in the context of slice health in these types of experiments.
When employing this protocol, it is important to keep in mind that the application time for agonist to produce a response will vary as a function of receptor availability. For a given concentration of agonist, the application time will have to be longer if receptors have scaled down, and shorter if receptors have scaled up, for the drug to reach an adequate concentration in the tissue to activate receptors sufficiently to produce a Ca2+ response. Therefore, drug application times, and potentially their concentrations, may have to be adjusted depending upon the intended direction of the scaling. For example, the agonist concentration may need to be lowered in the case of TTX to avoid saturating responses, and increased after incubating slices in high K+ to even see a response. Specifically, the DHPG concentration was shifted from 5-15 µM after TTX treatment to 30-50 µM after 5.0 mM K+ treatment in order to study Ca2+ response patterns, as 5-15 µM was often too low to produce reliable responses in astrocytes after scaling down of group I mGluRs.
Recording of astrocyte Ca2+ activity provides no direct evidence of receptor insertion or internalization to or from the plasma membrane. However, based on the remarkable similarity of the data with data from previous studies in vitro which examined the direct relationship between Gq GPCR expression levels and spontaneous and evoked Ca2+ transients21-24, the most logical interpretation of the changes in Ca2+ signaling is that the astrocyte surface receptor expression levels have changed. A complementary approach may be an important consideration if one wants to provide additional evidence about the locus of the effect on Ca2+ activity. A strategy we used was to examine the effect of TTX incubation on hippocampal slices from astrocytic MrgA1R mice. These transgenic mice express a foreign Gq GPCR (the MrgA1R) only in astrocytes. Because this receptor is not native to the brain, there is no endogenous neurotransmitter present to change its activity levels. Previous work suggested that this receptor engages the same intracellular signaling molecules as endogenous group I mGluRs in the same astrocytes34. After long-term incubation of slices from MrgA1R mice in TTX, no differences in agonist-evoked MrgA1R responses compared to littermate control incubated slices would provide evidence that the effect on astrocyte Ca2+ activity is due to changes localized to the surface receptor, especially if group I mGluR responses are still significantly enhanced in the same astrocytes. An alternative, though perhaps more involved strategy would be to isolate astrocytes from the slices for Western blot analysis, as long as a membrane fraction could be analyzed for changes in surface receptor expression levels. Fluorescence Activated Cell Sorting (FACS) or flow cytometry may be helpful here.
The possible applications of this technique to the study of neurons, astrocytes and astrocyte-neuronal interactions are many. In our experiments, only DHPG-evoked group I mGluR astrocytic Ca2+ responses were studied, in isolated acute hippocampal slices from juvenile mice. This preparation not only has the intact afferents (Schaffer collaterals), but also the neurons that give rise to them (CA3 pyramidal cells), making it possible to manipulate the firing rates of these glutamatergic neurons onto the postsynaptic cells (CA1 pyramidal cells) and the astrocytes in stratum radiatum whose processes associate with these synapses. The acute hippocampal slice may not be the best preparation for manipulating firing rates of other types of neurons, however, as many afferents are severed from the neurons that give rise to them. Nevertheless, it may be possible in certain slice preparations to observe plasticity of other astrocytic Gq GPCR subtypes. For example, slices could be prepared with basal forebrain cholinergic neurons and their projections to hippocampus intact. Incubation of these slices in TTX or elevated K+ would affect basal firing rates of cholinergic neurons, leading to scaling of mAchRs in astrocytes of stratum oriens, which receive a significant portion of the cholinergic input1. An alternative yet untested approach to study astrocytic receptor scaling within a specific area of the brain, with all of the connections intact while scaling occurs, could be to use an in vivo model where a sustained release of TTX is achieved by implantation of a plastic polymer Elvax 40W above the region of interest35. This approach has been used previously in a study of neuronal scaling but should also be applicable to astrocytic scaling. Finally, with the proper readout, future studies could examine other GPCR families, including changes in Gs or Gi GPCRs. One might predict astrocytic GABAB Gi GPCRs to be affected following inhibition of firing in locally projecting GABA interneurons within any slice preparation. Development of new indicators targeting other signaling molecules, such as a real-time indicator of the second messenger cAMP, would open up a whole new area of research on neuron-to-astrocyte receptor communication.
Bidirectional scaling of astrocytic mGluRs by manipulation of basal neuron firing rates provides a measure of the sensitivity of astrocytes to AP-mediated release of neurotransmitter. Astrocytes can apparently sense spontaneous APs and glutamate release at Schaffer collateral-CA1 pyramidal cell synapses even when extracellular K+ is within a physiological range. While acute application of TTX does not reduce the frequency of spontaneous astrocyte Ca2+ activity18,36,37, the Ca2+ activity among the astrocytes in the population becomes decorrelated36, providing evidence that astrocytic receptors are AP detectors. This suggests that astrocytes sense spontaneous neuronal APs with no affect on their overall Ca2+ activity. It is widely accepted that intracellular concentrations of IP3 need to reach a threshold level to stimulate IP3Rs sufficiently to lead to a detectable Ca2+ elevation. Could spontaneous neuronal APs activate astrocytic GPCRs without producing measurable Ca2+ elevations? Future studies could utilize Fluorescence Resonance Energy Transfer (FRET) or a similar technique (such as BRET) to examine the relationship between G protein coupling to the receptor (a measure of receptor activation) and Ca2+ release from internal stores. BRET has been used extensively in vitro to detect G protein-to-GPCR coupling38, although it may be some time before this technology becomes available for use in intact tissue preparations. It is possible that astrocytic Gq GPCRs are being activated much more frequently than can be recorded using the currently available Ca2+ imaging tools. In addition to sensing action potentials, astrocytic Gq GPCRs may also be able to detect miniature quantal release of neurotransmitter as reported in a recent study39. The bidirectional scaling method described here may be used in future studies to provide a measure of the extent to which astrocytic Gq GPCRs detect quantal vesicular release of neurotransmitter, by including bafilomycin A1 in the incubation protocol.
Thus far, the scaling protocols have only been used in hippocampal slices from juvenile mice (p12-p18). Therefore, it is currently unknown if astrocytic receptor scaling could also be induced in tissue obtained from adult mice. A compelling recent study suggests that group I mGluR expression in astrocytes diminishes considerably after the first week of age and continues to decline until adulthood, with very low levels of receptor expression in adult astrocytes40. It would therefore be interesting to determine if astrocytic mGluRs scale up following long-term inhibition of neuronal firing in adult mouse hippocampal slices to levels approaching those seen in astrocytes from juvenile mice. This finding would suggest that astrocytic receptor expression is not static at a given age but can change rapidly depending upon levels of neuronal activity. In contrast to reduced expression of group I mGluRs in adult mice, evidence is emerging that adrenergic receptors, including α1A, α2A, and β1 subtypes, are predominantly expressed by astrocytes in the adult brain3,4. The α1A adrenergic Gq GPCR may be an attractive target for future studies of neuron-to-astrocyte communication, including whether these receptors are sensitive to changes in adrenergic neuron firing rates.
Disclosures
The authors wish to disclose that the tetrodotoxin used in these studies was purchased from Abcam. Abcam had no involvement in the hypotheses, design, or collection of data. All communication regarding sponsorship of the work by Abcam occurred after the peer review process was complete.
Acknowledgments
The authors would like to acknowledge UC Riverside’s Center for Glial-Neuronal Interactions for valuable discussion of the scaling protocols and data. The authors would also like to give a sincere thank you to Abcam for sponsoring the publication of their work.
References
- Araque A, Martin ED, Perea G, Arellano JI, Buno W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J. Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete M, Araque A. Endocannabinoids mediate neuron-astrocyte communication. Neuron. 2008;57:883–893. doi: 10.1016/j.neuron.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Bekar LK, He W, Nedergaard M. Locus coeruleus alpha-adrenergic-mediated activation of cortical astrocytes in vivo. Cereb. Cortex. 2008;18:2789–2795. doi: 10.1093/cercor/bhn040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L, Lovatt D, Goldman SA, Nedergaard M. Adrenoceptors in brain: cellular gene expression and effects on astrocytic metabolism and Ca2. Neurochem. Int. 2010;57:411–420. doi: 10.1016/j.neuint.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J. Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardinelli Y, et al. Astrocytes display complex and localized calcium responses to single-neuron stimulation in the hippocampus. J. Neurosci. 2011;31:8905–8919. doi: 10.1523/JNEUROSCI.6341-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, et al. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146:785–798. doi: 10.1016/j.cell.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 2006;9:816–823. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, McCarthy KD. Sorting out astrocyte physiology from pharmacology. Annu. Rev. Pharmacol. Toxicol. 2009;49:151–174. doi: 10.1146/annurev.pharmtox.011008.145602. [DOI] [PubMed] [Google Scholar]
- Agulhon C, et al. Calcium Signaling and Gliotransmission in Normal vs Reactive Astrocytes. Front. Pharmacol. 2012;3 doi: 10.3389/fphar.2012.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Verkhratsky A. Artifact versus reality--how astrocytes contribute to synaptic events. Glia. 2012;60:1013–1023. doi: 10.1002/glia.22288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizar K, et al. In vivo stimulus-induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J. Neurosci. 2013;33:8411–8422. doi: 10.1523/JNEUROSCI.3285-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, et al. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Xie AX, et al. Bidirectional scaling of astrocytic metabotropic glutamate receptor signaling following long-term changes in neuronal firing rates. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0049637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Camacho P, Lechleiter JD, Herman B. Measurement of intracellular calcium. Physiol. Rev. 1999;79:1089–1125. doi: 10.1152/physrev.1999.79.4.1089. [DOI] [PubMed] [Google Scholar]
- Petravicz J, Fiacco TA, McCarthy KD. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J. Neurosci. 2008;28:4967–4973. doi: 10.1523/JNEUROSCI.5572-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nett WJ, Oloff SH, McCarthy KD. Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J. Neurophysiol. 2002;87:528–537. doi: 10.1152/jn.00268.2001. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Milos RI, Konnerth A. Targeted bulk-loading of fluorescent indicators for two-photon brain imaging in vivo. Nat Protoc. 2006;1:380–386. doi: 10.1038/nprot.2006.58. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat. Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Prezeau L, et al. Changes in the carboxyl-terminal domain of metabotropic glutamate receptor 1 by alternative splicing generate receptors with differing agonist-independent activity. Mol. Pharmacol. 1996;49:422–429. [PubMed] [Google Scholar]
- Shao Y, McCarthy KD. Quantitative relationship between alpha 1-adrenergic receptor density and the receptor-mediated calcium response in individual astroglial cells. Mol. Pharmacol. 1993;44:247–254. [PubMed] [Google Scholar]
- Wang SS, Thompson SH. Measurement of changes in functional muscarinic acetylcholine receptor density in single neuroblastoma cells using calcium release kinetics. Cell Calcium. 1994;15:483–496. doi: 10.1016/0143-4160(94)90112-0. [DOI] [PubMed] [Google Scholar]
- Ostasov P, Krusek J, Durchankova D, Svoboda P, Novotny J. Ca2+ responses to thyrotropin-releasing hormone and angiotensin II: the role of plasma membrane integrity and effect of G11alpha protein overexpression on homologous and heterologous desensitization. Cell Biochem. Funct. 2008;26:264–274. doi: 10.1002/cbf.1453. [DOI] [PubMed] [Google Scholar]
- Shelton MK, McCarthy KD. Hippocampal astrocytes exhibit Ca2+-elevating muscarinic cholinergic and histaminergic receptors in situ. J. Neurochem. 2000;74:555–563. doi: 10.1046/j.1471-4159.2000.740555.x. [DOI] [PubMed] [Google Scholar]
- Hermans E, Challiss RA. Structural signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. Biochem. J. 2001;359:465–484. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur Nieden R, Deitmer JW. The Role of Metabotropic Glutamate Receptors for the Generation of Calcium Oscillations in Rat Hippocampal Astrocytes In Situ. Cortex. 2005;16(5):676–687. doi: 10.1093/cercor/bhj013. [DOI] [PubMed] [Google Scholar]
- de Ligt RA, Kourounakis AP, AP IJ. Inverse agonism at G protein-coupled receptors: (patho)physiological relevance and implications for drug discovery. Br. J. Pharmacol. 2000;130:1–12. doi: 10.1038/sj.bjp.0703311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, et al. Sulforhodamine 101 induces long-term potentiation of intrinsic excitability and synaptic efficacy in hippocampal CA1 pyramidal neurons. Neuroscience. 2010;169:1601–1609. doi: 10.1016/j.neuroscience.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Shigetomi E, Looger LL, Khakh BS. Genetically encoded calcium indicators and astrocyte calcium microdomains. Neuroscientist. 2013;19:274–291. doi: 10.1177/1073858412468794. [DOI] [PubMed] [Google Scholar]
- Akerboom J, et al. Optimization of a GCaMP Calcium Indicator for Neural Activity Imaging. J. Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, et al. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol. 2013;141:633–647. doi: 10.1085/jgp.201210949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortinski PI, et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, et al. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron. 2007;54:611–626. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Echegoyen J, Neu A, Graber KD, Soltesz I. Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLoS One. 2007;2 doi: 10.1371/journal.pone.0000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E. Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J. Neurosci. 2002;22:9430–9444. doi: 10.1523/JNEUROSCI.22-21-09430.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata N, Hirase H. Cortical layer 1 and layer 2/3 astrocytes exhibit distinct calcium dynamics in vivo. PLoS ONE. 2008;3 doi: 10.1371/journal.pone.0002525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahpour A, et al. BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front. Endocrinol. 2012;3(105) doi: 10.3389/fendo.2012.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Castro MA, et al. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 2011;14:1276–1284. doi: 10.1038/nn.2929. [DOI] [PubMed] [Google Scholar]
- Sun W, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult. Science. 2013;339:197–200. doi: 10.1126/science.1226740. [DOI] [PMC free article] [PubMed] [Google Scholar]