

Background: The enzyme responsible for the saturation step of the sphingosine 1-phosphate (S1P) metabolic pathway remained unidentified.
Results: The trans-2-enoyl-CoA reductase TER, which functions in very long-chain fatty acid (VLCFA) synthesis, catalyzes the saturation step.
Conclusion: TER is involved in both VLCFA synthesis and sphingosine degradation within sphingolipids.
Significance: Our findings reveal further details of the S1P metabolic pathway.
Keywords: Fatty Acid, Lipid, Lipid Metabolism, Lysophospholipid, Phospholipid, Sphingolipid, Sphingosine 1-Phosphate (S1P), Yeast, Very Long-chain Fatty Acid
Abstract
The sphingolipid metabolite sphingosine 1-phosphate (S1P) functions as a lipid mediator and as a key intermediate of the sole sphingolipid to glycerophospholipid metabolic pathway (S1P metabolic pathway). In this pathway, S1P is converted to palmitoyl-CoA through 4 reactions, then incorporated mainly into glycerophospholipids. Although most of the genes responsible for the S1P metabolic pathway have been identified, the gene encoding the trans-2-enoyl-CoA reductase, responsible for the saturation step (conversion of trans-2-hexadecenoyl-CoA to palmitoyl-CoA) remains unidentified. In the present study, we show that TER is the missing gene in mammals using analyses involving yeast cells, deleting the TER homolog TSC13, and TER-knockdown HeLa cells. TER is known to be involved in the production of very long-chain fatty acids (VLCFAs). A significant proportion of the saturated and monounsaturated VLCFAs are used for sphingolipid synthesis. Therefore, TER is involved in both the production of VLCFAs used in the fatty acid moiety of sphingolipids as well as in the degradation of the sphingosine moiety of sphingolipids via S1P.
Introduction
Besides being the building blocks of membranes, lipids play many important functions such as acting as lipid mediators and controlling protein activities and cellular signaling by forming lipid microdomains or by covalently or non-covalently binding to specific proteins (1–4). A balance between synthesis and degradation of each lipid class has to be maintained in these functions to avoid disorders. In the degradation pathways, fatty acids (FA),3 which are components of most lipids such as glycerophospholipids, triglycerides, sphingolipids, cholesterol esters, and wax esters, are recycled to form other lipids or are degraded via β-oxidation in the mitochondria.
The sphingolipid backbone ceramide is composed of a FA and an amide bond-linked long-chain base (LCB) (5). The most abundant LCB in mammals is sphingosine (SPH), which contains two hydroxyl groups at C1 and C3, one amino group at C2, and one trans double bond between C4 and C5. Because only sphingolipids contain LCBs, they must be converted to other metabolizable compounds by separating the LCB-specific amino and hydroxyl groups for recycling to other lipids. LCBs are metabolized to acyl-CoAs through several reactions, then for the most part converted to glycerophospholipids (6–8).
In the first step of the SPH/LCB to glycerophospholipid metabolism, SPH/LCB is phosphorylated at the C1 position by SPH kinases to produce sphingosine 1-phosphate (S1P)/long-chain base 1-phosphate (LCBP) (5, 9). S1P is a well known lipid mediator. Extracellular S1P binds to the cell surface receptors (S1P1–5) and induces several cellular responses, such as proliferation, cell migration, adherent junction assembly, and cytoskeletal remodeling (5, 7). In immune systems, S1P plays a pivotal role in the egress of T cells from the thymus and secondary lymphoid tissues. This function has already been utilized as a therapeutic agent (fingolimod) for treating multiple sclerosis (10, 11). S1P also plays an important role as an intermediate in the SPH to glycerophospholipid metabolic pathway within cells, a role that originated earlier in evolution. Sphingolipids are found in all eukaryotes, and the SPH/LCB to glycerophospholipid metabolic pathway via LCBPs is conserved from yeast to humans (8). However, S1P has emerged as a lipid mediator only in vertebrates and chordates (12).
In the SPH to glycerophospholipid metabolic pathway (referred to as S1P metabolic pathway hereafter), S1P is cleaved between C2 and C3, producing the fatty aldehyde trans-2-hexadecenal (8). In this reaction, the hydroxyl group at C1 and the amino group at C2 are removed as phosphoethanolamine. This step is the first, irreversible step of the S1P metabolism and is catalyzed by the S1P lyase SPL (SGPL1) (13). Spl knock-out mice die approximately 1 month after birth as a result of conditions such as aberrant lipid homeostasis in the liver and brain, myeloid cell hyperplasia, and enhanced pro-inflammatory response, as well as lesions in lung, heart, urinary tract, and bone (14–18). This indicates the importance of sphingolipid homeostasis.
Although the presence of the S1P metabolic pathway was reported over 40 years ago (19), the reactions that occur after the S1P lyase-catalyzing step and the involved genes had not been identified until recently. In 2012, we newly described the S1P metabolic pathway: S1P is metabolized to palmitoyl-CoA via trans-2-hexadecenal, trans-2-hexadecenoic acid, and trans-2-hexadecenoyl-CoA (Fig. 1A) (6). We also identified the fatty aldehyde dehydrogenase ALDH3A2 and some members of acyl-CoA synthetases (ACSL1–6, ACSBG1, and ACSVL1 and -4) as genes responsible for the oxidation of trans-2-hexadecenal and addition of CoA to trans-2-hexadecenoic acid, respectively (6, 20). Mutations in the ALDH3A2 gene cause the neurocutaneous disorder Sjögren-Larsson syndrome (21), which implies a link between S1P metabolism and this condition (6, 8). However, the gene involved in the saturation step of the S1P metabolism (conversion of trans-2-hexadecenoyl-CoA to palmitoyl-CoA) has not yet been identified.
FIGURE 1.
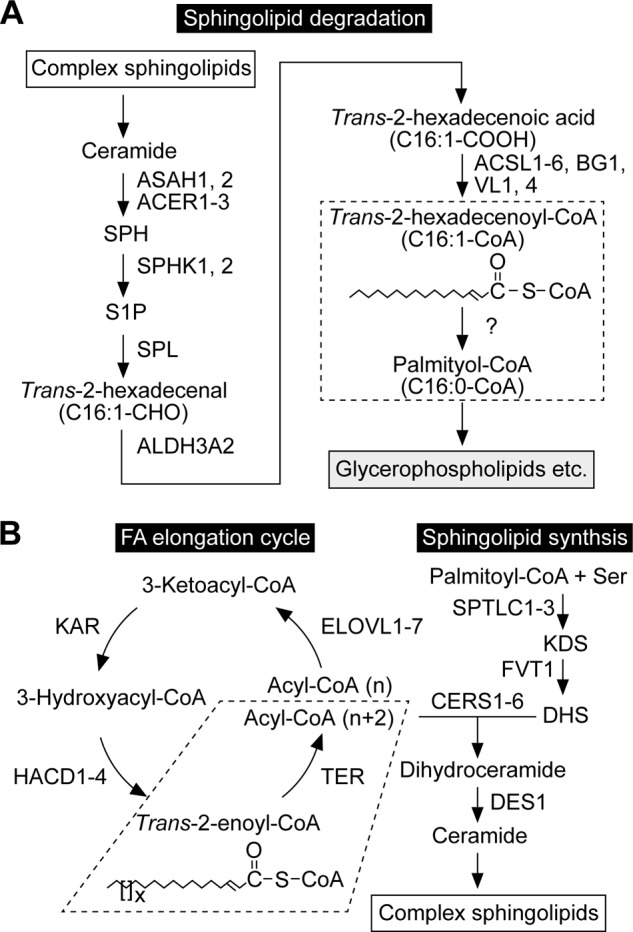
The pathways of sphingolipid degradation and synthesis. A, sphingolipid degradation pathway and involved enzymes. Complex sphingolipids are degraded to SPH by lysosomal degrading enzymes. After SPH is phosphorylated, the resulting S1P is metabolized to palmitoyl-CoA via the S1P metabolic pathway. In this pathway, S1P is first cleaved to trans-2-hexadecenal and phosphoethanolamine. Trans-2-hexadecenal is then oxidized to trans-2-hexadecenoic acid, followed by CoA addition. The produced trans-2-hexadecenoyl-CoA is finally saturated to palmitoyl-CoA by a trans-2-enoyl-CoA reductase (dashed box; subject of this study). The corresponding enzyme was unknown at the beginning of this study but identified as TER in the process. Palmitoyl-CoA is mainly active in glycerophospholipid synthesis, but may to some extent be metabolized to other lipids or degraded via β-oxidation. B, pathways of sphingolipid biosynthesis and FA elongation, and enzymes involved in each step. In the de novo sphingolipid biosynthetic pathway, palmitoyl-CoA and serine are condensed to 3-ketodihydrosphingosine (KDS), which is then reduced to DHS. DHS reacts with acyl-CoA to generate dihydroceramide. Acyl-CoA with ≥C18 is produced through FA elongation cycle. The last step of FA elongation cycle (dashed box) is catalyzed by the trans-2-enoyl-CoA reductase TER. The LCB moiety of dihydroceramide is converted from DHS to SPH by introducing a trans double bond between C4 and C5 to produce ceramide. Ceramide is converted to complex sphingolipids by receiving phosphocholine (sphingomyelin) or sugars (glycosphingolipids). The simplest glycosphingolipids are the monohexosylceramides glucosylceramide and galactosylceramide, which contain one glucose and one galactose residue, respectively. The addition of galactose to the glucose residue of glucosylceramide creates lactosylceramide. More than 100 glycosphingolipids originate from lactosylceramide, including globo-series Gb3 and Gb4 and ganglio-series GM3.
In the present study, we identified the trans-2-enoyl-CoA reductase TER as the missing gene in the S1P metabolic pathway. TER has been known as the gene involved in the FA elongation cycle (22), where palmitic acid synthesized by FA synthase or FAs taken from foods are elongated to very long-chain FAs (VLCFAs) with carbon chain lengths greater than 20 (>C20). The FA elongation cycle consists of four reactions (condensation, reduction, dehydration, and reduction) (23, 24), and TER is responsible for the fourth reaction (22) (Fig. 1B). Saturated and monounsaturated VLCFAs, especially C24 VLCFAs (C24:0 and C24:1), are major FA constituents of sphingolipids (23, 24). Hence, TER is involved in both the production of VLCFAs for sphingolipid synthesis and the degradation of SPH in sphingolipids through the S1P metabolic pathway.
EXPERIMENTAL PROCEDURES
Yeast Strains and Media
The yeast strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (25) was obtained from Open Biosystems (Huntsville, AL). YRF50 cells (BY4741, pTSC13::KanMX4-tTA-ptetO7) were constructed by replacing the promoter of the TSC13 gene (pTSC13) with tetO7 promoter (ptetO7) using the KanMX4-tTA-ptetO7 cassette from the pCM225 plasmid, which was obtained from EUROSCARF, as described elsewhere (26). ABY83 (BY4741, tsc13Δ::LEU2/pTW6 (TSC13)) and ABY80 (BY4741, tsc13Δ::LEU2/pAB119 (3xFLAG-CERS5)) cells were constructed by deletion of the TSC13 gene in BY4741 cells bearing the pTW6 or pAB119 plasmid, respectively, using a tsc13Δ::LEU2 fragment by homologous recombination. Cells were grown in either YPD medium (1% yeast extract, 2% bactopeptone, and 2% d-glucose) or in synthetic complete medium (SC; 0.67% yeast nitrogen and 2% d-glucose) containing nutritional supplements. Uracil auxotrophy was determined by culturing cells on a plate of 5-fluoroorotic acid (5-FOA; Wako Pure Chemical Industries, Osaka, Japan) containing SC medium, which includes 1 mg/ml of 5-FOA and 0.05 mg/ml of uracil.
Cell Culture, RNA Interference, and Transfection
HeLa and HepG2 cells were cultured in DMEM (D6049; Sigma) containing 10% FCS. Rat intestinal epithelial cells IEC-6 were cultured in DMEM (D6429; Sigma) containing 10% FCS supplemented with 4 μg/ml of recombinant human insulin (Wako Pure Chemical Industries). PC12 cells were cultured in DMEM (D6049) containing 10% FCS and 10% horse serum (Invitrogen). Nerve growth factor (50 ng/ml; TOYOBO, Osaka, Japan) was added to the medium 24 h before labeling to make PC12 cells differentiate into neuronal cells. All culture media contained 100 units/ml of penicillin and 100 μg/ml of streptomycin (Sigma).
The control siRNA was purchased from Qiagen (Hilden, Germany) and used at 16 nm. Two siRNAs for TER (siTER-1 and siTER-2) were prepared, and a mixture of siTER-1 and siTER-2 (each at 8 nm) was used. The nucleotide sequences of TER siRNAs were 5′-GUCACUCAUUCCACUACAUCA-3′ (siTER-1, sense), 5′-AUGUAGUGGAAUGAGUGACAG-3′ (siTER-1, antisense), 5′-CGCUCGCCAUCUUUGUGAUCU-3′ (siTER-2, sense), and 5′-AUCACAAAGAUGGCGAGCGCC-3′ (siTER-2, antisense). The corresponding duplex siRNAs were purchased from Sigma. Four days prior to the experiments, HeLa cells were transfected with the appropriate siRNA using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen), according to the manufacturer's instructions.
Plasmids
The pRS316 plasmid is a yeast centromere (CEN) vector with a URA3 marker (27). The pAKNF313 (CEN, HIS3 marker), pAKNF315 (CEN, LEU2 marker), and pAK158 (2 μ, URA3 marker) plasmids are yeast expression vectors designed to produce an N-terminal 3xFLAG-tagged protein (pAKNF313 and pAKNF315) or an N-terminal 2xHA-tagged protein (pAK158), all under control of the GAPDH promoter. The human TER and CERS5 cDNAs were amplified by PCR using human liver cDNA (Clontech/TAKARA Bio, Shiga, Japan) and primers (for TER, 5′-AGGATCCATGAAGCATTACGAGGTGGAGATTCTG-3′ and 5′-TTCAGAGCAGGAAGGGGATGATGGGC-3′; for CERS5, 5′-GGGATCCATGGCGACAGCAGCGCAGGGACC-3′ and 5′-TTACTCTTCAGCCCAGTAGCTGCCTCCC-3′; BamHI sites are underlined). The amplified fragments were cloned into the pGEM-T Easy vector (Promega, Madison, WI), producing the pGEM-TER and pGEM-CERS5 plasmids. The pTW8 (3xFLAG-TER) and pAB128 (2xHA-TER) plasmids were constructed by cloning the BamHI-NotI fragment of pGEM-TER into the BamHI-NotI site of the pAKNF315 or pAK158 vectors, respectively. Similarly, the BamHI-NotI fragment of pGEM-CERS5 was cloned into the BamHI-NotI site of the pAKNF313 vector, producing the pAB119 plasmid.
The yeast TSC13 gene with its promoter and the 3′-UTR region was amplified using yeast genomic DNAs and primers 5′-AACCCGGGTTACCTCTAGCAATGTAAACACCG-3′ and 5′-GACGAAGATGACGTTGTCAGCTCAAGC-3′ and cloned into the pGEM-T Easy vector, producing the pGEM-TSC13-1 plasmid. The yeast TSC13 gene without its promoter but containing the 3′-UTR was amplified using yeast genomic DNAs and primers 5′-AGGATCCATGCCTATCACCATAAAAAGCCGCTC-3′ (BamHI site underlined) and 5′-GACGAAGATGACGTTGTCAGCTCAAGC-3′ and cloned into pGEM-T Easy vector, creating the pGEM-TSC13-2 plasmid. The pTW6 (TSC13) and pAB134 (3xFLAG-TSC13) plasmids were constructed by cloning the NotI-NotI fragment of the pGEM-TSC13-1 or the BamHI-NotI fragment of the pGEM-TSC13-2 plasmid into the NotI site of the pRS316 vector or the BamHI-NotI site of the pAKNF313 vector, respectively.
[3H]Lipid Labeling Assay
Yeast cells were labeled for 2 h at 30 °C with [11,12-3H]SPH (6), [4,5-3H]dihydrosphingosine (DHS; 50 Ci/mmol; American Radiolabeled Chemicals, St. Louis, MO), or [9,10-3H]palmitic acid (40 Ci/mmol; American Radiolabeled Chemicals). After labeling, cells were collected by centrifugation and suspended in ethanol, water, diethyl ether, pyridine, 15 n ammonia (15:15:5:1:0.018, v/v). Lipids were extracted by incubation for 15 min at 60 °C. Cell debris and extracted lipids were separated by a 2-min centrifugation at 2,000 × g. Radioactivity was measured using a liquid scintillation system (LSC-3600; Aloka, Tokyo, Japan), and samples containing lipids of equal radioactivity were used for further study. The lipids were dried, suspended in chloroform/methanol/water (5:4:1, v/v), and separated by TLC on Silica Gel 60 high performance TLC plates (Merck) with chloroform, methanol, 4.2 n ammonia (9:7:2, v/v) as the solvent system. The lipids were visualized by spraying the TLC plates with a fluorographic reagent (2.8 mg/ml of 2,5-diphenyl-oxazole (Sigma) in 2-methylnaphthalene/1-buthanol (1:3.3, v/v)) and exposing them to x-ray film at −80 °C.
For the double bond cleavage assay, lipids extracted as described above were dried and subjected to trans-methylation, where lipids suspended in 400 μl of 1 m methanolic HCl (Supelco/Sigma) and 5% 2,2-dimethoxypropane (Sigma) were incubated for 1 h at 80 °C. Generated FA methyl esters (FAMEs) were separated from methanol with 400 μl of hexane. Note that most of the other lipids were retained in the methanol phase. Lipids in the hexane phase were dried, suspended in 100 μl of 90% acetic acid, and divided into two portions (50 μl each). Lipids were treated with 75 μl of 0.5% KMnO4 and 0.5% KIO4 (or 75 μl water for control) for 1 h at 37 °C, reduced with 15 μl of 20% sodium bisulfite (or 15 μl water for control), and neutralized with 50 μl of ammonia. Lipids were extracted by adding 250 μl of chloroform. After phase separation by centrifugation, the organic phase was recovered and dried. Lipids were suspended in 20 μl of chloroform, then separated by reverse-phase TLC on LKC18 Silica Gel 60 TLC plates (Whatman, Kent, UK) with chloroform/methanol/water (5:15:1, v/v).
Mammalian cells were incubated with 1 ml of medium without FBS for 30 min at 37 °C, then labeled with [11,12-3H]SPH, [4,5-3H]DHS, or [9,10-3H]palmitic acid for 4 or 24 h at 37 °C. Culture medium was removed from cells, which were then washed with 1 ml of PBS, detached from the dish with scrapers, and resuspended in 200 μl of PBS. Lipids were extracted by mixing with successive additions of 3.75 volumes of chloroform/methanol/HCl (100:200:1, v/v), 1.25 volumes of chloroform, and 1.25 volumes of 1% KCl. Phases were then separated by centrifugation, and the organic phase was recovered. In the case of alkaline treatment, lipids were incubated with 0.32 volumes of 0.5 m NaOH in methanol (or methanol only for the control) for 1 h at 37 °C. After neutralizing with 0.16 volumes of 1 m HCl in methanol (or 0.032 volumes of 5 m NaCl in methanol for the control), lipids were extracted by mixing with successive additions of 0.61 volumes of methanol and 0.95 volumes of 1% KCl. Phases were then separated by centrifugation, and the organic phase was recovered. Lipids were dried and suspended in 20 μl of chloroform/methanol (2:1, v/v) for normal phase TLC separation or in 123 μl of methanol for methyl esterification. Lipids were resolved by normal phase TLC on Silica Gel 60 high-performance TLC plates (Merck) with 1-butanol/acetic acid/water (3:1:1, v/v), followed by detection/quantification using the bioimaging analyzer BAS-2500 (Fuji Photo Film, Tokyo, Japan) and/or detection by autoradiography, as described above. The identities of the lipid bands on TLCs were determined by comparing the mobilities of radiolabeled lipid bands with those of standards, as described previously (6). Furthermore, alkaline treatment confirmed the validity of the determined lipid species.
Methyl esterification was performed as follows. Alkaline-treated lipids dissolved in 123 μl of methanol were mixed with 67 μl of 3 m methanolic HCl and 10 μl of 2,2-dimethoxypropane and incubated for 1 h at 100 °C. The generated FAMEs were extracted from methanol with 300 μl of hexane twice, dried, suspended in 20 μl of chloroform, separated by reverse-phase TLC on Silica Gel 60 RP-18 F254 TLC plates (Merck) with chloroform/methanol/water (15:30:2, v/v), and detected by autoradiography.
Preparation of Total Lysates and Total Membrane Fractions
HeLa cells transfected with control siRNA or siTER were washed with PBS, suspended in buffer A (50 mm HEPES-NaOH (pH 7.4), 150 mm NaCl, 10% glycerol, 1 mm DTT, 1 mm PMSF, and a 1× CompleteTM protease inhibitor mixture (EDTA-free; Roche Diagnostics), and lysed by sonication. After removal of cell debris by centrifugation (2,000 × g, 3 min, 4 °C), the supernatant was recovered as total lysates. Total lysates were centrifuged at 100,000 × g for 30 min at 4 °C, and the resulting pellet (total membrane fraction) was suspended in buffer A.
In Vitro S1P Metabolism Assay
Total membrane fractions were incubated with 0.05 μCi of [11,12-3H]S1P for 1 h at 37 °C in a reaction mixture (buffer A containing 2 mm MgCl2, 1 mm CaCl2, 5 μm fumonisin B1 (Cayman Chemical, Ann Arbor, MI), 1 mm glycerol 3-phosphate (Wako Pure Chemical Industries), 5 mm β-glycerophosphate (Sigma), 5 mm NaVO4 (Wako Pure Chemical Industries), and 250 μm pyridoxal phosphate (Sigma)) in the presence or absence of 200 μm CoA, 5 mm ATP, and/or 1 mm NADPH. [11,12-3H]S1P was prepared as described previously (6). After the reaction, lipids were extracted by mixing with successive additions of 3.75 volumes of chloroform/methanol/HCl (100:200:1, v/v), 1.25 volumes of chloroform, and 1.25 volumes of 1% KCl. Phases were then separated by centrifugation, and the organic phase was recovered and dried. Lipids were subjected to trans-methylation and the double bond cleavage assay as described above.
In Vitro S1P Lyase Assay
32P-Labeled S1P was used as a substrate for the in vitro S1P lyase assay. The 32P-labeled S1P was prepared by incubating 30 μm SPH (Sigma), complexed with 0.4 mg/ml of FA-free BSA (Sigma), with 13 μCi of [γ-32P]ATP (3,000 Ci/mmol; PerkinElmer Life Sciences) and 62.5 ng of purified 3xFLAG-Lcb4 (28) at 37 °C for 1 h in 100 μl of buffer A containing 5 mm MgCl2. Produced S1P was extracted by mixing with successive additions of 600 μl of chloroform/methanol (1:2, v/v), 7.5 μl of 15 m ammonia, 400 μl of chloroform, and 400 μl of 1% KCl. Phases were then separated by centrifugation, and the upper (water) phase was recovered and mixed with 600 μl of chloroform and 77 μl of 5 m HCl. Phases were separated again by centrifugation, and the lower (organic) phase was recovered, dried, and dissolved in ethanol.
The in vitro S1P lyase assay was performed by incubation of total membrane fractions with 5 μm [32P]S1P (10 μCi of [32P]S1P plus cold S1P (Sigma)), complexed with 2 mg/ml of FA-free BSA, at 37 °C for 10 min in a 50-μl reaction mixture (buffer A containing 5 mm β-glycerophosphate, 5 mm NaVO4, and 250 μm pyridoxal phosphate). After the reaction, the 7-μl reaction mixture was mixed with 33 μl of chloroform/methanol/water/HCl (100:200:30:1, v/v), and 5 μl of the resultant mixture was spotted on Silica Gel 60 high-performance TLC plates and separated with 1-butanol/acetic acid/water (2:1:1, v/v) as the solvent system. Produced [32P]phosphoethanolamine was detected and quantified using bioimaging analyzer BAS-2500 (Fuji Photo Film).
In Vitro SPH Kinase Assay
In vitro SPH kinase assay was performed as described elsewhere (29). Total lysates were incubated with [3-3H]SPH (0.04 μCi; 20 μm) ([3-3H]SPH (20 Ci/mmol; American Radiolabeled Chemicals) plus cold SPH (BIOMOL, Plymouth Meeting, PA)), complexed with 4 mg/ml of FA-free BSA, for 15 min at 37 °C in a reaction mixture (50 mm HEPES-NaOH (pH 7.4), 10 mm KCl, 15 mm MgCl2, 0.005% Triton X-100, 1 mm DTT, 1 mm PMSF, 1× CompleteTM protease inhibitor mixture, 15 mm NaF, 40 mm β-glycerophosphate, 5 mm NaVO4, 500 μm 4-deoxypyridoxine (Sigma), and 1 mm ATP). After the reaction, lipids were extracted by mixing with successive additions of 3.75 volumes of chloroform/methanol/HCl (100:200:1, v/v), 1.25 volumes of chloroform, and 1.25 volumes of 1% KCl. The phases were then separated by centrifugation, and the organic phase was recovered and dried. Lipids were suspended in 20 μl of chloroform/methanol (2:1, v/v), and resolved by TLC on Silica Gel 60 high performance TLC plates (Merck) with 1-butanol/acetic acid/water (3:1:1, v/v), followed by detection and quantification using a bioimaging analyzer BAS-2500 (Fuji Photo Film).
RT-PCR
Total RNA was isolated from HeLa cells transfected with control siRNA or siTER using a NucleoSpin RNA II kit (Machery-Nagel, Düren, Germany). RT-PCR was performed using SuperScript One-Step RT-PCR with Platinum Taq (Invitrogen) and the following primers: TER, 5′-TTCAGAGCAGGAAGGGGATGATGGGC-3′ and 5′-GTCACTCATTCCACTACATCAAGCG-3′; SPHK1, 5′-TGCTGATGTGGACCTAGAGAGTGAG-3′ and 5′-CAGGGGTCATAAGGGCTCTTCTGGC-3′; SPHK2, 5′-TTGGGCAGTGCCCGCTTCACACTGG-3′ and 5′-GATGGCCAACATGAGCACAAAGTCC-3′; SPL, 5′-TCAGTGGGGTTTTGGAGAACCATTCATCTG-3′ and 5′-AACTATCAGTTCTTCGTCGATACAGATTGG-3′; and GAPDH, 5′-CCAAGGTCATCCATGACAACTTTGG-3′ and 5′-GGTCCACCACCCTGTTGCTGTAGCC-3′.
RESULTS
Substantial Quantities of SPH Are Metabolized to Ester-linked Glycerophospholipids
SPH is metabolized to complex sphingolipids after conversion to ceramide or other lipids, mainly ester-linked glycerophospholipids (phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), and phosphatidylserine (PS)) after conversion to palmitoyl-CoA via the S1P metabolic pathway (6, 19). In the latter pathway, the generated palmitoyl-CoA can also be used for the synthesis of triglyceride (TG) and ether-linked glycerophospholipids or degraded to CO2 by β-oxidation (19, 30). However, quantitative analyses determining what extent of SPH is metabolized via the S1P metabolic pathway using cultured cells have not been performed. Therefore, we labeled HeLa cells with [11,12-3H]SPH for 2 and 24 h and quantitatively analyzed the SPH metabolites. Radioactivity in the medium decreased from 2 to 24 h, which opposed the increasing trend in radioactivity observed in cells (Fig. 2A), reflecting time-dependent absorption of SPH (Fig. 2B). Total radioactivity (medium plus cells) decreased slightly from 2 to 24 h (Fig. 2A), but not statistically significantly. Thus, only a small amount of SPH, if any, was metabolized to CO2 from 2 to 24 h.
FIGURE 2.

A high proportion of SPH is metabolized to glycerophospholipids. A–C, HeLa cells were labeled with 0.065 μCi of [11,12-3H]SPH for 2 or 24 h. Lipids were extracted from the cells and culture medium. A, the radioactivity associated with the cells and culture medium was quantified using liquid scintillation counter LSC-3600, and data are expressed as the mean ± S.D. from three independent experiments. Statistically significant differences are indicated (t test; *, p < 0.05; **, p < 0.01). B, extracted lipids were treated with or without alkaline solution and separated by normal-phase TLC with 1-butanol/acetic acid/water (3:1:1, v/v), followed by detection with bioimaging analyzer BAS-2500. Cer, ceramide; HexCer, monohexosylceramide; LacCer, lactosylceramide; GSL, glycosphingolipids other than monohexosylceramides and lactosylceramide; SM, sphingomyelin. The glycosphingolipid positioned between S1P and SM and below SM may be globosides Gb3 and Gb4, respectively, considering that these globosides are major glycosphingolipids in HeLa cells (52). The asterisk indicates unidentified lipids. C, the radioactivity associated with each lipid in B was quantified, and data are expressed as the mean ± S.D. relative to the radioactivity in total lipids from three independent experiments. Statistically significant differences are indicated (t test; *, p < 0.05, **, p < 0.01). Inset, radioactivity of total glycerolipids (GLs) and sphingolipids (SLs). D, HeLa cells were labeled with 0.16 μCi of [11,12-3H]SPH for 24 h. Lipids were extracted, treated with alkaline, and subjected to methyl esterification. The generated FAMEs were separated by reverse-phase TLC with chloroform/methanol/water (15:30:2, v/v) and detected by autoradiography. As standards, [9,10-3H]palmitic acid and [1-14C]stearic acid (both from American Radiolabeled Chemicals) were similarly methyl-esterified. E, PC12, HepG2, and IEC-6 cells were labeled with 0.065 μCi of [11,12-3H]SPH for 24 h. Lipids were extracted from the cells, treated with or without alkaline solution, separated by normal-phase TLC with 1-butanol/acetic acid/water (3:1:1, v/v), and detected by autoradiography. Plas-E, plasmanylethanolamine/plasmenylethanolamine; GPE, glycerophosphoethanolamine. Arrowheads represent the breakdown product of Plas-E, 1-alkyl/alkenyl-glycerophosphoethanolamine.
At 2 h, ceramide and monohexosylceramides (glucosylceramide and galactosylceramide) were the major SPH metabolites (Fig. 2, B and C), although their levels decreased at 24 h. Instead, more complex sphingolipids, such as sphingomyelin, lactosylceramide, and other glycosphingolipids, increased. Substantial amounts of SPH were also metabolized to glycerolipids (ester-linked glycerophospholipids and TG) at 2 h (17.0% of total radioactivity) (Fig. 2, B and C). These glycerolipids could be distinguished from sphingolipids by their sensitivity to mild alkaline. Alkaline cleaves the ester bonds connecting FAs and the glycerol backbone, liberating FAs. Total glycerolipid levels increased to 23.4% at 24 h, although TG levels decreased from 4.9 (2 h) to 1.4% (24 h) (Fig. 2, B and C). We observed little conversion of SPH to ether-linked glycerophospholipids. Although SPH is metabolized to glycerolipids via the S1P metabolic pathway (6), only little or no S1P was detected in cells and medium (Fig. 2B). These results indicate that S1P is a transient intermediate of SPH to glycerolipid conversion and most of the generated S1P is rapidly degraded without being released into the extracellular space surrounding the HeLa cells.
We next examined the FA species in glycerolipids that are derived from SPH. Labeled lipids were treated with alkaline, and the FAs released from glycerolipids were then methyl-esterified and separated by reverse-phase TLC. We found that the majority of the FA species of the SPH metabolites were C16:0 (palmitic acid) (Fig. 2D). Thus, most of the palmitoyl-CoA generated via the S1P metabolic pathway was directly used for glycerolipid synthesis without elongation.
We also examined the metabolism of SPH in other cell types (PC12 neuronal cells, HepG2 hepatocytes, and IEC-6 intestinal epithelial cells) by labeling these cells with [3H]SPH for 24 h (Fig. 2E). Again, large amounts of ester-linked glycerophospholipids were produced irrespective of cell type (PC12, 32%; HepG2, 21%; and IEC-6, 42%). Low amounts of ether-linked ethanolamine glycerophospholipids (plasmanylethanolamine/1-alkyl-2-acyl-glycero-3-phosphoethanolamine or plasmenylethanolamine/ethanolamine plasmalogen/1-alkenyl-2-acyl-glycero-3-phosphoethanol), which were converted to 1-alkyl/alkenyl-glycerophosphoethanolamine by alkaline treatment (Fig. 2E, arrowheads), were also detected (PC12, 3.0%; and HepG2, 5.2%). Again, little S1P was observed. In conclusion, 20–45% of SPH was metabolized via the S1P metabolic pathway and the ester-linked glycerophospholipids were the major metabolites of this pathway.
Ectopic Expression of Human Trans-2-Enoyl-CoA Reductase TER in Yeast TER Homolog Tsc13-lowered Cells Causes Recovery in the Deficient S1P Metabolic Pathway
The gene involved in the saturation step of the S1P metabolic pathway, the conversion of trans-2-hexadecenoyl-CoA to palmitoyl-CoA, has not yet been identified. Because trans-2-hexadecenoyl-CoA is classified as a trans-2-enoyl-CoA, the enzyme involved is trans-2-enoyl-CoA reductase. All known enzymes involved in the S1P metabolic pathway are localized in the endoplasmic reticulum (20, 31–33). Therefore, we expected the candidate trans-2-enoyl-CoA reductase to be localized in the endoplasmic reticulum. Because the trans-2-enoyl-CoA reductase TER catalyzes the conversion of long-chain or very long-chain trans-2-enoyl-CoA to saturated acyl-CoA in the FA elongation cycle in the endoplasmic reticulum (22), we decided to examine its involvement in the S1P metabolic pathway.
To analyze TER, we first utilized a genetically tractable yeast system. The genes involved in the S1P metabolic pathway and the FA elongation cycle are well conserved between yeast and mammals. Although SPH is not a natural LCB for yeast, we previously demonstrated that exogenously added SPH was metabolized to palmitoyl-CoA and incorporated into glycerophospholipids via S1P as in mammals (6). The yeast homolog of TER is Tsc13. Because VLCFA production is essential for yeast growth, the TSC13 gene cannot be deleted (34). We therefore utilized a controllable tetracyclin (doxycycline; DOX)-dependent promoter (Tet-TSC13 cells). To minimize nonspecific effects caused by growth inhibition, we used DOX-treated Tet-TSC13 cells at the beginning of growth inhibition (13 h after adding DOX). As reported previously (6), exogenously added [11,12-3H]SPH was mainly metabolized to glycerophospholipids PC, PE, PI, and PS and partly to complex sphingolipids (inositol phosphorylceramide, mannosylinositol phosphorylceramide, and mannosyldiinositol phosphorylceramide) in wild-type cells both in the presence and absence of DOX (Fig. 3B). The metabolism of SPH in the absence of DOX was similar in Tet-TSC13 and wild-type cells (Fig. 3B). However, DOX treatment caused a reduction in the metabolism of SPH to glycerophospholipids in Tet-TSC13 cells, suggesting that Tsc13 is involved in the S1P metabolism. DOX treatment also affected sphingolipid metabolism in Tet-TSC13 cells (Fig. 3B). The FA moiety of yeast sphingolipids is predominantly C26 VLCFA (35). The impairment of VLCFA production caused by lowered Tsc13 levels therefore affected the sphingolipid metabolism, as was reported for mutations in the other genes involved in VLCFA production (36, 37).
FIGURE 3.

Yeast Tsc13 and mammalian TER are involved in the S1P metabolic pathway. A, metabolic pathways of SPH, DHS, and palmitic acid to sphingolipids and glycerophospholipids. Most of the enzymes are common to metabolisms of SPH and DHS (reactions 1–4). The reaction specific to the metabolism of SPH is saturation of trans-2-hexadecenoyl-CoA to palmitoyl-CoA (reaction 5; dashed box; subject of this study). The enzymes responsible for the numbered reactions are: 1, SPH kinases SPHK1 and SPHK2; 2, S1P lyase SPL; 3, fatty aldehyde dehydrogenase ALDH3A2; 4, acyl-CoA synthetases ACSL1–6, ACSBG1, ACSVL1, and ACSVL4; 5, trans-2-enoyl-CoA reductase TER identified in this study. C16:1-CHO, trans-2-hexadecenal; C16:0-CHO, hexadecanal; C16:1-COOH, trans-2-hexadecenoic acid; C16:0-COOH, palmitic acid; C16:1-CoA, trans-2-hexadecenoyl-CoA; C16:0-CoA, palmitoyl-CoA; DHS1P, DHS 1-phosphate. B–D, BY4741 (wild type) cells bearing the pAKNF315 (vector) plasmid (BY4741/pAKNF315), YRF50 (pTet-TSC13)/pAKNF315, and YRF50/pTW8 (3xFLAG-TER) were grown for 13 h at 30 °C in SC medium lacking leucine in the presence or absence of 10 μg/ml of DOX. Cells were then labeled with 0.07 μCi of [11,12-3H]SPH (B), 0.07 μCi of [4,5-3H]DHS (C), or 0.07 μCi of [9,10-3H]palmitic acid (D) for 2 h. Lipids were extracted, separated by normal-phase TLC with chloroform, methanol, 4.2 n ammonia (9:7:2, v/v), and detected by autoradiography. IPC, inositol phosphorylceramide; MIPC, mannosylinositol phosphorylceramide; M(IP)2C, mannosyldiinositol phosphorylceramide.
As a control, we also conducted [3H]DHS labeling. DHS is a saturated LCB, existing both in mammals and yeast. The only difference between DHS and SPH is the existence of a trans double bond in SPH. The metabolism of DHS to glycerophospholipids is almost identical to that of SPH, except that the saturation step present in the S1P metabolic pathway is omitted (6) (Fig. 3A). Therefore, Tsc13 was not anticipated to be directly involved in the DHS metabolism. Examining the DHS metabolism was expected to serve as a nice control to determine to what extent steps in the S1P metabolic pathway minus the Tsc13-catalyzing step were affected by the indirect growth retardation effect caused by the lowered Tsc13. DHS to glycerophospholipid metabolism was almost normal in Tet-TSC13 cells in the presence of DOX (Fig. 3C). The metabolism of DHS to sphingolipid was impaired, due to the direct effect of lowered VLCFAs.
To further exclude the possibility that lowered Tsc13 levels indirectly affect glycerophospholipid synthesis, [3H]palmitic acid labeling was conducted. The metabolism of [3H]palmitic acid to glycerophospholipids was similar in Tet-TSC13 cells to that in wild-type cells, regardless of the presence or absence of DOX (Fig. 3D). Thus, Tsc13 seems to be involved in S1P metabolism directly rather than through the indirect growth effect.
The effect of the ectopic expression of human TER in Tet-TSC13 cells was examined. Expression of TER resulted in the recovery of the deficient S1P metabolic pathway as well as sphingolipid metabolism (Fig. 3, B and C). These results suggest that TER is involved in the S1P metabolism.
Creation of the tsc13Δ Yeast Cells and Restoration of the Deficient S1P Metabolism by TER
Under normal conditions, the TSC13 gene cannot be deleted due to the essential functions of VLCFAs (34). In yeast, VLCFAs are almost exclusively C26 (35). They are mainly used for sphingolipid synthesis, although a fraction takes part in glycosylphosphatidylinositol-anchor biosynthesis (35, 38). The lethality of VLCFA-deficient mutations might thus be attributable to essential functions of VLCFA-containing sphingolipids. However, a recent report indicated that the introduction of cotton ceramide synthase into yeast leads to a shift of C26 sphingolipids to C18 sphingolipids without severe growth defects (39). From this observation, we speculated that VLCFA-containing sphingolipids are not essential for growth but that VLCFAs are needed for the production of sphingolipids due to substrate specificity of yeast ceramide synthases Lag1 and Lac1. To test this possibility, we examined whether the TSC13 gene could be deleted in yeast cells if they instead expressed the human ceramide synthase CERS5, which is known to generate C16 and C18 ceramides like cotton ceramide synthase (40). The tsc13Δ cells harboring the plasmid encoding the TSC13 gene and URA3 as a marker did not grow on a plate containing 5-FOA, which kills URA3+ cells (Fig. 4A). This result indicates that tsc13Δ cells could not lose the TSC13-encoding plasmid and still survive. However, the introduction of a CERS5-encoding plasmid into tsc13Δ cells bearing the TSC13/URA3 plasmid enabled the cells to grow on the 5-FOA plate (Fig. 4A). These results indicate that if CERS5 is expressed, tsc13Δ yeast is able to grow. The tsc13Δ/CERS5 cells exhibited a small retardation in growth compared with the wild-type control, demonstrating that these cells can be used to eliminate the indirect growth effects. Furthermore, sphingolipid metabolism was expected to be unaffected by the tsc13Δ mutation, because the ceramide synthase CERS5 produces C16/C18 sphingolipids in place of yeast ceramide synthases Lag1 and Lac1, which cannot use C16/C18-CoAs as substrates.
FIGURE 4.

Blockage of the S1P metabolic pathway in tsc13Δ cells is recovered by expression of mammalian TER. A, ABY83 (tsc13Δ cells bearing the pTW6 plasmid encoding TSC13 (pTSC13) and URA3; ABY83/pTW6) were transfected with the pAKNF313 (HIS3 vector, 1), pAB119 (3xFLAG-CERS5 (pCERS5), 2) or pAB134 (3xFLAG-TSC13 (pTSC13), 3) plasmids. Cells were grown on a plate of SC medium lacking uracil and histidine (SC-His-Ura) or SC-His containing 5-FOA at 30 °C for 2 days. B–E, BY4741 (wild type)/pAB119 (3xFLAG-CERS5) cells and ABY80 (tsc13Δ cells/pAB119) cells were grown in SC-His medium and labeled with 0.07 μCi of [11,12-3H]SPH (B and E), 0.07 μCi of [4,5-3H]DHS (C), or 0.07 μCi of [9,10-3H]palmitic acid (D) at 30 °C for 2 h. B–D, lipids were extracted, separated by normal-phase TLC with chloroform, methanol, 4.2 n ammonia (9:7:2, v/v), and detected by autoradiography. CER, ceramide; DHS1P, DHS 1-phosphate; IPC, inositol phosphorylceramide; MIPC, mannosylinositol phosphorylceramide; M(IP)2C, mannosyldiinositol phosphorylceramide. E, lipids were extracted and subjected to trans-methylation. The generated FAMEs were isolated by hexane/methanol phase separation, treated with or without KMnO4/KIO4, and separated by reverse-phase TLC with chloroform/methanol/water (5:15:1, v/v). F, double bond cleavage reactions in E for palmitic acid methyl ester (C16:0 FAME), trans-2-hexadecenoic acid methyl ester (trans-2-C16:1 FAME), and palmitoleic acid (cis-9-hexadecenoic acid) methyl ester (cis-9-C16:1 FAME). KMnO4/KIO4 treatment causes a double bond cleavage and generation of two carboxylic acids. Note that two tritium atoms in palmitoleic acid methyl ester are removed during the oxidation reactions. Dots represent tritium atoms. Me, methyl group. G, BY4741 (wild type)/pAB119 (pCERS5)/pAK158 (vector), ABY80 (tsc13Δ/pCERS5)/pAK158, and ABY80 (tsc13Δ/pCERS5)/pAB128 (2xHA-TER) cells grown in SC-His-Leu medium were labeled with 0.07 μCi of [11,12-3H]SPH for 2 h. Lipids were extracted, separated by normal-phase TLC with chloroform, methanol, 4.2 n ammonia (9:7:2, v/v), and detected by autoradiography.
The same labeling experiments performed on the Tet-TSC13 cells were carried out on the tsc13Δ/CERS5 cells. The metabolism of [3H]DHS and [3H]palmitic acids was almost indistinguishable between wild-type control and tsc13Δ/CERS5 cells (Fig. 4, C and D). In contrast, SPH to glycerophospholipid metabolism was largely impaired in tsc13Δ/CERS5 cells, accompanied by an increase in the metabolism of SPH to sphingolipids (Fig. 4B). Because the only difference between the DHS and SPH to glycerophospholipid metabolic pathways is the presence of the saturation step for SPH, our results signify that the tsc13Δ mutation affected the saturation step.
Although the SPH to glycerophospholipid metabolic pathway was impaired to a large degree in tsc13Δ/CERS5 cells, it was not completely blocked. This may have been caused by the presence of another trans-2-enoyl-CoA reductase in yeast. Alternatively, the accumulated trans-2-hexadecenoyl-CoA may have been incorporated into glycerophospholipids without saturation. To further investigate these effects, FAs incorporated into glycerophospholipids were methyl-esterified and treated with KIO4 and KMnO4, which lead to cleavage of the double bonds and converted them to two carboxylic acids (Fig. 4F). These were separated by reverse-phase TLC. The FAs produced from SPH in wild-type cells were mostly C16:0 FA (palmitic acid), as the corresponding C16:0 FAME was resistant to KIO4/KMnO4 (Fig. 4E). These results are consistent with our previous studies (6). Some C16:1 FA (cis-9-C16:1 FA; palmitoleic acid) was also produced through conversion of palmitoyl-CoA to palmitoleoyl-CoA by the yeast Δ9 desaturase. The corresponding cis-9-C16:1 FAME was sensitive to KIO4/KMnO4, and tritium atoms at positions C9 and C10 were removed (Fig. 4F). On the other hand, the FAs generated in tsc13Δ/CERS5 cells were trans-2-hexadecenoic acid and its cis-Δ9 derivatives (Fig. 4E), because C14:0 FA was detected by KIO4/KMnO4 treatment (Fig. 4F). These results indicate that the tsc13Δ mutation almost prevented the conversion of SPH to palmitoyl-CoA, but some of the accumulated trans-2-hexadecenoyl-CoA was incorporated into glycerophospholipids without saturation. Introduction of TER into tsc13Δ/CERS5 cells restored the deficient S1P metabolic pathway (Fig. 4G). Thus, TER was found to be a trans-2-hexadecenoyl-CoA reductase functioning in the S1P metabolic pathway.
Knockdown of TER in HeLa Cells Causes Reductions in Both SPH and DHS to Glycerophospholipid Metabolisms
We examined the involvement of TER in the S1P metabolic pathway using HeLa cells. HeLa cells were treated with control siRNA or siRNA for TER (siTER). It can be seen from the RT-PCR results that siTER caused a large decrease in TER mRNA (Fig. 5A). Labeling experiments were performed using [3H]SPH. Treatment of siTER caused ∼50% reduction in the metabolism of SPH to glycerophospholipids (Fig. 5, B and C). The double bond cleavage assay and separation by reverse phase TLC revealed that SPH was incorporated into glycerophospholipids as palmitic acid but not as trans-2-hexadecenoic acid (data not shown). Sphingolipid metabolism was also affected by siTER treatment due to a high proportion of C24 VLCFAs (C24:0 and C24:1) in HeLa sphingolipids (41). Monohexosylceramide levels decreased, but ceramide levels were increased (Fig. 5B).
FIGURE 5.

Knockdown of TER in HeLa cells leads to a reduction in SPH kinase activity. A–H, HeLa cells were transfected with 16 nm control siRNA or siTER and grown at 37 °C for 4 days. A, total RNAs were prepared from the transfected cells and subjected to RT-PCR using primers specific to TER, SPHK1, SPHK2, SPL, and GAPDH. Amplified DNAs were separated by agarose gel and stained with ethidium bromide. B, D, and F, cells were labeled with 0.09 μCi of [11,12-3H]SPH (B), 0.09 μCi of [4,5-3H]DHS (D), or 0.09 μCi of [9,10-3H]palmitic acid (F) for 4 h. Lipids were extracted, separated by normal-phase TLC with 1-butanol/acetic acid/water (3:1:1, v/v), and detected by autoradiography. Autoradiograms with a shorter exposure to x-ray film than used in the right panels are presented as the left panels in B and D, because PC bands are saturated in the right panels. Cer, ceramide; HexCer, monohexosylceramide; SM, sphingomyelin. C, E, and G, radioactivities associated with glycerophospholipids in B, D, and F were quantified using a bioimaging analyzer and expressed as the mean ± S.D. relative to those of total lipids from three independent experiments. Statistically significant differences are indicated (t test; *, p < 0.05; **, p < 0.01). Note that radioactivities associated with PC in B and D could not be quantified due to proximity of PC and sphingomyelin bands. H, for SPH kinase assay, total lysates (30 μg) prepared from the siRNA-treated HeLa cells were incubated with [3-3H]SPH (0.04 μCi, 20 μm) and 1 mm ATP at 37 °C for 15 min. Lipids were extracted and separated by normal-phase TLC with 1-butanol/acetic acid/water (3:1:1, v/v). The radioactivities associated with [3-3H]S1P were quantified using a bioimaging analyzer. For S1P lyase assay, total membrane fractions (10 μg) prepared from siRNA-treated HeLa cells were incubated with [32P]S1P (10 μCi, 5 μm) at 37 °C for 10 min. The reaction products were separated by normal-phase TLC with 1-butanol/acetic acid/water (2:1:1, v/v). The radioactivities associated with [32P]phosphoethanolamine were quantified using a bioimaging analyzer. Values represent the mean ± S.D. of the percent of the radioactivities associated with the reaction products to those in control cells from three independent experiments. Statistically significant differences are indicated (t test; *, p < 0.05).
[3H]DHS and [3H]palmitic acid labeling experiments were also performed. In contrast to the case of yeast cells where TSC13 mutations had little effect on DHS to glycerophospholipid metabolism (Figs. 3C and 4C), knockdown of TER in HeLa cells caused a reduction in the DHS metabolism (Fig. 5, D and E), although palmitic acid was normally metabolized to glycerophospholipids (Fig. 5, F and G). These results suggest that a specific enzyme common to SPH and DHS to glycerophospholipid metabolisms was inhibited by knockdown of TER. We therefore investigated the activities of S1P synthesizing (SPH kinase) and S1P degrading (S1P lyase) enzymes, which are required for both types of metabolism (Fig. 3A). SPH kinase activity was reduced in HeLa cells treated with siTER; however, S1P lyase activity was not reduced (Fig. 5H). The mRNA levels of SPH kinases SPHK1 and SPHK2 and S1P lyase SPL were almost unaffected by the TER knockdown (Fig. 5A). These results suggest that a decrease in VLCFA production caused by reduced TER may cause a concomitant reduction in signaling to the S1P metabolic pathway. It is possible that the drop in S1P/DHS 1-phosphate synthesis was so great that the additional effects of the reduced enzyme activity of TER were unobservable in the SPH to glycerophospholipid metabolism as compared with the DHS to glycerophospholipid metabolism.
Knockdown of TER in HeLa Cells Causes Decreased S1P Metabolism in Vitro
To circumvent the regulatory mechanisms that occur within the cells, we directly examined the effects of TER knockdown using an in vitro assay, where fixed amounts of [11,12-3H]S1P could be used as starting material. For this purpose, we first established the assay system, reproducing the S1P metabolism in vitro. Total membrane fractions prepared from HeLa cells were incubated with [3H]S1P in the presence of pyridoxal phosphate, the cofactor of the S1P lyase SPL, and NAD+, the cofactor of the fatty aldehyde dehydrogenase ALDH3A2 (Fig. 6A). Almost all of the S1P was metabolized to trans-2-hexadecenoic acid, as evidenced by the TLC position of its corresponding FAME and its conversion to C14:0 FA upon treatment with KIO4/KMnO4 (Fig. 6B). Inclusion of ATP and CoA, which are needed for the following acyl-CoA synthetase-catalyzing reactions, resulted in the production of trans-2-hexadecenoyl-CoA and C14:0-CoA (Fig. 6B). C14:0-CoA might be an artifact of the in vitro assay, which was generated through partial β-oxidation of the accumulated trans-2-hexadecenoyl-CoA. In β-oxidation, trans-2-enoyl-CoA is converted to acyl-CoA with two fewer carbon units through hydration, NAD+-dependent oxidation, and cleavage (42). The generated C14:0-CoA was not subjected to further β-oxidation, because our in vitro assay did not include FAD, which was required for the first step (conversion of acyl-CoA to trans-2-enoyl-CoA) of the next round of β-oxidation (42). The conversion of trans-2-hexadecenoyl-CoA to palmitoyl-CoA required the inclusion of NADPH, which was consistent with the fact that the cofactor of TER is NADPH (Fig. 6B) (22, 43). In conclusion, the S1P metabolic pathway was reproduced in vitro in a form that allowed control of each step by inclusion or omission of the appropriate cofactors.
FIGURE 6.
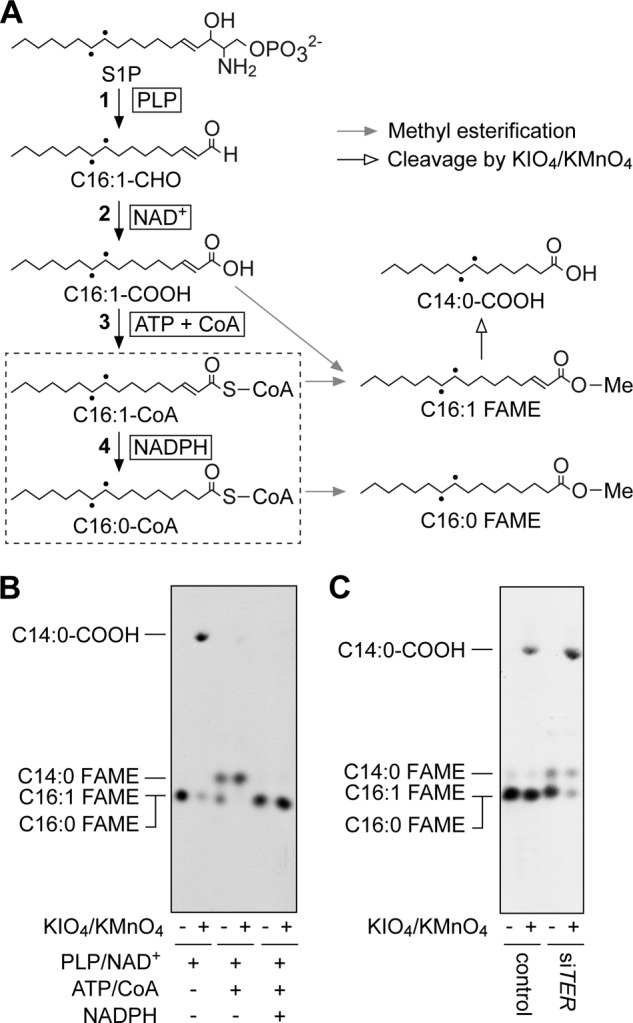
The S1P metabolic pathway is impaired in membrane fractions prepared from TER knockdown HeLa cells. A, reactions and cofactors in the S1P metabolic pathway as well as the expected products of the S1P metabolites by methyl esterification and KIO4/KMnO4 treatment. Dashed box and dots represent the TER-mediated reaction and tritium atoms, respectively. The following enzymes catalyze each reaction: 1, S1P lyase SPL; 2, fatty aldehyde dehydrogenase ALDH3A2; 3, acyl-CoA synthetases ACSL1–6, ACSBG1, ACSVL1, and ACSVL4; 4, trans-2-enoyl-CoA reductase TER. C16:1-CHO, trans-2-hexadecenal; C16:1-COOH, trans-2-hexadecenoic acid; C16:1-CoA, trans-2-hexadecenoyl-CoA; C16:0-CoA, palmitoyl-CoA; C14:0-COOH, myristic acid; Me, methyl group; PLP, pyridoxal phosphate. B, total membrane fractions (40 μg) prepared from HeLa cells were incubated with [11,12-3H]S1P at 37 °C for 1 h in the presence or absence of the indicated cofactors. Lipids were extracted and subjected to trans-methylation. The generated FAMEs were isolated by hexane/methanol phase separation, treated with or without KMnO4/KIO4, separated by reverse-phase TLC with chloroform/methanol/water (5:15:1, v/v), and detected by autoradiography. C, HeLa cells were transfected with 16 nm control siRNA or siTER and grown at 37 °C for 4 days. Total membrane fractions (30 μg) prepared from the cells were incubated with [11,12-3H]S1P at 37 °C for 1 h in the presence of pyridoxal phosphate, NAD+, ATP, CoA, and NADPH. Reaction products were processed as in B.
Using the established in vitro assay system, we examined the involvement of TER. We subjected membrane fractions prepared from siTER-treated HeLa cells to the assay. Although the reaction mixture included all the necessary cofactors, conversion of trans-2-hexadecenoyl-CoA to palmitoyl-CoA was largely impaired, and only a small amount of palmitoyl-CoA was produced (Fig. 6C). Instead, trans-2-hexadecenoyl-CoA was the main product, and C14:0-CoA was also detected (Fig. 6C). These results obtained from the in vitro assays clearly demonstrate that TER was the trans-2-enoyl-CoA reductase involved in the S1P metabolic pathway.
DISCUSSION
The S1P metabolic pathway is the only route for the conversion of the LCB moiety of sphingolipids to palmitoyl-CoA, which is further metabolized to mainly ester-linked glycerophospholipids. Therefore, this pathway is evidently quite important for sphingolipid homeostasis. Although the existence of the pathway was discovered in the late 1960s (19), its detailed reactions and the involved enzymes remained unidentified for a long time. In a study we published in 2012 (6), the reactions in the S1P metabolic pathway have been described and most of the genes involved identified, with the exception of the gene involved in the saturation step. In the present study, we show that the trans-2-enoyl-CoA reductase TER gene is responsible for the reaction. We quantitatively analyzed the metabolism of SPH using HeLa cells. Although palmitoyl-CoA is the common precursor for several lipids such as ester- and ether-linked glycerophospholipids, TG, and sphingolipids, and can be also converted to CO2 by β-oxidation for energy production, most palmitoyl-CoA derived from SPH was metabolized to ester-linked glycerophospholipids without elongation (Fig. 2, B–D). Little ether-linked glycerophospholipids were generated from SPH in HeLa cells, whereas small quantities were produced in PC12 and IEC-6 cells (Fig. 2E). SPH can be metabolized to fatty alcohols for ether-linked glycerophospholipid synthesis via two mechanisms. First, the S1P lyase product fatty aldehyde (hexadecenal) is reduced to fatty alcohol. This reduction takes place when the activity of fatty aldehyde dehydrogenase is low, as we previously demonstrated using ALDH3A2-deficient CHO-K1 cells (6). Alternatively, palmitoyl-CoA derived from SPH is subjected to the general plasmalogen biosynthetic pathway, where palmitoyl-CoA is converted to a fatty alcohol by fatty acyl-CoA reductase (44).
When HeLa cells were labeled with [3H]SPH, there was a tendency for the total radioactivity (medium plus cells) from 2 to 24 h to decrease slightly (Fig. 2A), although this was not statistically significant. Thus, only a small quantity of SPH, if any, was metabolized to CO2 from 2 to 24 h. The radioactivity of TG was reduced from 4.9 (2 h) to 1.4% (24 h) (Fig. 2, B and C). Because TG is conserved for energy production, it is possible that the decrease in TG was responsible for any potential slight decrease in total radioactivity (Fig. 2A).
Because palmitoyl-CoA is a starting material for sphingolipid synthesis (Fig. 1B), some of the palmitoyl-CoA derived from [3H]SPH via the S1P metabolic pathway may also be used for sphingolipid synthesis. However, we speculate that only a small amount is used for this, considering that the majority of [3H]palmitic acid was metabolized to glycerophospholipids, with only a small portion (<5%) being metabolized to sphingolipids (Fig. 5F).
Substantial amounts of SPH (20–45%) were metabolized to glycerolipids, irrespective of the cell type examined (HeLa, PC12, HepG2, and IEC-6 cells in this study (Fig. 2) and CHO-K1 and F9 cells in the previous study (6)), suggesting that the S1P metabolism is an active pathway that occurs ubiquitously and continuously in cells. The conversion of SPH to glycerolipids was especially high in IEC-6 intestinal epithelial cells (Fig. 2E), which may be of importance from a nutritional perspective. Dietary sphingolipids are absorbed by the small intestine after conversion to SPH (45). We speculate that the amount of dietary SPH in the small intestine is much higher than is required for sphingolipid synthesis. Therefore, the majority of SPH may be converted to glycerolipids via the S1P metabolic pathway in this tissue.
TER was originally identified as a component of the FA elongation machinery, which is responsible for the production of VLCFAs (22). Although cellular amounts of VLCFAs are much less abundant compared with those of long-chain FAs (C11-C20), VLCFAs play an important role and cannot be replaced by long-chain FAs (23, 24). For example, saturated and monounsaturated C24 VLCFAs (C24:0 and C24:1) are predominantly used for sphingolipid synthesis (C24 sphingolipids) and are important for myelin and liver functions. Gene knock-out for mouse CerS2, which is involved in C24 ceramide synthesis by catalyzing amide bond formation between LCB and C24-CoA, resulted in myelin sheath defects, cerebellar degeneration, hepatopathy, and high-frequency hepatocarcinomas (46, 47). The myelin sheath is a lipid-rich layer that surrounds the axon of a neuron. Lipid molecules keep the myelin sheath insulated and thus allow high-speed impulses to propagate along the myelinated fiber. C24 sphingolipids such as sphingomyelins, galactosylceramides, and sulfatides are abundant and important myelin lipids (48, 49). A point mutation in TER causing a substitution of Pro-182 to Leu was recently found in patients with non-syndromic mental retardation (50). This was a relatively weak mutation and caused only a slight reduction in C24 sphingolipids (43), suggesting that the requirement for C24 sphingolipids is especially high in neural systems, and that even a small decrease in C24 sphingolipids leads to neural disorders.
The use of saturated and monounsaturated VLCFAs is characteristic of sphingolipids, whereas polyunsaturated VLCFAs such as docosahexaenoic acid are predominantly used for glycerophospholipid synthesis (23, 24). The abundance of very long-chain sphingolipids (C24 and C22 sphingolipids) varies among tissues. Very long-chain sphingomyelin levels are highest in the liver (70% of total sphingomyelin levels) among the tissues examined, followed by white adipose tissue (57%), brown adipose tissue (53%), kidneys (53%), pancreas (52%), and lungs (51%) (24). Even in the testis, which possesses the lowest amount of very long-chain sphingomyelins, the ratio of very long-chain sphingomyelin levels to total sphingomyelin levels is ∼25% (24). VLCFAs can thus be concluded to be major components of sphingolipids, indicating that TER is closely related to sphingolipid synthesis. Indeed, monohexosylceramide synthesis was reduced by TER knockdown in HeLa cells (Fig. 5, B and D). Furthermore, TER also functions in the sphingolipid degradation pathway (S1P metabolic pathway), as revealed in this study (Figs. 3–6). TER is thus involved in both directions of the sphingolipid metabolism. In this study, we found that these two pathways are not independent but regulated. In TER-knockdown HeLa cells, SPH to glycerophospholipid metabolism and DHS to glycerophospholipid metabolism, in which TER is not directly involved, were affected (Fig. 5, B–E). This effect might be caused by a decrease in SPH kinase activity (Fig. 5H). SPH kinases catalyze the production of S1P/LCBP, which is the starting material of the sphingolipid to glycerophospholipid conversion. Blocking the generation of S1P/LCBP can thus lead to decreased sphingolipid degradation. The purpose of this regulation may be to prevent further reduction in sphingolipids caused by the decrease in VLCFAs for maintenance of the balance between biosynthesis and degradation of sphingolipids. The molecular mechanisms underlying the decreased SPH kinase activity in TER-knockdown HeLa cells is currently unclear. We hypothesize that the change in sphingolipid composition in the plasma membrane caused by lowered VLCFA production regulates SPH kinase activity. For example, because sphingolipids, especially very long-chain sphingolipids, modulate cellular signaling by forming lipid microdomains (51), it is possible that the change in the sphingolipid microdomain may affect SPH kinase activity.
In contrast to mammalian cells, neither lowered Tsc13 levels nor tsc13Δ affected DHS to glycerophospholipid metabolism in yeast (Figs. 3C and 4C). Because SPH is not a natural LCB in yeast, Tsc13 is normally involved only in VLCFA production. The lack of regulation between VLCFA production and LCBP metabolism in yeast is therefore reasonable.
Deletion of the yeast TSC13 gene is lethal (34), indicating that VLCFA has essential functions. Yeast sphingolipids normally contain C26:0 VLCFAs (35), and the impairment of VLCFA production leads to a large decrease in the levels of complex sphingolipids (Fig. 3, B and C). However, introduction of the mammalian ceramide synthase CERS5, which allows the production of long-chain ceramide, suppressed the lethality of the tsc13Δ cells, concomitant with production of complex sphingolipids (Fig. 4, B and C). These results indicate that the lethality of VLCFA-deficient cells can be attributed solely to a decrease in the functions of sphingolipids.
In summary, we revealed that TER is involved both in FA elongation and the S1P metabolic pathway. Our results also suggest that these two pathways are not independent, but rather mutually interdependent. Further studies are needed to determine the regulatory mechanism linking these two pathways.
Acknowledgments
We are grateful to Dr. Y. Ohno for useful input during discussions, and R. Fujimaki and Dr. K. Obara for the YRF50 yeast strain. The pCM225 plasmid and IEC-6 cells were provided by EUROSCARF, Institute of Molecular Biosciences, Johann Wolfgang Goethe-University, Frankfurt, and the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan, respectively.
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (25116701) and a Grant-in-Aid for Scientific Research (A) (26251010) (to A. K.) from the Japan Society for the Promotion of Science (JSPS).
- FA
- fatty acid
- DHS
- dihydrosphingosine
- DOX
- doxycycline
- FAME
- fatty acid methyl ester
- 5-FOA
- 5-fluoroorotic acid
- LCB
- long-chain base
- LCBP
- long-chain base 1-phosphate
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PI
- phosphatidylinositol
- PS
- phosphatidylserine
- SC
- synthetic complete
- SPH
- sphingosine
- S1P
- sphingosine 1-phosphate
- TG
- triglyceride
- VLCFA
- very long-chain fatty acid.
REFERENCES
- 1. Day C. A., Kenworthy A. K. (2009) Tracking microdomain dynamics in cell membranes. Biochim. Biophys. Acta 1788, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murakami M. (2011) Lipid mediators in life science. Exp. Anim. 60, 7–20 [DOI] [PubMed] [Google Scholar]
- 3. Resh M. D. (2012) Targeting protein lipidation in disease. Trends Mol. Med. 18, 206–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ernst A. M., Brügger B. (2014) Sphingolipids as modulators of membrane proteins. Biochim. Biophys. Acta 1841, 665–670 [DOI] [PubMed] [Google Scholar]
- 5. Kihara A., Mitsutake S., Mizutani Y., Igarashi Y. (2007) Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 46, 126–144 [DOI] [PubMed] [Google Scholar]
- 6. Nakahara K., Ohkuni A., Kitamura T., Abe K., Naganuma T., Ohno Y., Zoeller R. A., Kihara A. (2012) The Sjögren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol. Cell 46, 461–471 [DOI] [PubMed] [Google Scholar]
- 7. Hla T., Dannenberg A. J. (2012) Sphingolipid signaling in metabolic disorders. Cell Metab. 16, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kihara A. (2014) Sphingosine 1-phosphate is a key metabolite linking sphingolipids to glycerophospholipids. Biochim. Biophys. Acta 1841, 766–772 [DOI] [PubMed] [Google Scholar]
- 9. Spiegel S., Milstien S. (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 4, 397–407 [DOI] [PubMed] [Google Scholar]
- 10. Kihara A., Igarashi Y. (2008) Production and release of sphingosine 1-phosphate and the phosphorylated form of the immunomodulator FTY720. Biochim. Biophys. Acta 1781, 496–502 [DOI] [PubMed] [Google Scholar]
- 11. Groves A., Kihara Y., Chun J. (2013) Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 328, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hla T. (2004) Physiological and pathological actions of sphingosine 1-phosphate. Semin. Cell Dev. Biol. 15, 513–520 [DOI] [PubMed] [Google Scholar]
- 13. Zhou J., Saba J. D. (1998) Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem. Biophys. Res. Commun. 242, 502–507 [DOI] [PubMed] [Google Scholar]
- 14. Weber C., Krueger A., Münk A., Bode C., Van Veldhoven P. P., Gräler M. H. (2009) Discontinued postnatal thymocyte development in sphingosine 1-phosphate-lyase-deficient mice. J. Immunol. 183, 4292–4301 [DOI] [PubMed] [Google Scholar]
- 15. Vogel P., Donoviel M. S., Read R., Hansen G. M., Hazlewood J., Anderson S. J., Sun W., Swaffield J., Oravecz T. (2009) Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS One 4, e4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bektas M., Allende M. L., Lee B. G., Chen W., Amar M. J., Remaley A. T., Saba J. D., Proia R. L. (2010) Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 285, 10880–10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Allende M. L., Bektas M., Lee B. G., Bonifacino E., Kang J., Tuymetova G., Chen W., Saba J. D., Proia R. L. (2011) Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J. Biol. Chem. 286, 7348–7358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hagen-Euteneuer N., Lütjohann D., Park H., Merrill A. H., Jr., van Echten-Deckert G. (2012) Sphingosine 1-phosphate (S1P) lyase deficiency increases sphingolipid formation via recycling at the expense of de novo biosynthesis in neurons. J. Biol. Chem. 287, 9128–9136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stoffel W., Sticht G. (1967) Metabolism of sphingosine bases: I. degradation and incorporation of [3-14C]erythro-DL-dihydrosphingosine and [7-3H2]erythro-DL-sphingosine into sphingolipids of rat liver. Hoppe Seylers Z. Physiol. Chem. 348, 941–943 [PubMed] [Google Scholar]
- 20. Ohkuni A., Ohno Y., Kihara A. (2013) Identification of acyl-CoA synthetases involved in the mammalian sphingosine 1-phosphate metabolic pathway. Biochem. Biophys. Res. Commun. 442, 195–201 [DOI] [PubMed] [Google Scholar]
- 21. Rizzo W. B. (2007) Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol. Genet. Metab. 90, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moon Y. A., Horton J. D. (2003) Identification of two mammalian reductases involved in the two-carbon fatty acyl elongation cascade. J. Biol. Chem. 278, 7335–7343 [DOI] [PubMed] [Google Scholar]
- 23. Kihara A. (2012) Very long-chain fatty acids: elongation, physiology and related disorders. J. Biochem. 152, 387–395 [DOI] [PubMed] [Google Scholar]
- 24. Sassa T., Kihara A. (2014) Metabolism of very long-chain fatty acids: genes and pathophysiology. Biomol. Ther. (Seoul) 22, 83–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 26. Bellí G., Garí E., Aldea M., Herrero E. (1998) Functional analysis of yeast essential genes using a promoter-substitution cassette and the tetracycline-regulatable dual expression system. Yeast 14, 1127–1138 [DOI] [PubMed] [Google Scholar]
- 27. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kihara A., Kurotsu F., Sano T., Iwaki S., Igarashi Y. (2005) Long-chain base kinase Lcb4 is anchored to the membrane through its palmitoylation by Akr1. Mol. Cell. Biol. 25, 9189–9197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Billich A., Bornancin F., Dévay P., Mechtcheriakova D., Urtz N., Baumruker T. (2003) Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J. Biol. Chem. 278, 47408–47415 [DOI] [PubMed] [Google Scholar]
- 30. Stoffel W., LeKim D., Heyn G. (1970) Metabolism of sphingosine bases: XIV. sphinganine (dihydrosphingosine), an effective donor of the alk-1′-enyl chain of plasmalogens. Hoppe Seylers Z. Physiol. Chem. 351, 875–883 [DOI] [PubMed] [Google Scholar]
- 31. Ikeda M., Kihara A., Igarashi Y. (2004) Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem. Biophys. Res. Commun. 325, 338–343 [DOI] [PubMed] [Google Scholar]
- 32. Ashibe B., Hirai T., Higashi K., Sekimizu K., Motojima K. (2007) Dual subcellular localization in the endoplasmic reticulum and peroxisomes and a vital role in protecting against oxidative stress of fatty aldehyde dehydrogenase are achieved by alternative splicing. J. Biol. Chem. 282, 20763–20773 [DOI] [PubMed] [Google Scholar]
- 33. Soupene E., Kuypers F. A. (2008) Mammalian long-chain acyl-CoA synthetases. Exp. Biol. Med. 233, 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kohlwein S. D., Eder S., Oh C. S., Martin C. E., Gable K., Bacikova D., Dunn T. (2001) Tsc13p is required for fatty acid elongation and localizes to a novel structure at the nuclear-vacuolar interface in Saccharomyces cerevisiae. Mol. Cell. Biol. 21, 109–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ejsing C. S., Sampaio J. L., Surendranath V., Duchoslav E., Ekroos K., Klemm R. W., Simons K., Shevchenko A. (2009) Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 106, 2136–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oh C. S., Toke D. A., Mandala S., Martin C. E. (1997) ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J. Biol. Chem. 272, 17376–17384 [DOI] [PubMed] [Google Scholar]
- 37. Kihara A., Sakuraba H., Ikeda M., Denpoh A., Igarashi Y. (2008) Membrane topology and essential amino acid residues of Phs1, a 3-hydroxyacyl-CoA dehydratase involved in very long-chain fatty acid elongation. J. Biol. Chem. 283, 11199–11209 [DOI] [PubMed] [Google Scholar]
- 38. Reggiori F., Canivenc-Gansel E., Conzelmann A. (1997) Lipid remodeling leads to the introduction and exchange of defined ceramides on GPI proteins in the ER and Golgi of Saccharomyces cerevisiae. EMBO J. 16, 3506–3518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Epstein S., Castillon G. A., Qin Y., Riezman H. (2012) An essential function of sphingolipids in yeast cell division. Mol. Microbiol. 84, 1018–1032 [DOI] [PubMed] [Google Scholar]
- 40. Cerantola V., Vionnet C., Aebischer O. F., Jenny T., Knudsen J., Conzelmann A. (2007) Yeast sphingolipids do not need to contain very long chain fatty acids. Biochem. J. 401, 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mizutani Y., Kihara A., Chiba H., Tojo H., Igarashi Y. (2008) 2-Hydroxy-ceramide synthesis by ceramide synthase family: enzymatic basis for the preference of FA chain length. J. Lipid Res. 49, 2356–2364 [DOI] [PubMed] [Google Scholar]
- 42. Eaton S., Bartlett K., Pourfarzam M. (1996) Mammalian mitochondrial β-oxidation. Biochem. J. 320, 345–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abe K., Ohno Y., Sassa T., Taguchi R., Çalışkan M., Ober C., Kihara A. (2013) Mutation for nonsyndromic mental retardation in the trans-2-enoyl-CoA reductase TER gene involved in fatty acid elongation impairs the enzyme activity and stability, leading to change in sphingolipid profile. J. Biol. Chem. 288, 36741–36749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng J. B., Russell D. W. (2004) Mammalian wax biosynthesis: I. identification of two fatty acyl-coenzyme A reductases with different substrate specificities and tissue distributions. J. Biol. Chem. 279, 37789–37797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kono M., Dreier J. L., Ellis J. M., Allende M. L., Kalkofen D. N., Sanders K. M., Bielawski J., Bielawska A., Hannun Y. A., Proia R. L. (2006) Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J. Biol. Chem. 281, 7324–7331 [DOI] [PubMed] [Google Scholar]
- 46. Imgrund S., Hartmann D., Farwanah H., Eckhardt M., Sandhoff R., Degen J., Gieselmann V., Sandhoff K., Willecke K. (2009) Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J. Biol. Chem. 284, 33549–33560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pewzner-Jung Y., Brenner O., Braun S., Laviad E. L., Ben-Dor S., Feldmesser E., Horn-Saban S., Amann-Zalcenstein D., Raanan C., Berkutzki T., Erez-Roman R., Ben-David O., Levy M., Holzman D., Park H., Nyska A., Merrill A. H., Jr., Futerman A. H. (2010) A critical role for ceramide synthase 2 in liver homeostasis: II. insights into molecular changes leading to hepatopathy. J. Biol. Chem. 285, 10911–10923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Laviad E. L., Albee L., Pankova-Kholmyansky I., Epstein S., Park H., Merrill A. H., Jr., Futerman A. H. (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 283, 5677–5684 [DOI] [PubMed] [Google Scholar]
- 49. Potter K. A., Kern M. J., Fullbright G., Bielawski J., Scherer S. S., Yum S. W., Li J. J., Cheng H., Han X., Venkata J. K., Khan P. A., Rohrer B., Hama H. (2011) Central nervous system dysfunction in a mouse model of FA2H deficiency. Glia 59, 1009–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Çalışkan M., Chong J. X., Uricchio L., Anderson R., Chen P., Sougnez C., Garimella K., Gabriel S. B., dePristo M. A., Shakir K., Matern D., Das S., Waggoner D., Nicolae D. L., Ober C. (2011) Exome sequencing reveals a novel mutation for autosomal recessive non-syndromic mental retardation in the TECR gene on chromosome 19p13. Hum. Mol. Genet. 20, 1285–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iwabuchi K., Nakayama H., Iwahara C., Takamori K. (2010) Significance of glycosphingolipid fatty acid chain length on membrane microdomain-mediated signal transduction. FEBS Lett. 584, 1642–1652 [DOI] [PubMed] [Google Scholar]
- 52. Fujitani N., Furukawa J., Araki K., Fujioka T., Takegawa Y., Piao J., Nishioka T., Tamura T., Nikaido T., Ito M., Nakamura Y., Shinohara Y. (2013) Total cellular glycomics allows characterizing cells and streamlining the discovery process for cellular biomarkers. Proc. Natl. Acad. Sci. U.S.A. 110, 2105–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]