

Background: P-glycoprotein (P-gp) may alternate between the inward-facing (open) or outward-facing (closed) conformations during the reaction cycle.
Results: Cysteines introduced into extracellular loops 1 and 4 cross-linked spontaneously and inhibited drug efflux and ATPase activity.
Conclusion: P-gp activity was inhibited when P-gp was trapped in an open conformation.
Significance: P-gp can be blocked by multiple mechanisms involving extracellular loops.
Keywords: ABC Transporter, Drug Resistance, Membrane Enzyme, Membrane Protein, Protein Cross-linking
Abstract
P-glycoprotein (P-gp) is an ATP-binding cassette drug pump that protects us from toxic compounds and confers multidrug resistance. The protein is organized into two halves. The halves contain a transmembrane domain (TMD) with six transmembrane segments and a nucleotide-binding domain (NBD). The drug- and ATP-binding sites reside at the TMD1/TMD2 and NBD1/NBD2 interfaces, respectively. ATP-dependent drug efflux involves changes between the open inward-facing (NBDs apart, extracellular loops (ECLs) close together) and the closed outward-facing (NBDs close together, ECLs apart) conformations. It is controversial, however, whether the open conformation only exists transiently in intact cells because of the presence of high levels of ATP. To test for the presence of an open conformation in intact cells, reporter cysteines were placed in extracellular loops 1 (A80C, N half) and 4 (R741C, C half). The rationale was that cysteines A80C/R741C would only come close enough to form a disulfide bond in an open conformation (6.9 Å apart) because they are separated widely (30.4 Å apart) in the closed conformation. It was observed that the mutant A80C/R741C cross-linked spontaneously (>90%) when expressed in cells. In contrast to previous reports showing that trapping P-gp in a closed conformation highly activated ATPase activity, here we show that A80C/R741C cross-linking inhibited ATPase activity and drug efflux. Both activities were restored when the cross-linked mutant was treated with a thiol-reducing agent. The results show that an open conformation can be readily detected in cells and that cross-linking of cysteines placed in ECLs 1 and 4 inhibits activity.
Introduction
The P-glycoprotein drug pump (P-gp,2 ABCB1) mediates the ATP-dependent efflux of a wide range of hydrophobic compounds (such as hydrophobic drugs, steroids, peptides, detergents, and lipids) (1, 2). An important physiological role of P-gp is to protect us from toxins found in the diet (3). P-gp is clinically important because it can affect the bioavailability of drugs. For example, P-gp expression at the blood-brain barrier can reduce the efficacy of agents used to treat epilepsy, infections (such as HIV),and brain tumors (4, 5).
Because of its vital physiological role and clinical relevance, it is important to understand the structure, mechanism, and inhibition of this drug pump. The 1280 amino acids of human P-gp (6) are organized as two tandem repeats. Each repeat consists of an NH2-terminal transmembrane (TM) domain (TMD) containing six TM segments followed by a hydrophilic domain containing a nucleotide-binding domain (NBD).
There is no high-resolution crystal structure of human P-gp, but homology models have been constructed of the drug pump in open (7) and closed (8) (9) conformations on the basis of the crystal structures of the ABC Sav1866 drug pump (10) or P-gps from mouse (9) or Caenorhabditis elegans (8). In both conformations, there is an interface between the two halves. One half is composed of NBD1 and TM segments 1, 2, 3, 6, 10, and 11. The other half is composed of NBD2 and TM segments 4, 5, 7, 8, 9, and 12.
Two ATP molecules bind at the interface between the NBDs, and ATP hydrolysis occurs by an alternating site mechanism (11–15). Drug substrates appear to bind by an induced fit mechanism (16, 17) within a cavity located at the interface between the TMDs (17–21).
During the reaction cycle it is predicted that P-gp alternates between inward-facing (open, NBDs far apart, and drug-binding cavity facing the cytoplasm) and outward-facing (closed, NBDs close together, and drug-binding cavity facing the outside of the cell) conformations (reviewed in Ref. 22). The extracellular loops (ECLs) also show large changes in distance between the open and closed conformations. For example, ECL1 (connecting TM segments 1 and 2) and ECL4 (connecting TM segments 7 and 8) are at least 7 and 30 Å apart in the open and closed conformations, respectively (Fig. 1B). We showed previously that disulfide cross-linking could trap P-gp in a closed conformation to activate its ATPase activity (23–25).
FIGURE 1.
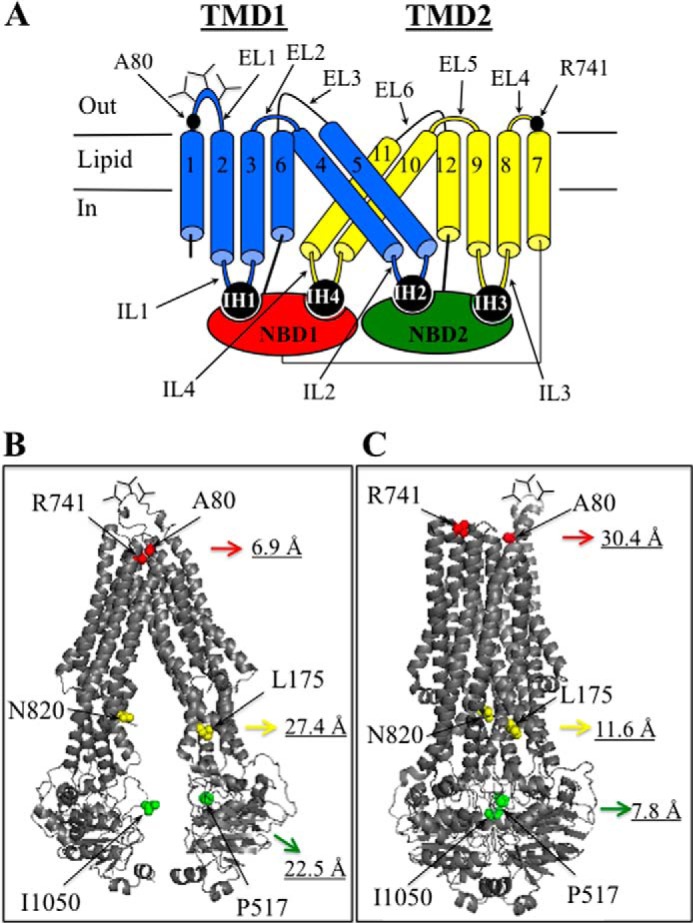
Distances between Ala-80 and Arg-741 in models of human P-gp in the open and closed conformations. A, secondary structure of P-gp showing the locations of Ala-80 and Arg-741 in extracellular loops ECL1 and ECL4. The branched lines represent the glycosylated sites, and the cylinders represent the TM segments. The TMDs interact with the NBDs through intracellular helices (IH) within intracellular loops (IL) connecting the TM segments. B, model of P-gp in the open conformation (9). C, model of human P-gp in the closed conformation ((7). The distances (in Ångstrom) are between the Cα carbons of amino acids Ala-80/Arg-741, Leu-175/Asn-820, and Pro-517/Ile-1050. The models were viewed using the PyMOL system (61).
P-gp has been crystallized only in the open conformation, in which the NBDs are widely separated (>30 Å apart) (8, 9). It is controversial, however, whether P-gp could adopt such a wide open conformation within the cell because the high levels of ATP may promote the formation of the closed structure (26). Therefore, the open conformation may exist transiently. It has been suggested that the open conformation may only be observed with solubilized P-gp in the absence of ATP (25–27) and that the NBDs in the cell might remain in constant contact throughout the reaction cycle (28).
In this study, we wanted to determine whether an open conformation could be detected in the cell. We constructed a mutant containing one cysteine in an extracellular loop of each half (ECL1 and ECL4) of P-gp that would only come close enough to directly cross-link when the molecule was in the open conformation. The mutant was used to test whether the cysteines would cross-link in intact cells and whether an extracellular disulfide bond affected P-gp drug efflux and ATPase activity.
EXPERIMENTAL PROCEDURES
Construction and Expression of Mutants
The seven endogenous cysteines at positions 137, 431, 717, 956, 1074, 1125, and 1227 were replaced with alanines to create a Cys-less P-gp. The mutant also contained an A52 epitope tag that helped distinguish it from endogenous P-gp expression (11). Mutations A80C, R741C, or A80C plus R741C were introduced into A52-tagged Cys-less P-gp or A52- or 10-histidine-tagged wild-type P-gps as described previously (25). The 10-histidine tag was added to the COOH-terminal end of P-gp to facilitate purification by nickel chelate chromatography (29).
The cDNAs were transiently expressed in HEK 293 cells by a calcium phosphate precipitation approach as described previously (30). Briefly, 20 μl of 2.5 m CaCl2 was added to 180 μl of H2O containing 4 μg of DNA, followed by addition of 200 μl of BES solution (50 mm N,N-bis(2-hydroyethyl)-2-aminoethanesulfonic acid, 280 mm NaCl, and 1.5 mm Na2HPO4 (pH 6.96)). After 10 min at room temperature, 4 ml of HEK 293 cells (about 100,000 cells/ml) in DMEM with high glucose (supplemented with nonessential amino acids, 4 mm l-glutamine, 10 IU/ml penicillin, 10 μg/ml streptomycin, and 10% (v/v) bovine calf serum) was added, and 1.5 ml of the mixture was added to duplicate wells of 6-well culture plates. After 5 h at 37 °C, the medium was replaced with fresh medium with or without 5 μm cyclosporine A. Approximately 16 h later, the cells were harvested and washed with PBS, and then the cell pellets were suspended in 200 μl of 2× SDS sample buffer (125 mm Tris-HCl (pH 6.8), 4% (w/v) SDS, and 50 mm EDTA) containing no thiol-reducing agent. Samples (equivalent to about 7000 cells) were subjected to electrophoresis on 7% SDS-PAGE gels (minigels, 1.5-mm spacers, 15 wells). The gels were electroblotted onto a sheet of nitrocellulose, and P-gp was detected using A52 monoclonal antibody, horseradish peroxidase-conjugated anti-mouse secondary antibody, and enhanced chemiluminescence. An equivalent amount of the sample was loaded onto 10% (v/v) SDS-PAGE gels and subjected to immunoblot analysis with a monoclonal antibody against GAPDH (Santa Cruz Biotechnology, Dallas, TX) (internal control).
Purification of P-gp and Measurement of ATPase Activity
Histidine-tagged P-gps were expressed in HEK 293 cells and then isolated by nickel chelate chromatography as described previously (29). Recovery of P-gp was monitored by immunoblot analysis with rabbit anti-P-gp polyclonal antibody (31). A sample of the isolated histidine-tagged P-gp was mixed with an equal volume of 10 mg/ml sheep brain phosphatidylethanolamine (Type II-S, Sigma) that had been washed and suspended in TBS (pH 7.4). Samples of the P-gp/lipid mixtures were assayed for ATPase activity by addition of an equal volume of 2× ATPase buffer (100 mm Tris-HCl (pH 7.5), 100 mm NaCl, 20 mm MgCl2, and 10 mm ATP) containing 1.2 mm verapamil. The samples were incubated for 30 min at 37 °C, and the amount of inorganic phosphate released was determined by the method of Chifflet et al. (32).
Drug Resistance Assays
Baby hamster kidney (BHK) cells expressing A52-tagged wild-type P-gp or mutant A80C/R741C were generated as described previously (33). Briefly, BHK cells were cotransfected with A52-tagged P-gp cDNA and pWL-neo (Stratagene, Cedar Creek, TX) at a ratio of 10:1 using the calcium phosphate method described above. After 5 h, the medium was replaced with fresh medium containing 1 mg/ml G418 (BioShop, Burlington, Ontario, Canada). G418-resistant colonies were isolated after 9 days and expanded. Clones expressing P-gp were identified by immunoblot analysis of whole cell SDS extracts using the monoclonal antibody A52.
Control BHK cells or BHK cells expressing A52-tagged P-gp were then incubated in the presence of various concentrations of colchicine (0–50 μm) or vinblastine (0–200 nm) for 6 days at 37 °C in the absence or presence of 1.1 mm DTT. The medium containing DTT was replaced daily with fresh medium containing the substrate and 1.1 mm DTT. Cell death was then determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (34).
Cross-linking of Mutant A80C/R741C in the Presence or Absence of ATP
HEK 293 cells were transiently transfected with the A52-tagged mutant A80C/R741C (in a Cys-less background) and then grown for 48 h at 30 °C to promote maturation of P-gp. The cells were then harvested and washed with PBS (pH 7.4) containing 5 mm DTT to reduce the spontaneously formed disulfide bond. Membranes were prepared as described previously (23) and suspended in TBS (pH 7.4) with or without 5 mm ATP plus 10 mm MgCl2 in an ice bath (0 °C). The samples were then treated for 5 min at 0 °C with or without 1 mm copper phenanthroline. The reactions were stopped by addition of 2× SDS sample buffer containing 50 mm EDTA and no thiol-reducing agent. The samples were then subjected to immunoblot analysis.
To test the effect of vanadate trapping of nucleotides on cross-linking, the membranes (prepared as above) were incubated for 5 min at 20 °C with 5 mm ATP, 10 mm MgCl2 in the absence or presence of 0.2 mm sodium orthovanadate. The samples were chilled for 5 min in an ice-bath and then treated with 1 mm copper phenanthroline for 5 min (on ice). The reactions were stopped as above, and the samples were subjected to immunoblot analysis.
RESULTS
Cysteines Introduced into ECL 1 (A80C) and ECL 4 (R741C) of Cys-less P-gp Cross-link Spontaneously
The major goal of the study was to use disulfide cross-linking to test whether we could detect the open conformation of P-gp in intact cells and whether locking P-gp in an open conformation would have an effect on its activity. Because high levels of glutathione would reduce any intracellular P-gp disulfide bonds, the best location to achieve cross-linking of P-gp cysteines in intact cells would be in the six extracellular loops that link the 12 TM segments (Fig. 1A). It would also be advantageous if the cysteines were able to come close enough (α carbons within 6–7 Å) (35) during the reaction cycle to spontaneously form a disulfide bond. This would avoid the complications of having to utilize thiol-reactive cross-linkers.
Models of human P-gp in the open and closed conformations on the basis of the crystal structures of mouse P-gp (9) or Sav1866 (7) are shown in Fig. 1, B and C, respectively. Ala-80 in ECL1 and Arg-741 in ECL4 (6.9 Å, all distances represent predicted α-α carbon distances) are extracellular amino acids predicted to be close to each other and could potentially cross-link the halves of P-gp in the open conformation (9). Direct cross-linking of cysteines at positions 80 and 741 would trap P-gp in the open conformation (Fig. 1B) only because these residues are widely separated (30.4 Å) in the closed conformation (Fig. 1C).
To test whether cysteines introduced at positions 80 and 741 would cross-link, mutants A80C, R741C, and A80C/R741C were constructed in a Cys-less P-gp background. We showed previously that alanine replacements of the seven endogenous cysteines yielded an active Cys-less P-gp (36). Cross-linking can be readily detected because disulfide bonds between the halves will cause the protein to migrate more slowly on SDS-PAGE gels (23).
The A52-tagged mutants were transiently expressed in HEK 293 cells in the absence or presence of cyclosporine A. Cyclosporine A acts as a pharmacological chaperone to promote maturation of Cys-less P-gp (37). P-gp is glycosylated at three sites in ECL1 (Fig. 1A) to yield a 150-kDa core-glycosylated protein in the endoplasmic reticulum. The protein is subsequently modified in the Golgi to yield a 170-kDa mature protein that is delivered to the cell surface (38). Whole cell SDS extracts of the transfected cells were then subjected to electrophoresis on a 7% SDS-PAGE gel, followed by immunoblot analysis. Mutants A80C, R741C, and A80C/R741C resembled the Cys-less parent because the 150-kDa immature protein was the major product when expressed in the absence of cyclosporine A (Fig. 2A). Cyclosporine A promoted maturation of the Cys-less, A80C, and R741C mutants to yield mature 170-kDa P-gp as the major product (Fig. 2A). By contrast, mutant A80C/R741C in the presence of cyclosporine A yielded a protein that migrated with slower mobility than the mature 170-kDa P-gp (Fig. 2A). Furthermore, there was very little (<5%) of the mature 170-kDa protein present. This result suggested that the protein was cross-linked spontaneously. The presence of a disulfide bond was supported by the observation that treatment of mutant A80C/R741C grown in cyclosporine A with 10 mm DTT converted the slow migrating P-gp protein into the 170-kDa protein (Fig. 2B). The results suggest that mature 170-kDa A80C/R741C protein efficiently cross-links when expressed in the presence of cyclosporine A.
FIGURE 2.
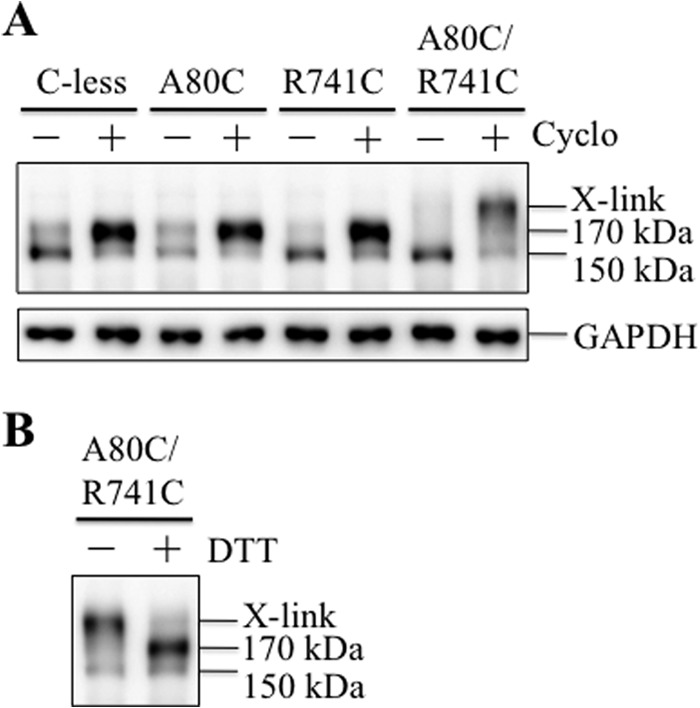
Cysteines A80C/R741C in a Cys-less background cross-link when the mutant was rescued with a drug substrate. A, A52-tagged Cys-less P-gp (C-less) and mutants A80C, R741C, or A80C/R741C were transiently expressed in HEK 293 cells in the absence (−) or presence (+) of 5 μm cyclosporine A (Cyclo). Whole cell SDS extracts (containing no thiol-reducing agent) were subjected to immunoblot analysis. B, whole cells SDS extracts of mutant A80C/R741C grown in the presence of cyclosporine A were treated without (−) or with (+) 10 mm DTT prior to immunoblot analysis. The positions of cross-linked (X-link), mature (170-kDa), and immature (150-kDa) P-gps are indicated.
Cysteines A80C (ECL1) and R741C (ECL4) Can Cross-link Spontaneously in the Absence of Drug Substrates
Because maturation of mutant A80C/R741C in the Cys-less background required the presence of cyclosporine A, we wanted to determine whether this mutant, when introduced into a wild-type background, would cross-link spontaneously in the absence of cyclosporine A. Maturation of wild-type P-gp is more efficient than the Cys-less P-gp (see below). Accordingly, mutations A80C, R741C, and A80C/R741C were introduced into A52-tagged wild-type P-gp. The mutants were then transiently expressed in HEK 293 cells. Immunoblot analysis of whole cell SDS extracts showed that wild-type P-gp matures efficiently to yield the 170-kDa mature protein as the major product in the absence of drug substrate (Fig. 3). The expression of mutants A80C and R741C also resembled wild-type P-gp in that the major product was the 170-kDa protein. The major product in mutant A80C/R741C, however, was a protein that migrated with slower mobility than the mature 170-kDa slower-migrating protein (Fig. 3). Treatment of the slowly migrating A80C/R741C protein with 10 mm DTT yielded the 170-kDa protein (Fig. 3). This result suggested that the slow-migrating protein contained a disulfide bond.
FIGURE 3.
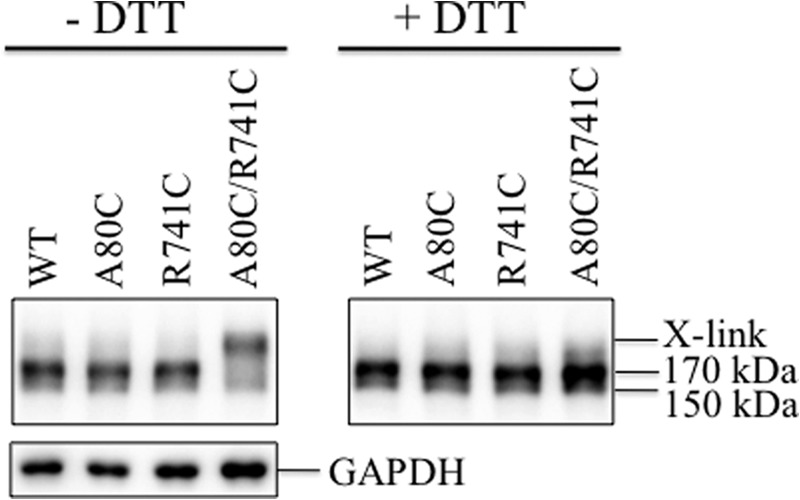
Cysteines A80C/R741C in a wild-type background cross-link in the absence of drug substrates. A52-tagged WT P-gp and mutants A80C, R741C, or A80C/R741C were transiently expressed in HEK 293 cells in the absence of drug substrates. Whole cell SDS extracts (containing no thiol-reducing agent) were subjected to immunoblot analysis before (−DTT) or after (+DTT) treatment with 10 mm DTT. The positions of cross-linked (X-link), mature (170-kDa), and immature (150-kDa) P-gps are indicated.
It is possible that mutant A80C/R741C showed efficient cross-linking because it was transiently expressed in HEK 293 cells. We then tested whether mutant A80C/R741C was also efficiently cross-linked if it was stably expressed in BHK cells. Accordingly, BHK cells were cotransfected with A52-tagged wild-type P-gp or mutant A80C/R741C (in a wild-type background) cDNAs and a plasmid containing a neomycin marker (pWL-neo). G418-resistanct colonies were selected, and stable cell lines expressing wild-type or mutant P-gp were identified by immunoblot analysis. We did not directly select for P-gp expression by using cytotoxic P-gp substrates. This was to avoid selection pressure that could favor cells expressing P-gp mutants with altered drug resistance profiles.
Immunoblot analysis of SDS extracts of BHK cells expressing P-gp showed that the major product in wild-type P-gp was the 170-kDa protein (Fig. 4, left panel). By contrast, the major products in mutant A80C/R741C were the slowly migrating cross-linked and the 150-kDa proteins. It appeared that the mature 170-kDa A80C/R741C protein was cross-linked efficiently because little (<5% of total P-gp) of the mature 170-kDa protein was present (Fig. 4, right panel).
FIGURE 4.
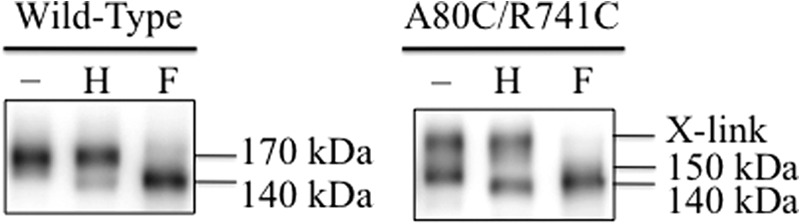
Endoglycosidase digestion of wild-type and mutant A80C/R741C P-gp expressed in BHK cells. Whole cell SDS extracts of BHK cells expressing A52-tagged wild-type P-gp or mutant A80C/R741C (wild-type background) that were grown without drug substrates were treated without (−) or with (+) endoglycosidase Hf (H) or PNGase F (F) and subjected to immunoblot analysis. The positions of cross-linked (X-link), mature (170-kDa), immature (150-kDa), and unglycosylated (140-kDa) P-gps are indicated.
We then determined whether the cross-linked P-gp was a result of cross-linking of the mature 170-kDa or the immature 140-kDa P-gp by using endoglycosidases. Whole cell SDS extracts from cells expressing wild-type P-gp or mutant A80C/R741C were treated with endoglycosidases that cleaved only core sugars (endoglycosidase H) or all carbohydrate (PNGase F). Fig. 4 (left panel) shows that the mature 170-kDa wild-type protein was sensitive only to PNGase F and yielded a 140-kDa unglycosylated protein. By contrast, the 150-kDa A80C/R741C immature protein, but not the cross-linked product, was sensitive to endoglycosidase H. The cross-linked A80C/R741C protein was sensitive only to PNGase F (Fig. 4, right panel). These results suggested that the slow-migrating product was the result of spontaneous cross-linking of only the mature 170-kDa protein. The results also show that cross-linked deglycosylated protein did not migrate very differently from the 140-kDa protein in 7% SDS-PAGE gels.
Cysteines A80C (ECL1) and R741C (ECL4) Can Cross-link Spontaneously After Synthesis and Maturation of P-gp
Disulfide bonds are formed in the endoplasmic reticulum of eukaryotic cells (reviewed in Ref. 39). It is possible that A80C/R741C may form a disulfide bond during synthesis in the endoplasmic reticulum but that the cross-linked protein did not run with altered mobility in SDS-PAGE gels so that it was not detected. A way to overcome this problem was to test whether the disulfide bond in mutant A80C/R741C could reform if synthesis of new P-gp was blocked and cross-linked P-gp at the cell surface was reduced. Accordingly, BHK cells expressing A52-tagged A80C/R741C P-gp were pretreated for 1 h with cycloheximide to inhibit protein synthesis (40). The cells were then treated briefly (1 min) with 5 mm DTT to reduce the A80C/R741C disulfide bond at the cell surface. DTT was then removed, and the cells were incubated for various times in fresh medium containing cycloheximide but no DTT. Whole cell SDS extracts were then subjected to immunoblot analysis. It was found that treatment of the cells with 5 mm DTT for 1 min was sufficient to reduce the disulfide bond to yield the 170-kDa protein as the major product (Fig. 5, +DTT, 0 min). In the absence of DTT, there was a time-dependent increase in cross-linked protein. About 50% of mutant A80C/R741C was cross-linked by 18 min and, at 54 min, the amount of cross-linked P-gp resembled that of the untreated control (Fig. 5). These results indicated that the disulfide bond could be formed after synthesis in the endoplasmic reticulum.
FIGURE 5.

Mature A80C/R741C P-gp cross-links after treatment with DTT. BHK cells expressing A52-tagged mutant A80C/R741C (wild-type background) were incubated for 1 h in the presence of 0.5 mg/ml cycloheximide. Cells were then treated without (−) or with (+) with 5 mm DTT for 1 min at 20 °C. The cells were washed to remove DTT and then incubated in the presence of 0.5 mg/ml cycloheximide for the indicated times. Whole cell SDS extracts were then subjected to immunoblot analysis. The positions of the cross-linked (X-link) and mature (170-kDa) P-gps are shown.
The A80C/R741C Disulfide Bond Reduces P-gp Drug-stimulated ATPase Activity
To test whether the A80C/R741C mutant was active, histidine-tagged wild-type P-gp or mutant A80C/R741C were transiently expressed in HEK 293 cells. The mutants were isolated by nickel chelate chromatography, mixed with lipid, and assayed for verapamil-stimulated ATPase activity in the absence or presence of 10 mm DTT. Activation of P-gp ATPase activity in the presence of drug substrates is a good measure of P-gp-drug interactions because there is a good correlation with drug transport (41, 42). Verapamil was selected because it is transported by P-gp (43) and because it highly stimulates P-gp ATPase activity (over 10-fold) (44).
Cross-linking of A80C and R741C reduced activity severely. In the absence of DTT, the cross-linked mutant showed only about 15% of the verapamil-stimulated ATPase activity of wild-type P-gp (Fig. 6). It was found however, that the verapamil-stimulated ATPase activity of mutant A80C/R741C was not significantly different from wild-type P-gp when assayed in the presence of DTT (Fig. 6). These results indicate that trapping P-gp in an open conformation inhibits ATPase activity and that residues Ala-80 or Arg-741 were not essential for activity.
FIGURE 6.
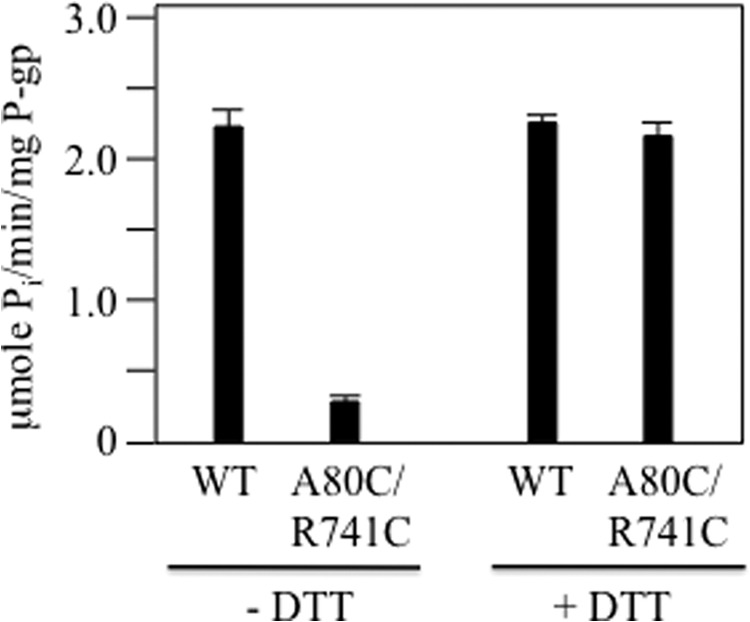
Cross-linking of mutant A80C/R741C inhibits verapamil-stimulated ATPase activity. Histidine-tagged P-gp (WT) or mutant A80C/R741C (in a wild-type background) was transiently expressed in HEK 293 cells. The P-gps were isolated by nickel chelate chromatography, mixed with lipid, and assayed for ATPase activity in the presence of verapamil before (− DTT) or after (+ DTT) treatment with 10 mm DTT. The results are mean ± S.D. from three different transfections.
The A80C/R741C Disulfide Bond Inhibits Drug Efflux
We then tested whether mutant A80C/R741C could confer drug resistance. Conferral of drug resistance would indicate that the mutant P-gp is capable of transporting cytotoxic substrates out of the cell. The classic cytotoxic P-gp substrates are vinblastine and colchicine (45). We first tested whether the A80C/R741C disulfide bond would affect the ability of P-gp to confer drug resistance on BHK cells. BHK control cells and cells expressing equivalent levels of wild-type or mutant A80C/R741C P-gp were incubated with various concentrations of vinblastine or colchicine to determine the concentration needed to inhibit cell growth. Growth of control BHK cells was inhibited by 50% (LD50) at 0.67 + 0.11 nm vinblastine and 0.53 + 0.11 μm colchicine, respectively. For cells expressing wild-type P-gp, the concentration of vinblastine and colchicine required to inhibit growth of cells (LD50) was 58.3 and 14.4 nm (86- and 21-fold increase in resistance) and 34.0 and 7.0 μm (34- and 7.0-fold increase in resistance), respectively (Fig. 7). By contrast, for cells expressing mutant A80C/R741C, the LD50s occurred at 2.3 and 0.6 nm (3-fold-increase in resistance) and 1.8 and 0.2 μm (about 3-fold increase in resistance) for vinblastine and colchicine, respectively. These values were very similar to those of control BHK cells (Fig. 7). These results suggest that the presence of the A80C/R741C disulfide bond inhibited drug efflux.
FIGURE 7.
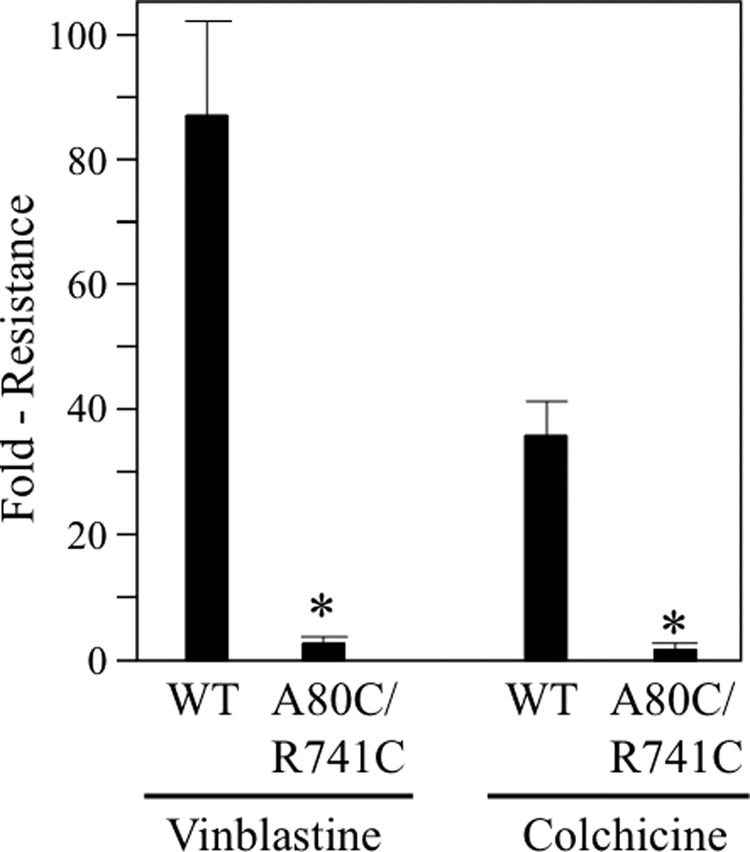
Cross-linking of A80C/R741C inhibits P-gp-mediated drug resistance. BHK cells expressing no P-gp, A52-tagged wild-type P-gp (WT) or mutant A80C/R741C (in a wild-type background) were incubated in the presence of various concentrations of vinblastine or colchicine to determine the LD50 concentration. The fold resistance represents the LD50 of the P-gp-expressing cell lines relative to cells expressing no P-gp. The results (mean ± S.D.) were obtained from three different cell lines expressing similar levels of wild-type or mutant P-gps. *, p < 0.05; Student's t test; compared with cells expressing wild-type P-gp.
Reduction of the A80C/R741C Disulfide Bond Promotes Drug Efflux
Would reduction of the A80C/R741C disulfide bond increase the ability of P-gp to confer drug resistance? Mammalian cells are very sensitive to thiol-reducing compounds because they contain important extracellular proteins at the cell surface that require critical extracellular disulfide bonds (46). In preliminary studies we found that growth of BHK cells was inhibited severely when the β-mercaptoethanol concentration in the medium was higher than 0.3 mm (data not shown). We also found that significant reduction of the A80C/R741C bond required 100-fold (30 mm) higher levels of β-mercaptoethanol.3
An alternative approach to reduce the A80C/R741C disulfide bond was be to use DTT. An advantage of DTT is that it is less toxic than β-mercaptoethanol. Accordingly, BHK cells expressing A52-tagged mutant A80C/R741C were incubated with medium containing various concentrations of DTT. After 18 h, the medium was removed, and the cells were washed with phosphate-buffered saline (pH 7.4). Whole cell SDS extracts were then subjected to immunoblot analysis. Fig. 8A shows that the cross-linked P-gp was almost completely reduced at 1.1 mm DTT so that the major product was the mature 170-kDa protein. At DTT concentrations greater than 1.1 mm, there was significant inhibition of cell growth, and this was reflected in the GAPDH blot. The level of GAPDH decreased when the cells were treated with 3.3 or 10 mm DTT (Fig. 8A).
FIGURE 8.
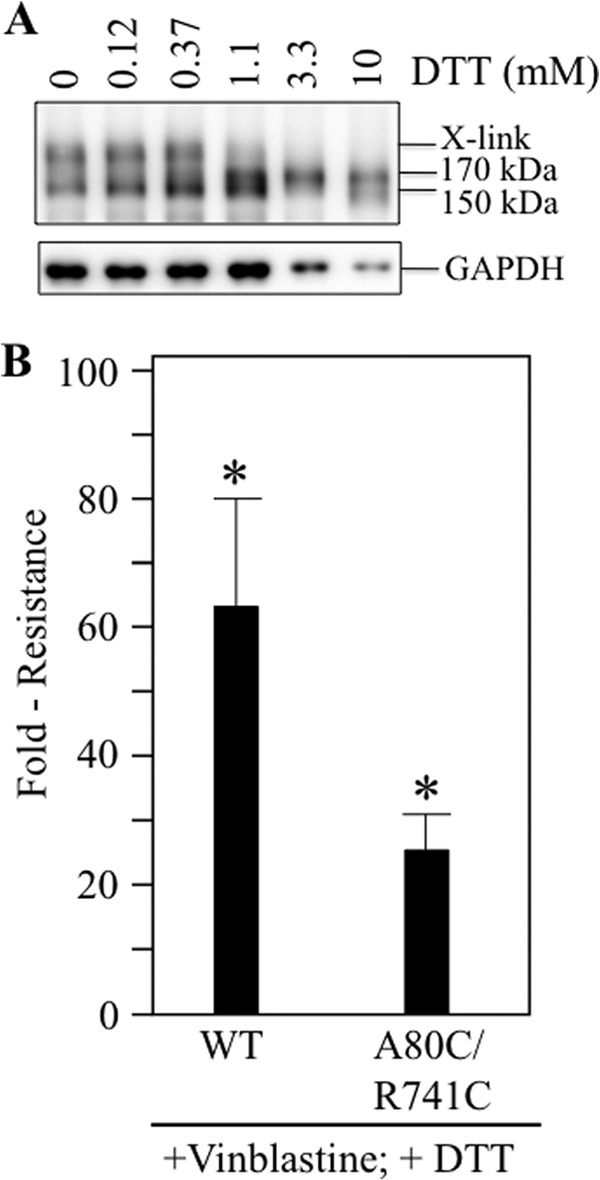
Expression of cells with DTT enhances the ability of mutant A80C/R741C to confer drug resistance. A, BHK cells expressing A52-tagged mutant A80C/R741C (in a wild-type background) were incubated for 18 h in the presence of various concentrations of DTT. Whole cell SDS extracts were subjected to immunoblot analysis. The positions of cross-linked (X-link), mature (170-kDa), and immature (150-kDa) P-gps are shown. B, BHK cells expressing A52-tagged wild-type P-gp (WT), mutant A80C/R741C, or no P-gp were incubated in the presence of various concentrations of vinblastine and 1.1 mm DTT to determine the LD50 concentration. The fold resistance represents the LD50 of the P-gp-expressing cell lines relative to cells not expressing P-gp. The results (mean ± S.D.) were obtained from three different cell lines expressing similar levels of wild-type or mutant P-gps. *, p < 0.05; Student's t test; compared with control cells.
We then tested whether mutant A80C/R741C was able to confer resistance to vinblastine when incubated in the presence of DTT. Accordingly, control BHK cells and BHK cells expressing equivalent levels of wild-type or mutant A80C/R741C P-gp were incubated in the presence of various concentrations of vinblastine and 1.1 mm DTT. Because this assay required about 6 days, it was necessary to change the medium daily because the half-life of DTT is only several hours (47) and because the reduced A80C/R741C mutant could readily reform the cross-link (Fig. 5). It was found that wild-type P-gp conferred a similar level of resistance to vinblastine in the presence of DTT (about 60-fold) (Fig. 8B), whereas mutant A80C/R741C showed an approximately 25-fold increase in the ability to confer resistance (10-fold increase compared with the absence of DTT). The results indicate that mutant A80C/R741C can confer relatively high levels of drug resistance when the disulfide bond is reduced.
Cysteines A80C/R741C Cross-link rapidly in the Absence or Presence of ATP
Spontaneous cross-linking of mutant A80C/R741C after treatment of the cells with 5 mm DTT was relatively slow because about 50% cross-linking occurred after 18 min at 37 °C (Fig. 5). During this period, P-gp would be expected to have cycled over 100 times in the absence of drug substrate (41). This suggests that ATP hydrolysis might be required for efficient cross-linking or that spontaneous cross-linking was inefficient and occurred only during a rare conformational change not associated with function.
We first tested whether ATP hydrolysis was required for spontaneous cross-linking of A80C/R741C. Accordingly, the catalytic carboxylates (Glu-556 in NBD1 and Glu-1201 in NBD2) were mutated to glutamine. These catalytic carboxylates have been shown previously to be essential for ATP hydrolysis because mutating both Glu-556 and Glu-1201 to glutamine completely abolished ATPase activity (48, 49). HEK 293 cells were then transfected with A52-tagged wild-type P-gp, mutant A80C/R741C (in a wild-type background), or A80C/R741C/E556Q/E1201Q (in a wild-type background). The cells were then suspended in SDS sample buffer with or without DTT, and the samples were subjected to immunoblot analysis. It was observed that the presence of E556Q/E1201Q mutations did not inhibit cross-linking (Fig. 9A). Both the A80C/R741C and A80C/R741C/E556Q/E1201Q mutants yielded cross-linked P-gp as the major product. The cross-linked product could be reduced by the presence of DTT so that the mature 170-kDa protein was the major product (Fig. 9A). These results indicate that ATP hydrolysis was not essential for cross-linking mutant A80C/R741C.
FIGURE 9.
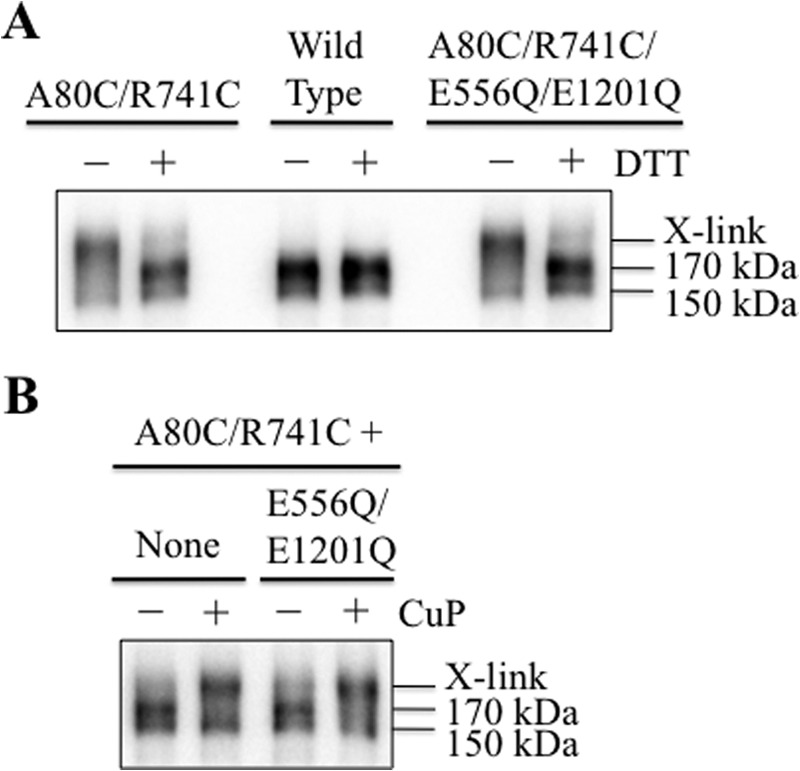
Cross-linking of mutant A80C/R741C does not require ATP hydrolysis and is rapid. A, HEK 293 cells were transfected with A52-tagged wild-type P-gp, mutants A80C/R741C (in a wild-type background), or A80C/R741C/E556Q/E1201Q. The cells were then suspended in 2× SDS sample buffer containing 50 mm EDTA and without (−) or with (+) 10 mm DTT. Samples were then subjected to immunoblot analysis. B, HEK 293 cells expressing A52-tagged mutants A80C/R741C or A80C/R741C/E556Q/E1201Q (in a wild-type background) were treated with 10 mm DTT for 3 min. The cells were washed with PBS (pH 7.4) and then incubated in the absence (−) or presence (+) of 1 mm copper phenanthroline (CuP) for 1 min at 20 °C. Samples of whole cell SDS extracts (None) were subjected to immunoblot analysis. The positions of cross-linked (X-link), mature (170-kDa), and immature (150-kDa) P-gps are shown.
We then determined whether spontaneous cross-linking of A80C/R741C and A80C/R741C/E556Q/E1201Q mutants was inefficient by testing the effect of an oxidant (copper phenanthroline) to catalyze formation of the disulfide bond at a reduced temperature. HEK 293 cells were transfected with the A52-tagged mutants A80C/R741C and A80C/R741C/E556Q/E1201Q (in a wild-type background). The cells were treated with 10 mm DTT to reduce the disulfide bond. The cells were then washed with PBS (pH 7.4) and treated with or without 1 mm copper phenanthroline for 1 min at 20 °C. The reaction was stopped by addition of SDS sample buffer containing 50 mm EDTA and no reducing agent. Samples were then subjected to immunoblot analysis. Fig. 9B shows that treatment of DTT-treated cells with oxidant almost completely cross-linked the mature 170-kDa protein in both mutants, A80C/R741C and A80C/R741C/E556Q/E1201Q, within 1 min. These results suggest that spontaneous cross-linking was inefficient and that conformational changes that bring extracellular loops 1 and 4 close together are not rare and do not require ATP hydrolysis.
Next, we tested whether ATP binding was required for cross-linking of mutant A80C/R741C. This required the use of membranes prepared from cells expressing mutant A80C/R741C in the Cys-less background. A Cys-less background was used because the endogenous Walker A cysteines (Cys-431 and Cys-1074) can be cross-linked when membranes are treated with copper phenanthroline (50). Accordingly, HEK 293 cells were transfected with the A52-tagged mutant A80C/R741C. The cells were incubated at 30 °C for 48 h to promote maturation of the mutant. The cells were then treated with 10 mm DTT to reduce the disulfide bond, washed with PBS (pH 7.4), and membranes were prepared. Samples of the membrane were then incubated in the absence or presence of ATP and an oxidant (copper phenanthroline) in an ice bath (to inhibit ATP hydrolysis) for 5 min. The reactions were stopped by addition of 2× SDS sample buffer containing 50 mm EDTA and no reducing agent. It was observed that about 50% of the mature 170-kDa protein was cross-linked in the absence or presence of ATP (Fig. 10A). These results show that ATP was not required for cross-linking.
FIGURE 10.
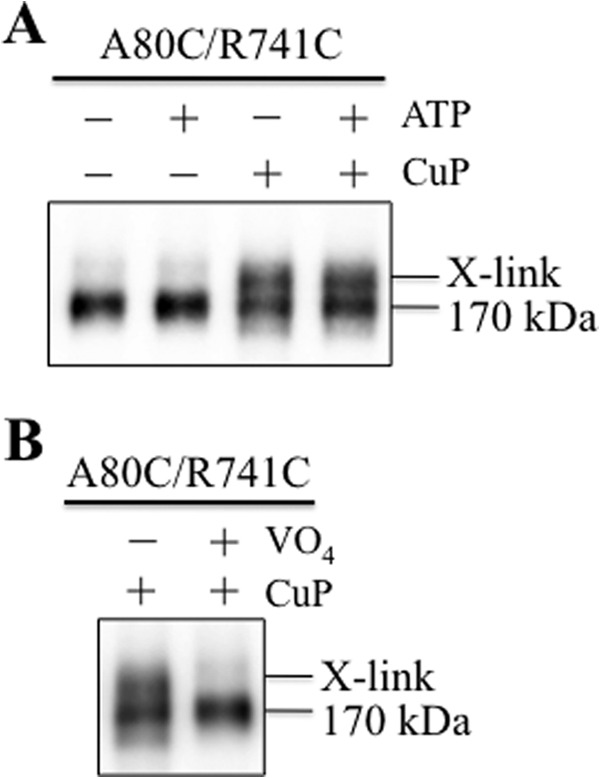
ATP is not required for cross-linking mutant A80C/R741C. A, HEK 293 cells expressing A52-tagged mutant A80C/R741C (in a Cys-less background) were treated with 10 mm DTT for 3 min. The cells were then washed with PBS (pH 7.4), and membranes were prepared. Samples of the membranes were incubated without (−) or with (+) 5 mm ATP plus 10 mm MgCl2 (ATP) in the absence (−) or presence (+) of 1 mm copper phenanthroline (CuP) for 5 min in an ice bath. The reactions were stopped by addition of 2× SDS sample buffer containing 50 mm EDTA and no reducing agent. The samples were then subjected to immunoblot analysis. B, membranes prepared above were incubated with 5 mm ATP, 10 mm MgCl2 in the absence (−) or presence (+) of 0.2 mm sodium orthovanadate (VO4) for 5 min at 20 °C. The samples were chilled in an ice bath for 5 min and then treated with (+) 1 mm copper phenanthroline (CuP) for 5 min on ice. The reactions were stopped by addition of 2× SDS sample buffer containing 50 mm EDTA and no reducing agent. The samples were then subjected to immunoblot analysis. The positions of cross-linked (X-link) and mature (170-kDa) P-gps are shown.
P-gp can be trapped in a transition state during ATP hydrolysis using vanadate (15). Vanadate traps ADP at either NBD by mimicking the transition state of the γ-phosphate of ATP during ATP hydrolysis. Therefore, we tested whether vanadate trapping of nucleotides could inhibit cross-linking of mutant A80C/R741C. Membranes were prepared from HEK 293 cells expressing A52-tagged A80C/R741C that were treated with DTT. The membranes were incubated for 5 min at 20 °C in the presence of 5 mm ATP, 10 mm MgCl2, and 0.2 mm sodium orthovanadate; chilled in an ice-bath for 5 min; and then treated with 1 mm copper phenanthroline for 5 min. The reactions were stopped by addition of 2× SDS sample buffer containing no thiol-reducing agent. Samples were then subjected to immunoblot analysis. Fig. 10B shows that vanadate trapping of nucleotides inhibits cross-linking of mutant A80C/R741C.
DISCUSSION
We found that linking the P-gp halves by direct cross-linking of extracellular cysteines A80C/R741C inhibited ATPase activity. By contrast, cross-linking of the intracellular cysteines resulted in stimulation of ATPase activity. Direct cross-linking of the cysteines L175C and N820C (23) or cross-linking of mutant P571C/I1050C with the short 1,4-butanediyl bismethanethiosulfonate cross-linker (7.8 Å) (24) highly activated P-gp ATPase activity over 10-fold in the absence of drug substrates. Cysteines L175C/N820C are located in the intracellular loops (Fig. 1, C and D), whereas cysteines P517C/I1050C are located in the NBDs (Fig. 1, C and D).
An explanation for the difference is that linkage of the homologous halves at the L175C/N820C or P517C/I1050C sites would tend to favor the closed conformation (Fig. 1C) to bring the NBDs close together. Two ATP molecules bind at the interface between the NBDs, and ATP hydrolysis occurs by an alternating site mechanism (11–15). In the closed conformation, the NBDs may come together more frequently to align the ATP γ-phosphate and hydrolytic water within the Walker B, LSGGQ, Q-loop, D-loop, and H-loop network for ATP hydrolysis (51).
Direct linkage of A80C and R741C would favor the open conformation (Fig. 1B). In the open conformation, ATPase activity would be reduced because the NBDs would come together less frequently. Our studies show that P-gp can adopt the open conformation in intact cells. It is likely that P-gp can exist in both open and closed conformations in cells because double electron-electron resonance spectroscopy studies (52) and molecular dynamics simulations (52, 53) of mouse P-gp show that the NBDs could adopt a wide range separation distances of at least 20 Å between the open and closed states. The wide range of conformations was observed in both the presence and absence of Mg-ATP and when P-gp was incorporated into lipid. In agreement with the mouse P-gp studies, we found that ATP did not cause any detectable changes in A80C/R741C cross-linking efficiency (Fig. 10).
The closed conformation was also observed in EM projection images (27). It has been reported that in the absence of nucleotide or substrate, a central chamber separated the two halves of lipid-bound P-gp. Addition of nucleotides and/or substrate closed the central cavity (54). Mouse P-gp has also been found to undergo spontaneous cross-linking when cysteines were placed at the C-terminal ends of the NBDs (55). The cross-linked mutant exhibited low drug-stimulated ATPase activity that suggested that the C-terminal ends of the two NBDs did not undergo a significant conformational change during ATP hydrolysis.
A clinical goal has been to devise strategies to turn off P-gp during chemotherapy to improve treatment (56). Inhibition of P-gp by targeting extracellular regions would be an attractive target because it would avoid the complications of requiring the inhibitor to diffuse into the cell that could possibly interfere with other metabolic pathways. Therefore, it may be possible to inhibit P-gp by trapping it at an intermediate step in the catalytic cycle using an extracellular target.
One approach to inhibit P-gp from outside of the cell has been to use inhibitory antibodies such as MRK16 (57) and UIC2 (58). The mechanisms of P-gp inhibition by MRK16 and UIC2 show some differences (59). It has been found that the UIC2 interaction with P-gp was influenced by the presence of drug substrates, whereas that of MRK16 was not. ATP hydrolysis has also been reported to influence UIC2 interactions with P-gp because ATP-depleting agents or mutational inactivation of both NBDs increased its reactivity. The characteristics of UIC2 interactions with P-gp suggested that it trapped P-gp in a transient conformation of the catalytic cycle (59).
Another study, in which P-gp was incorporated into lipid nanodiscs, suggested that the mechanisms of MRK16 and UIC2 inhibition were similar and did not involve capture of a specific conformation (60). It has been suggested that UIC2 or MRK16 did not trap P-gp in a specific conformation because they inhibited drug efflux but not ATPase activity. Drug substrates still activated P-gp ATPase activity in the presence of either antibody. Instead, the antibodies were predicted to uncouple ATPase activity from drug efflux.
Inhibition of P-gp by cross-linking of A80C/R741C was different from antibody inhibition because both ATPase activity and drug efflux were inhibited. Cross-linking likely traps P-gp in an open conformation of the catalytic cycle. Therefore, the extracellular domain of P-gp is an attractive target to develop inhibitors. It appears that P-gp activity can be blocked by multiple mechanisms involving extracellular loops.
This work was supported by a grant from the Canadian Institutes of Health Research (to D. M. C.).
T. W. Loo and D. M. Clarke, unpublished observations.
- P-gp
- P-glycoprotein
- TM
- transmembrane
- TMD
- transmembrane domain
- NBD
- nucleotide-binding domain
- ABC
- ATP-binding cassette
- ECL
- extracellular loop
- BHK
- baby hamster kidney.
REFERENCES
- 1. Ambudkar S. V., Dey S., Hrycyna C. A., Ramachandra M., Pastan I., Gottesman M. M. (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39, 361–398 [DOI] [PubMed] [Google Scholar]
- 2. Eckford P. D., Sharom F. J. (2009) ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 109, 2989–3011 [DOI] [PubMed] [Google Scholar]
- 3. Cordon-Cardo C., O'Brien J. P., Casals D., Rittman-Grauer L., Biedler J. L., Melamed M. R., Bertino J. R. (1989) Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. U.S.A. 86, 695–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim R. B., Fromm M. F., Wandel C., Leake B., Wood A. J., Roden D. M., Wilkinson G. R. (1998) The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J. Clin. Invest. 101, 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller D. S., Bauer B., Hartz A. M. (2008) Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 60, 196–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen C. J., Chin J. E., Ueda K., Clark D. P., Pastan I., Gottesman M. M., Roninson I. B. (1986) Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 47, 381–389 [DOI] [PubMed] [Google Scholar]
- 7. Globisch C., Pajeva I. K., Wiese M. (2008) Identification of putative binding sites of P-glycoprotein based on its homology model. Chem. Med. Chem. 3, 280–295 [DOI] [PubMed] [Google Scholar]
- 8. Jin M. S., Oldham M. L., Zhang Q., Chen J. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J., Jaimes K. F., Aller S. G. (2014) Refined structures of mouse P-glycoprotein. Protein Sci. 23, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 11. Loo T. W., Clarke D. M. (1995) Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug- stimulated ATPase activity. J. Biol. Chem. 270, 22957–22961 [DOI] [PubMed] [Google Scholar]
- 12. Sauna Z. E., Kim I. W., Nandigama K., Kopp S., Chiba P., Ambudkar S. V. (2007) Catalytic cycle of ATP hydrolysis by P-glycoprotein: evidence for formation of the E.S reaction intermediate with ATP-γ-S, a nonhydrolyzable analogue of ATP. Biochemistry 46, 13787–13799 [DOI] [PubMed] [Google Scholar]
- 13. Siarheyeva A., Liu R., Sharom F. J. (2010) Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: implications for catalysis. J. Biol. Chem. 285, 7575–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Delannoy S., Urbatsch I. L., Tombline G., Senior A. E., Vogel P. D. (2005) Nucleotide binding to the multidrug resistance P-glycoprotein as studied by ESR spectroscopy. Biochemistry 44, 14010–14019 [DOI] [PubMed] [Google Scholar]
- 15. Urbatsch I. L., Sankaran B., Weber J., Senior A. E. (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 270, 19383–19390 [DOI] [PubMed] [Google Scholar]
- 16. Loo T. W., Clarke D. M. (2001) Cross-linking of human multidrug resistance P-glycoprotein by the substrate, Tris-(2-maleimidoethyl)amine, is altered by ATP hydrolysis: evidence for rotation of a transmembrane helix. J. Biol. Chem. 276, 31800–31805 [DOI] [PubMed] [Google Scholar]
- 17. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein: direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 278, 13603–13606 [DOI] [PubMed] [Google Scholar]
- 18. Dey S., Ramachandra M., Pastan I., Gottesman M. M., Ambudkar S. V. (1997) Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 94, 10594–10599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lugo M. R., Sharom F. J. (2005) Interaction of LDS-751 and rhodamine 123 with P-glycoprotein: evidence for simultaneous binding of both drugs. Biochemistry 44, 14020–14029 [DOI] [PubMed] [Google Scholar]
- 20. Loo T. W., Bartlett M. C., Clarke D. M. (2009) Identification of residues in the drug-translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis. J. Biol. Chem. 284, 24074–24087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 278, 39706–39710 [DOI] [PubMed] [Google Scholar]
- 22. Gutmann D. A., Ward A., Urbatsch I. L., Chang G., van Veen H. W. (2010) Understanding polyspecificity of multidrug ABC transporters: closing in on the gaps in ABCB1. Trends Biochem. Sci. 35, 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loo T. W., Clarke D. M. (2014) Identification of the distance between the homologous halves of P-glycoprotein that Triggers the high/low ATPase activity switch. J. Biol. Chem. 289, 8484–9492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loo T. W., Bartlett M. C., Detty M. R., Clarke D. M. (2012) The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J. Biol. Chem. 287, 26806–26816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loo T. W., Bartlett M. C., Clarke D. M. (2010) Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochem. Biophys. Res. Commun. 395, 436–440 [DOI] [PubMed] [Google Scholar]
- 26. Gottesman M. M., Ambudkar S. V., Xia D. (2009) Structure of a multidrug transporter. Nat. Biotechnol. 27, 546–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee J. Y., Urbatsch I. L., Senior A. E., Wilkens S. (2002) Projection structure of P-glycoprotein by electron microscopy: evidence for a closed conformation of the nucleotide binding domains. J. Biol. Chem. 277, 40125–40131 [DOI] [PubMed] [Google Scholar]
- 28. Jones P. M., George A. M. (2013) Mechanism of the ABC transporter ATPase domains: catalytic models and the biochemical and biophysical record. Crit. Rev. Biochem. Mol. Biol. 48, 39–50 [DOI] [PubMed] [Google Scholar]
- 29. Loo T. W., Clarke D. M. (1995) Rapid purification of human P-glycoprotein mutants expressed transiently in HEK 293 cells by nickel-chelate chromatography and characterization of their drug-stimulated ATPase activities. J. Biol. Chem. 270, 21449–21452 [DOI] [PubMed] [Google Scholar]
- 30. Loo T. W., Bartlett M. C., Clarke D. M. (2008) Processing mutations disrupt interactions between the nucleotide binding and transmembrane domains of P-glycoprotein and the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 283, 28190–28197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loo T. W., Clarke D. M. (1995) P-glycoprotein: associations between domains and between domains and molecular chaperones. J. Biol. Chem. 270, 21839–21844 [DOI] [PubMed] [Google Scholar]
- 32. Chifflet S., Torriglia A., Chiesa R., Tolosa S. (1988) A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases. Anal. Biochem. 168, 1–4 [DOI] [PubMed] [Google Scholar]
- 33. Loo T. W., Bartlett M. C., Clarke D. M. (2005) Rescue of ΔF508 and other misprocessed CFTR mutants by a novel quinazoline compound. Mol. Pharm. 2, 407–413 [DOI] [PubMed] [Google Scholar]
- 34. Loo T. W., Bartlett M. C., Clarke D. M. (2005) The dileucine motif at the COOH terminus of human multidrug resistance P-glycoprotein is important for folding but not activity. J. Biol. Chem. 280, 2522–2528 [DOI] [PubMed] [Google Scholar]
- 35. Sowdhamini R., Srinivasan N., Shoichet B., Santi D. V., Ramakrishnan C., Balaram P. (1989) Stereochemical modeling of disulfide bridges: criteria for introduction into proteins by site-directed mutagenesis. Protein Eng. 3, 95–103 [DOI] [PubMed] [Google Scholar]
- 36. Loo T. W., Clarke D. M. (1995) Membrane topology of a cysteine-less mutant of human P-glycoprotein. J. Biol. Chem. 270, 843–848 [DOI] [PubMed] [Google Scholar]
- 37. Loo T. W., Clarke D. M. (2014) Locking intracellular helices 2 and 3 together inactivates human P-glycoprotein. J. Biol. Chem. 289, 229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loo T. W., Clarke D. M. (2013) Drug rescue distinguishes between different structural models of human P-glycoprotein. Biochemistry 52, 7167–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sevier C. S., Kaiser C. A. (2002) Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol. 3, 836–847 [DOI] [PubMed] [Google Scholar]
- 40. Loo T. W., Bartlett M. C., Clarke D. M. (2013) Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharmacol. 86, 612–619 [DOI] [PubMed] [Google Scholar]
- 41. Ambudkar S. V., Cardarelli C. O., Pashinsky I., Stein W. D. (1997) Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein. J. Biol. Chem. 272, 21160–21166 [DOI] [PubMed] [Google Scholar]
- 42. Gannon M. K., 2nd, Holt J. J., Bennett S. M., Wetzel B. R., Loo T. W., Bartlett M. C., Clarke D. M., Sawada G. A., Higgins J. W., Tombline G., Raub T. J., Detty M. R. (2009) Rhodamine inhibitors of P-glycoprotein: an amide/thioamide “switch” for ATPase activity. J. Med. Chem. 52, 3328–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Omote H., Al-Shawi M. K. (2002) A novel electron paramagnetic resonance approach to determine the mechanism of drug transport by P-glycoprotein. J. Biol. Chem. 277, 45688–45694 [DOI] [PubMed] [Google Scholar]
- 44. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Methanethiosulfonate derivatives of rhodamine and verapamil activate human P-glycoprotein at different sites. J. Biol. Chem. 278, 50136–50141 [DOI] [PubMed] [Google Scholar]
- 45. Ueda K., Cardarelli C., Gottesman M. M., Pastan I. (1987) Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc. Natl. Acad. Sci. U.S.A. 84, 3004–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fass D. (2012) Disulfide bonding in protein biophysics. Annu. Rev. Biophys. 41, 63–79 [DOI] [PubMed] [Google Scholar]
- 47. Stevens R., Stevens L., Price N. C. (1983) The stabilities of various thiol compounds used in protein purifications. Biochem. Educ. 11, 70 [Google Scholar]
- 48. Tombline G., Bartholomew L. A., Tyndall G. A., Gimi K., Urbatsch I. L., Senior A. E. (2004) Properties of P-glycoprotein with mutations in the “catalytic carboxylate” glutamate residues. J. Biol. Chem. 279, 46518–46526 [DOI] [PubMed] [Google Scholar]
- 49. Loo T. W., Bartlett M. C., Clarke D. M. (2007) Nucleotide binding, ATP hydrolysis, and mutation of the catalytic carboxylates of human P-glycoprotein cause distinct conformational changes in the transmembrane segments. Biochemistry 46, 9328–9336 [DOI] [PubMed] [Google Scholar]
- 50. Loo T. W., Clarke D. M. (2000) Drug-stimulated ATPase activity of human P-glycoprotein is blocked by disulfide cross-linking between the nucleotide-binding sites. J. Biol. Chem. 275, 19435–19438 [DOI] [PubMed] [Google Scholar]
- 51. Jones P. M., George A. M. (2012) Role of the D-loops in allosteric control of ATP hydrolysis in an ABC transporter. J. Phys. Chem. A 116, 3004–3013 [DOI] [PubMed] [Google Scholar]
- 52. Wen P. C., Verhalen B., Wilkens S., Mchaourab H. S., Tajkhorshid E. (2013) On the origin of large flexibility of P-glycoprotein in the inward-facing state. J. Biol. Chem. 288, 19211–19220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wise J. G. (2012) Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry 51, 5125–5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee J. Y., Urbatsch I. L., Senior A. E., Wilkens S. (2008) Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J. Biol. Chem. 283, 5769–5779 [DOI] [PubMed] [Google Scholar]
- 55. Verhalen B., Wilkens S. (2011) P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. J. Biol. Chem. 286, 10476–10482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Szakács G., Paterson J. K., Ludwig J. A., Booth-Genthe C., Gottesman M. M. (2006) Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 5, 219–234 [DOI] [PubMed] [Google Scholar]
- 57. Mickisch G. H., Pai L. H., Gottesman M. M., Pastan I. (1992) Monoclonal antibody MRK16 reverses the multidrug resistance of multidrug-resistant transgenic mice. Cancer Res. 52, 4427–4432 [PubMed] [Google Scholar]
- 58. Mechetner E. B., Roninson I. B. (1992) Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc. Natl. Acad. Sci. U.S.A. 89, 5824–5828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mechetner E. B., Schott B., Morse B. S., Stein W. D., Druley T., Davis K. A., Tsuruo T., Roninson I. B. (1997) P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc. Natl. Acad. Sci. U.S.A. 94, 12908–12913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ritchie T. K., Kwon H., Atkins W. M. (2011) Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J. Biol. Chem. 286, 39489–39496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. DeLano W. L. (2002) The PyMOL Molecular Graphics System, MacPyMoL version 1.3, DeLano Scientific LLC, San Carlos, CA [Google Scholar]