

Background: Ihh is required for chondrocyte differentiation with redundant functions on multiple differentiation steps.
Results: Ihh induces collagen type X expression and promotes its transcription through Gli1/2 cooperating with Runx2/Smads on a specific promoter region.
Conclusion: Ihh signaling plays an important role in Col X expression and mineralization.
Significance: This is the first detailed description of the molecular mechanism by which Ihh signaling controls late chondrocyte differentiation.
Keywords: Calcification, Chondrocyte, Differentiation, Hedgehog Signaling Pathway, Transcription, Col10a1, Gli1/Gli2-responsive Element, Runx2, Smads, Maturation
Abstract
Indian hedgehog (Ihh) is essential for chondrocyte differentiation and endochondral ossification and acts with parathyroid hormone-related peptide in a negative feedback loop to regulate early chondrocyte differentiation and entry to hypertrophic differentiation. Independent of this function, we and others recently reported independent Ihh functions to promote chondrocyte hypertrophy and matrix mineralization in vivo and in vitro. However, the molecular mechanisms for these actions and their functional significance are still unknown. We recently discovered that Ihh overexpression in chondrocytes stimulated the expression of late chondrocyte differentiation markers and induced matrix mineralization. Focusing on collagen type X (Col10α1) expression and transcription, we observed that hedgehog downstream transcription factors GLI-Krüppel family members (Gli) 1/2 increased COL10A1 promoter activity and identified a novel Gli1/2 response element in the 250-bp basic promoter. In addition, we found that Ihh induced Runx2 expression in chondrocytes without up-regulating other modulators of chondrocyte maturation such as Mef2c, Foxa2, and Foxa3. Runx2 promoted Col10α1 expression in cooperation with Ihh. Further analyses using promoter assays, immunofluorescence, and binding assays showed the interaction of Gli1/2 in a complex with Runx2/Smads induces chondrocyte differentiation. Finally, we could demonstrate that Ihh promotes in vitro matrix mineralization using similar molecular mechanisms. Our data provide an in vitro mechanism for Ihh signaling to positively regulate Col10α1 transcription. Thus, Ihh signaling could be an important player for not only early chondrocyte differentiation but maturation and calcification of chondrocytes.
Introduction
The primary function of the vertebrate skeleton is to provide strength, support, and protection. The skeleton begins forming during embryogenesis and continues to develop after birth to accommodate postnatal growth. Most of the skeleton forms through endochondral ossification, a process in which a cartilage anlage produced by chondrocytes forms a template that is then replaced by bone. Chondrocytes proliferate within the growth plate and differentiate into large hypertrophic cells. Eventually, the fully differentiated hypertrophic chondrocytes die and are replaced by osteoblasts that produce mineralized bone to replace the cartilaginous template. The process by which chondrocytes pass from the proliferative to the hypertrophic stage is tightly regulated by many factors that ensure the proper development and maintenance of the growth plate to produce the final skeletal elements (1, 2).
Indian hedgehog (Ihh)2 is a well known marker for prehypertrophic chondrocytes. Mutations in the human gene are reported to cause brachydactyly type A-1 and acrocapitofemoral dysplasia (3, 4). Our laboratory previously established the existence of an Ihh/parathyroid hormone-related peptide (PTHrP) negative feedback loop that regulates the rate of chondrocyte differentiation (5, 6). It has been suggested that Ihh can also act in a PTHrP-independent manner (7–9). We and others have recently demonstrated PTHrP-independent roles for Ihh in promoting chondrocyte hypertrophy and calcification (10, 11). However, a potential role of Ihh in extracellular matrix maturation after chondrocytes have progressed to the hypertrophic stage has not yet been established.
Hypertrophic chondrocytes express collagen type X (Col10α1), which is a required precursor to matrix mineralization. Runt-related transcription factor 2 (Runx2) was found to regulate expression of Col10α1 and matrix calcification in chondrocytes (12–15). In addition to Runx2, Col10α1 expression is also activated by myocyte enhancer factor-2c (Mef2c) through several conserved sequences in the promoter resembling MEF2-binding sites, which are antagonized by a co-repressor histone deacetylase 4 (Hdac4) (16, 17). Recent investigation using the chick Col Xb2 enhancer identified FoxA factors as additional regulators of hypertrophic chondrocyte (18). Interestingly, we have previously found that Ihh overexpression by adenoviruses in chondrocytes induced Col10α1 expression in vitro. However, the molecular mechanism used by Ihh to promote expression and transcription of Col10α1 had not been determined. In this study, we investigated how Ihh signaling acts on the expression and transcription of Col10α1 in chondrocytes. We can now demonstrate that hedgehog signal transcription factors Gli1/2 increase the COL10A1 promoter activity and directly interact with the COL10A1 basic promoter through a newly identified Gli-binding site. In addition, we found that there is cross-talk between Ihh signaling and Runx2/Smads in regulating Col10α1 expression and that Ihh stimulates in vitro matrix mineralization of primary chondrocytes utilizing the same mechanisms. These data reveal the molecular mechanisms of Ihh signaling in chondrocytes that directly regulate transcription and expression of Col10α1 and, thus, chondrocyte calcification.
EXPERIMENTAL PROCEDURES
Cell Cultures and Reagents
ATDC5 cells were cultured in DMEM/F-12 supplemented with 10% FBS and 1% penicillin/streptomycin solution. HEK293A, COS7, C3H10T1/2, and HTB94 human chondrosarcoma cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. All cells were maintained in a 37 °C, 5% CO2 incubator. Primary chondrocytes were isolated from the ribs of P3 pups as described previously (10). After the confluence in pre-culture in 10-cm dishes, primary chondrocytes were seeded on collagen-coated plates (StemCell Technologies) for subsequent experiments. Media were changed every day and infected with adenoviruses as described previously (10). For the mineralization assay, cultured primary chondrocytes were stimulated with 5 mm β-glycerophosphate (Sigma) starting on the 5th day. Upon visualization of calcification nodules, samples were subjected to Alizarin red staining.
Cyclopamine was purchased from Enzo Life Sciences and used at 5 μm. Noggin was purchased from R&D Systems and used at 100 ng/ml. Dorsomorphin was purchased from EMD Millipore and used at 5 μm. Ihh N-terminal protein was purchased from R&D Systems and used at 4 μg/ml.
Adenoviral Production and Infection
Construction of adenovirus for GFP, Ihh, Runx2, and a dominant-negative form of Runx2 was described previously (10, 24, 25, 45). We amplified viruses using HEK293A cells and purified them through repeated freeze and thaw. ATDC5 cells or pre-cultured primary rib chondrocytes were seeded, and media were then replaced by one containing each virus the next day. Virus infection was performed for 24 h as described previously (10). A virus carrying GFP was used as control in all experiments.
Quantitative RT-PCR Analysis
Total RNA was isolated from cultured cells using an RNeasy mini kit (Qiagen) according to the manufacturer's protocol. cDNA was synthesized using the QuantiTect reverse transcription kit (Qiagen). A reaction mixture was prepared using SYBR Green master mix (Roche Applied Science), following manufacturer instructions. Samples were then subjected to real time PCR analysis using an iCycler (Bio-Rad). The specific primers for gene expression analysis are listed in Table 1. Data calculated through the 2−ΔΔCT method are presented as fold change relative to control samples (n = 3 or 4).
TABLE 1.
Sequences of primers used in this study
| Genes | Forward primers | Reverse primers |
|---|---|---|
| GAPDH | ACTGAGGACCAGGTTGTC | TGCTGTAGCCGTATTCATTG |
| Gli1 | CCAAGCCAACTTTATGTCAGGG | AGCCCGCTTCTTTGTTAATTTGA |
| Gli2 | CATGGTATCCCTAGCTCCTC | GATGGCATCAAAGTCAATCT |
| Ptch1 | GGAAGGGGCAAAGCTACAGT | TCCACCGTAAAGGAGGCTTA |
| Col10a1 | TTCTGCTGCTAATGTTCTTGACC | GGGATGAAGTATTGTGTCTTGGG |
| ALP | CACGGCCATCCTATATGGTAA | GGGCCTGGTAGTTGTTGTGA |
| MMP13 | TGATGAAACCTGGACAAGCA | TCCTCGGAGACTGGTAATGG |
| Vegfa | CCTGGTAATGGCCCCTCCTC | CCCCATTGCTCTGTGCCTTG |
| Runx2 | TCCACAAGGACAGAGTCAGATTACAG | CAGAAGTCAGAGGTGGCAGTGTCATC |
| Mef2c | ATCCCGATGCAGACGATTCAG | AACAGCACACAATCTTTGCCT |
| Foxa2 | CCCTACGCCAACATGAACTCG | GTTCTGCCGGTAGAAAGGGA |
| Foxa3 | AACCCACTCAGCTCTCCCTAC | CCTTTGCCATCTCTTTTCCAT |
Alizarin Red Staining
Mouse cultured primary chondrocytes were rinsed with PBS, fixed in 10% buffered neutral formalin, and stained with 1% Alizarin red solution (Sigma) for 10 min.
Construction of Plasmids
Full-length and deletion mutant luciferase vectors of the human COL10A1 promoter were constructed through ligation of fragments amplified by PCR into a pGL4.14(luc2/Hygro) vector (Promega) using KpnI/HindIII sites. Templates were kindly gifted by Dr. Klaus von der Mark (Germany). The Gli1 expression vector was constructed by insertion of the cDNA into a pcDNA3 expression vector (Invitrogen) using HindIII/XbaI sites. Myc-tagged Gli1 or Runx2 was constructed by insertion of the cDNA into a pcDNA3 vector using EcoRI(MfeI)/XbaI site. Myc-tagged Gli2 was purchased from Addgene (catalog no. 17648). HA-tagged Sox9, FLAG-tagged Smad1, and FLAG-tagged Smad4 were kindly provided by Dr. Riko Nishimura (Osaka University, Japan). Site-directed mutagenesis to insert point mutations into the 250-bp COL10A1 luciferase vector was performed using the kit purchased from Agilent Technologies according to the manufacturer's protocol. The inserted mutations were confirmed by sequencing.
Transfection of Plasmids
PolyJet in vitro DNA transfection reagent (SignaGen Laboratories) was used for transient transfections. Transfection was performed according to the manufacturer's instructions using a ratio of reagent/DNA of 3:1.
Western Blotting
Preparation for cell lysates and Western blotting was performed as described previously (46). Primary antibodies were used with the following concentrations: rabbit or goat anti-Runx2 antibody (M-70 or S-19, 1:500 dilution, Santa Cruz Biotechnology, Inc.); mouse anti-Runx2 antibody (1:500 dilution, MBL International Corp.); rabbit anti-Col X antibody (1:200 dilution, Abcam); rabbit anti-Mef2c antibody (1:500 dilution, Cell Signaling); mouse anti-β-actin antibody (1:2000 dilution, Sigma); mouse anti-Myc antibody (9E10, 1:500 dilution, Santa Cruz Biotechnology, Inc.); mouse anti-FLAG antibody (M2, 1:1000 dilution, Sigma); and rabbit anti-Gli1 antibody (H-300, 1:80 dilution, Santa Cruz Biotechnology, Inc.).
Immunoprecipitation
Cell lysates were incubated with an anti-FLAG antibody (M2, 2 μg in 1 ml of lysate) overnight and then reacted with protein-G agarose beads (20 μl in 1 ml of lysate, Santa Cruz Biotechnology, Inc.) for 2 h. After three washes with lysis buffer, the precipitated samples were boiled in 20 μl of 2× SDS sample buffer for 5 min and subjected to Western blotting with anti-Myc antibody (9E10).
Immunofluorescence
C3H10T1/2 cells were transfected with expression vectors as indicated in the figures. The following day, the cells were treated with or without 100 ng/ml BMP2 for 24 h. Subsequently, the cells were rinsed by PBS, fixed with 10% buffered neutral formalin, and incubated in blocking solution containing 5% goat serum, 2% BSA, and 0.05% saponin for 1 h. The samples were reacted with primary antibodies for 3 h as described: mouse anti-Myc antibody (9E10, 1:100 dilution, Santa Cruz Biotechnology, Inc.), rabbit anti-phospho-Smad1/5/8 (1:200 dilution, Cell Signaling). After several washes with PBS, the cells were incubated with the following secondary antibodies for 1 h: Alexa Fluor 488 or 568 F(ab′)2 fragment of goat anti-mouse IgG and Alexa Fluor 488 F(ab′)2 fragment of goat anti-rabbit IgG (Invitrogen). Cell nucleus was stained by incubation with DAPI reagent (Invitrogen) for 30 min. Then the slides were mounted using Fluoromount-G (Southern Biotech) and observed with a confocal microscope (Olympus FV1000). ATDC5 cells or primary chondrocytes were also stained as above using the following primary antibodies: mouse anti-Gli2 antibody (C-10, 1:40 dilution, Santa Cruz Biotechnology, Inc.) and rabbit anti-FoxA2 antibody (1:50 dilution, Cell Signaling). More than five cells were recorded, and the data in the figures are representative of a typical observation.
Oligo Pulldown Assay
Protein lysates were prepared from HEK293A transfected with the expression vectors as indicated in each figure. Lysates were preincubated with streptavidin-agarose (Calbiochem, Thermo Scientific) for 1 h and 30 min and incubated with the biotinylated double-stranded DNA oligonucleotide that encodes the human COL10A1 basic promoter for 3 h. Three probes of 10, 29, and 65 bp in length were used as documented in each figure, and the sequences are described in Fig. 2D. Competition was performed in the presence of a 40-fold amount of nonbiotinylated oligonucleotide. Subsequently, lysates were incubated with streptavidin-agarose for 1 h. After three washes with lysis buffer, the precipitated samples containing streptavidin-agarose were boiled in 40 μl of 2× SDS sample buffer and subjected to Western blotting.
FIGURE 2.

Identification of the Gli-binding site in the 250-bp basic promoter of COL10A1. A, 250- or 2500-bp luciferase constructs of COL10A1 promoter were transfected into ATDC5 with either empty vectors, Gli1, or Gli2 expression vectors, and the luciferase/Renilla value was measured 48 h after transfection. **, p < 0.01; *, p < 0.05 (versus control vector) (n = 3). B, 250-bp luciferase constructs of COL10A1 promoter were transfected into ATDC5 with either empty vectors or different amounts of Gli2 expression vectors, and the luciferase/Renilla value was measured 48 h after transfection. **, p < 0.01; *, p < 0.05 (versus control vector). #, p < 0.05 (versus Gli2–0.25 ng group) (n = 3). C, 1800-bp luciferase constructs of COL10A1 promoter were used in a similar luciferase assay described in A to confirm reaction of Gli2 on COL10A1 promoter in ATDC5 cells. *, p < 0.05 (versus control vector) (n = 3). D, sequence analysis of the 250-bp basic promoter region of COL10A1. The suggested Gli-binding site in this study is highlighted in red. Previously reported Runx2-binding site is shown in blue. Conventional Smad-binding elements are indicated in green. Underlined areas show either the long probes (65 bp) or short probes (29 bp) used in the oligonucleotide pulldown assay. Arrows harbor the fragments amplified in CHIP assay. E, oligonucleotide pulldown assay using 29-bp probe of WT and mutants. F, sequence comparing the WT and mutants of suggested Gli-binding site in the COL10A1 promoter. The 29-bp probes containing either Mut1 or Mut2 were used in E, and 250-bp luciferase constructs having the insertion of point mutations as either Mut3 or Mut4 were used in H. G, oligonucleotide pulldown assay using a 10-bp probe of the suggested Gli-binding site showing interaction with Gli1. H, luciferase assay was performed in COS7 cells using either WT or mutant 250-bp COL10A1 promoter as mentioned under “Experimental Procedures.” The data were shown as fold change when compared with the increase using a WT promoter vector. **, p < 0.01 (versus WT vector) (n = 3). IB, immunoblot.
Luciferase Assay
The luciferase reporter construct for the COL10A1 gene promoter and Renilla construct were co-transfected with expression vectors into COS7, ATDC5, or C3H10T1/2 cells using PolyJet in vitro DNA transfection reagent (SignaGen Laboratories). After 48 h, cells were lysed, and the luciferase/Renilla activity was measured using a Dual-Luciferase reporter assay system on a luminometer according to the manufacturer's instructions (Promega). Renilla was used to normalize the transfection efficiency (n = 3 or 4).
Chromatin IP Experiments (CHIP)
CHIP assay was performed using an enzymatic chromatin IP kit according to manufacturer's protocols using HTB94 human chondrosarcoma cells (Cell Signaling). The following antibodies were used for the CHIP assay; rabbit anti-Gli1 antibody and rabbit anti-Runx2 antibody (Santa Cruz Biotechnology, Inc.); rabbit anti-phospho-Smad1/5/8 antibody (Cell Signaling); and mouse anti-Gli2 antibody (Santa Cruz Biotechnology, Inc.). Final products were amplified by PCR using a primer pair specific to the COL10A1 basic promoter region, forward 5′-GGTATCATCATTCCACCGTGA-3′ and reverse 5′-CCTCCCCTCAAAGTTGGTAAG-3′.
Cell Proliferation Assay
This assay was performed using WST-1 according to the manufacturer's protocol (Roche Applied Science). The cultured cells were incubated with the reagent at a 1:10 final dilution at 37 °C for 2 h, and the absorbance of the samples was measured using a microplate reader (absorbance at 450 nm, reference at 620 nm).
Statistical Analysis
Data were analyzed using the Student's t test and multiple comparisons (Tukey procedure) of one- or two-way analysis of variance. Data are presented as the mean ± S.D.
RESULTS
Indian Hedgehog Promotes Chondrocyte Maturation and Calcification in Vitro
To address the direct function of Ihh signaling in late chondrocyte differentiation, we first examined the effect of Ihh overexpression in a chondrogenic cell line, ATDC5, using an adenovirus delivery system. Reagents normally used to induce chondrocyte differentiation of ATDC5 were omitted, and the cells were simply incubated with an Ihh adenovirus in the presence or absence of a hedgehog inhibitor, cyclopamine. We observed that the cells treated with Ihh showed markedly increased expression of downstream hedgehog signaling molecules GLI-Krüppel family members Gli1, Gli2, and Patched 1 (Ptch1) (Fig. 1A). We could block expression of these molecules by adding cyclopamine, confirming the activity of the adenovirus. Similarly, immunofluorescence to detect Gli2 proteins indicated the cells treated with Ihh showed increased nuclear Gli2 expression that could be inhibited by cyclopamine (supplemental Fig. 1). Interestingly, we observed a dramatic induction of Col10α1 expression in transfected ATDC5 cells, which could also be attenuated by cyclopamine (Fig. 1A). This change in Col10α1 mRNA expression was also confirmed on the protein level using Western blotting (Fig. 1C). In addition to the use of the Ihh virus, we applied the Ihh N-terminal protein and confirmed induced expressions of hedgehog signaling molecules and Col10α1 upon treatment (Fig. 1D). Moreover, Ihh overexpression induced additional late chondrocyte differentiation markers such as alkaline phosphatase (Alp), matrix metallopeptidase 13 (Mmp13), and vascular endothelial growth factor A (Vegfa), which could all be inhibited by cyclopamine as well (Fig. 1A). Possible cytotoxic effects of cyclopamine were excluded by performing a cell proliferation assay that resulted in minor suppression of cell proliferation by cyclopamine itself, but no significant change in the group treated with both Ihh virus and cyclopamine could be detected (Fig. 1B).
FIGURE 1.

Gene expression profile induced by Ihh in chondrogenic cells. A and B, Ihh was overexpressed in ATDC5 cells with or without treatment of 5 μm cyclopamine. Isolated RNA was subjected to qPCR analysis using primers as shown in the figures (A). **, p < 0.01 (versus GFP-treated group). ##, p < 0.01 (versus Ihh-treated group) (n = 3 or 4). Cell proliferation was assessed using WST-1 reagent (B). **, p < 0.01 (versus GFP-treated group) (n = 4). C, changes in gene expression of Col10a1 is shown in A and were confirmed at the protein level using Western blotting. IB, immunoblot. D, changes in mRNA expression of hedgehog signal molecules and Col10a1 shown in A were also confirmed upon treatment of cells with Ihh protein instead of Ihh virus. (n = 3).
To further test the role of Ihh in chondrocyte calcification, we performed in vitro mineralization assays using primary chondrocytes in combination with the adenoviruses. We found that transfection with Ihh adenoviruses resulted in a marked increase in Alizarin red-positive matrix mineralization that was completely inhibited by cyclopamine (Fig. 8A). These data indicate that Ihh positively regulates maturation and calcification in chondrocytes.
FIGURE 8.

Ihh induces matrix mineralization in primary chondrocytes interacting with BMP/Runx2/Smads pathway. A–D, Ihh was overexpressed in primary chondrocytes with or without treatment by additional reagents (A, B, and D) or viruses (C) as shown in the figures, and an in vitro mineralization assay was performed under stimulation by β-glycerophosphate and assessed by Alizarin red staining (n = 3 or 4). E, an in vitro mineralization assay was performed in the presence with Runx2 overexpression with or without cyclopamine (n = 3 or 4). F, diagram showing how Ihh downstream signaling molecules Gli1/2 together with Runx2/Smads regulate transcription and expression of Col X.
Identification of an Ihh-responsive Element in the COL10A1 Promoter Activated by Hedgehog Downstream Signaling Molecules Gli1/2
We next aimed to identify the molecular mechanisms responsible for the strong induction of Col10α1 expression in ATDC5 cells by Ihh in vitro. Because we found that the expression of Gli1 and Gli2 was markedly increased in Ihh-treated cells, we determined whether these molecules directly regulate Col10α1 transcription by performing promoter analyses assays (19). We first constructed a vector that includes 2.5 kb of the human COL10A1 promoter. Transfection with either Gli1 or Gli2 alone increased the transcriptional activity, and co-transfection with both further enhanced this effect (Table 2). In contrast, co-transfection with Sox9, a key transcription factor for early chondrocyte differentiation, blocked Gli actions (Table 2) (20). We then proceeded to identify the Gli1/2-responsive element in the COL10A1 promoter. For this purpose, we generated various promoter deletion constructs as shown in Table 3. A 2.5-kb sequence of the COL10A1 promoter contains a reported enhancer region between −2410 and −1875 bp (19). Gli1 increased promoter activity with the h1800- and h2500-bp constructs, but not with the h900-bp construct alone or ligated with the distal enhancer region in COS7 cells (Table 3). Interestingly, we found the opposite effect with Gli2. These results would suggest that the distal enhancer region does not contain the response element for Gli; however, both Gli1 and Gli2 induced a high response when transfected with the basic 250-bp promoter segment in both COS7 (Table 3) and ATDC5 chondrogenic cells (Fig. 2A), suggesting that Gli1/2 interacts with this region of the COL10A1 promoter. The dose-dependent effect of Gli2 on this basic promoter in ATDC5 cells was confirmed using different amounts of Gli2 vector (Fig. 2B). In addition, Gli2 could increase the activity of h1800-bp COL10A1 promoter in ATDC5 (Fig. 2C).
TABLE 2.
Transcriptional activity of COL10A1 promoter stimulated by Gli factors
Human COL10A1 promoter construct (2.5 kb) was transfected into COS7 cells with or without transcription factors as described in the table, and the luciferase/Renilla value was measured 48 h after transfection. Gli1 or Gli2 alone increased the activity, and co-transfection of both Gli proteins further up-regulated it. In contrast, Sox9 inhibited COL10A1 promoter activity. n = 4 or 8.
| Expression vectors | Average fold changes of luciferase/Renilla | S.D. | Statistics |
|---|---|---|---|
| Empty vector | 1.00 | 0.12 | |
| Gli1 | 1.45 | 0.24 | a |
| Gli2 | 1.98 | 0.36 | a |
| Gli1+Gli2 | 3.31 | 0.33 | b |
| Sox9 | 0.60 | 0.01 | a |
| Gli1+Sox9 | 0.61 | 0.02 | b |
| Gli2+Sox9 | 0.85 | 0.04 | b |
a p < 0.01 when compared with the empty vector.
b p < 0.01 when compared with Gli1 or Gli2 alone.
TABLE 3.
Promoter activity of COL10A1 deletion mutants stimulated by Gli factors
Average fold changes of luciferase/Renilla when compared with transfection with only empty vector are described. Deletion mutant constructs of COL10A1 luciferase reporter were transfected into COS7 cells with either empty vectors, Gli1, or Gli2 expression vectors, and the luciferase/Renilla value was measured 48 h after transfection. The minimum 250-bp constructs show response to Gli factors. There was no significant change between h250 and h2500 in Gli1 group. n = 4 or 8.
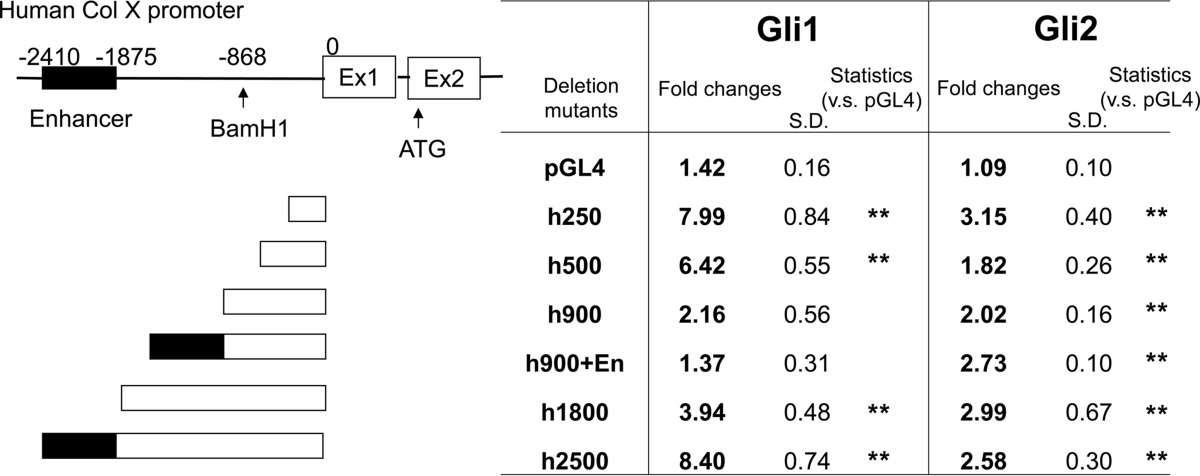
**, p < 0.01.
An analysis of the sequence of this region led to the discovery of a short sequence (GCCTACCCA) (Fig. 2D) that closely resembles the conventional Gli-binding site (GACCACCCA) (21). Our analysis also revealed a Runx2-binding site (TGAGGG) and three Smad-binding elements (GTCT/AGAC) in this region (Fig. 2D) (12, 22). Oligo pulldown assays were then performed to confirm specific Gli1/2 binding to this region. We first designed 29-bp short probes containing this binding site and found that Gli1 exhibited strong binding with a biotin-labeled probe that could be markedly decreased by the addition of the nonlabeled probe (Fig. 2E, lanes 2 and 3). Furthermore, mutations (Mut1 and Mut2) introduced into this site (Fig. 2F) efficiently prevented binding (Fig. 2E, lanes 4 and 5). In addition, Gli1 exhibited strong binding with a 10-bp biotin-labeled probe representing this binding site, which could be modulated by competition with a nonlabeled oligonucleotide (Fig. 2G). Similarly, point mutations inserted into this suggested Gli-binding site (Fig. 2F) reduced the activity of the h250-bp COL10A1 promoter in COS7 cells (Fig. 2H). We also performed CHIP assays, which indicated endogenous interaction between Gli1/2 and the basic promoter (Fig. 7G). Taken together, these results demonstrate that Gli1/2 directly regulate COL10A1 transcriptional activity by binding to this 250-bp region of the basic promoter.
FIGURE 7.

Involvement of Smads in Col X regulation conducted by hedgehog signal. A, 2.5-kb luciferase construct of the COL10A1 promoter was transfected into COS7 with expression vectors as indicated in the figure, and the luciferase/Renilla value was measured after 48 h. **, p < 0.01 (versus control). ##, p < 0.01 (versus Gli1 or Smad1/4) (n = 3). B, 2.5-kb luciferase construct of COL10A1 promoter was transfected into C3H10T1/2 cells with expression vectors as indicated followed by 100 ng/ml recombinant BMP2, and the luciferase/Renilla value was measured after 48 h. *, p < 0.05 (versus control). #, p < 0.05 (versus Gli2 or Smad1) (n = 3). C and D, physical interaction between Gli1/2 and Smad1 was assessed by co-IP. Samples were immunoprecipitated (IP) by FLAG and immunoblotted (IB) by Myc. E and F, physical interaction between Gli1/2, Runx2, and Smad1 was assessed by co-IP in which the samples were immunoprecipitated by FLAG and immunoblotted by Myc. Simultaneous interaction of Gli1/Runx2/Smad1 or Gli2/Runx2/Smad1 was seen in the last lane of E or F. G, CHIP on COL10A1 promoter was performed using HTB94 human chondrosarcoma cells. Sonicated and purified chromatin was incubated with antibodies as shown in the figure. Endogenous binding of Gli, Runx2, and pSmads was detected in the COL10A1 basic promoter region. An α-histone antibody was used as a positive control, and rabbit or mouse IgGs were used as a negative control. The detection was confirmed in three independent experiments. The representative data are shown here.
Ihh Signaling Interacts with Runx2/Smads to Regulate Col10α1 Transcription and Calcification
We next addressed the molecular mechanisms for the regulation of Col10α1 expression and transcription by Ihh signaling. Ihh overexpression in mouse primary chondrocytes induced expression of both Col10α1 and a key hypertrophic factor, Runx2, at the mRNA and protein levels (Fig. 3, A and C). In addition to Runx2/3, previous studies have shown that chondrocyte hypertrophy is also regulated by Mef2c/d and Foxa2/3 (16, 18, 23). However, our data show that Ihh did not induce Mef2c, Foxa2, or Foxa3 mRNA expression (Fig. 3B). By Western blotting, we observed that Ihh down-regulated Mef2c protein expression (Fig. 3C). Immunofluorescence assays showed that a few but not all primary chondrocytes expressed Foxa2 protein in their nucleus in the control group (Fig. 3D). Similar results were obtained in the Ihh-treated group (Fig. 3D). The fact that a Runx2-binding site was found close to the identified Gli-binding site and the induction of Runx2 expression by Ihh encouraged us to investigate how Ihh and Runx2 interact to regulate Col10α1 expression. We then performed in vitro experiments in which we treated chondrogenic cells with adenoviruses carrying Runx2 or a dominant-negative form of Runx2 (Fig. 4). DN-Runx2 is an N-terminal fragment of Runx2 that is capable of DNA binding but has no transcriptional activity (24, 25). We found that Ihh and Runx2 synergize to induce Col10α1 expression in ATDC5 cells (Fig. 4, A and B), whereas DN-Runx2 suppresses Ihh-induced Col10α1 expression (Fig. 4, C and D). After demonstrating synergy between Ihh and Runx2, we then determined whether Ihh can act as a mediator of the Runx2 function in regulating Col10α1 expression. Runx2-induced Col10α1 expression in ATDC5 cells was inhibited when cells were treated with cyclopamine (Fig. 4E).
FIGURE 3.

Ihh induces Runx2 expression but does not up-regulate other hypertrophic modulators. A and B, Ihh was overexpressed in primary chondrocytes. RNA was collected on the 2nd or 4th day of cultures and subjected to qPCR analyses of chondrogenic markers as mentioned in the figure. **, p < 0.01 (versus GFP-treated group). n = 3. C, cell lysates of primary chondrocytes treated with GFP or Ihh viruses were collected in the 4th days of cultures and subjected to Western blotting to observe the changes at protein levels. IB, immunoblot. D, primary chondrocytes treated with GFP or Ihh viruses were fixed and immunostained with anti-Foxa2 antibody in the 4th days of cultures. IF, immunofluorescence.
FIGURE 4.

Ihh positively controls Col10α1 expression in cooperation with Runx2 in chondrocytes. A and C, Runx2 or DN-Runx2 was overexpressed in ATDC5 as confirmed by the protein level using Western blotting. IB, immunoblot. B, Ihh was overexpressed in ATDC5 cells with or without Runx2 co-infection, and Col10α1 expression was measured by qPCR analyses. **, p < 0.01 (versus GFP-treated group). ##, p < 0.01 (versus Runx2 or Ihh-treated group) (n = 3). D, Ihh was overexpressed in ATDC5 with or without DN-Runx2 co-infection, and Col10α1 expression was measured by qPCR analyses. *, p < 0.05 (versus GFP-treated group). ##, p < 0.01 (versus Ihh-treated group) (n = 3). E, Runx2 was overexpressed in ATDC5 with or without treatment of cyclopamine (Cyclo), and Col10α1 expression was measured by qPCR analyses. **, p < 0.01 (versus GFP-treated group). ##, p < 0.01 (versus Runx2-treated group) (n = 3).
Furthermore, we investigated the molecular interaction between hedgehog transcription factors Gli1/2 and Runx2. It has been shown previously that Gli2 and Runx2 interact (25), so we focused our analyses on possible interaction between Gli1 and Runx2 using promoter assays, co-IP, and immunofluorescence. Gli1 was found to up-regulate the COL10A1 promoter activity in cooperation with Runx2 and that it could be efficiently immunoprecipitated with Runx2 (Fig. 5, A and B). Furthermore, we could show that Gli1 co-localizes with Runx2 in the nucleus of C3H10T1/2 cells (Fig. 5C). These data support molecular interaction between Gli1 and Runx2. We then performed an oligonucleotide pulldown assay using a COL10A1 biotin-labeled 65-bp probe containing both the Gli- and the Runx2-binding sites (Fig. 2D). Gli2 and Runx2 both bind to the probe (Fig. 5, D, lane 3, and E, lane 3 in an upper panel), and binding could be blocked for both using a competitive probe (Fig. 5, D, lane 4, and E, lane 4 in an upper panel). When we incubated both Gli1/2 and Runx2 proteins with the biotin-labeled probe, we found the physical interaction between Runx2 and Gli1 or Gli2 on this probe (Fig. 5E, lanes 5 and 6 in an upper panel). Moreover, endogenous binding of Runx2 in conjunction with Gli1/2 was detected on a COL10A1 basic promoter in a CHIP assay (Fig. 7G). These data demonstrate that Ihh positively regulates expression and transcription of Col10α1 through the interaction of Gli1/2 with Runx2.
FIGURE 5.

Interaction of Gli1 and Runx2 in the regulation of Col X. A, 250-bp luciferase construct of COL10A1 promoter was transfected into COS7 with expression vectors as indicated in the figure, and the luciferase/Renilla value was measured after 48 h. **, p < 0.01 (versus control). ##, p < 0.01 (versus Runx2 or Gli1) (n = 4). B, physical interaction between Gli1 and Runx2 was assessed by co-IP. Samples were immunoprecipitated by FLAG and immunoblotted by Myc. C, Myc-Gli1 and Dsred-Runx2 were co-transfected into C3H10T1/2 cells, and cells were immunostained by α-Myc antibody. Nucleus staining was performed with DAPI. D and E, oligonucleotide pulldown assay using a 65-bp COL10A1 promoter probe. The representative data are shown here. IB, immunoblot.
The possibility of cross talk between Ihh and BMP signaling in chondrocyte proliferation and differentiation has been previously suggested (26, 27). This is consistent with our finding because Runx2 is a known downstream target for BMP signaling (28). Also, Smads and Runx2 have been reported to interact in the transcription of collagen type X (18, 29, 30). Interestingly, we identified three conventional Smad-binding sites in the 250-bp basic COL10A1 promoter (Fig. 2D). These factors led us to hypothesize that Smads participate in Ihh-induced chondrocyte differentiation. We explored this possibility using immunofluorescence, a promoter assay, and co-IP to detect Gli/Smads interactions. C3H10T1/2 cells were transfected with Gli1/2 expression vectors and treated with 100 ng/ml BMP2. Transfected Gli1 or Gli2 localized in the nucleus of the cells (Fig. 6, A and B, upper panels). Upon treatment with BMP2, Gli1 or Gli2 was found to co-localize with the phosphorylated Smad1/5/8 (pSmads) assembly in the nucleus (Fig. 6, A and B, lower panels). In addition, Gli1 cooperated with Smad1/4 to increase COL10A1 promoter activity in COS7 cells (Fig. 7A). A similar assay was performed using BMP2-treated C3H10T1/2 cells. The results show that Gli2 and Smad1 each increased the promoter activity, whereas co-transfection further increased it (Fig. 7B). Furthermore, a co-IP assay showed physical interaction between Smad1 and Gli1 or Gli2 (Fig. 7, C and D, last lane). We also performed a co-IP assay using three proteins: Gli, Runx2, and Smad1. Binding between Smad1/Runx2 could be observed as reported previously (Fig. 7, E, lane 4, and F, lane 3) (31). co-IP using all three proteins indicated binding among Gli1/Runx2/Smad1 and Gli2/Runx2/Smad1 (Fig. 7, E and F, last lane). Finally, CHIP assay analyses demonstrated that Gli1/2, Runx2, and pSmads formed a transcriptional complex on the COL10A1 basic promoter (Fig. 7G). These data demonstrate that Ihh signaling regulates Col10α1 by interacting with Runx2 and Smads (Fig. 8F).
FIGURE 6.

Co-localization of Gli1/2 and pSmads in BMP2-treated C3H10T1/2 cells. A and B, Myc-Gli1 or Gli2 were transfected into C3H10T1/2 cells that are followed by 100 ng/ml BMP2 stimulation. The samples were immunostained by α-Myc and α-pSmad antibody, and the nucleus was stained with DAPI. The representative data are shown here.
We confirmed our observations with in vitro mineralization assays using primary chondrocytes (Fig. 8). Ihh-induced calcification was inhibited by Noggin (an antagonist for BMP), DN-Runx2, and dorsomorphin (an inhibitor for Smad1/5/8 phosphorylation) (Fig. 8, B–D) (32). In contrast, Ihh, in conjunction with Runx2, dramatically enhanced mineralization (Fig. 8C). Finally, Runx2-induced calcification was reduced by treatment with cyclopamine (Fig. 8E). These data indicate that Ihh promotes matrix mineralization by cross-talk with the BMP/Runx2/Smad pathway (Fig. 8F).
DISCUSSION
Gain of function analyses using Ihh adenoviruses in chondrogenic cells revealed up-regulation of maturation markers and matrix mineralization. Our study focused on the regulation of Col10α1 expression by Ihh signaling, whereupon we identified a new Gli1/2-response element in the 250-bp basic promoter sequence of the COL10A1 promoter. Furthermore, we show that a previously reported enhancer region located further upstream is not involved in this process. This new Gli-binding site is located in an area that also includes a Runx2-binding site and three Smad-binding sites. Ihh can up-regulate Runx2 expression without inducing the expression of other modulators of hypertrophic conversion such as Mef2c and Foxa2/3. Detailed analyses demonstrated that Ihh, coupled with Runx2, stimulates Col10α1 expression. We could demonstrate that Gli1/2 physically interacts with Runx2 and Smads to further promote transcriptional activity. Moreover, a Gli1/2-linked transcription complex, including Runx2 and pSmads was found to bind the endogenous COL10A1 promoter by CHIP assay. Finally, in vitro mineralization assays indicated that Ihh stimulated matrix mineralization of primary chondrocytes using a similar mechanism.
We, and others, previously reported that Ihh acts in a PTHrP-independent manner to positively regulate chondrocyte maturation and mineralization (10, 11). In this study, we demonstrate how Ihh signaling directly regulates Col10α1 expression by an interaction between Ihh downstream transcription factors Gli1/2 and a Runx2-Smad complex (Fig. 8F). This mechanism is also used by Ihh to promote chondrocyte calcification.
Gli1-Gli3 are known mediators for hedgehog signaling (33), but the role of Gli1 during endochondral ossification was unclear because mice homozygous for the Gli1zfd allele appeared normal or, at most, exhibited a mild phenotype in osteogenesis (26, 34–36). Gli1/Gli2 double homozygous mutants, however, presented a more severe phenotype, suggesting an overlap in functionality (26, 34–36). Unlike Gli1zfd/zfd mice, Gli2zfd/zfd mice exhibited shortened limbs and delayed endochondral ossification (37, 38). Col2-Cre;R26ΔNGli2/+ mice expressing a constitutively active form of Gli2, however, do exhibit chondrocyte maturation and could partially rescue the phenotype of Ihh−/− mice (39). These studies suggest a role for Gli2 as an activator in skeletal Ihh signaling. However, the mechanism for these hedgehog signaling molecules, Gli, to control late chondrocyte differentiation, especially Col X transcription, has not been elucidated so far. Our in vitro results show that Gli1 functions similarly as Gli2 to interact with Runx2/Smads to promote COL10A1 transcriptional activity. Moreover, transfecting cells with both Gli1 and Gli2 increased the promoter activity more than either alone, and both factors bind to the endogenous COL10A1 promoter in CHIP assay analysis. Therefore, our data provide a novel insight into Gli1/Gli2 as activators in regulating Col10α1 transcription. In contrast, Gli3 was reported to act as a repressor downstream of Ihh in chondrocyte differentiation (40, 41). We could not detect any changes in the level of Gli3 expression upon Ihh overexpression in chondrogenic cells (data not shown) suggesting that Gli3 might be less involved in the regulation of Col10α1 expression.
We observed inhibition of COL10A1 promoter activity by Sox9, and a recent paper suggested that Sox9 suppresses Col10α1 transcription in cooperation with Gli2/3 as repressors acting on the upstream enhancer (42). In our study, Gli1/2 as activators enhanced Col10α1 transcription by interacting Runx2/Smad and binding in the 250-bp basic promoter. Based on these reports, hedgehog signaling may have the opposite function in the presence of Sox9, interacting with Runx2/Smad but on different promoter sites for the same gene. It has also been speculated that hedgehog signaling controls the balance between transactivation and repression by changing its partners for Col10α1 transcription. Antagonism between Sox9 and Runx2 is a well established mechanism in skeletogenesis (43). Hedgehog signaling might function as both an enhancer and a repressor, depending on which of these two is present, to bridge early to late chondrocyte differentiation, which in turn regulates balanced chondrocyte differentiation.
Previous studies have suggested that Runx2/3, Smads, Mef2c transcription factor, and FoxA family members act as enhancers for Col10α1 expression and subsequent induction of hypertrophic differentiation (15, 16, 18, 23, 29, 30). The COL10A1 distal enhancer region is highly conserved among mammals, including humans, cows, and mice (19). The tissue specificity for Col10α1 has been studied extensively, and the b2 enhancer region has been discovered in the avian system (13, 19, 29, 30, 44). This study is the first detailed investigation of the COL10A1 basic promoter region. We were unable to detect consistent activation of the human enhancer region in the COL10A1 promoter using Gli1/2 as activators. Instead, we found that Gli1/2 activated the 250-bp basic promoter region where we identified a sequence located in the cluster of Runx2- and Smad-binding sites that was similar to the conventional Gli-binding site. Apart from COL10A1 enhancement, overexpression of Ihh also stimulated Runx2 expression. However, it failed to induce expression of other important transcription factors such as Mef2c or Foxa2/3, indicating that these genes do not play a significant role in this system. In addition, we could show that Ihh and Runx2 synergistically induce Col10α1 expression. However, the presence of two MEF2-binding sites in the basic Col10α1 promoter region suggests that future studies of possible functional interactions between Ihh signaling and MEF2c/FoxA factors during chondrocyte differentiation and skeletal development might be worthwhile (16).
Supplementary Material
Acknowledgments
We thank Drs. Andrew Lassar (Harvard Medical School) and Bjorn Olsen (Harvard School of Dental Medicine) for helpful discussions. We are grateful to Drs. Roland Baron, Hiroaki Saito, and William Addison (Harvard School of Dental Medicine) for sharing resources and ideas. We also thank Drs. Tadotoshi Sato, Takenobu Ishii, and Noriko Ide (Harvard School of Dental Medicine) and Mikihiko Kogo and Toshiyuki Yoneda (Osaka University) for general help.
This work was supported, in whole or in part, by National Institutes of Health Grant R01AR050560. This work was also supported by institutional funds from Harvard School of Dental Medicine (to B. L.), postdoctoral fellowships for research abroad from the Japan Society for the Promotion of Science (to K. A.), and research fellowship for study abroad from the Uehara Memorial Foundation (to K. A.).

This article contains supplemental Fig. S1
- Ihh
- Indian hedgehog
- Col10α1
- collagen type X
- Gli
- GLI-Krüppel family members
- PTHrP
- parathyroid hormone-related peptide
- oligo
- oligonucleotide
- qPCR
- quantitative
- DN-Runx2
- dominant-negative form of Runx2.
REFERENCES
- 1. de Crombrugghe B., Lefebvre V., Nakashima K. (2001) Regulatory mechanisms in the pathways of cartilage and bone formation. Curr. Opin. Cell Biol. 13, 721–727 [DOI] [PubMed] [Google Scholar]
- 2. Kronenberg H. M. (2003) Developmental regulation of the growth plate. Nature 423, 332–336 [DOI] [PubMed] [Google Scholar]
- 3. Gao B., Guo J., She C., Shu A., Yang M., Tan Z., Yang X., Guo S., Feng G., He L. (2001) Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat. Genet. 28, 386–388 [DOI] [PubMed] [Google Scholar]
- 4. Hellemans J., Coucke P. J., Giedion A., De Paepe A., Kramer P., Beemer F., Mortier G. R. (2003) Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am. J. Hum. Genet. 72, 1040–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lanske B., Karaplis A. C., Lee K., Luz A., Vortkamp A., Pirro A., Karperien M., Defize L. H., Ho C., Mulligan R. C., Abou-Samra A. B., Jüppner H., Segre G. V., Kronenberg H. M. (1996) PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 273, 663–666 [DOI] [PubMed] [Google Scholar]
- 6. Vortkamp A., Lee K., Lanske B., Segre G. V., Kronenberg H. M., Tabin C. J. (1996) Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273, 613–622 [DOI] [PubMed] [Google Scholar]
- 7. Karp S. J., Schipani E., St-Jacques B., Hunzelman J., Kronenberg H., McMahon A. P. (2000) Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone-related protein-dependent and -independent pathways. Development 127, 543–548 [DOI] [PubMed] [Google Scholar]
- 8. Kobayashi T., Soegiarto D. W., Yang Y., Lanske B., Schipani E., McMahon A. P., Kronenberg H. M. (2005) Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J. Clin. Invest. 115, 1734–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maeda Y., Schipani E., Densmore M. J., Lanske B. (2010) Partial rescue of postnatal growth plate abnormalities in Ihh mutants by expression of a constitutively active PTH/PTHrP receptor. Bone 46, 472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amano K., Ichida F., Sugita A., Hata K., Wada M., Takigawa Y., Nakanishi M., Kogo M., Nishimura R., Yoneda T. (2008) MSX2 stimulates chondrocyte maturation by controlling Ihh expression. J. Biol. Chem. 283, 29513–29521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mak K. K., Kronenberg H. M., Chuang P. T., Mackem S., Yang Y. (2008) Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 135, 1947–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Higashikawa A., Saito T., Ikeda T., Kamekura S., Kawamura N., Kan A., Oshima Y., Ohba S., Ogata N., Takeshita K., Nakamura K., Chung U. I., Kawaguchi H. (2009) Identification of the core element responsive to runt-related transcription factor 2 in the promoter of human type X collagen gene. Arthritis Rheum. 60, 166–178 [DOI] [PubMed] [Google Scholar]
- 13. Li F., Lu Y., Ding M., Napierala D., Abbassi S., Chen Y., Duan X., Wang S., Lee B., Zheng Q. (2011) Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. J. Bone Miner. Res. 26, 2899–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ueta C., Iwamoto M., Kanatani N., Yoshida C., Liu Y., Enomoto-Iwamoto M., Ohmori T., Enomoto H., Nakata K., Takada K., Kurisu K., Komori T. (2001) Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J. Cell Biol. 153, 87–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng Q., Zhou G., Morello R., Chen Y., Garcia-Rojas X., Lee B. (2003) Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Biol. 162, 833–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arnold M. A., Kim Y., Czubryt M. P., Phan D., McAnally J., Qi X., Shelton J. M., Richardson J. A., Bassel-Duby R., Olson E. N. (2007) MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 12, 377–389 [DOI] [PubMed] [Google Scholar]
- 17. Kozhemyakina E., Cohen T., Yao T. P., Lassar A. B. (2009) Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol. Cell. Biol. 29, 5751–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ionescu A., Kozhemyakina E., Nicolae C., Kaestner K. H., Olsen B. R., Lassar A. B. (2012) FoxA family members are crucial regulators of the hypertrophic chondrocyte differentiation program. Dev. Cell 22, 927–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gebhard S., Pöschl E., Riemer S., Bauer E., Hattori T., Eberspaecher H., Zhang Z., Lefebvre V., de Crombrugghe B., von der Mark K. (2004) A highly conserved enhancer in mammalian type X collagen genes drives high levels of tissue-specific expression in hypertrophic cartilage in vitro and in vivo. Matrix Biol. 23, 309–322 [DOI] [PubMed] [Google Scholar]
- 20. Akiyama H., Chaboissier M. C., Martin J. F., Schedl A., de Crombrugghe B. (2002) The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sasaki H., Hui C., Nakafuku M., Kondoh H. (1997) A binding site for Gli proteins is essential for HNF-3β floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124, 1313–1322 [DOI] [PubMed] [Google Scholar]
- 22. Morikawa M., Koinuma D., Miyazono K., Heldin C. H. (2013) Genome-wide mechanisms of Smad binding. Oncogene 32, 1609–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yoshida C. A., Yamamoto H., Fujita T., Furuichi T., Ito K., Inoue K., Yamana K., Zanma A., Takada K., Ito Y., Komori T. (2004) Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18, 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hesse E., Saito H., Kiviranta R., Correa D., Yamana K., Neff L., Toben D., Duda G., Atfi A., Geoffroy V., Horne W. C., Baron R. (2010) Zfp521 controls bone mass by HDAC3-dependent attenuation of Runx2 activity. J. Cell Biol. 191, 1271–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimoyama A., Wada M., Ikeda F., Hata K., Matsubara T., Nifuji A., Noda M., Amano K., Yamaguchi A., Nishimura R., Yoneda T. (2007) Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol. Biol. Cell 18, 2411–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Minina E., Kreschel C., Naski M. C., Ornitz D. M., Vortkamp A. (2002) Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev. Cell 3, 439–449 [DOI] [PubMed] [Google Scholar]
- 27. Minina E., Wenzel H. M., Kreschel C., Karp S., Gaffield W., McMahon A. P., Vortkamp A. (2001) BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 128, 4523–4534 [DOI] [PubMed] [Google Scholar]
- 28. Lee K. S., Kim H. J., Li Q. L., Chi X. Z., Ueta C., Komori T., Wozney J. M., Kim E. G., Choi J. Y., Ryoo H. M., Bae S. C. (2000) Runx2 is a common target of transforming growth factor β1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol. Cell. Biol. 20, 8783–8792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Drissi M. H., Li X., Sheu T. J., Zuscik M. J., Schwarz E. M., Puzas J. E., Rosier R. N., O'Keefe R. J. (2003) Runx2/Cbfa1 stimulation by retinoic acid is potentiated by BMP2 signaling through interaction with Smad1 on the collagen X promoter in chondrocytes. J. Cell. Biochem. 90, 1287–1298 [DOI] [PubMed] [Google Scholar]
- 30. Leboy P., Grasso-Knight G., D'Angelo M., Volk S. W., Lian J. V., Drissi H., Stein G. S., Adams S. L. (2001) Smad-Runx interactions during chondrocyte maturation. J. Bone Joint Surg. Am. 83, S15–S22 [PubMed] [Google Scholar]
- 31. Nishimura R., Hata K., Harris S. E., Ikeda F., Yoneda T. (2002) Core-binding factor α1 (Cbfa1) induces osteoblastic differentiation of C2C12 cells without interactions with Smad1 and Smad5. Bone 31, 303–312 [DOI] [PubMed] [Google Scholar]
- 32. Yu P. B., Hong C. C., Sachidanandan C., Babitt J. L., Deng D. Y., Hoyng S. A., Lin H. Y., Bloch K. D., Peterson R. T. (2008) Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 4, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang J., Hui C. C. (2008) Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hojo H., Ohba S., Taniguchi K., Shirai M., Yano F., Saito T., Ikeda T., Nakajima K., Komiyama Y., Nakagata N., Suzuki K., Mishina Y., Yamada M., Konno T., Takato T., Kawaguchi H., Kambara H., Chung U. I. (2013) Hedgehog-Gli activators direct osteo-chondrogenic function of bone morphogenetic protein toward osteogenesis in the perichondrium. J. Biol. Chem. 288, 9924–9932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hojo H., Ohba S., Yano F., Saito T., Ikeda T., Nakajima K., Komiyama Y., Nakagata N., Suzuki K., Takato T., Kawaguchi H., Chung U. I. (2012) Gli1 protein participates in Hedgehog-mediated specification of osteoblast lineage during endochondral ossification. J. Biol. Chem. 287, 17860–17869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park H. L., Bai C., Platt K. A., Matise M. P., Beeghly A., Hui C. C., Nakashima M., Joyner A. L. (2000) Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127, 1593–1605 [DOI] [PubMed] [Google Scholar]
- 37. Miao D., Liu H., Plut P., Niu M., Huo R., Goltzman D., Henderson J. E. (2004) Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp. Cell Res. 294, 210–222 [DOI] [PubMed] [Google Scholar]
- 38. Mo R., Freer A. M., Zinyk D. L., Crackower M. A., Michaud J., Heng H. H., Chik K. W., Shi X. M., Tsui L. C., Cheng S. H., Joyner A. L., Hui C. (1997) Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123 [DOI] [PubMed] [Google Scholar]
- 39. Joeng K. S., Long F. (2009) The Gli2 transcriptional activator is a crucial effector for Ihh signaling in osteoblast development and cartilage vascularization. Development 136, 4177–4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hilton M. J., Tu X., Cook J., Hu H., Long F. (2005) Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development 132, 4339–4351 [DOI] [PubMed] [Google Scholar]
- 41. Koziel L., Wuelling M., Schneider S., Vortkamp A. (2005) Gli3 acts as a repressor downstream of Ihh in regulating two distinct steps of chondrocyte differentiation. Development 132, 5249–5260 [DOI] [PubMed] [Google Scholar]
- 42. Leung V. Y., Gao B., Leung K. K., Melhado I. G., Wynn S. L., Au T. Y., Dung N. W., Lau J. Y., Mak A. C., Chan D., Cheah K. S. (2011) SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 7, e1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou G., Zheng Q., Engin F., Munivez E., Chen Y., Sebald E., Krakow D., Lee B. (2006) Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 19004–19009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zheng Q., Keller B., Zhou G., Napierala D., Chen Y., Zabel B., Parker A. E., Lee B. (2009) Localization of the cis-enhancer element for mouse type X collagen expression in hypertrophic chondrocytes in vivo. J. Bone Miner. Res. 24, 1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seriwatanachai D., Densmore M. J., Sato T., Correa D., Neff L., Baron R., Lanske B. (2011) Deletion of Zfp521 rescues the growth plate phenotype in a mouse model of Jansen metaphyseal chondrodysplasia. FASEB J. 25, 3057–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Amano K., Hata K., Sugita A., Takigawa Y., Ono K., Wakabayashi M., Kogo M., Nishimura R., Yoneda T. (2009) Sox9 family members negatively regulate maturation and calcification of chondrocytes through up-regulation of parathyroid hormone-related protein. Mol. Biol. Cell 20, 4541–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.