

Background: Gene regulation of the bone repressor sclerostin (SOST) is only poorly understood.
Results: SOST gene suppression by parathyroid hormone is partially mediated by HDAC5 inhibiting MEF2, and SOST gene expression requires class I HDAC activity.
Conclusion: SOST gene expression is negatively regulated by HDAC5 and positively by class I HDACs.
Significance: Class I HDAC inhibitors represent a novel approach for bone forming osteoporosis therapies.
Keywords: Gene Regulation, Histone Deacetylase (HDAC), Parathyroid Hormone, Transcription Corepressor, Transcriptional Coactivator, Sclerostin
Abstract
Adult bone mass is controlled by the bone formation repressor sclerostin (SOST). Previously, we have shown that intermittent parathyroid hormone (PTH) bone anabolic therapy involves SOST expression reduction by inhibiting myocyte enhancer factor 2 (MEF2), which activates a distant bone enhancer. Here, we extended our SOST gene regulation studies by analyzing a role of class I and IIa histone deacetylases (HDACs), which are known regulators of MEF2s. Expression analysis using quantitative PCR (qPCR) showed high expression of HDACs 1 and 2, lower amounts of HDACs 3, 5, and 7, low amounts of HDAC4, and no expression of HDACs 8 and 9 in constitutively SOST-expressing UMR106 osteocytic cells. PTH-induced Sost suppression was associated with specific rapid nuclear accumulation of HDAC5 and co-localization with MEF2s in nuclear speckles requiring serine residues 259 and 498, whose phosphorylations control nucleocytoplasmic shuttling. Increasing nuclear levels of HDAC5 in UMR106 by blocking nuclear export with leptomycin B (LepB) or overexpression in transient transfection assays inhibited endogenous Sost transcription and reporter gene expression, respectively. This repressor effect of HDAC5 did not require catalytic activity using specific HDAC inhibitors. In contrast, inhibition of class I HDAC activities and expression using RNA interference suppressed constitutive Sost expression in UMR106 cells. An unbiased comprehensive search for involved HDAC targets using an acetylome analysis revealed several non-histone proteins as candidates. These findings suggest that PTH-mediated Sost repression involves nuclear accumulation of HDAC inhibiting the MEF2-dependent Sost bone enhancer, and class I HDACs are required for constitutive Sost expression in osteocytes.
Introduction
Sclerostin (SOST),2 encoded by the SOST gene, is a crucial inhibitor of bone formation that is exclusively secreted by osteocytes in adult bone (1). SOST deficiency leads to drastic high bone mass disorders as illustrated in the human loss-of-function hereditary disorders van Buchem disease and sclerosteosis. Similarly, Sost knock-out mouse models display enormously elevated bone mass and strength due to increased bone formation in the entire skeleton and throughout adult growth (2, 3). Conversely, transgenic mice overexpressing SOST have low bone mass and strength due to a decrease in bone forming osteoblasts (4, 5). SOST inhibits bone formation by antagonizing canonical Wnt signaling, which is required for normal osteoblastogenesis and control of osteoclastogenesis (6–9). It does so by binding to the Wnt co-receptors Lrp5 and -6 preventing their interaction with the Wnt-Frizzled receptor complex, which triggers Wnt signaling in target cells. We and others have shown that suppression of SOST expression is a key mechanism as to how intermittent parathyroid hormone (PTH) treatment leads to bone mass elevation (3, 10, 11). Furthermore, we and others have shown that SOST is a direct target gene of PTH (12), (11) and that PTH exerts its repressive effect by inhibiting myocyte enhancer factor 2 (MEF2) transcription factors, which are binding to a distant downstream SOST gene enhancer that is required for SOST expression in adult bone (4, 13). In agreement with an important role of MEF2s in SOST gene control and, thus, adult bone metabolism MEF2C was recently identified as one of 20 loci affecting bone mineral density in a meta-analysis of five genome-wide association studies of femoral neck and lumbar spine bone mineral density (14).
MEF2s are widely expressed regulators of cell differentiation and organogenesis, and are well known for their role in the development of skeletal muscle, heart, vasculature, neurons, and T-cells (15). Their role in the control of adult bone metabolism is only poorly understood so far. In vertebrates, there are four genes MEF2A, -B, -C, and -D, which are highly homologous in their MADS and MEF2 domains mediating DNA binding, dimerization, and co-factor recruitment, but are highly divergent in their C-terminal transcriptional activation domains. Our previous quantitative real-time RNA expression analyses indicated that Mef2a, -c, and -d, but not b are expressed in adult bone (13). Mef2c plays a crucial role in bone development by controlling chondrocyte hypertrophy during endochondral ossification via activation of Collagen 10α1 and Runx2 genes (16). The activity of MEF2s is controlled by a variety of signaling pathways including acetylation by p300, phosphorylation by mitogen-activated protein kinases, association with calcineurin-dephosphorylated NFAT, sumoylation by SUMO2 and -3, and interaction with class IIa histone deacetylases (HDACs) whose nuclear to cytoplasmic distribution is controlled by calcium-regulated protein kinases (15, 17–19). The first three mechanisms stimulate MEF2 activity, whereas the latter two are inhibitory. Inhibition of MEF2 activity by class IIa HDACs is well known in regulating cardiomyocyte hypertrophy (20). The inhibitory effect of class IIa HDACs on MEF2s does not involve their catalytic activity, but is due to the formation of a transcriptional repressor protein-protein complex involving the recruitment of class I HDACs such as HDAC3, which deacetylate MEF2s (21). The transcription inhibition activity of class IIa HDACs is regulated by nucleocytoplasmic shuttling (22). Upon stimulation by a variety of physiological signals, three conserved serine residues are phosphorylated leading to binding of the 14-3-3 chaperone protein, which induces nuclear export of HDACs and thereby derepression of target genes such as MEF2s.
The role of HDACs in bone metabolism is not well understood. There are 11 mammalian HDACs, which are grouped into four classes based on protein domain arrangement similarities (23). Class I comprises HDAC1, -2, -3, and -8, class IIa HDAC4, -5, -7, and -9, class IIb HDAC6 and -10, and class IV HDAC11. In vitro experiments using HDAC inhibitors (HDIs) have shown inhibition of osteoclasts differentiation (24–26) and stimulation of osteoclast apoptosis (27) suggesting that HDACs promote bone resorption. However, suppression of class I HDAC3 and class IIa HDAC7 in bone marrow stromal cells showed opposite effects on osteoclastogenesis indicating a possible functional difference between class I and class IIa HDACs in osteoclast differentiation (28). Moreover, HDIs were shown to stimulate osteoblast differentiation inducing Type I collagen, osteopontin, bone sialoprotein, and osteocalcin gene expression (29–31). A role of HDACs in osteocytes, the most abundant bone cell, has not been described so far. Overall, in vitro experiments suggest that HDACs inhibit bone formation and stimulate bone resorption although there may be opposing effects of HDAC isoforms. Complex effects of HDACs have also been described in bone metabolism in vivo. Rodent studies using the two clinically approved HDIs valproate and suberoylanilide hydroxamic acid showed negative effects on bone mass with reductions in bone mineral content, trabecular bone volume, bone formation, and osteoblast number (31). However, local increases in mineral apposition rates were observed in suberoylanilide hydroxamic acid-treated mice confirming in vitro studies that HDIs stimulate osteoblasts activity, but decrease osteoblast number and/or formation. Clinical findings with the two HDIs show a similar picture as seen in animals. Valproate, which is used a long time for the treatment of epilepsy and bipolar disease, has been associated with bone mineral density decrease in several studies (31). However, it is currently unclear how HDIs affect human bone remodeling leading to bone mass reductions due to inconsistent results of serum and urinary biomarkers for bone resorption and formation.
Here, we show roles of HDACs in regulating the expression of the key bone formation inhibitor SOST using the validated osteocytic UMR106 cell model. PTH-mediated Sost inhibition was accompanied by specific nuclear import of class IIa HDAC5 and association with MEF2 transcription factors. HDAC5 inhibited the SOST bone enhancer in a MEF2-dependent but catalytically independent manner. Finally, we report that class I HDACs 1, 2, and 3 are required for Sost expression suggesting opposing roles and a complex interplay of class IIa and class I HDACs in regulating Sost expression.
EXPERIMENTAL PROCEDURES
Reagents and Cloning
Human parathyroid hormone (hPTH)-(1–34) was synthesized at Novartis. Leptomycin B, phorbol 12-myristate 13-acetate (PMA), trichostatin A (TSA), and apicidin were purchased from Sigma. Forskolin was from Biomol. The class II HDAC inhibitor compound 2 (32) was re-synthesized and its specificity validated at Novartis (Table 1). Human HDAC expression plasmids (pcDNA3.1 HDAC1, -2, -3, -4, -5, -6, and -10) were a generous gift from R. Vega (Novartis). All C-terminal green fluorescent protein (GFP)-tagged HDACs were in pEGFP-N1 (Clontech) except HDAC7, which was in pEGFP-N2 (Clontech). Luciferase reporter plasmids with the human SOST promoter alone or in combination with the SOST bone enhancer element have been described (13). siRNAs against rat Hdac2 and -3 and non-targeting negative control siRNAs were purchased from Dharmacon and siRNA against Hdac1 was bought from Qiagen. Cloning of the serine 259 and serine 498 to alanine HDAC5 double mutant was done by GenScript from our wild-type (wt) human HDAC5 plasmids. All other mutants were cloned according to standard procedures. V5-tagged HDAC5 expression plasmid (pHDAC5-V5) was generated at Solvias by replacing the EGFP coding region of pHDAC5-EGFP with a V5-encoding oligonucleotide primer pair using XhoI and XbaI restriction sites and the following oligonucleotide pair: TCGAGGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGTAAAGCGGCGGCCCGCGACT (forward) and CTAGAGTCGCGGGCCGCCGCTTTACGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCC (reverse).
TABLE 1.
HDAC inhibition specificity of compound 2
Shown are average IC50 (μm).
| Novartis | Ontoria et al. (32) | |
|---|---|---|
| HDAC1 | 2.78 | 2.7 |
| HDAC3 | 2.7 | |
| HDAC4 | 0.039 | 0.024 |
| HDAC5 | 0.007 | |
| HDAC6 | 0.76 | 0.23 |
Cell Culture and Gene Expression Analyses
The UMR106 cell line was bought from ATCC and maintained in DMEM/F-12 (Invitrogen) containing 10% FCS (AMIMED), 1% non-essential amino acids (Invitrogen), 1% Glutamax (Invitrogen), and antibiotics. Total RNA was isolated using RNeasy (Qiagen) and reverse-transcribed into cDNA using the high-capacity cDNA archive kit (Applied Biosystems) according to the manufacturer's recommendations. Quantitative real-time reverse transcription-PCR (qPCR) expression analysis was performed on an ABI Prism 7900 HT sequence detection system using TaqMan assays and the Universal PCR Master mixture all from Applied Biosystems. For Sost gene expression analysis following compound treatments UMR106 cells were seeded in 96-well plates (Nunc) at a density of 10,000 cells per well. The next day the culture medium was replaced with fresh culture medium containing the desired compounds and the cells were incubated for 4 h. Subsequently, Sost mRNA gene expression determinations were done using the FastLane One-Step QuantiTect Multiplex RT-PCR kit (Qiagen). The following rat TaqMan® assays were used: β2m (Rn00560865_m1) for normalization, Sost (Rn00577971_m1), Hdac1 (Rn01519307_g1), Hdac2 (Rn01193634_g1), Hdac3 (Rn01405468_m1), Hdac4 (Rn01427053_m1), Hdac5 (Rn01464251_g1), Hdac7 (Rn01533232_m1), Hdac8 (Rn01419050_m1), and Hdac9 (Rn01769547_m1 and Rn01769541_m1).
High Content Analysis (HCA)
UMR106 cells were seeded in 96-well plates (PerkinElmer Life Sciences) at a density of 5,000 cells per well. The next day, cells were transfected with 40 ng of GFP-HDAC or FLAG-tagged HDAC expression plasmids using FuGENE6 (Roche Applied Science) and OptiMEM (Invitrogen). 24 h later, cells were treated with compounds (n = 3–4) before fixation with 3.7% formalin (Sigma). Nuclei were stained with DAPI (Sigma). Intracellular HDAC distribution was measured using a Cellomics Array Scan II system with a ×20 objective and the cytoplasm to nucleus translocation BioApplication. The individual ratios of the nuclear to cytoplasmic signal (CircRingAvgIntenRatioCh2) were determined for transfected cells (n > 100) and averages were calculated. FLAG-tagged HDAC immunostaining was performed using mouse anti-FLAG M2 monoclonal antibody (Sigma) at 1:1,000 dilution and Alexa Fluor® 488 goat anti-mouse IgG (H+L) (Invitrogen) at 1:500 dilution. HCA of FLAG-tagged HDACs was performed as with GFP-tagged samples.
Confocal Microscopy
UMR106 cells were seeded in 8-well μ-slide (ibidi GmbH) at a density of 15,000 cells per well. Transfection and treatments was done as for HCA. Cells were permeabilized with phosphate-buffered saline (PBS) containing 0.2% Triton X-100 (Fluka) before blocking with the same solution containing 5% goat serum (Jackson ImmunoResearch). The cells were then first incubated with rabbit polyclonal anti-MEF2 antibody (Santa Cruz sc-10794 at 0.2 μg/μl) at 1:100 in blocking solution and then with TRITC-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch) at 1:200 in blocking solution containing DAPI (Sigma). Confocal microscopy was done with a LSM710 Axio observer inverted confocal microscope using ZEN 2009 Light Edition software (Zeiss).
Chromatin Immunoprecipitation
UMR106 cells were grown in 15-cm cell culture dishes until 80–90% confluence. After medium change, cells were transfected overnight with HDAC5-V5 expression plasmid using Lipofectamine LTX Plus (Invitrogen) followed by stimulation of one dish with 100 nm hPTH or solvent control for 1 h in fresh growth medium. ChIP was basically done using the SimpleChIP Enzymatic Chromatin IP kit with magnetic beads (Cell Signaling Technology). Chromatin shearing was performed using an S-220 ultrasonicator (Covaris). Samples were immunoprecipitated with a polyclonal rabbit anti-V5 tag antibody (Abcam) or control rabbit IgG (Cell Signaling Technology) and bound Sost enhancer DNA was quantified by quantitative real-time PCR using the following primers: CTCTGATGTTCCCCAAACC (forward) and AGAAACTGGGCCAGTGCT (reverse).
Reporter Gene Assays
UMR106 cells were seeded in 48-well plates (Nunc) at a density of 20,000 cells per well. The next day, cells were transfected with 40 ng of reporter plasmid, 2 ng of HDAC expression plasmid, and 50 ng of CMV β-Gal plasmid using FuGENE6 (Roche Applied Science) and OptiMEM (Invitrogen). After 24 h the cells were lysed with passive lysis buffer (Promega) and luciferase activity was measured using a Mithras CB 940 multimode reader (Berthold) and luciferase assay reagent (Promega).
HDAC Inhibition Assays
Compound dilutions in DMSO were incubated with human HDACs1, -3, -4, -5, and -6 catalytic domains (final protein concentration 1.65 ng/ml) in assay buffer (25 mm Tris/Cl, pH 8.0, 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2, 0.1 mg/ml of BSA), 10 μm Ac-Gly-Ala-Lys(TFA)-AMC substrate solution and incubated for 1 h at 37 °C. The reaction was stopped by adding a solution of 10 μm TSA and trypsin (0.02 mg/ml). The concentration of the cleaved AMC substrate was measured by fluorescence readings at 360 nm excitation and 460 nm emission.
siRNA Transfections
siRNA stock solutions of 12.5 ng/μl were prepared in siRNA dilution buffer (Qiagen). 10,000 cells per well in 96-well plates (Nunc) were transfected directly upon seeding with 18.75 ng of siRNA using HiPerFect transfection reagent (Qiagen) diluted in OptiMEM (Invitrogen). After 24 h, medium was exchanged and 48 h later gene expressions were determined using the FastLane One-Step QuantiTect Multiplex RT-PCR kit (Qiagen).
HDAC Assay
Total activity of HDAC class I and II enzymes was measured using the HDAC-Glo I/II Assay and Screening System (Promega). UMR106 cells were seeded at a density of 10,000 cells per well in 96-well plates. The next day, cells were incubated with 1 μm PTH or 10 μm apicidin for 60 min before luciferase activity was measured according to the manufacturer's instructions.
Whole Cell Acetylome Analysis
Global acetylation changes in response to the HDI apicidin were determined by stable-isotope labeling with amino acids in cell culture (SILAC) using the SILACTM Protein ID and Quantitation Media Kit (Invitrogen) according to the manufacturer's instructions. UMR106 cells were either grown in medium with unlabeled Lys and Arg (light sample) or in the presence of [13C6]Lys and [13C6,15N4]Arg (heavy sample). Cells grown in heavy medium served as control, whereas cells grown in light medium were treated with either 1 μm apicidin or 100 nm PTH for 4 h before lysis. Enrichment of acetyllysine peptides using a pan-acetyllysine antibody was performed as published (33) with minor modifications. Briefly, cells were lysed by sonication in a lysis buffer containing 20 mm Hepes, pH 8, and 8 m urea. After centrifugation, the lysate was diluted to 2 m with 20 mm Hepes and trypsinized overnight, using a trypsin to protein ratio of 1:50. Digestion was stopped by the addition of 1% trifluoroacetic acid, after checking by SDS-PAGE. After desalting on SepPak cartridges (Waters), acetyllysine-containing peptides were enriched by immunoaffinity chromatography as described (33). The enriched fraction was desalted on SepPak and lyophilized before further fractionation by hydrophilic interaction chromatography. Peptide mixtures were resuspended in 80% acetonitrile/H2O and separated on a TSK-Amide column (250 × 4 mm), using a linear gradient from 80% acetonitrile/H2O to 60/40 acetonitrile/H2O at a flow rate of 0.5 ml/min. Fractions of 0.5 ml were collected, lyophilized to dryness, and resuspended in 98% H2O, 2% acetonitrile + 0.1% formic acid for analysis by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). The peptide mixtures were injected onto a 15 cm × 75-μm ProteoPep 2 PicoFrit column (New Objectives), coupled to a LTQ-OrbiTrap XL mass spectrometer (Thermo). Buffer A consisted of H2O with 0.1% formic acid and Buffer B of 80% acetonitrile with 0.1% formic acid. Peptides were separated using a 90 min gradient from 2% B to 50% B. Data acquisition was done using a “Top 5 method,” where every full MS scan was followed by 5 data-dependent scans on the 5 most intense precursor ions. Full scans were performed in the Orbitrap at 60,000 resolution with target values of 5 E5 ions and 500-ms injection time, whereas MS/MS scans were done in the ion trap with 1E4 ions and 200 ms. The resulting data files were analyzed using the MaxQuant (58) software package (version 1.0.0.18) using the Mascot server and the rat IPI database (version 3.64), with trypsin as the enzyme, allowing 4 missed cleavages. MS/MS mass tolerances were set at 0.8 Da and identifications were filtered at a false discovery rate of 1% (MaxQuant) and finally at a Mascot score of 20. For visualization data tables from MaxQuant were exported to TIBCO Spotfire.
RESULTS
Expression of Class I and IIa HDACs in UMR106 Cells
Class IIa Hdacs and Hdac3 expressions were determined in UMR106 cells because these HDACs are known repressors of MEF2s (15, 17, 21), and, thus, we hypothesized that they could be involved in the inhibition of MEF2s by PTH for controlling Sost expression in osteocytes. UMR106 cells are a valid cell model to study Sost gene regulation by PTH in vitro because they reproduce SOST gene regulation by PTH as observed in osteocytes in vivo (4, 12, 13). Among class IIa Hdacs, Hdac5 showed the highest expression followed by Hdac7 showing about 2-fold lower expression and Hdac4 showing about 10-fold lower expression (Fig. 1). Hdac9 expression was not detected. In addition to class IIa Hdacs, we determined expression of class I Hdacs, because Hdac3 as a member of this family had also been described as MEF2 inhibitor. Hdac1 and -2 were about 2-fold stronger expressed than Hdac5, whereas Hdac3 showed a similar expression level as Hdac7 and Hdac8 was not detected like Hdac9. In summary, we detected strong expression of Hdacs 1, 2, and 5, medium expression of Hdacs 3 and 7, low expression of Hdac4, and no expression of Hdacs 8 and 9 in UMR106 cells.
FIGURE 1.
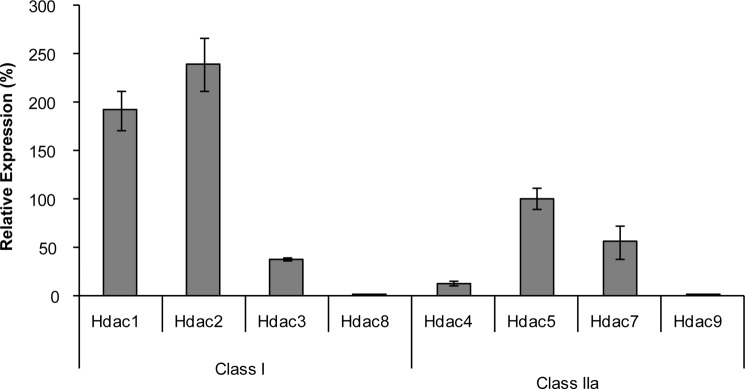
Expression of class I and IIa Hdacs in UMR106 cells. RNA expressions of class I and IIa Hdacs were determined by qPCR. Relative expression levels are expressed as the mean ± S.E. of two independent experiments. Expression of Hdac5 was arbitrarily taken as 100%.
PTH Stimulates Nuclear Localization of HDAC5 in UMR106 Cells
Repression of MEF2-mediated transcription by class IIa HDACs and HDAC3 is known to be regulated by nucleocytoplasmic shuttling, which controls their nuclear abundance (22, 34–37). Thus, we have analyzed whether PTH-mediated Sost gene suppression involves nuclear accumulation of these HDACs. UMR106 cells were transfected with expression plasmids for GFP-HDAC fusion proteins and the effect of PTH on their subcellular distributions was determined using fluorescence microscopy and HCA. Microscopic analysis of non-treated cells showed considerable cell-to-cell variation, but in general HDAC3 and -5 were predominantly nuclear, HDAC4 mostly in the cytoplasm and HDAC7 in both compartments (Fig. 2A). Quantitative analysis using HCA confirmed these differences as shown in Fig. 2B. Upon PTH treatment, only HDCA5 showed a clear change in subcellular distribution by further increasing nuclear localization to become exclusively nuclear (Fig. 2, A and B). Time course analysis using HCA showed that PTH rapidly induced nuclear accumulation of HDCA5 reaching a plateau after about 20 min (Fig. 2D). In contrast, nucleocytoplasmic distributions of HDAC3, -4, and -7 were only marginally affected by PTH in agreement with non-quantitative microscopic analysis. HDAC4 and -7 showed a continuous small nuclear increase, whereas HDAC3 slowly increased in the cytoplasm. Intracellular distribution was also analyzed for HDAC1, -2, -6, and -10. HDAC1 was predominantly nuclear, whereas HDACs 2, 6, and 10 were predominantly cytoplasmic (Fig. 2C). Neither showed marked changes in subcellular distribution upon PTH treatment, which is consistent with the fact that these HDACs have not been reported to shuttle between the cytoplasm and nucleus. HDACs are well known as transcription regulators in the nucleus. However, functional roles have also been described in the cytoplasm such as regulation of translation (38). In addition to inducing nuclear localization of HDCA5 PTH led to the appearance of many distinct nuclear spots suggesting further compartmentalization of HDCA5 within the nucleus (Fig. 2A). In summary, PTH lead to a specific rapid and strong nuclear localization of HDAC5 and the formation of distinct subnuclear speckles suggesting a role in repressing MEF2-mediated SOST gene expression.
FIGURE 2.

PTH specifically induces a rapid and strong nuclear localization of HDAC5. A, UMR106 cells were transfected with HDAC-GFP expression vectors for HDAC3, -4, -5, and -7 and their subcellular distributions were analyzed using fluorescence microscopy and HCA. Representative green fluorescence images are shown of transfected non-treated cells (upper panel) and transfected cells stimulated with 100 nm PTH for 1 h (lower panel). B, the ratio between nuclear and cytoplasmic localization (Nuc/Cyt ratio) was individually determined for transfected cell. Shown are the mean ± S.E. of at least four independent experiments relative to HDAC5, which was taken as 1. C, HCA of the subcellular distribution of FLAG-tagged HDAC1, -2, -5, -6, and -10 expressed in UMR106 cells in the absence or presence of 100 nm PTH. D, time courses of the effect of 100 nm PTH on HDACs nuclear to cytoplasmic localization. Shown are relative Nuc/Cyt ratios normalized to time 0. Each data point is the mean ± S.E. of at least three independent experiments.
LepB Induces Nuclear Accumulation of HDAC5 and Inhibits SOST Expression
Having shown that PTH-mediated Sost expression inhibition in UMR106 cells is associated with specific nuclear accumulation of the MEF2 inhibitor HDAC5 we analyzed whether there is a causal relationship. To this end, we induced nuclear accumulation of HDAC5 by inhibiting its nuclear export and analyzed the effect on endogenous Sost expression. HDAC5 is known to be exported by the specific export receptor CRM1 (39), which can be inhibited by the specific antagonist leptomycin B (40). Leptomycin B (LepB) treatment led to strong nuclear accretion of HDAC5 (Fig. 3A) surpassing PTH-induced nuclear accumulation. Addition of PTH to LepB showed a minimal further nuclear accumulation. Next, we analyzed the effect of blocking nuclear import on HDAC5 subcellular distribution. PMA blocks nuclear import by repressing the nuclear localization signal activity (41). Consistently, it led to strong nuclear depletion of HDAC5 (Fig. 3, A and B). Interestingly, PTH was not able to override the action of PMA demonstrating that import inhibition by PMA is dominant over nuclear accumulation by PTH. We also investigated whether a PTH-mediated nuclear increase of HDAC5 involved the cAMP pathway as previously shown to be involved in Sost repression (12). The cAMP stimulator forskolin fully mimicked strong nuclear accretion of HDAC5 as observed with PTH (Fig. 3A). Next, we analyzed the effect of LepB-induced HDAC5 nuclear accumulation on endogenous Sost expression in UMR106 cells. LepB inhibited Sost expression in a time- and dose-dependent manner (Fig. 3, C and D). LepB inhibited Sost expression with an IC50 of 1.2 nm corresponding to inhibition of CRM1 export receptor (42, 43). Maximal repression of about 60% was reached after 4 h. In summary, these data suggest a causal role of HDAC5 nuclear accumulation in PTH-mediated Sost repression.
FIGURE 3.
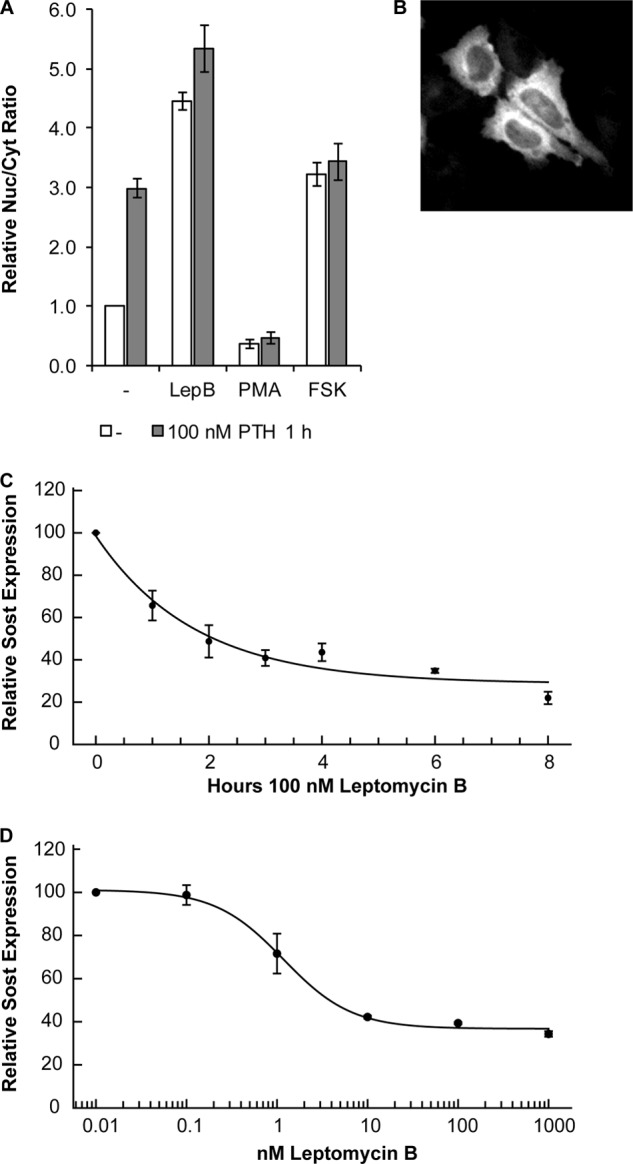
The nuclear export inhibitor LepB inhibits Sost expression. A, regulation of HDAC5-cytoplasmic shuttling by LepB, PMA, and forskolin. UMR106 cells were preincubated for 30 min with 40 nm LepB, 100 nm PMA, 10 μm forskolin or solvent control (−) and then stimulated with 100 nm PTH or solvent control for 1 h followed by HCA of HDAC5 nucleocytoplasmic distribution. Shown are mean ± S.E. of three independent experiments normalized to control. B, representative image of PMA-induced HDAC5-GFP nuclear export. Time (C)- and dose (D)-dependent inhibition of Sost expression by LepB. UMR106 cells were incubated with LepB as indicated in the figure and subsequently Sost expression was analyzed by qPCR. Sost expression levels were normalized to β2-microglobulin expression. Shown are relative Sost expression level mean ± S.E. of two independent experiments.
Mutational Analysis of HDAC5 Shuttling
Serine residues 259 and 498 are critically involved in the regulation of HDAC5 nuclear export (44). Thus, we have analyzed their role in the regulation of HDAC5 subcellular distribution by PTH in UMR106 cells. Mutation of serine 498 to alanine did not significantly change basal and PTH-induced nuclear accumulation compared with wt HDAC5 (Fig. 4A). In contrast, mutation of serine 259 to alanine resulted in an increased basal nuclear localization, which was increased by PTH to a similar level as seen with wt HDAC5. When both serine residues were mutated to alanines (S2A) full nuclear accumulation was observed of this mutant HDAC5 independent of PTH. Next, we analyzed whether MEF2 binding interferes with nucleocytoplasmic shuttling of HDAC5. A MEF2-binding deficient HDAC5 mutant (dMEF2) was created by mutating lysine 184 and arginine 186 to alanines as reported (45). In addition, we generated the double mutant S2A-dMEF2. As shown in Fig. 4A, basal and PTH-induced subcellular distributions of the dMEF2 mutant were not significantly different from those of wt HDAC5, whereas distributions of S2A-dMef2 were very similar to those of the S2A mutant suggesting that MEF2 binding is not involved in the control of nuclear export of HDAC5. Mutants were also tested for their behavior in blocking HDAC5 nuclear import by PMA, which we showed to be dominant over suppressing export by PTH (Fig. 3A). Both single serine mutants (S498A and S259A) were equally well prevented from nuclear import as wt HDAC5 (Fig. 4B). In contrast, the double mutant S2A was accumulated in the nucleus and this could not be inhibited by PMA. In summary, HDAC5 mutational analysis suggests that PTH-mediated export inhibition mainly involves serine 259, but in addition requires serine 498 for full effect. In contrast, one of the two serine residues is sufficient to confer suppression of nuclear import by PMA.
FIGURE 4.
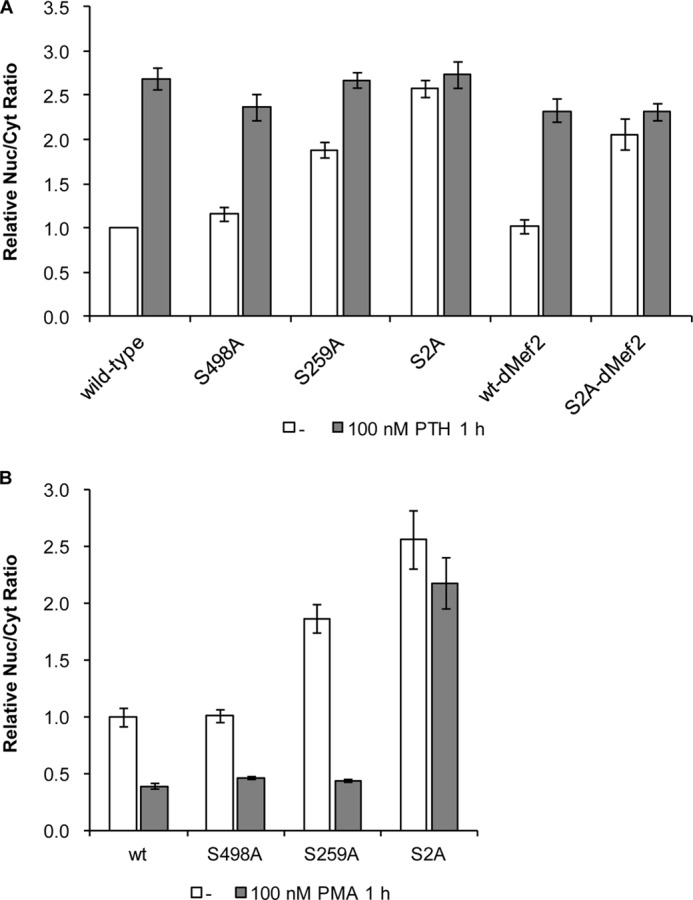
Effect of PTH and PMA on HDAC5 shuttling mutants. A, intracellular localizations of wt HDAC5-GFP and the mutants serine 498 to alanine (S498A), serine 259 to alanine (S259A), both serine to alanine (S2A), lysine 184 and arginine 186 to alanine (dMEF2), and all four residues to alanines (S2A-dMEF2) were analyzed after transfection of the corresponding expression plasmids into UMR106 cells using HCA. Transfected cells were incubated with solvent control, 100 nm PTH (A), or 100 nm PMA (B) for 1 h. Shown are the relative mean ± S.E. of at least four experiments normalized to untreated wt HDAC5.
Nuclear Accumulation of HDAC5 Leads to Speckle Formation and Co-localization with MEF2
Confocal microscopy was used to analyze the subnuclear localization of HDAC5 and association with MEF2. Endogenous MEF2 transcription factors and transfected HDAC5-GFP showed a diffuse nuclear distribution (Fig. 5A). HDAC5-GFP, but not MEF2, was also weakly detected in the cytoplasm. PTH treatment led to complete nuclear localization of HDAC5 and a drastic change in subnuclear localization in the form of speckles (Fig. 5B), which was already hinted at with conventional microscopy (Fig. 2A). Interestingly, PTH also induced speckle formation of MEF2 that co-localized with HDAC5 speckles. Nuclear accumulation of HDCA5 by LepB or using the S2A mutant produced the same nuclear speckle formation and co-association with MEF2 (Fig. 5, C and D). In contrast, the MEF2 binding-deficient HDAC5 mutant (dMEF2) formed PTH-induced nuclear speckles as wt HDAC5, but did not induce co-localization of MEF2 (Fig. 5E) suggesting that HDAC5 recruits MEF2 to nuclear speckle domains. Finally, we used ChIP to directly analyze the effect of PTH on Sost enhancer chromatin binding by HDAC5. UMR106 cells were transfected with V5-tagged HDAC5, followed by treatment with PTH or solvent for 1 h. Subsequently, we immunoprecipitated with anti-V5 or control IgG and used qPCR to measure HDAC5-V5 association with the Sost bone enhancer. PTH massively induced HDAC5-V5 chromatin binding (Fig. 5F). These results further support the conclusions that PTH induces nuclear accumulation of HDAC5 and binding to the Sost bone enhancer in association with MEF2.
FIGURE 5.

PTH-induced nuclear speckle formation of HDAC5 and association with MEF2 and Sost enhancer. UMR106 cells were transfected with wt HDAC5-GFP (A–C), S2A mutant (D), or dMEF2 mutant (E) expression plasmids. 24 h later, subnuclear localization of HDAC-GFP fusion proteins were analyzed by confocal microscopy (middle column) in control cells (A and D) or cells treated with 100 nm PTH (B and E) or 40 nm LepB (C) for 1 h. Localization of endogenous MEF2 transcription factors was done by immunostaining (red fluorescence, left column). Furthermore, an overlay of HDAC5-GFP and MEF2 fluorescence is shown in the right column. F, UMR106 cells were transfected with HDAC5-V5 expression plasmid and incubated with PTH (100 nm) or solvent control for 1 h followed by anti-V5 or control IgG ChIP and quantitative PCR for Sost enhancer. Results are shown as percentage bound relative to total input DNA.
HDAC5 Inhibits SOST Bone Enhancer Activity through MEF2
Previously, we have shown that MEF2 activates SOST gene enhancer activity that is required for SOST expression in adult bone (13). Thus, we have analyzed the effect of varying levels of nuclear HDAC5 on SOST enhancer activity using reporter gene assays. Overexpression of wt HDAC5 had little effect on activity of the SOST promoter, but decreased SOST enhancer activity by 39% (Fig. 6). The nuclear-enriched S2A HDAC5 mutant inhibited SOST enhancer activity much stronger (64%) and again affected SOST promoter activity only marginally. In contrast, HDAC5 mutants deficient of Mef2 binding (HDAC5-dMef2 and HDAC5-S2A_dMef2) only marginally inhibited SOST bone enhancer activity. These data suggest that nuclear HDAC5 inhibits SOST bone enhancer activity by masking MEF2.
FIGURE 6.
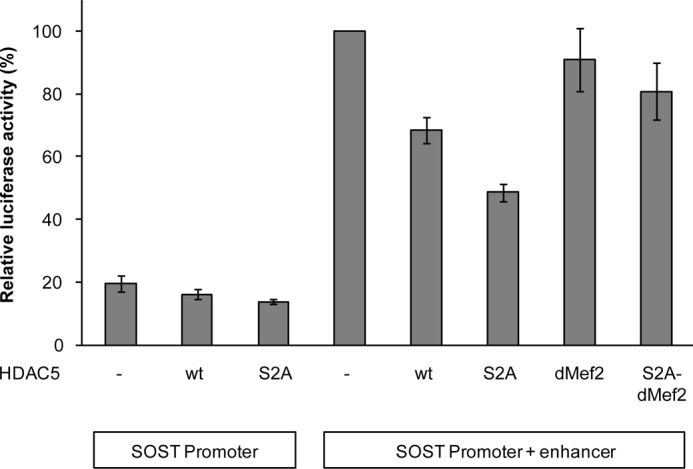
Nuclear HDAC5 inhibits SOST bone enhancer activity by masking MEF2. UMR106 cells were transiently transfected with control expression vector (−), expression plasmids for wt or mutant HDAC5s, and luciferase reporter plasmids as indicated in the figure. After 48 h, cells were lysed and luciferase activities were determined. Shown are mean ± S.E. of relative luciferase activities from four independent experiments.
Pharmacological Inhibition of HDACs
To investigate whether HDAC5 catalytic activity is involved in Sost gene regulation, UMR106 cells were treated with a series of HDIs analyzing their effect on Sost expression. First, we tested the specific and potent class II HDI compound 2 (32) inhibiting HDAC4 and -5 catalytic activities with IC50 values in the lower nanomolar range. Compound 2 was re-synthesized and its specificity confirmed (Table 1). It only weakly inhibited Sost expression at 10 μm (Fig. 7A) and did not relieve PTH-induced SOST repression (data not shown) indicating that HDAC5 catalytic activity is not required for its regulation of Sost expression. In contrast, the pan-HDI TSA (46, 47) very potently inhibited endogenous Sost expression >95% with an IC50 of 2.4 nm suggesting that Sost expression may depend on class I HDAC catalytic activity. To corroborate and further analyze the requirement of class I HDAC isoform activities for Sost expression we tested the specific class I HDAC inhibitor apicidin (46–48). Apicidin efficiently inhibited Sost expression >95% with an IC50 of 24 nm. Because controversial data have been reported on inhibition of HDAC1 by apicidin (46, 48), we further used HDI MS-275, which preferentially inhibits HDAC1 (46, 47, 49, 50). In contrast to TSA and apicidin, MS-275 did not inhibit SOST expression, but showed a bell-shaped about 2-fold stimulation with a maximum at 1 μm. A similar response was obtained with MGCD0103 (51, 52), another potent HDAC1 inhibitor (data not shown). As the class I HDAC8 is not expressed in UMR106 cells (Fig. 1), these data suggest that Sost expression requires HDAC2 and -3 catalytic activities. Overexpression of HDACs2 and -3 did not significantly affect SOST promoter and enhancer activities indicating that their activities are not limiting in UMR106 cells (data not shown). In summary, these data suggest that the catalytic activity of HDCA5 is not required for its role in PTH-mediated Sost repression. In contrast, catalytic activities of HDACs 2 and 3 appear to be required for Sost expression in UMR106 cells.
FIGURE 7.
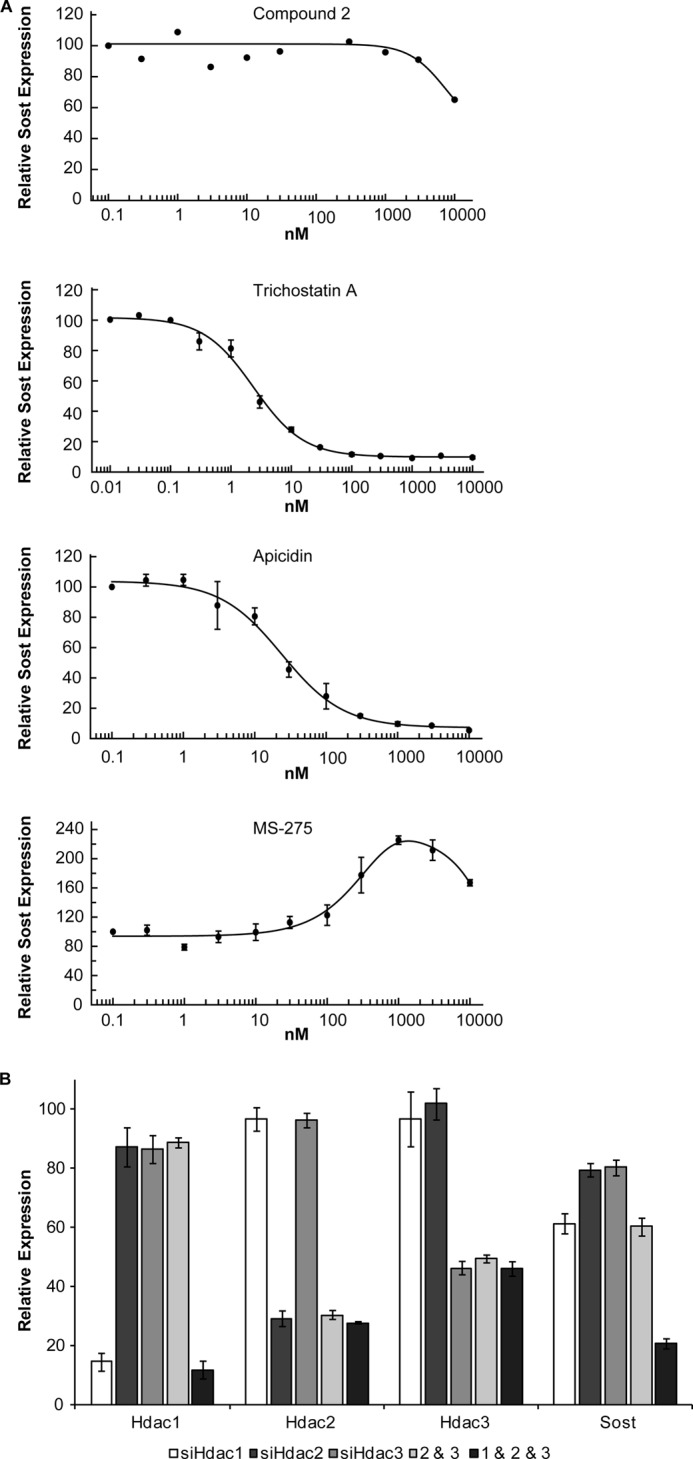
The class I HDACs 1, 2, and 3 are required for Sost expression. A, dose-response curves of HDIs compound 2, TSA, apicidin, and MS-275. UMR106 cells were incubated with increasing concentrations of HDIs for 4 h followed by Sost expression analysis using qPCR. Shown are mean ± S.E. of two independent experiments. B, siRNA-mediated knock-down of Hdacs 1, 2, and 3 and effect on Sost expression. UMR106 cells were transfected upon seeding with individual HDAC siRNAs or combinations thereof as indicated in the figure. After 72 h, gene expression levels of Hdac1, -2, -3, and Sost were determined using qPCR. Expressions were normalized to β2-microglobulin expression and non-targeting control siRNA, which was set at 100%. Shown are mean ± S.E. of at least 2 independent experiments.
siRNA-mediated Knock-down of Class I HDACs
Having shown that Sost expression requires class I HDAC catalytic activities we analyzed the role of class I HDAC isoforms in Sost expression by siRNA-mediated knock-down (Fig. 7B). UMR106 cells were transfected with siRNAs against class I Hdacs 1, 2, and 3, which are expressed in these cells (Fig. 1). Specific down-regulation of the corresponding genes was observed achieving reductions of 85% for Hdac1, 71% for Hdac2, and 54% for Hdac3 expressions. Hdac1, -2, and -3 knock-down led to SOST expression inhibitions of 39, 21, and 20%, respectively. A combination of Hdac2 and -3 siRNAs resulted in an additive Sost expression inhibition of about 40%, and a synergistic inhibition of 79% was observed when all three siRNAs were combined. These results suggest all three Hdac1, -2, and -3 isoforms are required for full Sost expression in UMR106 cells.
Acetylome Analysis
Having shown that HDAC2 and -3 catalytic activities are required for Sost expression in UMR106 cells we performed an acetylome analysis to elucidate potential target proteins using the specific HDI apicidin. It inhibited global cellular HDAC activity >97% (Fig. 8). In contrast, PTH did not significantly change overall cellular HDAC activity. To identify potential HDAC targets we preformed large scale mapping of lysine acetylation sites and measured acetylation changes in response to the HDAC inhibitor apicidin using immunoaffinity enrichment of acetyllysine peptides and SILAC-based quantitative mass spectrometry (33). As expected, apicidin treatment resulted in significantly increased acetylation levels (Fig. 9B), whereas there was little effect on overall protein levels judging from the distribution of all peptides, acetyllysine and non-acetyllysine-containing peptides, which co-purified (Fig. 9A). Because of the asymmetric distribution of the acetyllysine-peptides, the ratio distribution of all peptides was used to determine the cutoffs for significance (Fig. 9A). Based on these cutoffs, apicidin treatment triggered increased acetylation of 40 different peptides (Table 2). As expected and confirming inhibition of HDAC activities, many hyper-acetylated proteins were histones and histone-related proteins. These proteins are unlikely to be specifically involved in Sost gene regulation. The non-histone proteins with increased acetylation upon apicidin treatment are listed in Table 3. These 12 proteins are potential targets of HDAC2 and -3 in UMR106 cells that might play a role in Sost gene regulation. In summary, we identified a number of non-histone proteins as potential targets for HDAC2 and -3-mediated Sost gene expression stimulation.
FIGURE 8.
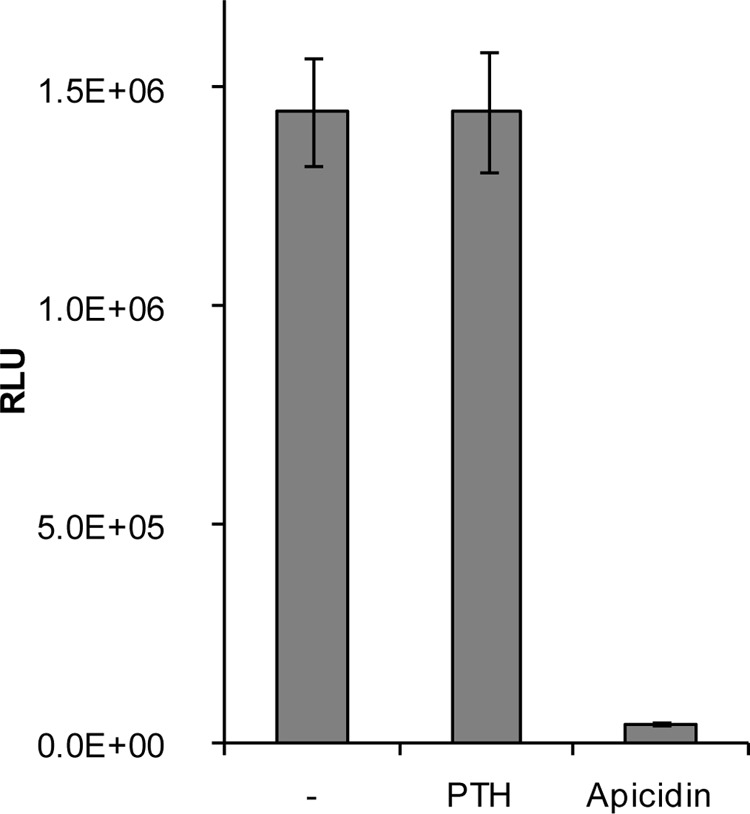
Global cellular HDAC activity is suppressed by apicidin but not by PTH. Overall relative HDAC activities were determined in UMR106 cells. Cells were treated with solvent control (-), 1 μm PTH, or 10 μm apicidin for 1 h before measuring total HDAC activities using a luciferase reporter kit. Shown are mean ± S.E. of a representative experiment.
FIGURE 9.

Analysis of whole cell protein acetylation in response to apicidin treatment of UMR106 cells. Protein acetylation was analyzed by SILAC-based quantitative mass spectrometry of immuno-enriched acetyllysine peptides. Shown are histogram distributions of the apicidin-treated/untreated ratios for all peptides in the acetyllysine-enriched sample (A) versus only acetyllysine-containing peptides (B). Vertical dotted lines indicate the 95% confidence interval determined for all peptides and applied to acetyllysine-containing peptides (average ± 2 S.D.).
TABLE 2.
Peptides with increased acetylation based on log2 ratio significance cutoffs −0.44 and 1.04 from all peptides
Acetyl (K) probabilities display the sequence with site assignment probabilities following each Lys residue (between 0 and 1). The sum of the probabilities equals the total number of acetyllysines in the peptide, i.e. the value in the column. Number of acetyl (Lys) shows the number of acetylated lysines in the peptide.
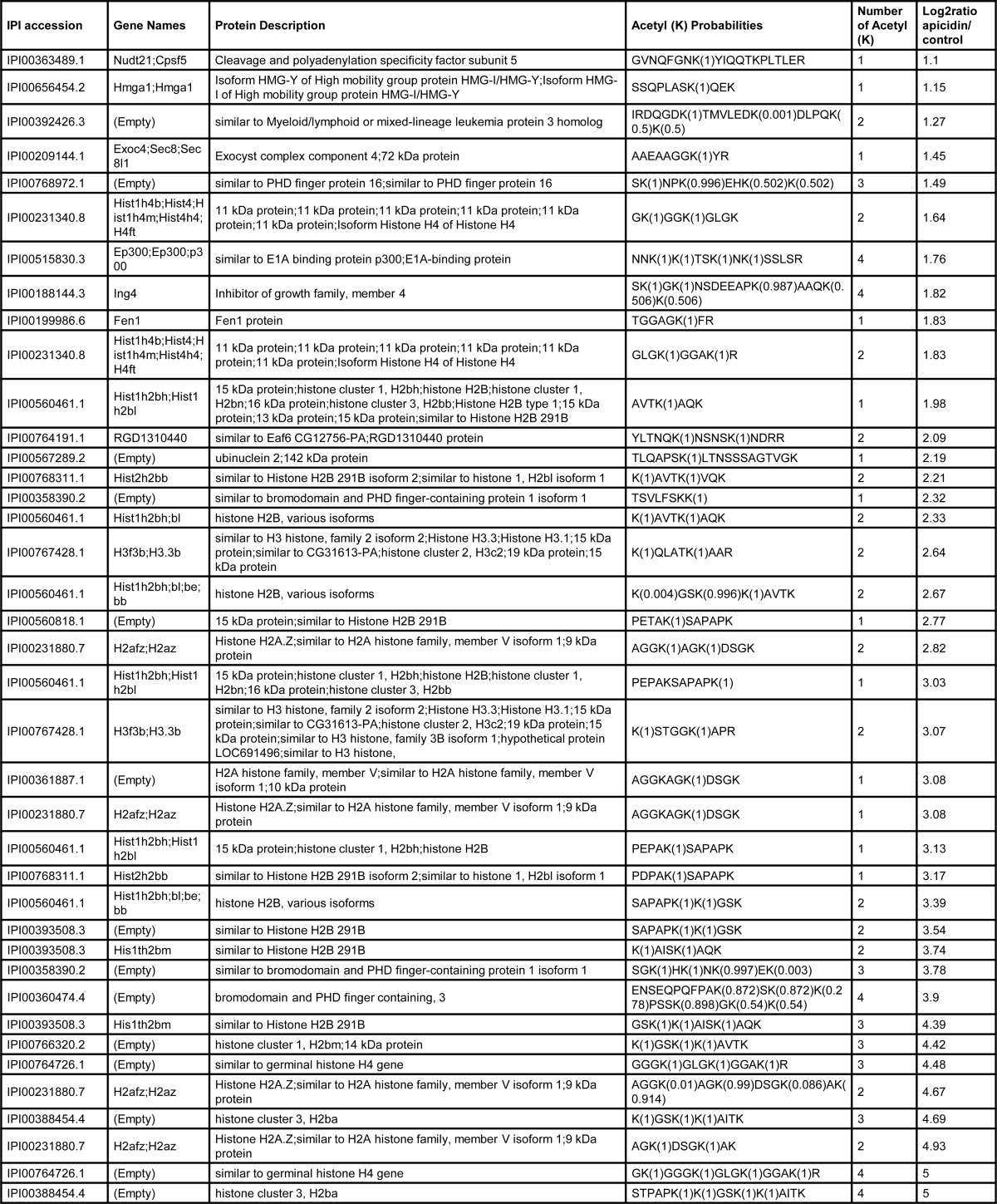
TABLE 3.
Non-histone proteins whose acetylation is increased more than 2-fold by apicidin treatment of UMR106 cells
| Symbol | Full name | Uniprot identifier | Peptide sequences | Ratio apicidin/Ctrl | Modifications |
|---|---|---|---|---|---|
| Brpf1 | Bromodomain and PHD finger-containing, 1 | D3ZIA0 | SGKHKNKEK | 13.71 | 3 Acetyl (K) |
| TSVLFSKK | 4.98 | Acetyl (K) | |||
| Brpf3 | Bromodomain and PHD finger containing, 3 | D3ZMD3 | ENSEQPQFPAKSKKPSSKGKK | 14.90 | 4 Acetyl (K) |
| Ep300 | E1A-binding protein p300 | Q5RJS0 | NNKKTSKNKSSLSR | 3.39 | 4 Acetyl (K) |
| Exoc4 | Exocyst complex component 4 | Q62824 | AAEAAGGKYR | 2.73 | Acetyl (K), Acetyl (Protein N-term) |
| Fen1 | Flap structure-specific endonuclease 1 | Q5XIP6 | TGGAGKFR | 3.55 | Acetyl (K) |
| Hmga1 | High mobility group AT-hook 1 | Q8K585 | SSQPLASKQEK | 2.22 | Acetyl (K) |
| Ing4 | Inhibitor of growth family, member 4 | A1L130 | SKGKNSDEEAPKAAQKK | 3.54 | 4 Acetyl (K) |
| SKGKNSDEEAPKAAQK | 1.90 | 3 Acetyl (K) | |||
| QIESSDYDSSSSKGKR | 1.61 | 2 Acetyl (K) | |||
| Meaf6 | MYST/Esa1-associated factor 6 | D3ZFLD | YLTNQKNSNSKNDR | 4.38 | 2 Acetyl (K) |
| Mll3 | Myeloid/lymphoid or mixed-lineage leukemia 3 | Q8BRH4 | SKNPKEHKK | 2.94 | 3 Acetyl (K) |
| IRDQGDKTMVLEDKDLPQKK | 2.41 | 2 Acetyl (K), Oxidation (M) | |||
| Nudt21 | Nudix (Nucleoside diphosphate-linked moiety X)-type motif 21 | Q4KM65 | GVNQFGNKYIQQTKPLTLER | 2.14 | Acetyl (K) |
| Rps12 | Ribosomal protein S12 | Q6PDW1 | AEEGIAAGGVMDVNTALQEVLK | 73.48 | Acetyl (Protein N-terminal), Oxidation (M) |
| Ubn2 | Ubinuclein 2 | D4A666 | TLQAPSKLTNSSSAGTVGK | 4.56 | Acetyl (K) |
DISCUSSION
Osteocyte-secreted sclerostin is a key repressor of bone formation. Its absence leads to massive bone overgrowth of the whole skeleton as observed in sclerosteosis and Van Buchem disease patients. On this basis, novel bone anabolic osteoporosis therapies are being developed, for example, by suppressing the activity of sclerostin with antibodies (53). With the exception of daily PTH injections there are currently no treatments available for the restoration of lost bone in severe osteoporosis patients. Interestingly, stimulation of bone formation by intermittent PTH involves suppression of Sost expression as an important mechanism further portending to the sclerostin pathway as a central control mechanism in adult bone metabolism (10, 12). Despite its significance in bone mass control regulation of SOST expression is only poorly understood. Previously, we have shown that PTH down-regulation of Sost expression is mediated by interfering with a crucial MEF2 binding site in the distant bone enhancer of the SOST gene (13). Our present results further elucidate the molecular mechanisms of SOST gene control by uncovering a PTH-induced repressive role of class IIa HDAC5 acting on MEF2 transcription factors and an unanticipated stimulatory role of class I HDACs catalytic activity for which we identified potential targets using a comprehensive acetylome analysis. Our study is the first report suggesting a role of HDACs in osteocytes to regulate adult bone metabolism via controlling SOST gene expression. In addition, we demonstrate opposite effects of class I and IIa HDACs on Sost gene regulation and consequently on adult bone mass control via the same pathway. Expression analysis by qPCR showed expression of several members of class I and IIa HDACs in osteocytic UMR106 cells, but only HDAC5 showed nuclear accumulation upon PTH treatment. In addition to nuclear buildup, PTH induced localization of HDAC5 to a multitude of small subnuclear bodies and recruitment of MEF2s to the same compartments. Similar structures called nuclear speckles have been described for the recruitment of HDAC4 and -5 to chromatin-bound MEF2C and nuclear receptors by the nuclear corepressor SMRTe (54). Whereas SMRTe similarly recruited both HDAC4 and -5 into the nucleus for repression of target transcription factors such as MEF2s, we observed selective employment of HDAC5 by PTH for Sost gene repression. HDAC4 was expressed at much lower levels in UMR106 cells, was mainly located in the cytoplasm, and was not imported into the nucleus by PTH. Furthermore, increasing nuclear levels of HDAC5 by overexpression or by blocking its nuclear export receptor CRM1 with LepB inhibited expression in SOST reporter gene assays and the endogenous Sost gene, respectively. Finally, we observed massive recruitment of HDAC5 to the MEF2 response element-containing Sost bone enhancer region using ChIP. These results strongly suggest a critical co-repressor role of HDAC5 in SOST gene regulation and, thus, bone mass control. This is also supported by the identification of HDAC5 as one of 20 bone mineral density loci in a meta-analysis of five genome-wide association studies of femoral neck and lumbar spine bone mineral density in almost 20,000 subjects (14) and by the discovery that microRNA-2861-mediated HDAC5 repression regulates osteoblast differentiation and bone mass in humans (55). Interestingly, HDAC4, but not other class IIa HDACs, are critically involved in limiting chondrocyte hypertrophy during bone development demonstrating a functional specification of class IIa HDACs in controlling bone development and mature bone homeostasis (56). The transcriptional activity of class IIa HDACs is regulated by dynamic nucleocytoplasmic shuttling (44). Our demonstration of increased nuclear HDAC5 levels by blocking its export receptor CRM1 with LepB and decreased nuclear HDCA5 levels by inhibiting nuclear import with PMA support such an active equilibrium of cytoplasmic and nuclear HDAC5 in UMR106 cells. This is further supported by the rapid disappearance of PTH-induced nuclear accretion of HDAC5 within 10 min when PTH was washed out (data not shown). Consistent with our earlier report that Sost gene repression by PTH involves the cAMP pathway, PTH-induced HDAC5 nuclear accumulation was fully mimicked by cAMP induction with forskolin suggesting an important role for nuclear HDAC5 in PTH-induced Sost gene repression. However, despite inducing higher nuclear accumulation of HDAC5 than with PTH LepB maximally suppressed SOST expression only by 60–70% (Fig. 3C) compared with 90% by PTH (12). Thus, there may be further mechanisms of PTH-mediated SOST gene repression independent of HDAC5 nuclear accumulation, which remain to be uncovered.
Two conserved serine residues in the N-terminal half of HDAC5 provide phosphorylation sites for protein kinase D and calcium/calmodulin-dependent protein kinase-mediated regulation of nucleocytoplasmic shuttling (17, 44). HDACs phosphorylated at these sites are bound by the chaperone protein 14-3-3, which frees HDAC-bound MEF2s and induces nuclear expulsion by uncovering a nuclear export sequence that is recognized by the export receptor CRM1. We have observed that the two serine residues are also critically involved in nuclear-cytoplasmic localization of HDAC5 in UMR106 cells. When both were mutated to alanines HDAC5 was fully accumulated in the nucleus as seen after PTH treatment. However, in contrast to the reported results obtained in COS cells (44) Ser-498 alone had little effect on shuttling, whereas Ser-259 alone was already responsible for about half of the effect suggesting that it plays a dominant role in PTH-mediated control of HDAC5 subcellular trafficking in osteocytes. A number of kinase pathways have been reported to be involved in the phosphorylation of class IIa HDACs in different tissues and in response to a variety of stimuli (22). In addition, myosin phosphatase and protein phosphatase 2A have been described to dephosphorylate class IIa HDACs. Despite this plethora of pathways there are examples of distinct pathways regulating specific HDACs suggesting high specificity for particular physiological signals in particular tissues. For example, protein kinase D and microtubule affinity-regulating kinases are specifically involved in HDAC-mediated regulation of cardiac growth and remodeling genes in pathological myocyte hypertrophy (20). Furthermore, CaMKII specifically phosphorylates HDAC4, but not HDAC5, -7, and -9, in this process. The signaling pathways involved in PTH-mediated regulation of HDAC5 nucleocytoplasmic shuttling in osteocytes remain to be elucidated. A likely pathway candidate is the one involved in PTH-related peptide-mediated inhibition of chondrocyte hypertrophy (57). It involves activation of protein phosphatase 2A, which dephosphorylates HDAC4 leading to repression of MEF2C-mediated collagen X expression. Activation of protein phosphatase 2A was proposed to occur by cAMP-stimulated PKA. However, we have not observed an inhibition of Sost repression by PTH using the specific PKA inhibitor H89 or the protein phosphatase 2A inhibitor calyculin A (data not shown). Thus, comprehensive genomics and proteomics approaches will be required to uncover the pathway that signals from PTH-induced cAMP to nuclear accumulation of HDAC5 in osteocytes. Class IIa HDACs nucleocytoplasmic shuttling is also regulated at nuclear import. PMA was shown to inhibit HDAC4 nuclear import by repressing nuclear localization signals also involving the chaperone protein 14-3-3 (41). Our results showing cytoplasmic accumulation of HDAC5 by PMA are consistent with this observation. Along the same line, elimination of the two crucial phosphoserine binding sites for 14-3-3 prohibited PMA action. In addition, we have investigated the individual contribution of the two serine residues and demonstrate that either of them is enough to confer full cytoplasmic retention by PMA. These data suggest that the binding requirements of 14-3-3 to HDACs are diverging between its roles in nuclear export and import. Moreover, we observed that HDAC5 nuclear import inhibition by PMA was dominant over PTH-induced nuclear accumulation. Obviously, inhibition of HDAC5 nuclear export by PTH cannot lead to nuclear accumulation if nuclear import of cytoplasm-produced HDAC5 is prevented in the first place. However, this result also suggests that if PTH stimulated nuclear import of HDAC5 it would have to act more upstream than PMA. The signaling pathways regulating class IIa HDACs nuclear import are only poorly understood. The action of PMA appears to be mediated by PKCδ and PKCϵ, but not PKCα, however, not acting directly on HDACs (41).
Our results using the specific class II HDI compound 2 (32) suggest that HDAC5 catalytic activity is not required for PTH-mediated inhibition of Sost expression. This is consistent with a large body of evidence showing that class IIa HDACs repress target transcription factors by direct protein-protein binding and recruitment of co-repressors such as SMRT/N-CoR independently of their inefficient catalytic activity (17, 22). Surprisingly, the pan-specific HDI TSA fully suppressed Sost expression in UMR106 cells like PTH indicating a positive effect of class I HDACs in Sost expression. HDAC I activities do not appear to be limiting in UMR106 cells as overexpression of HDACs1–3 did not increase Sost expression. Consistently, we found high expression levels of HDAC1 and -2. Further analysis using class I subtype-specific HDIs combined with our HDAC expression analysis suggest that the catalytic activity of HDAC2 and -3, but not 1 and 8 are required. Gene expression knock-down experiments using RNA interference confirmed the stimulatory role of HDAC2 and -3, but showed an additional role for HDAC1. These data suggest that all three class I HDACs expressed in UMR106 cells are required for Sost expression, but only the catalytic activity of HDAC2 and -3. Direct deacetylation of MEF2D by HDAC3 has been reported (21). However, this resulted in a repression of MEF2-dependent transcription and inhibition of myogenesis. Thus, it remains to be analyzed how HDAC1, -2, and -3 activate Sost transcription and whether it involves direct actions on MEF2s. To identify putative targets for HDAC1, -2, and -3 regulating Sost expression in osteocytes we did a comprehensive acetylome analysis. The specific HDAC2, -3, and -8 inhibitor apicidin almost completely suppressed HDAC activity in UMR106 cells. Because we could not detect HDAC8 expression in these cells this suggests that HDAC2 and -3 are responsible for by far most HDAC activities. PTH had no effect on overall HDAC activity. This is consistent with our findings that PTH-induced Sost gene repression by HDAC5 did not require catalytic activity of HDAC5. Furthermore, we did not observe differences in protein acetylation by PTH treatment. Analysis of acetylated proteins after apicidin treatment revealed a number of non-histone proteins as promising candidates for regulating Sost expression by HDAC2 and -3. Most of them are associated with transcription regulation. Brpf1, Brpf3, Ep300, Ing4, and Meaf6 are components of chromatin remodeling and transcription co-activator multiprotein complexes with histone acetyltransferase activities. Hmga1 and Nudt21 are involved in RNA processing, and Mll3 possesses histone methyltransferase activity.
A specific role of these candidates in Sost gene regulation remains to investigated. Interestingly, our acetylome analysis did not detect MEF2s as putative targets indicating that they are not involved in Sost gene regulation by HDAC2 and -3 actions. Thus, it will be interesting to elucidate the upstream signaling pathways and downstream transcription co-factors that regulate Sost gene expression by class I HDACs. Last but not least, class I HDIs and particularly, specific HDIs of HDAC2 and -3 represent a novel approach for the development of novel bone forming osteoporosis therapies.
Acknowledgments
We are grateful to Simone Degen and Heidi Jeker for expert technical help. We thank Andreas Huber and Stephan Ruetz for their advice on HCA, and Andrew W. Dunbar and Bruno Tigani for guidance using confocal microscopy. We also thank Olga Kolesow and Serge Summermatter for technical help and expert advice on ChIP, respectively. Finally, we thank Joachim Nozulak for re-synthesis and testing of the class II HDAC inhibitor compound 2.
Footnotes
- SOST
- sclerostin
- EGFP
- enhanced green fluorescent protein
- HCA
- high content analysis
- HDAC
- histone deacetylase
- HDI
- HDAC inhibitor
- LepB
- leptomycin B
- MEF2
- myocyte enhancer factor 2
- PMA
- phorbol 12-myristate 13-acetate
- PTH
- parathyroid hormone
- qPCR
- quantitative PCR
- SILAC
- stable-isotope labeling with amino acids in cell culture
- TSA
- trichostatin A
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1. Moester M. J., Papapoulos S. E., Löwik C. W., van Bezooijen R. L. (2010) Sclerostin: current knowledge and future perspectives. Calcif. Tissue Int. 87, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li X., Ominsky M. S., Niu Q. T., Sun N., Daugherty B., D'Agostin D., Kurahara C., Gao Y., Cao J., Gong J., Asuncion F., Barrero M., Warmington K., Dwyer D., Stolina M., Morony S., Sarosi I., Kostenuik P. J., Lacey D. L., Simonet W. S., Ke H. Z., Paszty C. (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 23, 860–869 [DOI] [PubMed] [Google Scholar]
- 3. Kramer I., Loots G. G., Studer A., Keller H., Kneissel M. (2010) Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J. Bone Miner. Res. 25, 178–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Loots G. G., Kneissel M., Keller H., Baptist M., Chang J., Collette N. M., Ovcharenko D., Plajzer-Frick I., Rubin E. M. (2005) Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 15, 928–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winkler D. G., Sutherland M. K., Geoghegan J. C., Yu C., Hayes T., Skonier J. E., Shpektor D., Jonas M., Kovacevich B. R., Staehling-Hampton K., Appleby M., Brunkow M. E., Latham J. A. (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 22, 6267–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glass D. A., 2nd, Bialek P., Ahn J. D., Starbuck M., Patel M. S., Clevers H., Taketo M. M., Long F., McMahon A. P., Lang R. A., Karsenty G. (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8, 751–764 [DOI] [PubMed] [Google Scholar]
- 7. Holmen S. L., Zylstra C. R., Mukherjee A., Sigler R. E., Faugere M. C., Bouxsein M. L., Deng L., Clemens T. L., Williams B. O. (2005) Essential role of β-catenin in postnatal bone acquisition. J. Biol. Chem. 280, 21162–21168 [DOI] [PubMed] [Google Scholar]
- 8. Johnson M. L., Kamel M. A. (2007) The Wnt signaling pathway and bone metabolism. Curr. Opin. Rheumatol. 19, 376–382 [DOI] [PubMed] [Google Scholar]
- 9. Glass D. A., 2nd, Karsenty G. (2007) Minireview: in vivo analysis of Wnt signaling in bone. Endocrinology 148, 2630–2634 [DOI] [PubMed] [Google Scholar]
- 10. Kramer I., Keller H., Leupin O., Kneissel M. (2010) Does osteocytic SOST suppression mediate PTH bone anabolism? Trends Endocrinol. Metab. 21, 237–244 [DOI] [PubMed] [Google Scholar]
- 11. Bellido T., Ali A. A., Gubrij I., Plotkin L. I., Fu Q., O'Brien C. A., Manolagas S. C., Jilka R. L. (2005) Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146, 4577–4583 [DOI] [PubMed] [Google Scholar]
- 12. Keller H., Kneissel M. (2005) SOST is a target gene for PTH in bone. Bone 37, 148–158 [DOI] [PubMed] [Google Scholar]
- 13. Leupin O., Kramer I., Collette N. M., Loots G. G., Natt F., Kneissel M., Keller H. (2007) Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J. Bone Miner. Res. 22, 1957–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rivadeneira F., Styrkársdottir U., Estrada K., Halldórsson B. V., Hsu Y. H., Richards J. B., Zillikens M. C., Kavvoura F. K., Amin N., Aulchenko Y. S., Cupples L. A., Deloukas P., Demissie S., Grundberg E., Hofman A., Kong A., Karasik D., van Meurs J. B., Oostra B., Pastinen T., Pols H. A., Sigurdsson G., Soranzo N., Thorleifsson G., Thorsteinsdottir U., Williams F. M., Wilson S. G., Zhou Y., Ralston S. H., van Duijn C. M., Spector T., Kiel D. P., Stefansson K., Ioannidis J. P., Uitterlinden A. G. (2009) Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet. 41, 1199–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Potthoff M. J., Olson E. N. (2007) MEF2: a central regulator of diverse developmental programs. Development 134, 4131–4140 [DOI] [PubMed] [Google Scholar]
- 16. Arnold M. A., Kim Y., Czubryt M. P., Phan D., McAnally J., Qi X., Shelton J. M., Richardson J. A., Bassel-Duby R., Olson E. N. (2007) MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 12, 377–389 [DOI] [PubMed] [Google Scholar]
- 17. McKinsey T. A., Zhang C. L., Olson E. N. (2002) MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 27, 40–47 [DOI] [PubMed] [Google Scholar]
- 18. Grégoire S., Yang X. J. (2005) Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol. Cell. Biol. 25, 2273–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma K., Chan J. K., Zhu G., Wu Z. (2005) Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol. Cell. Biol. 25, 3575–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKinsey T. A., Olson E. N. (2005) Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J. Clin. Investig. 115, 538–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grégoire S., Xiao L., Nie J., Zhang X., Xu M., Li J., Wong J., Seto E., Yang X. J. (2007) Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol. Cell. Biol. 27, 1280–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parra M., Verdin E. (2010) Regulatory signal transduction pathways for class IIa histone deacetylases. Curr. Opin. Pharmacol. 10, 454–460 [DOI] [PubMed] [Google Scholar]
- 23. Haberland M., Montgomery R. L., Olson E. N. (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim H. N., Ha H., Lee J. H., Jung K., Yang D., Woo K. M., Lee Z. H. (2009) Trichostatin A inhibits osteoclastogenesis and bone resorption by suppressing the induction of c-Fos by RANKL. Eur. J. Pharmacol. 623, 22–29 [DOI] [PubMed] [Google Scholar]
- 25. Nakamura T., Kukita T., Shobuike T., Nagata K., Wu Z., Ogawa K., Hotokebuchi T., Kohashi O., Kukita A. (2005) Inhibition of histone deacetylase suppresses osteoclastogenesis and bone destruction by inducing IFN-β production. J. Immunol. 175, 5809–5816 [DOI] [PubMed] [Google Scholar]
- 26. Rahman M. M., Kukita A., Kukita T., Shobuike T., Nakamura T., Kohashi O. (2003) Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood 101, 3451–3459 [DOI] [PubMed] [Google Scholar]
- 27. Yi T., Baek J. H., Kim H. J., Choi M. H., Seo S. B., Ryoo H. M., Kim G. S., Woo K. M. (2007) Trichostatin A-mediated upregulation of p21(WAF1) contributes to osteoclast apoptosis. Exp. Mol. Med. 39, 213–221 [DOI] [PubMed] [Google Scholar]
- 28. Pham L., Kaiser B., Romsa A., Schwarz T., Gopalakrishnan R., Jensen E. D., Mansky K. C. (2011) HDAC3 and HDAC7 have opposite effects on osteoclast differentiation. J. Biol. Chem. 286, 12056–12065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schroeder T. M., Westendorf J. J. (2005) Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 20, 2254–2263 [DOI] [PubMed] [Google Scholar]
- 30. Jung H. M., Song G. A., Lee Y. K., Baek J. H., Ryoo H. M., Kim G. S., Choung P. H., Woo K. M. (2010) Modulation of the resorption and osteoconductivity of α-calcium sulfate by histone deacetylase inhibitors. Biomaterials 31, 29–37 [DOI] [PubMed] [Google Scholar]
- 31. McGee-Lawrence M. E., Westendorf J. J. (2011) Histone deacetylases in skeletal development and bone mass maintenance. Gene 474, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ontoria J. M., Altamura S., Di Marco A., Ferrigno F., Laufer R., Muraglia E., Palumbi M. C., Rowley M., Scarpelli R., Schultz-Fademrecht C., Serafini S., Steinkühler C., Jones P. (2009) Identification of novel, selective, and stable inhibitors of class II histone deacetylases. Validation studies of the inhibition of the enzymatic activity of HDAC4 by small molecules as a novel approach for cancer therapy. J. Med. Chem. 52, 6782–6789 [DOI] [PubMed] [Google Scholar]
- 33. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 34. Walkinshaw D. R., Tahmasebi S., Bertos N. R., Yang X. J. (2008) Histone deacetylases as transducers and targets of nuclear signaling. J. Cell. Biochem. 104, 1541–1552 [DOI] [PubMed] [Google Scholar]
- 35. Longworth M. S., Laimins L. A. (2006) Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene 25, 4495–4500 [DOI] [PubMed] [Google Scholar]
- 36. Gao Z., He Q., Peng B., Chiao P. J., Ye J. (2006) Regulation of nuclear translocation of HDAC3 by IκBα is required for tumor necrosis factor inhibition of peroxisome proliferator-activated receptor γ function. J. Biol. Chem. 281, 4540–4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Escaffit F., Vaute O., Chevillard-Briet M., Segui B., Takami Y., Nakayama T., Trouche D. (2007) Cleavage and cytoplasmic relocalization of histone deacetylase 3 are important for apoptosis progression. Mol. Cell. Biol. 27, 554–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu X., Vatsyayan J., Gao C., Bakkenist C. J., Hu J. (2010) HDAC2 promotes eIF4E sumoylation and activates mRNA translation gene specifically. J. Biol. Chem. 285, 18139–18143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harrison B. C., Roberts C. R., Hood D. B., Sweeney M., Gould J. M., Bush E. W., McKinsey T. A. (2004) The CRM1 nuclear export receptor controls pathological cardiac gene expression. Mol. Cell. Biol. 24, 10636–10649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kudo N., Wolff B., Sekimoto T., Schreiner E. P., Yoneda Y., Yanagida M., Horinouchi S., Yoshida M. (1998) Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 242, 540–547 [DOI] [PubMed] [Google Scholar]
- 41. Nishino T. G., Miyazaki M., Hoshino H., Miwa Y., Horinouchi S., Yoshida M. (2008) 14–3-3 regulates the nuclear import of class IIa histone deacetylases. Biochem. Biophys. Res. Commun. 377, 852–856 [DOI] [PubMed] [Google Scholar]
- 42. Kwon Y. J., Genovesio A., Youl Kim N., Hi Chul K., Jung S., David-Watine B., Nehrbass U., Emans N. (2007) High-content classification of nucleocytoplasmic import or export inhibitors. J. Biomol. Screen. 12, 621–627 [DOI] [PubMed] [Google Scholar]
- 43. Zanella F., Rosado A., Blanco F., Henderson B. R., Carnero A., Link W. (2007) An HTS approach to screen for antagonists of the nuclear export machinery using high content cell-based assays. Assay. Drug Dev. Technol. 5, 333–341 [DOI] [PubMed] [Google Scholar]
- 44. McKinsey T. A., Zhang C. L., Lu J., Olson E. N. (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Urbich C., Rössig L., Kaluza D., Potente M., Boeckel J. N., Knau A., Diehl F., Geng J. G., Hofmann W. K., Zeiher A. M., Dimmeler S. (2009) HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood 113, 5669–5679 [DOI] [PubMed] [Google Scholar]
- 46. Khan N., Jeffers M., Kumar S., Hackett C., Boldog F., Khramtsov N., Qian X., Mills E., Berghs S. C., Carey N., Finn P. W., Collins L. S., Tumber A., Ritchie J. W., Jensen P. B., Lichenstein H. S., Sehested M. (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 409, 581–589 [DOI] [PubMed] [Google Scholar]
- 47. Bradner J. E., West N., Grachan M. L., Greenberg E. F., Haggarty S. J., Warnow T., Mazitschek R. (2010) Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huber K., Doyon G., Plaks J., Fyne E., Mellors J. W., Sluis-Cremer N. (2011) Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 286, 22211–22218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Soragni E., Xu C., Plasterer H. L., Jacques V., Rusche J. R., Gottesfeld J. M. (2012) Rationale for the development of 2-aminobenzamide histone deacetylase inhibitors as therapeutics for Friedreich ataxia. J. Child Neurol. 27, 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thomas E. A. (2014) Involvement of HDAC1 and HDAC3 in the pathology of polyglutamine disorders: therapeutic implications for selective HDAC1/HDAC3 inhibitors. Pharmaceuticals 7, 634–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fournel M., Bonfils C., Hou Y., Yan P. T., Trachy-Bourget M. C., Kalita A., Liu J., Lu A. H., Zhou N. Z., Robert M. F., Gillespie J., Wang J. J., Ste-Croix H., Rahil J., Lefebvre S., Moradei O., Delorme D., Macleod A. R., Besterman J. M., Li Z. (2008) MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 7, 759–768 [DOI] [PubMed] [Google Scholar]
- 52. Buglio D., Mamidipudi V., Khaskhely N. M., Brady H., Heise C., Besterman J., Martell R. E., MacBeth K., Younes A. (2010) The class-I HDAC inhibitor MGCD0103 induces apoptosis in Hodgkin lymphoma cell lines and synergizes with proteasome inhibitors by an HDAC6-independent mechanism. Br. J. Haematol. 151, 387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Padhi D., Jang G., Stouch B., Fang L., Posvar E. (2011) Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J. Bone Miner. Res. 26, 19–26 [DOI] [PubMed] [Google Scholar]
- 54. Wu X., Li H., Park E. J., Chen J. D. (2001) SMRTE inhibits MEF2C transcriptional activation by targeting HDAC4 and -5 to nuclear domains. J. Biol. Chem. 276, 24177–24185 [DOI] [PubMed] [Google Scholar]
- 55. Li H., Xie H., Liu W., Hu R., Huang B., Tan Y. F., Xu K., Sheng Z. F., Zhou H. D., Wu X. P., Luo X. H. (2009) A novel microRNA targeting HDAC5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J. Clin. Invest. 119, 3666–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vega R. B., Matsuda K., Oh J., Barbosa A. C., Yang X., Meadows E., McAnally J., Pomajzl C., Shelton J. M., Richardson J. A., Karsenty G., Olson E. N. (2004) Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119, 555–566 [DOI] [PubMed] [Google Scholar]
- 57. Kozhemyakina E., Cohen T., Yao T.-P., Lassar A. B. (2009) Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol. Cell. Biol. 29, 5751–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]