

Background: The antigen 85 complex represents three homologous mycolyltransferases that show promise as tuberculosis drug targets.
Results: Structures of antigen 85C (Ag85C) covalently modified at a conserved cysteine and Ag85C active site mutants exhibit disruption of the active site structure.
Conclusion: Structural dynamics are important for Ag85C function and inhibition.
Significance: Targeted thiol modification of the antigen 85 complex is a valid inhibitory mechanism for inhibitor design.
Keywords: Carbohydrate, Drug Discovery, Enzyme Inactivation, Mycobacterium tuberculosis, Protein Structure, Antigen 85 Complex
Abstract
The rise of multidrug-resistant and totally drug-resistant tuberculosis and the association with an increasing number of HIV-positive patients developing tuberculosis emphasize the necessity to find new antitubercular targets and drugs. The antigen 85 (Ag85) complex from Mycobacterium tuberculosis plays important roles in the biosynthesis of major components of the mycobacterial cell envelope. For this reason, Ag85 has emerged as an attractive drug target. Recently, ebselen was identified as an effective inhibitor of the Ag85 complex through covalent modification of a cysteine residue proximal to the Ag85 active site and is therefore a covalent, allosteric inhibitor. To expand the understanding of this process, we have solved the x-ray crystal structures of Ag85C covalently modified with ebselen and other thiol-reactive compounds, p-chloromercuribenzoic acid and iodoacetamide, as well as the structure of a cysteine to glycine mutant. All four structures confirm that chemical modification or mutation at this particular cysteine residue leads to the disruption of the active site hydrogen-bonded network essential for Ag85 catalysis. We also describe x-ray crystal structures of Ag85C single mutants within the catalytic triad and show that a mutation of any one of these three residues promotes the same conformational change observed in the cysteine-modified forms. These results provide evidence for active site dynamics that may afford new strategies for the development of selective and potent Ag85 inhibitors.
Introduction
Tuberculosis (TB)2 continues to be a major global health problem; nearly 9 million new TB cases and 1.3 million TB deaths were reported in 2012 (1). The long treatment regimen (6 months minimum) as well as the appearance of multidrug-resistant, extensively drug-resistant, and totally drug-resistant TB contribute to the difficulties in treating this bacterial infection (1, 2). In addition, co-infection with HIV is a constant challenge because most of the antiretroviral drugs used to treat HIV are not compatible with certain TB treatments (3–5). Taken together, these facts stress the need to discover new drug targets and identify new antitubercular compounds. Mycobacterium tuberculosis is the bacterium responsible for most of the TB cases. The complex mycobacterial cell wall is essential for the viability of M. tuberculosis, and the biosynthesis of various cell wall components represents the major target of current anti-TB treatment. Penetrating the mycobacterial cell wall and the associated mycobacterial outer membrane (mycomembrane) with anti-TB drugs is a major challenge in TB treatment because the hydrophobic nature of the mycomembrane constitutes an impermeable barrier that prevents the entry of potential antibiotics into the bacterial cell (6). The cell wall of M. tuberculosis consists of three main covalently linked components: peptidoglycan, arabinogalactan, and mycolic acids. Together, these form the mycolyl-arabinogalactan-peptidoglycan complex, or mAGP (7–9). Mycolic acids are long, α-alkyl, β-hydroxy fatty acid chains either esterifying the end of the arabinogalactan or forming the lipophilic tail of non-covalently bound glycolipids (10).
A prominent strategy used over the past 2 decades has been to study the enzymes that play roles in the biosynthesis of the different cell wall components (11–14). Several compounds targeting enzymes involved in the biosynthesis of cell wall subunits or proteins that transport those subunits are currently in clinical trials (SQ109, OPC-67683, PA-824, and BTZ043) (15–18).
The antigen 85 complex (Ag85) consists of three secreted enzymes (Ag85A/B/C), with Ag85B being the major secreted protein in M. tuberculosis (19). The three homologous enzymes catalyze the transfer of mycolic acids from trehalose monomycolate (TMM) to either another TMM molecule to generate trehalose dimycolate (TDM) or to arabinose to mycolate the AG complex (20, 21). A deletion mutant of fbpC, the gene encoding Ag85C, reduces by 40% the cell wall-bound mycolic acids but does not affect the production of non-covalently linked mycolates, implying that Ag85C transfers mycolic acid to the AG complex in vivo (22). Additionally, knockout of the fbpA or fbpB genes, encoding Ag85A and Ag85B, respectively, leads to a decrease in the production of TDM (23, 24). Solving the crystal structures of the different enzymes, Ronning et al. (25, 26) and Anderson et al. (27) established that all three Ag85 enzymes possess a conserved active site and therefore likely share the same mycolic acid donor TMM. The structures also support the hypothesis that the transesterification reaction catalyzed by the Ag85 enzymes follows a ping-pong mechanism via the formation of an acyl-enzyme intermediate (25).
Recently, the selenazole compound ebselen was found to inhibit the Ag85 complex using a mechanism not previously considered for these enzymes (28). Ebselen reacts with a conserved cysteine residue (Cys-209 in Ag85C) located near the active site of the enzyme but not involved in the enzyme mechanism (Fig. 1). This covalent modification results in the formation of a selenenylsulfide bond, forcing an otherwise kinked α helix (α9) to adopt a relaxed or straightened conformation that disrupts the hydrogen-bonded network within the catalytic triad of the enzyme and inactivates Ag85C. The covalent modification and enzymatic inactivation were confirmed for each of the M. tuberculosis-encoded Ag85 enzymes using mass spectrometry and two different enzymatic assays, respectively. However, the x-ray crystal structure solved using Ag85C crystallized in the presence of ebselen (Ag85C-EBS, PDB accession code 4MQM) did not exhibit density representing the covalent modification (28).
FIGURE 1.
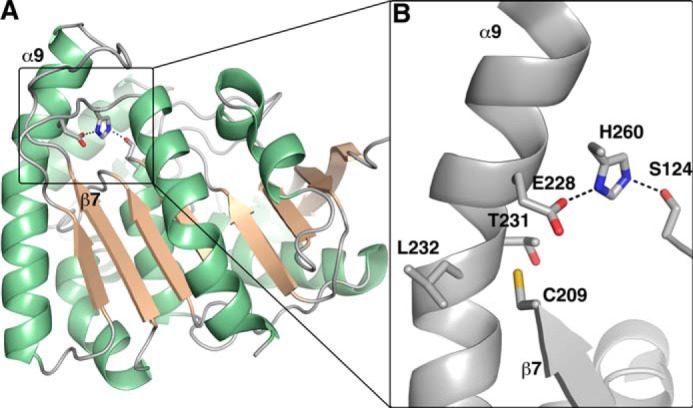
Native Ag85C structure (PDB 1DQZ). A, overall structure of Ag85C. The α helices are colored in green, and the β strands are colored in wheat, respectively. B, active site of the native Ag85C structure. The catalytic triad residues (Ser-124, Glu-228, and His-260), as well as Cys-209 and Thr-231/Leu-232, are represented in sticks. Cys-209 is positioned on strand β7 and interacts through van der Waals interactions with the peptide bond linking Thr-231 to Leu-232 located on helix α9; the interactions maintain the helix α9 in a bent conformation. Helix α9 also harbors Glu-228, one of the residues of the catalytic triad participating in the Ag85 mechanism.
Because the lack of visible ebselen was probably due to radiation damage during the diffraction experiment, this work describes the efforts to better understand how covalent modification or mutation at the cysteine residue is responsible for the inactive conformation adopted by the enzyme. Toward this aim, Ag85C was covalently modified with the thiol-reactive compounds iodoacetamide and p-chloromercuribenzoate; the resulting crystal structures exhibit the same disruption of the hydrogen-bonded network within the active site as the ebselen structure. A new crystal structure of Ag85C reacted with ebselen was also solved, with density observed for ebselen. Mutation of the cysteine to a glycine leads to an identical conformational change. Finally, because the conformation of helix α9 is highly sensitive to chemical changes near the active site, the effects of mutations in the catalytic triad were assessed using activity assays and by structure determination. These data provide further insights into novel mechanisms to efficiently inhibit the Ag85 enzymes.
EXPERIMENTAL PROCEDURES
Molecular Cloning
The segment of the M. tuberculosis fbpC that encodes the secreted form of Ag85C was cloned into a pET29-based vector (EMD Biosciences) using the restriction sites NdeI and XhoI (New England Biolabs) (29). The mutants C209G, S124A, E228Q, and H260Q were created by site-directed mutagenesis using the construct pET29-fbpC as the template. The following primers and their respective complements (Integrated DNA Technology) were used to carry out the site-directed mutagenesis: GGATCTGGGTGTACGGCGGTAACGGCACA (C209G) (28), AACGCGGCGGTGGGTCTTGCGATGTCGGGCGGTTCCGCG (S124A), GGCGAAGTTCCTGCAGGGCCTCACCCTGC (E228Q), and CCGCCCAACGGAACACAGTCGTGGCCC (H260Q). Nucleotide sequencing was performed by Eurofins MWG Operon to confirm the presence of the mutations.
Protein Purification of Ag85C and Mutants
The different plasmids were used to transform T7 express Escherichia coli cells (New England BioLabs). Bacterial cells were cultured at 37 °C in Luria-Bertani broth (Research Products International) until reaching an A600 nm of 0.6. Isopropyl β-d-1-thiogalactopyranoside (Gold Biotechnology) was added to induce protein expression. The bacterial cells were incubated at 16 °C and harvested after 24–36 h of induction using centrifugation. The pelleted cells were resuspended in a 20 mm Tris buffer at pH 8.0 and 5 mm β-mercaptoethanol. The protein variants were purified as described previously (29). Briefly, cell lysis was carried out by the addition of lysozyme (Hampton Research) and DNase I (Roche Applied Science) as well as a sonication step (Sonicator 3000, Misonix). Lysates were clarified by centrifugation for 20 min at 10,000 × g. Cobalt affinity chromatography and anion exchange chromatography were used to purify the samples. Both chromatography steps used a 20 mm Tris buffer, pH 8.0. Protein was eluted from the cobalt affinity column with an imidazole gradient from 0 to 150 mm (GE Healthcare). Protein was eluted from a HiTrap FF Q column using an NaCl gradient from 0 to 1.0 m (GE Healthcare). To concentrate the different protein samples prior to crystallization, the protein was subjected to ammonium sulfate precipitation (2.6 m), followed by dissolving the precipitated protein pellet in the crystallization buffer: 10 mm Tris, pH 7.5, 2 mm EDTA, and 0 or 1 mm dithiothreitol (DTT). The samples were then dialyzed overnight against a similar buffer. Absorbance spectroscopy at a wavelength of 280 nm was used to determine the enzyme concentration using the theoretical extinction coefficient (84,340 m−1 cm−1) as calculated by the ProtParam function from the ExPASY proteomics server (30).
Enzymatic Activity Assays
The enzymatic activity of the different enzymes was tested using a fluorescence-based assay described previously (28). Resorufin butyrate (Santa Cruz Biotechnology, Inc.) was used as an acyl donor for Ag85C and its modified forms, whereas trehalose was used as an acyl acceptor. Production of resorufin, a fluorescent compound (λex = 500 nm and λem = 593 nm), was monitored for 20 min following the addition of resorufin butyrate to the reaction. All reactions were carried out in a 50 mm sodium phosphate buffer, pH 7.5, using enzyme previously dialyzed against the assay buffer. All of the activity tests were performed at 37 °C on a Synergy H4 Hybrid Reader (BioTek) using 4 mm trehalose and 100 μm resorufin butyrate diluted from a stock solution of 10 mm resorufin butyrate dissolved in DMSO. All of the mutants were tested at an enzyme concentration of 500 nm, and the enzymatic activities were compared with the wild-type Ag85C activity.
The inhibition of Ag85C by p-chloromercuribenzoic acid was tested using the same fluorescence-based assay described above. The enzyme was reacted with p-chloromercuribenzoic acid at 2.5 μm final concentration. The enzymatic activity of the sample was tested after a 2 h incubation and an overnight incubation. A control reaction with an unmodified Ag85C enzyme was also performed.
Crystallization Studies
All crystals were grown using the hanging drop vapor diffusion method. Ag85C (6.2 mg/ml) was reacted with p-chloromercuribenzoic acid at a 1.4-fold molar excess for 2 h and was crystallized against a well solution of 0.1 m sodium acetate trihydrate, pH 4.5, and 25% (w/v) polyethylene glycol 3350. Ag85C (5.5 mg/ml) was incubated with iodoacetamide (IAA; 50 mm) at room temperature for 1 h in the dark. Ag85C-IAA was crystallized in 0.2 m lithium sulfate monohydrate, 0.1 m Bis-Tris, pH 5.5, and 25% (w/v) polyethylene glycol 3350. Ag85C (5.1 mg/ml) was reacted with 200 μm ebselen (10 mm stock solution in DMSO) for 2 h on ice. The Ag85C-ebselen complex was crystallized in 0.1 m sodium acetate trihydrate, pH 4.5, and 25% (w/v) polyethylene glycol 3350. These three samples lacked DTT in the crystallization buffer.
The crystals of Ag85C-C209G (5 mg/ml) were grown in 1.0 m ammonium sulfate, 0.1 m Bis-Tris, pH 5.5, and 1% (w/v) polyethylene glycol 3350. For cryoprotection, polyethylene glycol 3350 was added to a final concentration of 25% prior to flash cooling in liquid nitrogen. Ag85C-E228Q (5 mg/ml) and H260Q (6.5 mg/ml) were crystallized in 0.1 m sodium acetate trihydrate, pH 4.5, and 25 or 20% (w/v) polyethylene glycol 3350, respectively, in the presence of 13 mm hexaethylene glycol monooctyl ether or 6 mm CHAPSO (Hampton Research). Finally, Ag85C-S124A (2 mg/ml) mutant was crystallized against a well solution of 0.3 m lithium sulfate, 0.1 m Bis-Tris, pH 5.5, 0.1 mm d-glucose, and 23% polyethylene glycol 3350.
All crystals were flash-cooled in liquid nitrogen before data collection. Diffraction data were collected at the LS-CAT beamline (Ag85C-Hg, Ag85C-IAA, Ag85C-ebselen, Ag85C-C209G, Ag85C-E228Q, and Ag85C-S124A) or at the GM/CA-CAT beamline (Ag85C-H260Q) at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL).
Structure Determination
HKL2000 was used to index, integrate, and scale the diffraction data (31). Molecular replacement, when necessary, was carried out using EPMR (Evolutionary Program for Molecular Replacement) (32). PDB entry 4MQM (Ag85C-EBS) was used as the search model for each data set. Using the best solution from EPMR, rigid body refinement, simulated annealing, and positional and B-factor refinements were performed with the refine tool in PHENIX (33). The model was manually corrected using COOT (34). The eLBOW (electronic Ligand Builder and Optimization Workbench) tool was used to define the geometric restraints of the ligands in the Ag85C-ebselen, Ag85C-Hg, Ag85C-E228Q, and Ag85C-H260Q structures (35).
RESULTS
Modification of Ag85C by Thiol-reactive Compounds Imparts Structural Changes in the Ag85C Active Site
The addition of ebselen and two other thiol-reactive compounds, iodoacetamide and p-chloromercuribenzoic acid, was used to covalently modify Ag85C at position Cys-209. Iodoacetamide is an alkylating reagent commonly used to irreversibly label cysteines in biomolecules, and previous studies show that covalent modification by iodoacetamide at Cys-209 of Ag85C inactivates the enzyme (28). Additionally, p-chloromercuribenzoic acid is known to inhibit some enzymes requiring a cysteine residue for enzymatic activity and is commonly used to label cysteine residues for phasing x-ray crystal structures (36). All complexes were crystallized using a hanging drop vapor diffusion technique, and the corresponding x-ray crystal structures were solved. For clarity, the crystal structures will be referred to as Ag85C-ebselen (Ag85C modified with ebselen, PDB accession code 4QDU), Ag85C-IAA (iodoacetamide-modified Ag85C, PDB accession code 4QDT), and Ag85C-Hg (Ag85C modified with p-chloromercuribenzoic acid, PDB accession code 4QDO), respectively. The Ag85C-IAA and Ag85C-Hg covalent complexes were solved to 1.50 and 1.90 Å, respectively, whereas the Ag85C-ebselen covalent complex was solved to 1.40 Å (Table 1). All three structures are isomorphous with the previously published crystal structure of Ag85C incubated with ebselen (referred to as Ag85C-EBS; PDB accession code 4MQM). The low r.m.s. displacement values for the Cα atom positions (0.4 Å for Ag85C-ebselen, 0.3 Å for Ag85C-IAA, and 0.2 Å for Ag85C-Hg) when superimposing each structure onto the original Ag85C-EBS structure highlight the fact that all three structures are very similar to the ebselen-modified form of Ag85C, which is known to represent an inactivated form of Ag85C (28).
TABLE 1.
Data collection and refinement statistics
| Ag85C-IAA | Ag85C-Hg | Ag85C-ebselen | Ag85C-C209G | Ag85C-S124A | Ag85C-E228Q | Ag85C-H260Q | |
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| PDB ID | 4QDT | 4QDO | 4QDU | 4QDX | 4QEK | 4QDZ | 4QE3 |
| Space group | P212121 | P212121 | P212121 | P212121 | C2 | I222 | P212121 |
| Unit cell dimensions | |||||||
| a, b, c (Å) | 60.8, 68.0, 76.3 | 60.7, 68.0, 74.4 | 59.5, 67.8, 75.2 | 60.2, 68.4, 76.4 | 136.7, 67.9, 35.7 | 68.2,74.9, 136.1 | 60.7, 68.1, 76.3 |
| α, β, γ (degrees) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 94.4, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution range (Å) | 50.0–1.50 | 50.0–1.90 | 50.0–1.40 | 50.0–1.50 | 50.0–1.30 | 50.0–1.90 | 50.0–1.35 |
| Wavelength (Å) | 0.97872 | 0.97856 | 1.07818 | 0.97856 | 0.97872 | 0.97872 | 0.97936 |
| Rsym (highest shell) | 6.6 (37.0) | 9.7 (45.8) | 6.6 (30.4) | 7.1 (39.8) | 6.7 (39.4) | 9.3 (56.1) | 7.8 (51.3) |
| I/σI | 28.2 (5.7) | 27.7 (5.8) | 26.8 (5.4) | 35.7 (7.9) | 27.8 (3.7) | 31.5 (5.8) | 16.0 (2.8) |
| Completeness (%) (highest shell) | 99.9 (100.0) | 98.2 (96.2) | 98.5 (90.8) | 99.2 (100.0) | 99.9 (99.4) | 99.5 (98.8) | 97.9 (85.5) |
| Redundancy (highest shell) | 7.3 (7.3) | 13.1 (9.3) | 6.4 (4.2) | 13.9 (14.6) | 4.9 (4.8) | 13.1 (12.1) | 5.5 (3.9) |
| Refinement | |||||||
| Total reflections (unique) | 375222 (51419) | 320135 (24485) | 384079 (59726) | 695388 (50123) | 388902 (79792) | 371096 (28231) | 379104 (68542) |
| Rwork/Rfree | 14.7/17.5 | 19.3/24.0 | 14.6/18.2 | 17.9/19.3 | 12.7/14.3 | 16.3/18.7 | 17.1/19.8 |
| B-factors (Å2) | |||||||
| Protein | 20.8 | 25.1 | 20.0 | 20.6 | 13.6 | 26.7 | 14.9 |
| Water | 32.0 | 28.1 | 32.6 | 28.9 | 33.5 | 31.1 | 27.1 |
| Wilson B-factor | 16.4 | 20.2 | 14.9 | 18.1 | 10.3 | 24.1 | 11.5 |
| r.m.s. deviations | |||||||
| Bond lengths (Å) | 0.006 | 0.009 | 0.006 | 0.006 | 0.006 | 0.007 | 0.006 |
| Bond angles (degrees) | 1.075 | 1.405 | 1.095 | 1.063 | 1.106 | 1.126 | 1.115 |
| Ramachandran plot | |||||||
| Favored (%) | 96.32 | 95.49 | 96.14 | 97.00 | 96.9 | 96.24 | 96.59 |
| Outliers (%) | 0.00 | 0.38 | 0.70 | 0.37 | 0.00 | 0.38 | 0.76 |
Each structure exhibits a relaxation of helix α9, but some differences are present upon closer inspection of the active site. The covalent modification of Cys-209 promotes the observed conformational change that stems from disrupting the van der Waals interactions between Cys-209 and the kinked region of helix α9. Due to covalent modification of Cys-209 by these thiol-reactive compounds, the Cys-209 side chain can no longer interact with the kink in helix α9 as observed in the native structures (Fig. 1B), thereby altering the Ag85C active site structure.
In the Ag85C-IAA structure, His-260 interacts via hydrogen bonding with Ser-148 instead of the serine nucleophile Ser-124. Also, the Cα position for Glu-228 is shifted 6.2 Å in comparison with the native Ag85C structure, preventing the formation of a stable hydrogen bond between Glu-228 and His-260 as observed in the native structure. Clear difference density contiguous with Cys-209 is observed for the acetamide moiety (YCM residue) as shown in Fig. 2A.
FIGURE 2.

A, superposition of the native Ag85C (gray), Ag85C-EBS (orange), and Ag85C-IAA (green) structures. The difference density of a Fo − Fc omit map is shown contoured at 3σ (green); the side chain of the YCM residue was omitted during map calculation. B, superimposition of the native Ag85C (gray), Ag85C-EBS (orange), and Ag85C-Hg (blue) structures. The difference density of a Fo − Fc omit map is shown contoured at 3σ (blue); the side chain of the CHg residue was omitted during map calculation. CHg represents the Cys-209 residue modified with mercuribenzoic acid.
When compared with the Ag85C-EBS structure, the His-260 side chain in the Ag85C-Hg structure is disordered (Fig. 2B). The B-factor for His-260 Cγ in the Ag85C-Hg structure is 43.4 Å2, which is much higher than for previous Ag85C structures (14.7 Å2 on average). His-260 does not seem to interact strongly with either Ser-124 or Ser-148. Additionally, as in the Ag85C-ebselen and Ag85C-IAA structures, a hydrogen bond is lacking between Glu-228 and His-260 for the Ag85C-Hg structure (Fig. 2B).
Reaction of Ag85C with p-chloromercuribenzoic acid leads to inhibition of the enzymatic activity (Fig. 3). After a 2-h incubation, the modified enzyme displays about 60% activity (Fig. 3A), whereas after an overnight incubation, the enzyme retains about 30% activity (Fig. 3B).
FIGURE 3.
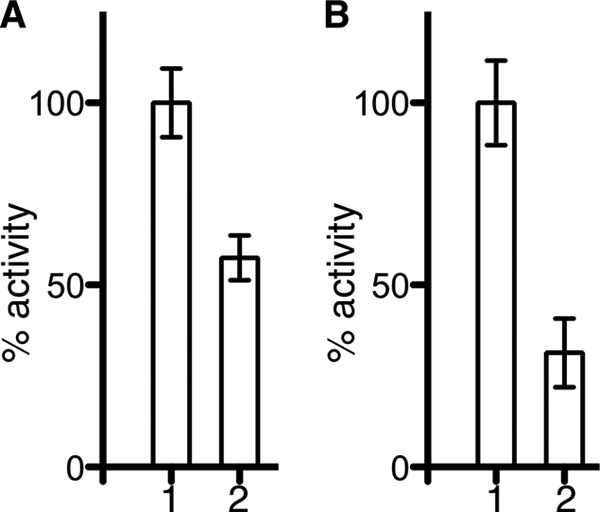
Inhibition of Ag85C by p-chloromercuribenzoic acid. The enzymatic activity of the different samples was tested using a fluorescence-based assay. The activity is normalized to the unmodified Ag85C enzyme, and the error bars are calculated from triplicate reactions. A, inhibition of Ag85C by p-chloromercuribenzoic acid after a 2-h incubation. Bar 1 corresponds to the unmodified Ag85C, whereas bar 2 relates to the Ag85C enzyme modified with p-chloromercuribenzoic acid. B, inhibition of Ag85C by p-chloromercuribenzoic acid after an overnight incubation. Bar 1 corresponds to the unmodified Ag85C, whereas bar 2 relates to the Ag85C enzyme modified with p-chloromercuribenzoic acid. Error bars correspond to S.D. from triplicate reactions.
A new crystal structure of Ag85C reacted with ebselen was also solved (Fig. 4A). The data were collected at a wavelength of 1.078 Å (11.5 keV) to decrease x-ray absorbance by the selenium atom and avoid oxidation that could break the selenenylsulfide bond. As anticipated, the new Ag85C-ebselen structure displays a conformational change similar to that previously observed. In contrast to the previously determined Ag85C-EBS structure (PDB accession code 4MQM), electron density is observed for the selenenylsulfide bond as well as one of the aromatic moieties of ebselen, whereas the second aromatic ring is not (Fig. 4B). To further confirm the presence and refine the location of the selenium atom in this structure, an anomalous difference map was calculated, and the peaks within this map clearly define the positions of the selenium atom as well as the sulfur atom in Cys-209 and each methionine residue (Fig. 4C).
FIGURE 4.
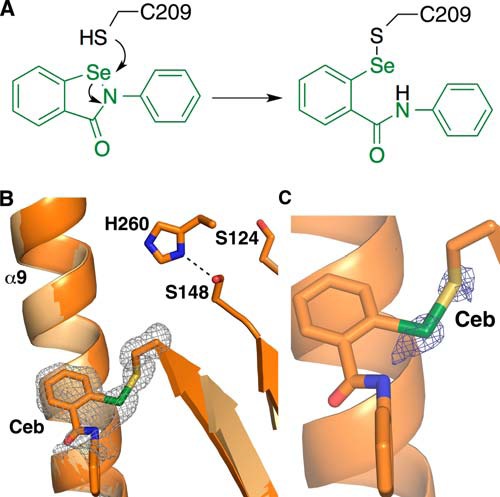
Ag85C-ebselen structure. A, proposed reaction mechanism of cysteine modification with ebselen. B, superposition of the new Ag85C-ebselen (dark orange) and Ag85C-EBS (orange) structures. The electron density of an Fo − Fc omit map is shown contoured at 3σ (gray); the side chain of the Ceb (Cys-209 modified by ebselen) residue was omitted during map calculation. Electron density was only observed for the exocyclic selenium atom, the first aromatic ring, and the amide linker. C, Ag85C-ebselen structure (bright orange). The electron density of an anomalous map is shown contoured at 4σ (blue).
Additionally, the loop connecting strand β7 and helix α9 (Lβ7-α9) composed of residues 210–222 is well ordered in this new crystal structure, contrary to the previously published structure. The conformation of Lβ7-α9 is similar to that observed in the Ag85C-C209S structure (PDB accession code 4MQL) (28). Moreover, the His-260 side chain is ordered in this Ag85C-ebselen and is forming a hydrogen bond with Ser-148 (Fig. 4B). Once again, the hydrogen-bonded network normally observed for the residues of the catalytic triad is disrupted.
The Ag85C-C209G Mutant Exhibits a Similar Inactive Conformation
Cys-209 was mutated to a glycine using site-directed mutagenesis. The Ag85C-C209G mutant displays less than 10% activity compared with the wild-type enzyme (28). To determine whether a similar structural change to the cysteine-modified forms of Ag85C is taking place in the mutant, the x-ray crystal structure of Ag85C-C209G was solved to 1.50 Å (Table 1). Again, the structure was isomorphous with Ag85C-EBS. When superimposing the Ag85C-C209G (PDB accession code 4QDX) and Ag85C-EBS structures, the r.m.s. displacement observed for the Cα atoms is 0.2 Å, indicating clearly that both structures are similar. Moreover, the Ag85C-C209G mutant structure is comparable with the Ag85C-C209S structure previously solved (PDB accession code 4MQL) (Fig. 5) (28). Superimposing these two structures provides a r.m.s. displacement value of 0.8 Å for the Cα atoms. The main difference between the two mutant structures is the extent of the shift in helix α9 when relaxed, as indicated by the relative position of the Glu-228 Cα atom; in the Ag85C-C209G structure, the Cα position of Glu-228 has shifted 6.2 Å compared with the wild-type enzyme, whereas the Cα of Glu-228 in the Ag85C-C209S structure has only shifted 3.5 Å. The His-260 side chain of the Ag85C-C209G structure interacts with Ser-148, whereas the hydrogen bond between Glu-228 and His-260 is disrupted.
FIGURE 5.
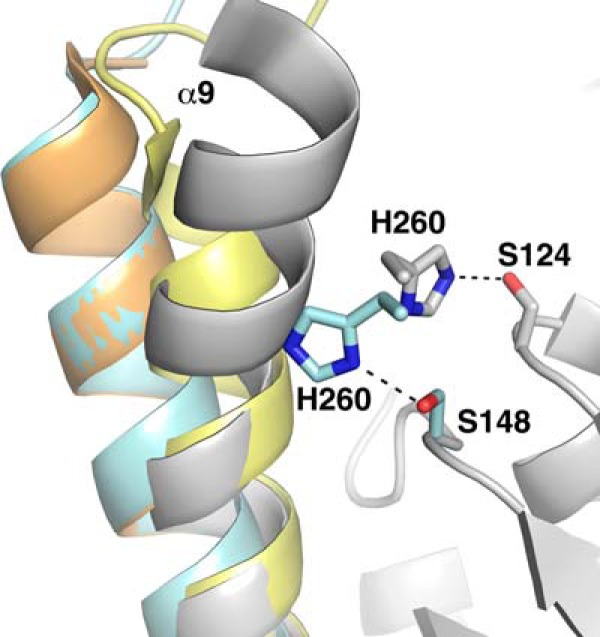
Superposition of Ag85C (gray), Ag85C-EBS (orange), Ag85C-C209S (yellow), and Ag85C-C209G (cyan) structures. The His-260 side chain interacts with the serine nucleophile Ser-124 in the native structure, whereas it interacts with a different serine residue (Ser-148) in the Ag85C-C209G structure, explaining the lack of activity displayed by the mutant. The mutation promotes the relaxation of helix α9, similar to the structural change observed in the Ag85C-C209S or Cys-209 covalently modified Ag85C structures.
Mutation of Any Catalytic Triad Residue Results in Loss of Enzymatic Activity and Relaxation of Helix α9
Using a radiometric mycolyltransferase assay, Belisle et al. (20) showed previously that mutation of the catalytic nucleophile Ser-124 to an alanine leads to an inactive Ag85C enzyme. Site-directed mutagenesis was carried out to make Ag85C variants possessing amino acid changes at the other residues of the catalytic triad; both Glu-228 and His-260 were mutated to glutamine. The resorufin butyrate-based assay (28) used to assess the C209G mutant was again used to test the enzymatic activity of the active site mutants. Ag85C-H260Q displays less than 10% of activity when compared with the wild-type enzyme, whereas Ag85C-E228Q retains 17% of the wild-type activity (Fig. 6).
FIGURE 6.
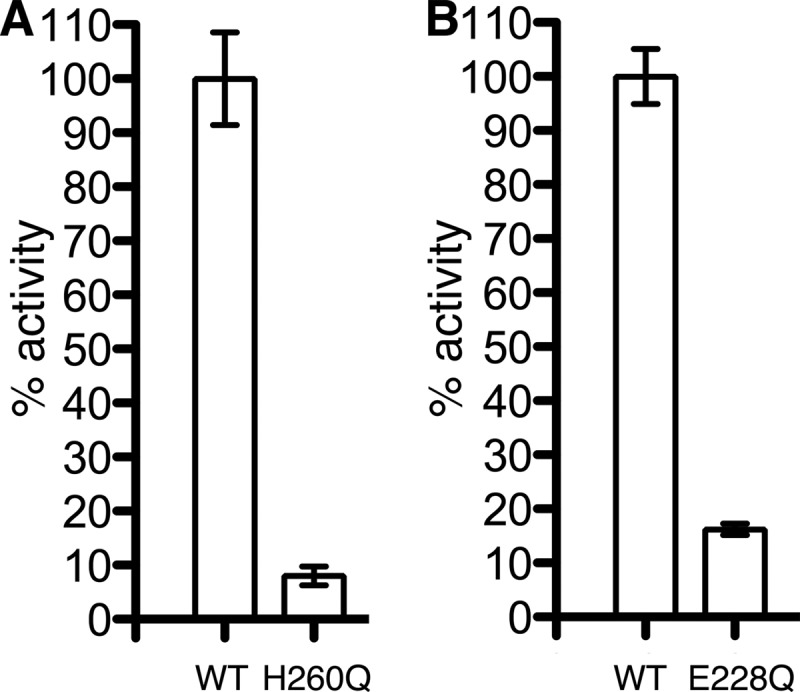
Effect of mutations among the catalytic triad residues. The enzymatic activity of the mutants was tested using a fluorometric assay. The activity was normalized to the wild-type enzyme Ag85C, and the error bars were calculated from triplicate reactions. A, the mutant Ag85C-H260Q exhibits less than 10% of activity compared with the wild-type Ag85C. B, Ag85C-E228Q retains 17% of wild-type activity. Error bars correspond to S.D. from triplicate reactions.
X-ray crystallography experiments were carried out to gain insight into the structures of the three mutants. The crystal structures of Ag85C-S124A (PDB accession code 4QEK) and Ag85C-H260Q (PDB accession code 4QE3) were solved to 1.30 and 1.35 Å, respectively, whereas Ag85C-E228Q (PDB accession code 4QDZ) was solved to 1.90 Å (Table 1). Ag85C-H260Q is the only structure among the three mutants to be isomorphous with the Ag85C-EBS, and the low r.m.s. displacement for the Cα atoms after superimposing both structures (0.4 Å) confirms their structural similarity (Fig. 7).
FIGURE 7.
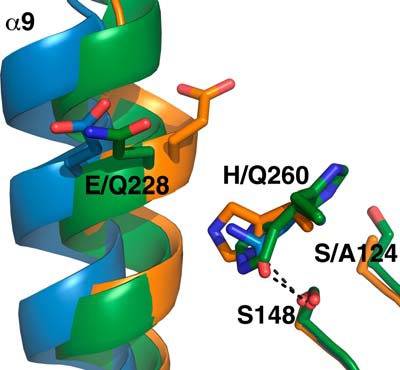
Superposition of Ag85C-S124A (orange), Ag85C-E228Q (green), and Ag85C-H260Q (blue) structures. The three mutants exhibit a relaxation of helix α9, accounting for the loss of enzymatic activity.
The Ag85C-S124A mutant structure (PDB accession code 4QEK), similarly to the phenomenon observed in the Ag85C-IAA, Ag85C-ebselen, and Ag85C-C209G structures, exhibits a shift of the N-terminal portion of helix α9 (Fig. 7). The Cα of Glu-228 in the Ag85C-S124A mutant has shifted 4 Å with respect to the equivalent atom in the native Ag85C structure (PDB accession code 1DQZ (25)). This shift, afforded solely by the loss of the hydrogen bond between residue 124 and His-260, again affords the formation of a hydrogen bond between His-260 and Ser-148. Contrary to the Ag85C-EBS structure and many of the other structures that possess a relaxed α9 helix, the loop Lβ7-α9 is well resolved in the Ag85C-S124A crystal structure. This loop adopts a conformation similar to the Ag85C-C209S structure (PDB accession code 4MQL) (28) and the previously published Ag85C-diethylphosphate structure (PDB accession code 1DQY (25)).
The Ag85C-E228Q structure (PDB accession code 4QDZ) also displays the same shift of the helix α9 as Ag85C-S124A (Fig. 7). However, the His-260 side chain is disordered in this structure; the B-factor for Cβ is 33.7 Å2. Indeed, the weak difference density suggests that residue His-260 may take two alternative conformations, but neither is well resolved. One of these two conformations probably interacts with the serine nucleophile Ser-124, whereas the other probably forms a hydrogen bond with Ser-148.
Finally, the Ag85C-H260Q structure (PDB accession code 4QE3) exhibits the same shift in helix α9 as that observed in the Ag85C-C209G mutant structure, preventing the formation of any hydrogen bond between residues 260 and Glu-228. Additionally, the shift in helix α9 repositions the side chain of residue 260 to form a hydrogen bond between Oϵ1 of the Gln-260 side chain and the side chain hydroxyl of Ser-148.
DISCUSSION
Ebselen, a small molecule containing a thiol-reactive selenium atom, has been shown to be an effective compound against both drug-sensitive and multidrug-resistant strains of M. tuberculosis exhibiting a minimum inhibitory concentration (MIC) of 20 μg/ml in each case (37). More recently, ebselen was demonstrated to be a potent inhibitor of the M. tuberculosis Ag85 complex. In particular, application of ebselen to M. tuberculosis cultures at the MIC exhibits inhibition of both TDM and mAGP biosynthesis, further suggesting that the Ag85 complex is the primary target of ebselen (28). The mechanism of Ag85 inhibition is that of a covalent, allosteric inhibitor that modifies the conserved Cys-209 residue buried in a hydrophobic pocket near the Ag85C active site. Although Cys-209 does not participate in the catalytic reaction performed by the enzymes, this cysteine side chain forms an important structural feature of each enzyme of the Ag85 complex. Specifically, the van der Waals interaction between the Cys-209 side chain and the peptide bond linking residue Thr-231 to residue Leu-232 stabilizes a kink in helix α9 (Fig. 1B). Because helix α9 harbors Glu-228, one of the residues of the catalytic triad important for Ag85C catalytic turnover, modification of Cys-209 has a profound impact on enzymatic activity. Indeed, the kink in helix α9 is required to form the hydrogen-bonded network between the three residues of the catalytic triad. When Cys-209 is mutated or covalently modified, this weakens the interaction between the side chain and helix α9 (28). Weakening of that interaction causes the straightening of helix α9 and the repositioning of Glu-228, which prevents the formation of a stable hydrogen bond with His-260. This disruption of the hydrogen-bonded network within the catalytic triad is observed in the structure of Ag85C crystallized in presence of ebselen (PDB accession code 4MQM) and appears to be the primary reason for lack of enzymatic activity in Ag85C when Cys-209 is modified by either mutation or chemical modification. Nevertheless, the absence of visible density for ebselen in this structure raises the question of whether the relaxation of helix α9 is solely responsible for the lack of enzymatic activity, so determining the location ebselen when covalently complexed with Ag85C is important for future drug development.
The lack of density for the covalent modification can be explained by radiation damage; solvent-exposed disulfide bonds can be easily oxidized when subjected to x-ray radiation (38–40), and a selenenylsulfide bond would probably be even less stable. To overcome this, a co-crystal structure of an Ag85C-ebselen complex was solved using x-rays at a wavelength of 1.07 Å (11.5 keV). Performing the x-ray diffraction experiment at a wavelength below the selenium K-edge (12.7 keV) decreases x-ray absorption by the selenium atom in the selenenylsulfide bond and therefore decreases the potential oxidation of that selenium atom. Contrary to the previously published Ag85C-EBS structure (PDB accession code 4MQM), the selenenylsulfide bond in the current structure exhibits strong difference density for both the selenium and sulfur atoms as well as the aromatic moiety of ebselen that harbors the now exocyclic selenium atom and the amide linkage to the phenyl moiety now distal to the selenenylsulfide bond (Fig. 4A). Nonetheless, the Fo − Fc omit map does not appear to account perfectly for the entire Ag85C-ebselen covalent complex (Fig. 4B). To further confirm the location of the selenium atom, an anomalous difference map was calculated. Although the data were collected at energy below the absorption edge of selenium, some anomalous scattering signal was observed, and the resulting difference map gives the position of the selenium (Fig. 4C). Despite the hydrophobic nature of ebselen, the molecule is oriented toward the surface of Ag85C. The amide moiety in ebselen forms a π-π interaction with the guanidinium moiety of Arg-239, although the side chain of the arginine is not entirely resolved due to slightly elevated B-factors between 30.4 and 39.4 Å2 for atoms of the guanidinium moiety. Additionally, a π-π stacking interaction is observed between the first aromatic ring of ebselen and Phe-254 of Ag85C. The rest of the protein component of the structure is identical to the previous Ag85C-EBS, as indicated by the low r.m.s. displacement values observed for the Cα atoms.
Ebselen has beneficial characteristics that make it an interesting lead compound for further drug development. First, the MIC of 20 μg/ml for drug-sensitive and drug-resistant M. tuberculosis strains suggests that ebselen would be an efficient compound that could be used against all M. tuberculosis strains (37). Additionally, ebselen has good oral bioavailability and is already approved by the United States Food and Drug Administration for ischemic stroke and bipolar disorder (41–43). However, the chemical reactivity of ebselen to any thiol could lead to off-target effects or a decrease in the concentration of ebselen in its active form. The latter point is one possible reason for the relatively modest MIC of ebselen against M. tuberculosis. Additionally, elevated selenium levels are toxic for many organisms. Because of these potential issues, it is important to determine whether other thiol-reactive compounds exhibit similar inhibitory effects on the enzymatic activity and to determine whether the mechanism of inhibition is similar to that of ebselen.
To determine whether any covalent modification of the cysteine at position 209 triggers a similar active site rearrangement, Ag85C was reacted with two other thiol-reactive compounds. The modification of Cys-209 by IAA is already known to cause the loss of Ag85C activity (28). The resulting thioether moiety is more stable than the selenenylsulfide bond formed when Ag85C is reacted with ebselen; thus, the thioether bond is effectively irreversible in vivo and is less sensitive to radiation damage during x-ray diffraction experiments.
The corresponding crystal structure, Ag85C-IAA, exhibits covalent modification at Cys-209, and difference density is observed for the acetamide moiety extending from Cys-209 (Fig. 2A). Similarly to the ebselen-modified Ag85C, the relatively bulky acetamide moiety added to Cys-209 by IAA promotes steric hindrance and prevents van der Waals interactions with the kinked form of helix α9, thereby promoting the relaxation of this helix to a straightened form. This phenomenon again alters the Ag85C active site structure and prevents the formation of a stable hydrogen-bonded network between the different catalytic triad residues: the His-260 side chain forms a hydrogen bond with Ser-148 rather than the serine nucleophile Ser-124, whereas the hydrogen bond between Glu-228 and His-260 is broken due to the straightening of helix α9, resulting in the movement of Glu-228 6.2 Å away from its position in the active form of the enzyme. Interestingly, the acetamide moiety is not positioned within the substrate binding site, suggesting that the inhibitor is not competing with substrate for access to the active site and that disruption of the catalytic triad network explains the observed inhibition of Ag85C.
The same conformational change is observed following mercury modification of Cys-209 (Fig. 2B). The activity of Ag85C in the presence of p-chloromercuribenzoic acid was tested. As expected from the Ag85C-Hg structure solved, this covalent complex exhibits a significant decrease in enzymatic activity (Fig. 3).
Because each of the chemical modifications increases the bulk of the residue at position 209 and published data show that single nucleotide changes in codon 209 have a universally detrimental effect on enzyme activity, it is interesting to determine whether Cys-209 mutations that encode residues with a smaller molecular volume also disrupt the catalytic triad hydrogen-bonded network.
To assess this, we determined the x-ray crystal structure of an Ag85C-C209G mutant. We have shown previously that the Ag85C-C209G mutant, although the side chain is a smaller hydrophobic residue, exhibits a very low level of enzymatic activity when compared with wild-type Ag85C (28). The crystal structure of Ag85C-C209G confirms that helix α9 relaxes and repositions residue Glu-228, resulting in the disruption of the hydrogen bond between Glu-228 and His-260, and His-260 is positioned to form a hydrogen bond with Ser-148. The overall structure is similar to that of the Ag85C-C209S mutant, which is also shown to be inactive in vitro (28). The C209S mutant confers a stronger polarity than the cysteine, whereas the C209G mutant imparts a lower polarity at that site and, as stated previously, has a much smaller volume than the corresponding cysteine. The C209G mutation probably results in much weaker van der Waals interactions between residue 209 and helix α9. Therefore, the low level of Ag85C-C209G enzymatic activity appears to be a consequence of active site structural alterations identical to that observed for the other Cys-209 mutants and the wild type enzyme covalently modified by thiol-reactive compounds; however, the rearrangement is a consequence of weakening interactions between the residue at position 209 rather than repulsion due to steric hindrance. Because both the Ag85C-C209G and Ag85C-C209S structures exhibit the same structural change when compared with wild-type Ag85C, this suggests that the van der Waals interactions between Cys-209 and helix α9 form an important core of interactions that maintain the strained kink in this helix and the hydrogen-bonded network between the three catalytic triad residues.
Combining the structural results of the different mutations at position 209 with that of Ag85C modified at position Ser-124 with the covalent serine protease inhibitor diethyl p-nitrophenyl phosphate suggests that helix α9 is highly sensitive to active site perturbations (25, 28). To further assess this hypothesis, experiments to measure the enzymatic activity and determine the x-ray crystal structures of Ag85C catalytic triad variants were performed. Each of the engineered Ag85C mutations (S124A, E228Q, and H260Q) exhibited either no enzymatic activity or a significant loss of activity (Fig. 6) (20). The lack of activity of the different mutants is readily explained by the chemical changes in the active site and the corresponding x-ray crystal structures of these three mutant enzymes.
Clearly, the lack of a nucleophile in the S124A mutant led to elimination of the enzymatic activity. In the H260Q mutant, the loss of the general base significantly decreased the activity. Similar to that observed for serine proteases, conversion of the third residue of the catalytic triad, in this case Glu-228, to the corresponding amide-containing side chain causes a decrease in activity greater than what would be expected if the role of Glu-228 is only to stabilize the imidazolium ion formed on His-260 during catalysis (44, 45).
The decrease in enzymatic activity of each of the Ag85C active site mutants can also be attributed to active site structural changes, because all three of the mutants exhibit the same conformational change observed in the previously described structures where Cys-209 was covalently modified. However, slight differences are apparent in the hydrogen-bonding pattern within the active sites of the three mutants. In both the Ag85C-S124A and Ag85C-H260Q, the residue 260 side chain interacts with Ser-148, and the hydrogen bond between residues Glu-228 and His-260 is disrupted. However, in the Ag85C-E228Q structure, the His-260 side chain is not well ordered and might be shifting between two different conformations. The conformation in which His-260 hydrogen bonds with the serine nucleophile would explain the higher residual activity observed for Ag85C-E228Q. Specifically, the activity of the Ag85C-E228Q variant probably reflects the confluence of two different factors. First, important components of the active site remain intact. Specifically, the serine nucleophile and oxyanion hole can still function to create and stabilize the transition state formed during catalysis. Second is the likelihood of the Ag85C-E228Q mutant enzyme transiently taking the wild-type structure, as indicated by the multiple conformations of His-260.
The propensity of helix α9 to transition from the kinked to the relaxed state is highlighted by two of the active site mutants. First, the comparison between the native form of Ag85C and the Ag85C-S124A structure suggests that the loss of a single hydrogen bond between His-260 and the residue at position 124 is sufficient to promote helix α9 relaxation. This suggests that the enzyme has evolved to undergo this structural change following the formation of the acyl-enzyme intermediate and the likely disruption of the hydrogen bond between His-260 and Ser-124. Furthermore, the rearrangement of Lβ7-α9, which is concomitant with helix α9 relaxation, can be hypothesized to play a role in substrate binding during the second half-reaction of the enzymatic mechanism (25). Second, the E228Q mutation does not eliminate any hydrogen bonds in the catalytic triad but simply replaces a carboxylate moiety with an amide. Based on the native structures of each Ag85 enzyme, this mutation would be expected to maintain a hydrogen bond with His-260 and the native structure of the enzyme. Because the lowest energy conformation observed in the Ag85C-E228Q structure contradicts that expectation, the structural change exhibited by helix α9 in the E228Q mutant further suggests that this helix may be central to Ag85C enzymatic function.
This hypothesis may explain some published data. The enzymes of the Ag85 complex appear to have redundant activity in vivo based on the proposed ability to use TMM as a mycolyl donor and subsequent transfer to a variety of substrates (21, 46). Additionally, Backus et al. (47) have shown that trehalose-based compounds can be amended at various positions and still function as acyl donors. The structures described here suggest that helix α9 relaxation maybe particularly important for selecting the mycolyl acceptor during the second half-reaction of the catalytic cycle. The Ag85B-trehalose crystal structure shows that the N terminus of helix α9 forms a portion of the trehalose-binding site using residues Pro-223 and Ala-224, which are conserved in the M. tuberculosis Ag85 enzymes. However, upon relaxation of helix α9, Pro-223 and Ala-224 move and are replaced by a portion of loop Lβ7-α9 in the crystal structures where the loop is resolved. It is possible that the reshaping of the carbohydrate-binding site upon formation of the acyl-enzyme intermediate promotes binding of the arabinan within the Ag85C active site.
Taken together, all of the structures described here confirm the inhibition mechanism of the Ag85 complex by ebselen. The cysteine Cys-209, despite not taking part in the catalytic reaction carried out by Ag85C, is of considerable structural importance. The position of Cys-209, as well as the interactions it makes with neighboring atoms, orders the active site structure in a conformation that promotes the first half-reaction of catalysis. Any modification or mutation of Cys-209 leads to either a dramatic decrease or complete loss of enzymatic activity, which indicates a low probability of developing resistance to a drug modifying the cysteine. Indeed, if the organism would mutate one of the Ag85 enzymes to generate resistance to a drug functioning similarly to ebselen, the mutant would probably display a low level of activity, whereas the two other enzymes would still be inhibited. The same would still be true if two enzymes would undergo mutation with the third one being inhibited.
The extreme sensitivity exhibited by Ag85C to minor disruption in the catalytic triad hydrogen-bonded network strongly suggests that helix α9 dynamics are important for enzyme function. Based on this information, it can be proposed that the native form affords transfer of mycolic acids to the trehalose of TMM to make TDM, whereas the relaxed form has an altered carbohydrate-binding site that may promote mycolyl transfer to the terminal arabinosyl moieties of the mAGP.
Taken together, these results support a strategy for inhibiting the Ag85 complex with mechanism-based inhibitors that first react with Ser-124 to promote relaxation of helix α9 and expose Cys-209 and, second, react with the Cys-209 side chain thiol to covalently modify this conserved residue. Such a bifunctional inhibitor would offer specificity while minimizing the probability of selecting for drug-resistant mutants.
This work was supported, in whole or in part, by National Institutes of Health (NIH) Grants AI089653 and AI105084. Use of the Advanced Photon Source, an Office of Science User Facility operated for the United States Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the DOE under Contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). GM/CA has been funded in whole or in part with Federal funds from NCI, NIH, Grant Y1-CO-1020 and NIGMS, NIH, Grant Y1-GM-1104.
- TB
- tuberculosis
- Ag85
- antigen 85
- TMM
- trehalose monomycolate
- TDM
- trehalose dimycolate
- IAA
- iodoacetamide
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid
- PDB
- Protein Data Bank
- r.m.s.
- root mean square
- MIC
- minimum inhibitory concentration.
REFERENCES
- 1. World Health Organization (2013) Global Tuberculosis Report 2013, World Health Organization, Geneva [Google Scholar]
- 2. Loewenberg S. (2012) India reports cases of totally drug-resistant tuberculosis. Lancet 379, 205. [DOI] [PubMed] [Google Scholar]
- 3. Vernon A., Burman W., Benator D., Khan A., Bozeman L. (1999) Acquired rifamycin monoresistance in patients with HIV-related tuberculosis treated with once-weekly rifapentine and isoniazid. Tuberculosis Trials Consortium. Lancet 353, 1843–1847 [DOI] [PubMed] [Google Scholar]
- 4. Weiner M., Benator D., Burman W., Peloquin C. A., Khan A., Vernon A., Jones B., Silva-Trigo C., Zhao Z., Hodge T. (2005) Association between acquired rifamycin resistance and the pharmacokinetics of rifabutin and isoniazid among patients with HIV and tuberculosis. Clin. Infect. Dis. 40, 1481–1491 [DOI] [PubMed] [Google Scholar]
- 5. Boulle A., Van Cutsem G., Cohen K., Hilderbrand K., Mathee S., Abrahams M., Goemaere E., Coetzee D., Maartens G. (2008) Outcomes of nevirapine- and efavirenz-based antiretroviral therapy when coadministered with rifampicin-based antitubercular therapy. JAMA 300, 530–539 [DOI] [PubMed] [Google Scholar]
- 6. Jarlier V., Nikaido H. (1994) Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol. Lett. 123, 11–18 [DOI] [PubMed] [Google Scholar]
- 7. Crick D. C., Mahapatra S., Brennan P. J. (2001) Biosynthesis of the arabinogalactan-peptidoglycan complex of Mycobacterium tuberculosis. Glycobiology 11, 107R–118R [DOI] [PubMed] [Google Scholar]
- 8. Song H., Sandie R., Wang Y., Andrade-Navarro M. A., Niederweis M. (2008) Identification of outer membrane proteins of Mycobacterium tuberculosis. Tuberculosis 88, 526–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaur D., Guerin M. E., Skovierová H., Brennan P. J., Jackson M. (2009) Chapter 2: Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69, 23–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marrakchi H., Lanéelle M. A., Daffé M. (2014) Mycolic acids: structures, biosynthesis, and beyond. Chem. Biol. 21, 67–85 [DOI] [PubMed] [Google Scholar]
- 11. Jackson M., McNeil M. R., Brennan P. J. (2013) Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Future Microbiol. 8, 855–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Favrot L., Ronning D. R. (2012) Targeting the mycobacterial envelope for tuberculosis drug development. Expert Rev. Anti Infect. Ther. 10, 1023–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banerjee A., Dubnau E., Quemard A., Balasubramanian V., Um K. S., Wilson T., Collins D., de Lisle G., Jacobs W. R., Jr. (1994) inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263, 227–230 [DOI] [PubMed] [Google Scholar]
- 14. Belanger A. E., Besra G. S., Ford M. E., Mikusová K., Belisle J. T., Brennan P. J., Inamine J. M. (1996) The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc. Natl. Acad. Sci. U.S.A. 93, 11919–11924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tahlan K., Wilson R., Kastrinsky D. B., Arora K., Nair V., Fischer E., Barnes S. W., Walker J. R., Alland D., Barry C. E., 3rd, Boshoff H. I. (2012) SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1797–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsumoto M., Hashizume H., Tomishige T., Kawasaki M., Tsubouchi H., Sasaki H., Shimokawa Y., Komatsu M. (2006) OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3, e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stover C. K., Warrener P., VanDevanter D. R., Sherman D. R., Arain T. M., Langhorne M. H., Anderson S. W., Towell J. A., Yuan Y., McMurray D. N., Kreiswirth B. N., Barry C. E., Baker W. R. (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405, 962–966 [DOI] [PubMed] [Google Scholar]
- 18. Makarov V., Manina G., Mikusova K., Möllmann U., Ryabova O., Saint-Joanis B., Dhar N., Pasca M. R., Buroni S., Lucarelli A. P., Milano A., De Rossi E., Belanova M., Bobovska A., Dianiskova P., Kordulakova J., Sala C., Fullam E., Schneider P., McKinney J. D., Brodin P., Christophe T., Waddell S., Butcher P., Albrethsen J., Rosenkrands I., Brosch R., Nandi V., Bharath S., Gaonkar S., Shandil R. K., Balasubramanian V., Balganesh T., Tyagi S., Grosset J., Riccardi G., Cole S. T. (2009) Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324, 801–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harth G., Lee B. Y., Wang J., Clemens D. L., Horwitz M. A. (1996) Novel insights into the genetics, biochemistry, and immunocytochemistry of the 30-kilodalton major extracellular protein of Mycobacterium tuberculosis. Infect. Immun. 64, 3038–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Belisle J. T., Vissa V. D., Sievert T., Takayama K., Brennan P. J., Besra G. S. (1997) Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 276, 1420–1422 [DOI] [PubMed] [Google Scholar]
- 21. Sanki A. K., Boucau J., Ronning D. R., Sucheck S. J. (2009) Antigen 85C-mediated acyl-transfer between synthetic acyl donors and fragments of the arabinan. Glycoconj. J. 26, 589–596 [DOI] [PubMed] [Google Scholar]
- 22. Jackson M., Raynaud C., Lanéelle M. A., Guilhot C., Laurent-Winter C., Ensergueix D., Gicquel B., Daffé M. (1999) Inactivation of the antigen 85C gene profoundly affects the mycolate content and alters the permeability of the Mycobacterium tuberculosis cell envelope. Mol. Microbiol. 31, 1573–1587 [DOI] [PubMed] [Google Scholar]
- 23. Armitige L. Y., Jagannath C., Wanger A. R., Norris S. J. (2000) Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infect Immun. 68, 767–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen L., Chinnapapagari S., Thompson C. J. (2005) FbpA-dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J. Bacteriol. 187, 6603–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ronning D. R., Klabunde T., Besra G. S., Vissa V. D., Belisle J. T., Sacchettini J. C. (2000) Crystal structure of the secreted form of antigen 85C reveals potential targets for mycobacterial drugs and vaccines. Nat. Struct. Biol. 7, 141–146 [DOI] [PubMed] [Google Scholar]
- 26. Ronning D. R., Vissa V., Besra G. S., Belisle J. T., Sacchettini J. C. (2004) Mycobacterium tuberculosis antigen 85A and 85C structures confirm binding orientation and conserved substrate specificity. J. Biol. Chem. 279, 36771–36777 [DOI] [PubMed] [Google Scholar]
- 27. Anderson D. H., Harth G., Horwitz M. A., Eisenberg D. (2001) An interfacial mechanism and a class of inhibitors inferred from two crystal structures of the Mycobacterium tuberculosis 30 kDa major secretory protein (antigen 85B), a mycolyl transferase. J. Mol. Biol. 307, 671–681 [DOI] [PubMed] [Google Scholar]
- 28. Favrot L., Grzegorzewicz A. E., Lajiness D. H., Marvin R. K., Boucau J., Isailovic D., Jackson M., Ronning D. R. (2013) Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nat. Commun. 4, 2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boucau J., Sanki A. K., Voss B. J., Sucheck S. J., Ronning D. R. (2009) A coupled assay measuring Mycobacterium tuberculosis antigen 85C enzymatic activity. Anal. Biochem. 385, 120–127 [DOI] [PubMed] [Google Scholar]
- 30. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server. in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press Inc., Totowa, NJ [Google Scholar]
- 31. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 27, 307–326 [DOI] [PubMed] [Google Scholar]
- 32. Kissinger C. R., Gehlhaar D. K., Fogel D. B. (1999) Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D. Biol. Crystallogr. 55, 484–491 [DOI] [PubMed] [Google Scholar]
- 33. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moriarty N. W., Grosse-Kunstleve R. W., Adams P. D. (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guzman Barron E. S., Singer T. P. (1945) Studies on biological oxidations: XIX: sulfhydryl enzymes in carbohydrate metabolism. J. Biol. Chem. 157, 221–240 [Google Scholar]
- 37. Lu J., Vlamis-Gardikas A., Kandasamy K., Zhao R., Gustafsson T. N., Engstrand L., Hoffner S., Engman L., Holmgren A. (2013) Inhibition of bacterial thioredoxin reductase: an antibiotic mechanism targeting bacteria lacking glutathione. FASEB J. 27, 1394–1403 [DOI] [PubMed] [Google Scholar]
- 38. Ravelli R. B., McSweeney S. M. (2000) The “fingerprint” that x-rays can leave on structures. Structure 8, 315–328 [DOI] [PubMed] [Google Scholar]
- 39. Weik M., Ravelli R. B., Kryger G., McSweeney S., Raves M. L., Harel M., Gros P., Silman I., Kroon J., Sussman J. L. (2000) Specific chemical and structural damage to proteins produced by synchrotron radiation. Proc. Natl. Acad. Sci. U.S.A. 97, 623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leiros H. K., McSweeney S. M., Smalås A. O. (2001) Atomic resolution structures of trypsin provide insight into structural radiation damage. Acta Crystallogr. D Biol. Crystallogr. 57, 488–497 [DOI] [PubMed] [Google Scholar]
- 41. Yamaguchi T., Sano K., Takakura K., Saito I., Shinohara Y., Asano T., Yasuhara H. (1998) Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial. Stroke 29, 12–17 [DOI] [PubMed] [Google Scholar]
- 42. Parnham M., Sies H. (2000) Ebselen: prospective therapy for cerebral ischaemia. Expert Opin. Investig. Drugs 9, 607–619 [DOI] [PubMed] [Google Scholar]
- 43. Singh N., Halliday A. C., Thomas J. M., Kuznetsova O. V., Baldwin R., Woon E. C., Aley P. K., Antoniadou I., Sharp T., Vasudevan S. R., Churchill G. C. (2013) A safe lithium mimetic for bipolar disorder. Nat. Commun. 4, 1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Craik C. S., Roczniak S., Largman C., Rutter W. J. (1987) The catalytic role of the active site aspartic acid in serine proteases. Science 237, 909–913 [DOI] [PubMed] [Google Scholar]
- 45. Sprang S., Standing T., Fletterick R. J., Stroud R. M., Finer-Moore J., Xuong N. H., Hamlin R., Rutter W. J., Craik C. S. (1987) The three-dimensional structure of Asn102 mutant of trypsin: role of Asp102 in serine protease catalysis. Science 237, 905–909 [DOI] [PubMed] [Google Scholar]
- 46. Puech V., Guilhot C., Perez E., Tropis M., Armitige L. Y., Gicquel B., Daffé M. (2002) Evidence for a partial redundancy of the fibronectin-binding proteins for the transfer of mycoloyl residues onto the cell wall arabinogalactan termini of Mycobacterium tuberculosis. Mol. Microbiol. 44, 1109–1122 [DOI] [PubMed] [Google Scholar]
- 47. Backus K. M., Boshoff H. I., Barry C. S., Boutureira O., Patel M. K., D'Hooge F., Lee S. S., Via L. E., Tahlan K., Barry C. E., 3rd, Davis B. G. (2011) Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat. Chem. Biol. 7, 228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]