

Background: Epithelial-mesenchymal transition (EMT) plays critical roles in tissue development and cancer biology.
Results: TIF1γ promotes sumoylation of SnoN1 and, thereby, regulates EMT.
Conclusion: A novel TIF1γ-SnoN1 sumoylation pathway is crucial for the suppression of EMT.
Significance: The identification of the TIF1γ-SnoN1 sumoylation signaling link advances our understanding of EMT.
Keywords: Cell Signaling, Epithelial-Mesenchymal Transition (EMT), Protein-Protein Interaction, Sumoylation, Transforming Growth Factor β (TGFβ), SnoN, TIF1γ
Abstract
Epithelial-mesenchymal transition (EMT) is a fundamental cellular process that contributes to epithelial tissue morphogenesis during normal development and in tumor invasiveness and metastasis. The transcriptional regulator SnoN robustly influences EMT in response to the cytokine TGFβ, but the mechanisms that regulate the fundamental role of SnoN in TGFβ-induced EMT are not completely understood. Here we employ interaction proteomics to uncover the signaling protein TIF1γ as a specific interactor of SnoN1 but not the closely related isoform SnoN2. A 16-amino acid peptide within a unique region of SnoN1 mediates the interaction of SnoN1 with TIF1γ. Strikingly, although TIF1γ is thought to act as a ubiquitin E3 ligase, we find that TIF1γ operates as a small ubiquitin-like modifier (SUMO) E3 ligase that promotes the sumoylation of SnoN1 at distinct lysine residues. Importantly, TIF1γ-induced sumoylation is required for the ability of SnoN1 to suppress TGFβ-induced EMT, as assayed by the disruption of the morphogenesis of acini in a physiologically relevant three-dimensional model of normal murine mammary gland (NMuMG) epithelial cells. Collectively, our findings define a novel TIF1γ-SnoN1 sumoylation pathway that plays a critical role in EMT and has important implications for our understanding of TGFβ signaling and diverse biological processes in normal development and cancer biology.
Introduction
Control of epithelial-mesenchymal transition (EMT)6 is essential in normal development and homeostasis (1). The cellular morphogenetic events of EMT are comprised of the loss of epithelial cuboidal morphology, loss of cell-cell contact, and the establishment of a fibroblastic mesenchymal shape (2, 3). Attendant with these morphogenetic changes in cells undergoing EMT, markers of epithelial cells such as E-cadherin are down-regulated, and mesenchymal proteins such as N-cadherin are up-regulated (4). EMT of malignant cells in epithelial tumors is thought to portend cancer invasiveness and metastasis (5). Therefore, elucidation of the molecular basis of EMT will advance our understanding of tissue development and cancer.
The cellular and molecular mechanisms that control EMT have been the subject of intense investigation. Much of what we have learned about EMT has come from standard tissue culture studies of epithelial cells. However, three-dimensional models of epithelial cells such as NMuMG mammary epithelial cells provide a more physiologically relevant system in which EMT manifests in the disruption of the normal morphogenesis of tubular acini (6–9).
An essential role for the cytokine TGFβ has been established in EMT, which provides the basis for the ability of TGFβ to promote the progression of epithelial tumors (1–3, 10). Progress has been achieved in our understanding of the signaling mechanisms by which TGFβ regulates cellular responses, including EMT. The TGFβ receptor activates the Smad signaling pathway, which leads to Smad-dependent alterations in gene expression and consequent cellular responses (11). The transcriptional regulator SnoN, which interacts with the transcription factors Smad2, Smad3, and Smad4, suppresses TGFβ-induced EMT (12, 13). Although SnoN robustly influences EMT, the mechanisms that regulate the fundamental role of SnoN in EMT are not completely understood.
Posttranslational modifications impact SnoN function in diverse biological settings in proliferating and postmitotic cells. Several ubiquitin ligases, including Cdh1-anaphase-promoting complex (Cdh1-APC), Smurf2, and Arkadia induce the ubiquitination and consequent proteasome-dependent degradation of SnoN (14–17). Ubiquitin-dependent degradation of SnoN influences cell cycle progression in proliferating cells and axon growth in postmitotic neurons (18, 19). SnoN also undergoes sumoylation at the distinct sites lysine 50 and lysine 383, which contributes to the ability of SnoN to regulate transcription (12, 20).
Recent studies suggest that SnoN exerts distinct biological functions in an isoform-specific manner (21). SnoN is the product of the Sno (Ski-related novel) gene. SnoN1 and SnoN2 represent the two major alternatively spliced isoforms of SnoN that are nearly identical except for a 46-amino acid region that is present in SnoN1 and absent in SnoN2 (22). In the nervous system, SnoN1 and SnoN2 play opposing roles in the control of neuronal migration (21), a biological process with parallels to EMT (23, 24). These studies have raised the fundamental question of whether SnoN might be regulated in an isoform-specific manner and whether such regulation might be of general significance, including in EMT.
In this study, we discovered a novel link between the signaling protein TIF1γ and the transcriptional regulator SnoN1 that plays a critical role in the control of EMT. Using an affinity capture mass spectrometry-based approach, we uncovered the protein TIF1γ as a specific high confidence interactor of SnoN1, but not SnoN2, in cells. In structure-function analyses, we identified a 16-amino acid, TIF1γ-interacting peptide (TIPtide) motif that resides within the unique 46-amino acid region in SnoN1. Strikingly, although TIF1γ reportedly acts as an E3 monoubiquitin ligase for the signaling protein Smad4 (25), we discovered that TIF1γ operates as a SUMO E3 ligase that triggers the sumoylation of SnoN1 at lysines 50 and 383. Importantly, TIF1γ-induced sumoylation of SnoN1 plays a critical role in the ability of SnoN1 to suppress TGFβ-induced EMT assayed by the disruption of the morphogenesis of acini in the three-dimensional model of NMuMG mammary epithelial cells. Collectively, our findings define a novel TIF1γ-SnoN1 sumoylation pathway that plays a critical role in EMT. Our study bears significant implications for our understanding of TGFβ signaling and diverse biological processes in normal development and cancer biology.
EXPERIMENTAL PROCEDURES
Plasmids and Antibodies
The TIF1γ, SnoN1, and SnoN2 RNAi plasmids were generated to target the sequences GGACCAAAGGAAATGTGAAC, AACCAGTAGAGAATTATACAGTT, and AAGGCAGAGACAAATTCATCAAT, respectively, as described previously (21, 26, 27). The FLAG-TIF1γ expression plasmid was provided by Dr. Frank J. Rauscher III. The HA- and GFP-TIF1γ expression plasmids were generated by cloning full-length human TIF1γ into pcDNA3 or pEGFP-C1, respectively. The SnoN1/2, SnoN1KdR, and SUMO-SnoN1 expression plasmids have been described previously (20, 21, 28, 29). The mutant TIF1γ and SnoN expression plasmids were generated by site-directed PCR mutagenesis. Rabbit TIF1γ (Bethyl and Santa Cruz Biotechnology), rabbit SnoN (Santa Cruz Biotechnology), rat HA (Roche), mouse FLAG (Sigma), rabbit GFP (Invitrogen), mouse GFP (NeuroMab), rabbit ERK1/2 (Cell Signaling Technology), mouse Cdc27 (Santa Cruz Biotechnology), and rabbit E-cadherin (Cell Signaling Technology) antibodies were used.
Proteomic Analysis of SnoN Complexes
293T cells expressing HA-FLAG-SnoN1 or SnoN2 were lysed in lysis buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40, and protease inhibitors) and processed for proteomic analysis as described previously (30–32). Briefly, cell lysates were subjected to immunoprecipitation with HA antibody resin (Sigma), and proteins were eluted with HA peptide (Sigma). Eluted proteins were precipitated with trichloroacetic acid and digested with trypsin at 37 °C for 4 h. Digested peptides were desalted with C18 resin (Empore, 3 m), and analyzed with LC-MS/MS using an LTQ linear ion trap mass spectrometer (Thermo Scientific). The resulting spectra were searched using SEQUEST, and the resulting list of identifications was loaded into CompPASS to facilitate a determination of the WD and Z scores (30).
Analysis of Sumoylation
Analysis of sumoylation was performed as described previously (28, 29), with modifications. Briefly, 293T cells cotransfected with expression plasmids for FLAG-TIF1γ, HA-SUMO1, and GFP-SnoN, as indicated, were lysed in 150 μl of denaturing buffer (150 mm NaCl, 50 mm Tris-HCl (pH 7.5), 1 mm EDTA, 1% Nonidet P-40, 1% SDS, 1 mm PMSF, 10 mm N-ethylmaleimide, 10 μg/ml aprotinin, 10 μg/ml pepstatin, 10 μg/ml leupeptin, 1 mm dithiothreitol, 50 mm NaF, and 1 mm Na3VO4) and sonicated. The lysate was diluted with 1350 μl of lysis buffer (150 mm NaCl, 50 mm Tris-HCl (pH 7.5), 1 mm EDTA, 1% Nonidet P-40, 1 mm PMSF, 10 mm N-ethylmaleimide, 10 μg/ml aprotinin, 10 μg/ml pepstatin, 10 μg/ml leupeptin, 1 mm dithiothreitol, 50 mm NaF, and 1 mm Na3VO4) and subjected to immunoprecipitation with GFP or SnoN antibodies at 4 °C. Immunoprecipitated protein and input samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibodies. Analysis of sumoylation was also performed using HepG2 cells or TIF1γ shRNA-expressing HepG2 cells that were prepared with blasticidin selection after transfection with the TIF1γ or control RNAi plasmid together with a blasticidin-resistant plasmid using Lipofectamine 2000 (Invitrogen).
Quantitative RT-PCR
DNase-treated TRIzol-extracted (Invitrogen) RNA from NMuMG cells was reverse-transcribed using SuperScript II transcriptase (Invitrogen) and oligo(dT)12–18 (Amersham Biosciences) (12, 26, 33, 34). The cDNAs were subjected to quantitative PCR of the following genes: Zeb1, 5′ TCGGAAGACAGAGAATGGAATG3′ (forward) and 5′CCTCTTACCTGTGTGCTCATATT3′ (reverse); Zeb2, 5′CTCATTCTGGGTCCTACAGTTC3′ (forward) and 5′GGGAAGAACCCGTCTTGATATT3′ (reverse); snail, CCACTCGGATG TGAAGAGATAC3′ (forward) and 5′CCAGACTCTTGG TGCTTGT3′ (reverse); matrix metalloproteinase 9 (MMP9), 5′CTGGAAC TCACACGACATCTT3′ (forward) and 5′TCCACCTT GTTCACCTCATTT3′ (reverse); plasminogen activator inhibitor 1 (PAI1), 5′TCTCAGAGGTGGAAAGAGCCAG3′ (forward) and 5′TGAAGTAGAGGGCATTCACCAGC3′ (reverse); and, as a housekeeping gene, GAPDH, 5′TCAACAGCAACTCCCACTCTTCCA3′ (forward) and 5′ACCCTGTTGCTGTAGCCGTATTCA (reverse), employed as an internal control and using a 2× SYBR Green mix (Bio-Rad) and Rotor-Gene thermocycler (Corbett Research). The specificity of the amplification products was confirmed using the melting curve method. Data were analyzed and expressed as described previously (34).
Three-dimensional NMuMG Cell Acini Formation Assay
Mouse mammary epithelial (NMuMG) cells were purchased from the ATCC and maintained in growth medium (DMEM containing 10% FBS and 10 μg/ml insulin (Invitrogen)) in standard culture dishes in a 5% CO2 humidified incubator at 37 °C (12). NMuMG cells were transfected with the indicated plasmids using Lipofectamine LTX with Plus reagent (Invitrogen) 48 or 72 h prior to the following procedure. Three-dimensional cultures of NMuMG cells were prepared in 8-well chamber slides (Millicell EZ Slide, Millipore). 8-well chamber slide wells were precoated with Matrigel (BD Biosciences) cushions, where 75 μl of ice-cold 30% growth factor-reduced Matrigel solution (3 mg/ml final concentration) in antibiotic-antimitotic-containing NMuMG growth medium was added to each well. The chamber slides were then transferred to a 5% CO2 humidified incubator at 37 °C for 1 h to allow setting of the Matrigel cushions. Next, 75 μl of 1.3 × 104 cells/ml of NMuMG cells trypsinized and resuspended in a 30% growth factor-reduced Matrigel-containing growth medium was overlaid on top of the Matrigel cushion within each well of the 8-well chamber slides and incubated in a 5% CO2 humidified incubator at 37 °C. The next day and every third day, growth medium alone or with 20 pm or 100 pm TGFβ1 was added until completion of the assay. Differential interference contrast (DIC) images of the live multicellular structures after 10 days in three-dimensional cultures were captured using an inverted microscope with a ×20 objective lens (Olympus IX70). The multicellular structures, including acini, were then fixed with 4% paraformaldehyde, permeabilized with 0.5% cold Triton X-100, blocked with 5% BSA in phosphate-buffered saline, and subjected to immunocytochemical analyses using a rabbit E-cadherin antibody (Cell Signaling Technology) as the primary antibody and Cy3-conjugated anti-rabbit IgG (Jackson Lab) as the secondary antibody, and incubated with the DNA dye bisbenzimide (Hoechst 33258) to visualize nuclei (Sigma). Images of multicellular colonies were captured using a fluorescent microscope with a ×40 objective lens (Zeiss Axiovert 200 M). For each experiment, exposure times for E-cadherin and Hoechst-derived signals were kept constant. A total of six colonies were assessed for each condition per experiment.
RESULTS
Identification of TIF1γ as an Interaction Partner of SnoN1
Recent studies suggest that the two isoforms of SnoN, SnoN1 and SnoN2, regulate cellular responses in an isoform-specific manner (21). To determine how SnoN might be regulated isoform-specifically, we performed affinity capture followed by mass spectrometry to identify specific SnoN1- and SnoN2-interacting proteins. We immunopurified SnoN1 or SnoN2 from 293T cells and subjected purified protein complexes to LC-MS/MS analyses to identify associated proteins. We next used the software platform Comparative Proteomics Analysis Software Suite (CompPASS) to compare the SnoN immunoprecipitation/MS dataset against a large number of unrelated parallel immunoprecipitation/MS datasets to distinguish high confidence interacting proteins (HCIPs) from the background (Fig. 1A) (30). CompPASS identifies HCIPs on the basis of the WD score, which incorporates the frequency with which they are identified within the stats table, the abundance as represented by total spectral counts when found, and the reproducibility of technical replicates (30). Proteins with WD scores of approximately >30 were considered as HCIPs (30). We identified the transcriptional regulatory proteins Smad2, Smad4, and Ski as HCIPs of both SnoN1 and SnoN2 (Fig. 1A), validating our proteomics approach because these proteins are known to interact with SnoN (13, 35). Strikingly, we uncovered the protein TIF1γ (also referred to as Trim33) as a robust and specific interactor of SnoN1 but not SnoN2 (Fig. 1A). TIF1γ was also of particular interest because, similar to SnoN, TIF1γ suppresses TGFβ-induced EMT (12, 36). These observations raised the fundamental question of whether TIF1γ and SnoN1 might represent components of a novel signaling link that regulates EMT.
FIGURE 1.
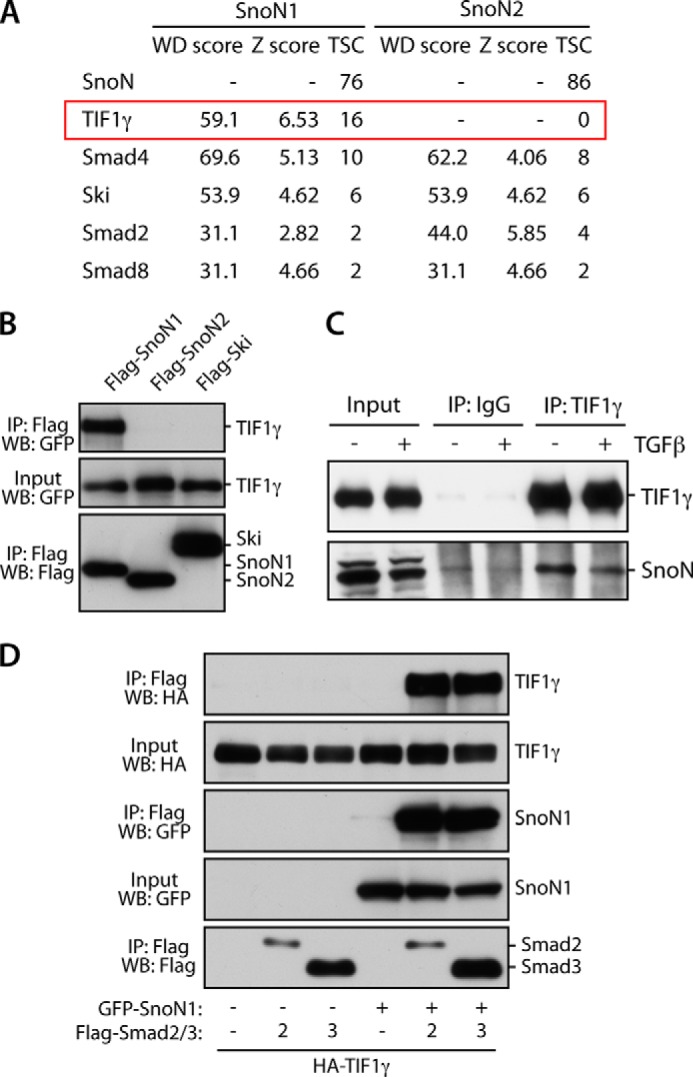
Identification of TIF1γ as an interaction partner of SnoN1. A, lysates of 293T cells stably expressing HA-SnoN1 or SnoN2 were immunoprecipitated with HA antibody and subjected to proteomic analysis using LC-MS/MS and CompPASS. TSC, total spectral count. B, lysates of 293T cells transfected with the GFP-TIF1γ expression plasmid together with an expression plasmid encoding FLAG-SnoN1, FLAG-SnoN2, or FLAG-Ski were immunoprecipitated (IP) using the FLAG antibody. WB, Western blot. C, lysates of HepG2 cells were immunoprecipitated with the TIF1γ antibody or control IgG. Endogenous SnoN formed a complex with endogenous TIF1γ in cells. Treatment of the cells with TGFβ (2 ng/ml, 1.5 h) did not affect the TIF1γ-SnoN interaction. D, lysates of 293T cells transfected with expression plasmids encoding HA-TIF1γ and GFP-SnoN1 or a control vector and FLAG-Smad2, FLAG-Smad3, or a control vector together with a plasmid expressing constitutively active TGFβR1 were immunoprecipitated with the FLAG antibody.
We first validated the interaction of TIF1γ with SnoN1 in cells. In coimmunoprecipitation analyses, SnoN1, but not SnoN2, formed a complex with TIF1γ in 293T cells (Fig. 1B). Like SnoN2, the SnoN-related protein Ski also failed to interact with TIF1γ (Fig. 1B). We also found that endogenous TIF1γ interacted with endogenous SnoN in cells (Fig. 1C). Together, these results confirm that TIF1γ and SnoN1 form a complex in cells.
TIF1γ has been suggested to associate with the SnoN1-interacting transcription factors Smad2 and Smad3 (37), raising the question of whether SnoN1 regulates TIF1γ-Smad2/3 association. To address this question, we examined the coimmunoprecipitation of TIF1γ by Smad2 or Smad3 in the absence or presence of expressed SnoN1. Interestingly, we found that, in the absence of SnoN1, TIF1γ failed to interact with Smad2/3 (Fig. 1D). These results suggest that SnoN1 may play a key role in assembling a protein complex containing Smad2 or Smad3 and TIF1γ, with potential functional implications for the TGFβ-Smad signaling pathway.
A 16-Amino Acid Region of SnoN1 Interacts with TIF1γ
We next performed structure-function analyses to determine the regions of SnoN1 that mediate the TIF1γ-SnoN1 interaction. SnoN1 and SnoN2 are nearly identical, except for a 46-amino acid insert present in SnoN1. The 46-amino acid region of SnoN1 (432–477) includes amino acid residues conserved across different species (Fig. 2A). The finding that SnoN1 selectively interacts with TIF1γ suggested that the 46-amino acid region of SnoN1 might play a key role in specifying the TIF1γ-SnoN1 interaction. In deletion analyses of the 46-amino acid region within SnoN1, removal of the N-terminal (Δ432–451) or C-terminal (Δ468–477) portion of the 46-amino acid region failed to diminish the ability of SnoN1 to form a complex with TIF1γ in cells (Fig. 2B). However, deletions that encroached on a 16-amino acid motif (452–467) profoundly reduced the ability of SnoN1 to interact with TIF1γ in cells (Fig. 2B). Alanine scanning mutagenesis within the 16-amino acid TIPtide motif revealed that almost the entire motif was required for robust interaction of SnoN1 with TIF1γ in cells (Fig. 2C). In other analyses, the 16-amino acid TIPtide motif within SnoN1 was sufficient to interact with TIF1γ in cells, albeit less effectively than full-length SnoN1 (Fig. 2D). Collectively, these studies suggest that SnoN1 forms a specific complex with TIF1γ in cells.
FIGURE 2.

A 16-amino acid region (TIPtide) mediates SnoN1 interaction with TIF1γ. A, schematic of SnoN1 and SnoN2, the alternatively spliced isoforms of the Sno gene. The dachshund homology domain (DHD), SAND domain, Smad2-interacting motif, and coiled-coil domains (CC) are indicated. Alignment of the 46-amino acid (aa) region from various species indicates conserved amino acids. B, lysates of 293T cells transfected with expression plasmids encoding HA-TIF1γ and wild-type GFP-SnoN1 or deletion mutants of GFP-SnoN1 were immunoprecipitated (IP) with the GFP antibody. WB, Western blot. C, lysates of 293T cells transfected with expression plasmids encoding HA-TIF1γ and wild-type GFP-SnoN1 or alanine mutants of GFP-SnoN1 were immunoprecipitated with the GFP antibody. D, lysates of 293T cells transfected with expression plasmids encoding HA-TIF1γ and wild-type GFP-SnoN1, GFP-TIPtide, or GFP were immunoprecipitated with the GFP antibody.
TIF1γ Acts as a Novel SnoN1 SUMO E3 Ligase
Having identified a specific interaction of TIF1γ with SnoN1, we next determined how TIF1γ might regulate SnoN1. TIF1γ has been reported to form a complex with the SnoN1-interacting transcription factors Smad2 and Smad3 in diverse cell types, including epithelial and embryonic stem cells (13, 37, 38). TIF1γ is classified as a member of the tripartite motif/ring-finger, two-B-boxes (B1 and B2) and a predicted a-helical coiled-coil domain (TRIM/RBCC) family of E3 ubiquitin ligases, containing an N-terminal really interesting new gene (RING) domain, tandem B-box motifs, and C-terminal plant homeodomain (PHD) and bromodomain (BRD) domains (39). Accordingly, TIF1γ appears to promote monoubiquitination of Smad4, which also forms a complex with SnoN1 (13). The TIF1γ-induced ubiquitination of Smad4 inhibits its interaction with Smad2 during embryonic germ layer specification (25, 40). Because SnoN is known to undergo ubiquitination (15–17), we first tested whether TIF1γ might induce ubiquitination of SnoN1. Surprisingly, TIF1γ failed to effectively stimulate ubiquitination of SnoN1 in cells (data not shown).
We reasoned that TIF1γ might induce a distinct lysine-directed modification of SnoN1. Because the TIF1γ-related protein TIF1β operates as a SUMO E3 ligase (41), we asked whether TIF1γ might induce the sumoylation of SnoN1. Remarkably, we found that TIF1γ stimulated the robust covalent modification of SnoN1 with SUMO in cells (Fig. 3A). Consistent with the specific interaction of TIF1γ with SnoN1, but not SnoN2, TIF1γ failed to induce the sumoylation of SnoN2 (Fig. 3A). Notably, SnoN undergoes sumoylation at lysines 50 and 383 (12, 20). Consistent with a key role for these lysines in TIF1γ-induced sumoylation, TIF1γ failed to induce the sumoylation of a SnoN1 mutant protein in which lysines 50 and 383 were replaced with arginine (SnoN1KdR) (Fig. 3A). Because lysines 50 and 383 are present in both SnoN1 and SnoN2, our results suggest that TIF1γ specifically stimulates the sumoylation of SnoN1, but not SnoN2, in cells because of its specific interaction with SnoN1.
FIGURE 3.

TIF1γ acts as a novel SnoN1 SUMO E3 ligase. A, lysates of 293T cells transfected with the FLAG-TIF1γ expression plasmid or control vector together with the HA-SUMO1 and GFP-SnoN1, SnoN2, or SnoN1KdR expression plasmid were immunoprecipitated (IP) with the GFP antibody. WB, Western blot. B, lysates of 293T cells transfected with expression plasmids encoding GFP-SnoN1 and wild-type FLAG-TIF1γ or deletion mutants of FLAG-TIF1γ were immunoprecipitated with the FLAG antibody. Vec, vector. C, lysates of 293T cells transfected with the FLAG-TIF1γ expression plasmid, deletion mutants of FLAG-TIF1γ, or control vector together with the HA-SUMO1 expression plasmid and GFP-SnoN1 were sonicated and immunoprecipitated with the GFP antibody. D, lysates of 293T cells transfected with FLAG-TIF1γ were subjected to a pulldown assay with GST-UBC9 or GST and immunoblotted with FLAG antibody or stained with Ponceau S dye. E, lysates of HepG2 cells stably transfected with an RNAi plasmid expressing shRNAs targeting TIF1γ or the control U6 plasmid and transiently transfected with the GFP-SnoN1 or GFP-SnoN1KdR expression plasmid together with the HA-SUMO1 expression plasmid or a control vector were sonicated and immunoprecipitated with the GFP antibody. F, lysates of HepG2 cells stably expressing shRNAs targeting TIF1γ or control cells were sonicated and immunoprecipitated with the IgG control or SnoN antibody, followed by immunoblotting with the SnoN and TIF1γ antibodies (bottom panel). Lysates (top panel) were immunoblotted with the TIF1γ or Cdc27 antibody, the latter serving as a loading control.
To further define the role of TIF1γ in SnoN1 sumoylation, we performed structure-function analyses of TIF1γ. Mutation of two conserved cysteines within the RING domain of TIF1γ (RING CS) disrupted the interaction of TIF1γ with SnoN1 (Fig. 3B). In contrast, deletion of the middle (Δmid) or the C-terminal region (ΔC-term) of TIF1γ did not impair its interaction with SnoN1 (Fig. 3B). These results suggest that the RING domain of TIF1γ associates with SnoN1. Consistent with these results, mutation of the RING domain blocked the ability of TIF1γ to induce the sumoylation of SnoN1 (Fig. 3C). In contrast, deletion of the middle region of TIF1γ had little or no effect on TIF1γ-induced sumoylation of SnoN1 (Fig. 3C). Notably, removal of the C-terminal region also blocked the ability of TIF1γ to induce the sumoylation of SnoN1 (Fig. 3C), suggesting that the C-terminal region is critical for the ability of TIF1γ to promote sumoylation independently of its association with SnoN1. In other experiments, we found that TIF1γ interacted with a recombinant form of the SUMO E2 enzyme Ubc9 (Fig. 3D), suggesting that TIF1γ may act as a SUMO E3 ligase. Finally, knockdown of TIF1γ substantially reduced the sumoylation of exogenously expressed SnoN1 or endogenous SnoN in cells (Fig. 3, E and F). Collectively, our data suggest that TIF1γ may act as a SUMO E3 ligase that promotes the sumoylation of SnoN1.
The TIF1γ-SnoN1 Sumoylation Link Controls Epithelial-Mesenchymal Transition
The identification of a function for TIF1γ in the sumoylation of SnoN1 led us next to determine the biological implications of the novel TIF1γ-SnoN1 signaling link. TIF1γ and sumoylated SnoN have been implicated in the suppression of TGFβ-induced EMT in standard two-dimensional cultures of epithelial cells (12, 36). These observations suggested that TIF1γ-induced SnoN1 sumoylation might play a critical role in the regulation of EMT. To address this question, we employed three-dimensional cultures of non-transformed NMuMG mammary epithelial cells in these analyses because these cultures provide a more physiologically relevant system for the study of biological processes, including EMT (6–8). Subjecting NMuMG cells in which SnoN1 shRNAs or SnoN2 shRNAs were expressed to immunoblotting with the SnoN antibody confirmed that NMuMG cells expressed the two isoforms SnoN1 and SnoN2 (Fig. 4A) (21). In the three-dimensional cultures, NMuMG cells underwent cell proliferation and cell-cell attachment and organized into acini with hollow centers (Fig. 4B), reflecting the apical-basal polarity nature of these structures and, thus, phenocopying the in vivo acinar nature of glandular epithelial tissue (6–8). Supporting this idea, immunofluorescence analyses of three-dimensional NMuMG cell cultures showed basolateral localization of the epithelial marker E-cadherin (Fig. 4C). Exposure of the three-dimensional NMuMG cell culture to TGFβ induced acinar lumen filling, outward protrusions at the basal surface, and deformation of the acinar structures (Fig. 4, B and C). The alterations in acinar morphology were accompanied by the down-regulation and loss of the basolateral localization of E-cadherin (Fig. 4C). Inhibition of the TGFβ-type I receptor kinase rescued normal acini morphology and E-cadherin abundance and localization in TGFβ-treated NMuMG cells (Fig. 4, B and C), suggesting that the canonical TGFβ pathway plays a critical role in the ability of TGFβ to induce dysregulation of acinar morphology and associated loss of E-cadherin levels in NMuMG cells. Collectively, these findings show that NMuMG cells form organized acini in three-dimensional cultures and that TGFβ triggers stereotypic alterations in acinar morphogenesis reflecting EMT.
FIGURE 4.
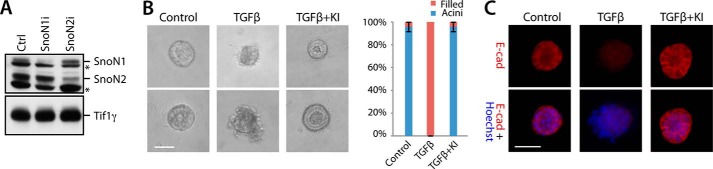
Three-dimensional acini formation of NMuMG cells. A, lysates of NMuMG cells transfected with a plasmid expressing shRNA against SnoN1 or SnoN2 were immunoblotted with the SnoN antibody. Ctrl, control. * a non-specific immunoreactive band. B, representative DIC images (left panel) and quantification of acini or filled colony morphology (right panel, mean ± S.E., n = 3) of NMuMG cells left untreated or incubated with 100 pm TGFβ for 10 days. TGFβ reduced the proportion of acini with hollow centers (ANOVA, p < 0.001). The TGFβ-specific receptor kinase inhibitor SB-431542 (KI) reversed the effect of TGFβ. C, three-dimensional NMuMG cultures as in B were subjected to immunocytochemistry using the E-cadherin (E-cad, red) antibody and Hoechst 33258 (blue). B and C, scale bar = 50 μm.
We compared the effect of wild-type SnoN1, a sumoylation gain-of-function SnoN1 in which SUMO is fused to SnoN1 (SUMO-SnoN1), or the SnoN1KdR loss of sumoylation mutant on the ability of TGFβ to disrupt the morphogenesis of acini in three-dimensional cultures of NMuMG cells. We found that SnoN1 and SUMO-SnoN1 suppressed the ability of TGFβ to induce lumen filling and disorganization of NMuMG cell acini (Fig. 5A). In contrast, expression of SnoN1KdR enhanced lumen filling of NMuMG acini (Fig. 5A). Immunocytochemical analyses showed that SnoN1 and SUMO-SnoN1 blocked, whereas SnoN1KdR promoted, the ability of TGFβ to down-regulate and disrupt the basolateral localization of E-cadherin (Fig. 5B). These data suggest that sumoylation of SnoN1 suppresses the ability of TGFβ to induce EMT in NMuMG cell acini.
FIGURE 5.

TIF1γ and SnoN1 control epithelial morphogenesis. A, representative DIC images (left panel) and quantification of acini or filled colony morphology (right panel, mean ± S.E., n = 3 or 4) of NMuMG cells transfected with vector control, wild-type SnoN1, SnoN1KdR, or SUMO-SnoN1-expressing plasmids that were left untreated or incubated with 20 pm or 100 pm TGFβ for 10 days. Wild-type SnoN1 and SUMO-SnoN1 significantly suppressed the ability of TGFβ to reduce the proportion of hollow acini (p < 0.05). SnoN1KdR decreased the proportion of hollow acini even in untreated three-dimensional cultures (p < 0.001). B, three-dimensional NMuMG cultures as in A were analyzed as in Fig. 4C. E-cad, E-cadherin. C, representative DIC images (left panel) and quantification of colony morphology (right panel) of NMuMG cells transfected with expression plasmids encoding GFP and wild-type FLAG-TIF1γ, FLAG-TIF1γ RING CS, or FLAG-TIF1γ ΔC-term that were left untreated or incubated with 20 pm or 100 pm TGFβ for 10 days. Wild-type TIF1γ significantly suppressed the ability of TGFβ to reduce the proportion of hollow acini (ANOVA, p < 0.001). Both TIF1γ mutants decreased the proportion of acini with hollow centers even in the absence of TGFβ addition (ANOVA, p < 0.001). D, three-dimensional NMuMG cultures as in C were analyzed as in Fig. 4C. A–C, scale bar = 50 μm.
We next asked whether TIF1γ regulates TGFβ-induced EMT in three-dimensional cultures of NMuMG cells in a SnoN1 sumoylation-dependent manner. Like SnoN1 and SUMO-SnoN1, TIF1γ antagonized the ability of TGFβ to induce the lumen filling and loss and mislocalization of E-cadherin in NMuMG cell acini (Fig. 5, C and D). Importantly, the TIF1γ RING CS mutant, which failed to interact with SnoN1 and induce its sumoylation (Fig. 3, B and C), failed to suppress and, instead, promoted the ability of TGFβ to disrupt acinar morphogenesis and to down-regulate E-cadherin (Fig. 5, C and D). Likewise, the TIF1γ ΔC-term mutant, which failed to induce SnoN1 sumoylation (Fig. 3C), failed to suppress and, instead, promoted the ability of TGFβ to disrupt acinar morphogenesis and to down-regulate E-cadherin in three-dimensional cultures of NMuMG cells (Fig. 5, C and D). Therefore, the phenotypes induced by the expression of the RING CS or ΔC-term TIF1γ mutant mimicked the phenotypes induced by SnoN1KdR in NMuMG cell acini. Interestingly, the RING CS and ΔC-term TIF1γ mutants and SnoN1KdR induced EMT-like alterations in NMuMG cell acini even in the absence of exogenous TGFβ (Fig. 5), suggesting that these mutants interfere dominantly with the function of endogenous TIF1γ. Consistent with these results, knockdown of endogenous TIF1γ in NMuMG cells triggered lumen filling and loss of E-cadherin in NMuMG cell acini in the absence of TGFβ (Figs. 6, A and B). These results suggest that endogenous TIF1γ regulates EMT in mammary cell acini.
FIGURE 6.

TIF1γ acts via SnoN1 sumoylation to control epithelial morphogenesis. A, representative DIC images (left panel) and quantification of colony morphology (right panel, mean ± S.E., n = 3 or 4) of NMuMG cells transfected with vector control, the RNAi plasmid encoding TIF1γ shRNAs, SUMO-SnoN1 expression plasmid, or the RNAi plasmid encoding TIF1γ shRNAs together with the SUMO-SnoN1 expression plasmid that were left untreated or incubated with 20 pm or 100 pm TGFβ for 10 days. TIF1γ RNAi decreased the proportion of acini with hollow centers even in the absence of TGFβ addition (ANOVA, p < 0.01). SUMO-SnoN1 reversed the ability of TIF1γ RNAi to reduce hollow acini under all three conditions (ANOVA, p < 0.05). B, three-dimensional NMuMG cultures as in A were analyzed as in Fig. 4C. E-cad, E-cadherin. C, representative DIC images (left panel) and quantification of colony morphology (right panel) of NMuMG cells transfected with the vector control, expression plasmid encoding wild type TIF1γ, SnoN1KdR, or TIF1γ together with SnoN1KdR that were left untreated or incubated with 20 pm or 100 pm TGFβ for 10 days. SnoN1KdR suppressed the ability of wild-type TIF1γ to maintain the proportion of hollow acini in the absence and presence of 20 pm TGFβ (ANOVA, p < 0.05). D, three-dimensional NMuMG cultures as in C were analyzed as in Fig. 4C. A–C, scale bar = 50 μm.
We also performed epistasis analyses to determine the relationship of TIF1γ and SnoN1 sumoylation in the control of EMT in mammary cell acini. Expression of SUMO-SnoN1 suppressed the ability of TIF1γ knockdown to induce the phenotype of lumen filling and loss of E-cadherin in NMuMG cell acini in the presence or absence of TGFβ (Fig. 6, A and B). In other experiments, we found that expression of the sumoylation-deficient SnoN1KdR mutant or knockdown of SnoN1 suppressed the ability of TIF1γ to inhibit TGFβ-induced acini filling and loss of E-cadherin in the three-dimensional cultures of NMuMG cells (Figs. 6, C and D, and 7, A and B). These data suggest that TIF1γ acts via sumoylation of SnoN1 to suppress EMT and the consequent disruption of acinar morphogenesis.
FIGURE 7.

SnoN1 operates downstream of TIF1γ to control epithelial morphogenesis. A, representative DIC images (left panel) and quantification of acini or filled colony morphology (right panel, mean ± S.E., n = 6) of NMuMG cells transfected with vector control, TIF1γ expression plasmid, the RNAi plasmid encoding SnoN1 shRNA, or TIF1γ expression plasmid together with the SnoN1 RNAi plasmid that were left untreated or incubated with 20 pm or 100 pm TGFβ for 10 days. TIF1γ did not reverse the ability of SnoN RNAi to reduce hollow acini (ANOVA, p < 0.001). B, three-dimensional NMuMG cultures as in A were analyzed as in Fig. 4C. E-cad, E-cadherin. B, scale bar = 50 μm.
TGFβ induces the expression of a number of transcription factors, including Zeb1, Zeb2, and snail, which, in turn, lead to repression of E-cadherin, a hallmark of EMT (1, 42). To gain further insight into the potential mechanism by which the TIF1γ-SnoN sumoylation axis controls EMT, we characterized the role of the TIF1γ-SnoN1 sumoylation pathway in TGFβ-up-regulation of Zeb1, Zeb2, and snail. In quantitative RT-PCR analyses, expression of the SUMO gain-of-function SnoN1, SUMO-SnoN1, or TIF1γ significantly suppressed the expression of Zeb1, Zeb2, and snail in TGFβ-treated NMuMG cells (Fig. 8, A and B). MMP9 and PAI-1 are extracellular genes that are induced by TGFβ and contribute to EMT (43, 44). Just as in the case of TGFβ-regulated transcription factors, SUMO-SnoN1 and TIF1γ suppressed the expression of MMP9 and PAI1 in TGFβ-treated NMuMG cells (Fig. 8, C and D). Collectively, our data define TIF1γ-SnoN1 sumoylation as a novel signaling link in the control of TGFβ-regulation of epithelial tissue morphogenesis.
FIGURE 8.
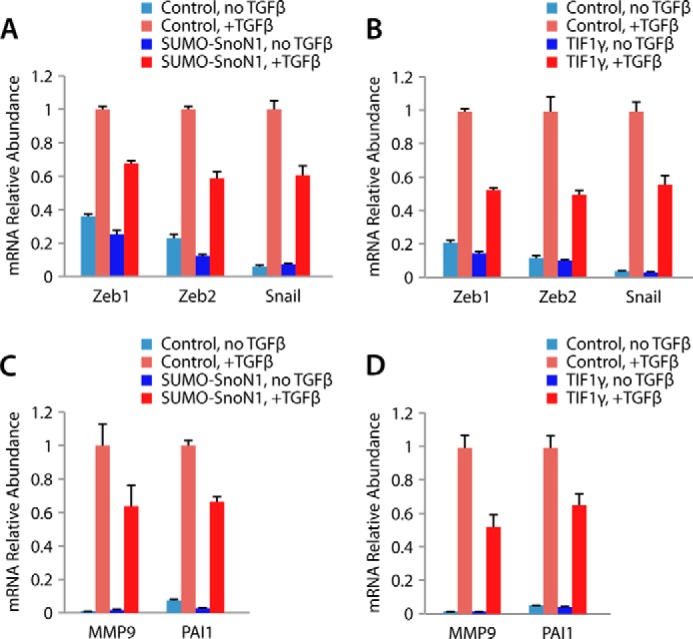
TIF1γ-SnoN sumoylation suppresses TGFβ-induced gene expression in EMT. A, quantitative RT-PCR analysis of mRNA extracted from NMuMG cells transfected with a vector control or a SUMO-SnoN1 expressing plasmid that were left untreated or incubated with 100 pm TGFβ for 24 h to measure the abundance of the mRNA of Zeb1, Zeb2, snail, and GAPDH, with the latter serving as the internal control for normalization. The mean mRNA abundance is presented relative to the expression of the plus TGFβ control, together with the mean ± S.E. from four independent experiments. SUMO-SnoN1 significantly suppressed the expression of Zeb1, Zeb2, and snail in TGFβ-treated NMuMG cells (ANOVA, p < 0.001). B, quantitative RT-PCR analysis of mRNA from NMuMG cells transfected with a vector control or a plasmid encoding FLAG-TIF1γ that were analyzed as described in A. TIF1γ significantly suppressed the expression of Zeb1, Zeb2, and snail in TGFβ-treated NMuMG cells (ANOVA, p < 0.001). C, quantitative RT-PCR analysis of MMP9 and PAI-1 as described in A. SUMO-SnoN1 suppressed the expression of MMP9 and PAI-1 in TGFβ-treated NMuMG cells (ANOVA, p < 0.05). D, quantitative RT-PCR analysis of MMP9 and PAI-1 as described in B. TIF1γ suppressed the expression of MMP9 and PAI-1 in TGFβ-treated NMuMG cells (ANOVA, p < 0.001).
DISCUSSION
In this study, we discovered a novel TIF1γ-SnoN1 sumoylation signaling mechanism that regulates EMT. Utilizing the CompPASS interaction proteomics platform (30), we identified the signaling protein TIF1γ as a novel and specific interactor of the transcriptional regulator protein SnoN1 but not the closely related isoform SnoN2. Structure-function analyses further revealed that a 16-amino acid peptide motif within a unique region of SnoN1 mediates its interaction with TIF1γ. Strikingly, whereas TIF1γ is thought to stimulate the ubiquitination of the transcription factor Smad4, we found that TIF1γ stimulates the sumoylation of SnoN1. Importantly, TIF1γ-induced SnoN1 sumoylation suppresses EMT, as assayed by disruption of the morphogenesis of acini in three-dimensional cultures of NMuMG mammary epithelial cells. Collectively, our findings define an intimate link between TIF1γ and SnoN1 that controls epithelial tissue morphogenesis.
The identification of a TIF1γ-SnoN1 sumoylation signaling link advances our understanding of the mechanisms that control epithelial tissue morphogenesis with implications for normal development and cancer biology. TGFβ-induced EMT plays a critical role in tissue morphogenesis in diverse systems during normal embryogenesis as well as during cancer invasiveness and metastasis (1–3). The finding that TIF1γ-induced sumoylation of SnoN1 suppresses TGFβ-induced EMT suggests that it will be interesting to determine whether the novel TIF1γ-induced SnoN1 sumoylation link might regulate normal epithelial tissue morphogenesis and the invasiveness and metastatic potential of epithelial tumors.
Our data suggest that the canonical Smad2/3 pathway contributes significantly to the ability of TGFβ to induce EMT, as evidenced by a complete reversal of TGFβ-induced acinar dysregulation by specific inhibition of the TGFβ type I receptor kinase. Other reports have suggested that TGFβ activation of other signaling proteins, such as Smad1/5/8 or ERK, may also contribute to EMT (42, 45). We found that TGFβ modestly and only transiently induced Smad1 phosphorylation and did not induce ERK phosphorylation in NMuMG cells (data not shown). Therefore, our data suggest that the TIF1γ-SnoN1 sumoylation axis suppresses EMT by disruption of the canonical TGFβ pathway.
In addition to TIF1γ, the SUMO E3 ligase PIAS1 acts as a SUMO E3 ligase for SnoN that regulates EMT (12). Therefore, TIF1γ and PIAS1 may cooperate in cells to stimulate the sumoylation of SnoN and, thereby, regulate EMT. In future studies, it will be important to determine whether PIAS1 stimulates sumoylation of both SnoN1 and SnoN2. In that scenario, it will be interesting to determine whether TIF1γ and PIAS1 have differential biological effects of SnoN.
The identification of a novel interaction between TIF1γ and SnoN1 also bears significant implications for our understanding of TGFβ-regulated signaling pathways. Intriguingly, TIF1γ is thought to regulate hematopoietic cell differentiation through formation of a complex with the SnoN1-interacting transcription factor Smad2/3 (37). TIF1γ also appears to induce the ubiquitination of Smad4, another SnoN1-interacting transcription factor (25, 37, 40). In our analyses, we identified a specific 16-amino acid peptide (TIPtide) within a unique region of SnoN1 that mediates the interaction of SnoN1 with TIF1γ. Notably, a distinct domain within the N-terminal region and the sp100, AIRS-1, NucP41/75, DEAF-1 (SAND) domain of SnoN mediates its interaction with the transcription factors Smad2/3 and Smad4, respectively (46). Consistent with the possibility that SnoN1 might mediate the interaction of TIF1γ and Smad proteins, in our experiments, TIF1γ failed to interact with Smad proteins in cells in the absence of SnoN1. Therefore, SnoN1 might facilitate the biological consequences attributed to TIF1γ-Smad interactions, such as the control of hematopoietic cell differentiation and germ layer specification during embryogenesis. Our findings may shed further light on the role of TIF1γ in regulating TGFβ signaling and biological responses.
Our findings also have implications for our understanding of SnoN1 functions beyond the control of EMT in epithelial tissues. In the nervous system, SnoN1 forms, isoform-specifically, a transcriptional repressor complex with the transcription factor FOXO1 and, thereby, regulates neuronal branching and migration in the developing mammalian brain (21). In addition, SnoN1 and SnoN2 promote axon growth in the developing brain (14, 18, 19). In future studies, it will be interesting to determine whether and how TIF1γ-induced sumoylation of SnoN1 might impact the key developmental events of axon growth, branching, and neuronal migration in the brain.
In summary, we identified a novel function for the signaling protein TIF1γ as a SnoN1 SUMO E3 ligase. The TIF1γ-SnoN1 sumoylation link plays a critical role in epithelial tissue morphogenesis. In addition to advancing our understanding of normal development, our findings may suggest potential new druggable targets for the treatment of malignant epithelial tumors.
Acknowledgments
We thank the members of the Bonni laboratory for helpful discussions and critical reading of the manuscript and Frank J. Rauscher III for the FLAG-TIF1γ plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grants NS041021 (to A. B.) and AG011085 (to J. W. H.). This work was also supported by the Canadian Institutes of Health Research, Alberta Innovates Health Solutions, and Canadian Breast Cancer Foundation-Prairies/Northwest Territories (to S. B.); by a JSPS postdoctoral fellowship for research abroad (to Y. I.);, and by an Alberta Cancer Foundation graduate studentship (to A. S. D.).
- EMT
- epithelial-mesenchymal transition
- SUMO
- small ubiquitin-like modifier
- TIPtide
- TIF1γ-interacting peptide
- DIC
- differential interference contrast
- HCIP
- high confidence interacting protein
- ANOVA
- analysis of variance
- NMuMG
- normal murine mammary gland
- RING
- really interesting new gene
- TSC
- total spectral count.
REFERENCES
- 1. Thiery J. P., Acloque H., Huang R. Y., Nieto M. A. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 [DOI] [PubMed] [Google Scholar]
- 2. Jakowlew S. B. (2006) Transforming growth factor-β in cancer and metastasis. Cancer Metastasis Rev. 25, 435–457 [DOI] [PubMed] [Google Scholar]
- 3. Kang Y., Massagué J. (2004) Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 118, 277–279 [DOI] [PubMed] [Google Scholar]
- 4. Thiery J. P., Sleeman J. P. (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 [DOI] [PubMed] [Google Scholar]
- 5. Thiery J. P. (2002) Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454 [DOI] [PubMed] [Google Scholar]
- 6. Shaw K. R., Wrobel C. N., Brugge J. S. (2004) Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J. Mammary Gland Biol. Neoplasia 9, 297–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Godde N. J., Galea R. C., Elsum I. A., Humbert P. O. (2010) Cell polarity in motion: redefining mammary tissue organization through EMT and cell polarity transitions. J. Mammary Gland Biol. Neoplasia 15, 149–168 [DOI] [PubMed] [Google Scholar]
- 8. Debnath J., Brugge J. S. (2005) Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 5, 675–688 [DOI] [PubMed] [Google Scholar]
- 9. Moreno-Bueno G., Peinado H., Molina P., Olmeda D., Cubillo E., Santos V., Palacios J., Portillo F., Cano A. (2009) The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat. Protoc. 4, 1591–1613 [DOI] [PubMed] [Google Scholar]
- 10. Zavadil J., Böttinger E. P. (2005) TGF-β and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774 [DOI] [PubMed] [Google Scholar]
- 11. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 12. Netherton S. J., Bonni S. (2010) Suppression of TGFβ-induced epithelial-mesenchymal transition like phenotype by a PIAS1 regulated sumoylation pathway in NMuMG epithelial cells. PLoS ONE 5, e13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stroschein S. L., Wang W., Zhou S., Zhou Q., Luo K. (1999) Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science 286, 771–774 [DOI] [PubMed] [Google Scholar]
- 14. Stegmüller J., Huynh M. A., Yuan Z., Konishi Y., Bonni A. (2008) TGFβ-Smad2 signaling regulates the Cdh1-APC/SnoN pathway of axonal morphogenesis. J. Neurosci. 28, 1961–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonni S., Wang H. R., Causing C. G., Kavsak P., Stroschein S. L., Luo K., Wrana J. L. (2001) TGF-β induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat. Cell Biol. 3, 587–595 [DOI] [PubMed] [Google Scholar]
- 16. Levy L., Howell M., Das D., Harkin S., Episkopou V., Hill C. S. (2007) Arkadia activates Smad3/Smad4-dependent transcription by triggering signal-induced SnoN degradation. Mol. Cell Biol. 27, 6068–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagano Y., Mavrakis K. J., Lee K. L., Fujii T., Koinuma D., Sase H., Yuki K., Isogaya K., Saitoh M., Imamura T., Episkopou V., Miyazono K., Miyazawa K. (2007) Arkadia induces degradation of SnoN and c-Ski to enhance transforming growth factor-β signaling. J. Biol. Chem. 282, 20492–20501 [DOI] [PubMed] [Google Scholar]
- 18. Ikeuchi Y., Stegmüller J., Netherton S., Huynh M. A., Masu M., Frank D., Bonni S., Bonni A. (2009) A SnoN-Ccd1 pathway promotes axonal morphogenesis in the mammalian brain. J. Neurosci. 29, 4312–4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stegmüller J., Konishi Y., Huynh M. A., Yuan Z., Dibacco S., Bonni A. (2006) Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron 50, 389–400 [DOI] [PubMed] [Google Scholar]
- 20. Hsu Y. H., Sarker K. P., Pot I., Chan A., Netherton S. J., Bonni S. (2006) Sumoylated SnoN represses transcription in a promoter-specific manner. J. Biol. Chem. 281, 33008–33018 [DOI] [PubMed] [Google Scholar]
- 21. Huynh M. A., Ikeuchi Y., Netherton S., de la Torre-Ubieta L., Kanadia R., Stegmüller J., Cepko C., Bonni S., Bonni A. (2011) An isoform-specific SnoN1-FOXO1 repressor complex controls neuronal morphogenesis and positioning in the mammalian brain. Neuron 69, 930–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pearson-White S., Crittenden R. (1997) Proto-oncogene Sno expression, alternative isoforms and immediate early serum response. Nucleic Acids Res. 25, 2930–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Itoh Y., Moriyama Y., Hasegawa T., Endo T. A., Toyoda T., Gotoh Y. (2013) Scratch regulates neuronal migration onset via an epithelial-mesenchymal transition-like mechanism. Nat. Neurosci. 16, 416–425 [DOI] [PubMed] [Google Scholar]
- 24. Rogers C. D., Saxena A., Bronner M. E. (2013) Sip1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. J. Cell Biol. 203, 835–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dupont S., Mamidi A., Cordenonsi M., Montagner M., Zacchigna L., Adorno M., Martello G., Stinchfield M. J., Soligo S., Morsut L., Inui M., Moro S., Modena N., Argenton F., Newfeld S. J., Piccolo S. (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 136, 123–135 [DOI] [PubMed] [Google Scholar]
- 26. Sarker K. P., Wilson S. M., Bonni S. (2005) SnoN is a cell type-specific mediator of transforming growth factor-β responses. J. Biol. Chem. 280, 13037–13046 [DOI] [PubMed] [Google Scholar]
- 27. Gaudilliere B., Shi Y., Bonni A. (2002) RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J. Biol. Chem. 277, 46442–46446 [DOI] [PubMed] [Google Scholar]
- 28. Shalizi A., Bilimoria P. M., Stegmüller J., Gaudillière B., Yang Y., Shuai K., Bonni A. (2007) PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. J. Neurosci. 27, 10037–10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shalizi A., Gaudillière B., Yuan Z., Stegmüller J., Shirogane T., Ge Q., Tan Y., Schulman B., Harper J. W., Bonni A. (2006) A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 311, 1012–1017 [DOI] [PubMed] [Google Scholar]
- 30. Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Litterman N., Ikeuchi Y., Gallardo G., O'Connell B. C., Sowa M. E., Gygi S. P., Harper J. W., Bonni A. (2011) An OBSL1-Cul7Fbxw8 ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 9, e1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang C., Mejia L. A., Huang J., Valnegri P., Bennett E. J., Anckar J., Jahani-Asl A., Gallardo G., Ikeuchi Y., Yamada T., Rudnicki M., Harper J. W., Bonni A. (2013) The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron 78, 986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eapen S. A., Netherton S. J., Sarker K. P., Deng L., Chan A., Riabowol K., Bonni S. (2012) Identification of a novel function for the chromatin remodeling protein ING2 in muscle differentiation. PLoS ONE 7, e40684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pot I., Patel S., Deng L., Chandhoke A. S., Zhang C., Bonni A., Bonni S. (2013) Identification of a novel link between the protein kinase NDR1 and TGFβ signaling in epithelial cells. PLoS ONE 8, e67178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cohen S. B., Zheng G., Heyman H. C., Stavnezer E. (1999) Heterodimers of the SnoN and Ski oncoproteins form preferentially over homodimers and are more potent transforming agents. Nucleic Acids Res. 27, 1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hesling C., Fattet L., Teyre G., Jury D., Gonzalo P., Lopez J., Vanbelle C., Morel A. P., Gillet G., Mikaelian I., Rimokh R. (2011) Antagonistic regulation of EMT by TIF1γ and Smad4 in mammary epithelial cells. EMBO Rep. 12, 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He W., Dorn D. C., Erdjument-Bromage H., Tempst P., Moore M. A., Massagué J. (2006) Hematopoiesis controlled by distinct TIF1γ and Smad4 branches of the TGFβ pathway. Cell 125, 929–941 [DOI] [PubMed] [Google Scholar]
- 38. Xi Q., Wang Z., Zaromytidou A. I., Zhang X. H., Chow-Tsang L. F., Liu J. X., Kim H., Barlas A., Manova-Todorova K., Kaartinen V., Studer L., Mark W., Patel D. J., Massagué J. (2011) A poised chromatin platform for TGF-β access to master regulators. Cell 147, 1511–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peng H., Feldman I., Rauscher F. J., 3rd (2002) Hetero-oligomerization among the TIF family of RBCC/TRIM domain-containing nuclear cofactors: a potential mechanism for regulating the switch between coactivation and corepression. J. Mol. Biol. 320, 629–644 [DOI] [PubMed] [Google Scholar]
- 40. Dupont S., Zacchigna L., Cordenonsi M., Soligo S., Adorno M., Rugge M., Piccolo S. (2005) Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 121, 87–99 [DOI] [PubMed] [Google Scholar]
- 41. Liang Q., Deng H., Li X., Wu X., Tang Q., Chang T. H., Peng H., Rauscher F. J., 3rd, Ozato K., Zhu F. (2011) Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 187, 4754–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu J., Lamouille S., Derynck R. (2009) TGF-β-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gordon G. M., Ledee D. R., Feuer W. J., Fini M. E. (2009) Cytokines and signaling pathways regulating matrix metalloproteinase-9 (MMP-9) expression in corneal epithelial cells. J. Cell Physiol. 221, 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gerwin B. I., Keski-Oja J., Seddon M., Lechner J. F., Harris C. C. (1990) TGF-β 1 modulation of urokinase and PAI-1 expression in human bronchial epithelial cells. Am. J. Physiol. 259, L262–L269 [DOI] [PubMed] [Google Scholar]
- 45. Chaudhury A., Howe P. H. (2009) The tale of transforming growth factor-β (TGFβ) signaling: a soigné enigma. IUBMB Life 61, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pot I., Ikeuchi Y., Bonni A., Bonni S. (2010) SnoN: bridging neurobiology and cancer biology. Curr. Mol. Med. 10, 667–673 [DOI] [PMC free article] [PubMed] [Google Scholar]