

Background: Mechanism(s) that regulate the phosphatase activity of PGAM5 are poorly understood.
Results: PGAM5 contains an N-terminal WDXNWD motif required for multimerization and maximal phosphatase activity.
Conclusion: PGAM5 is regulated by an intermolecular allosteric mechanism in which the assembly of PGAM5 into multimeric complexes results in robust phosphatase activity.
Significance: Our data suggest a mechanism for regulating the function of PGAM5.
Keywords: Allosteric Regulation, Enzyme Kinetics, Mitochondria, Protein Cross-linking, Protein Motif, Protein Self-assembly, Protein Serine/Threonine Phosphatase (PSP)
Abstract
Phosphoglycerate mutase 5 (PGAM5) is an atypical mitochondrial Ser/Thr phosphatase that modulates mitochondrial dynamics and participates in both apoptotic and necrotic cell death. The mechanisms that regulate the phosphatase activity of PGAM5 are poorly understood. The C-terminal phosphoglycerate mutase domain of PGAM5 shares homology with the catalytic domains found in other members of the phosphoglycerate mutase family, including a conserved histidine that is absolutely required for catalytic activity. However, this conserved domain is not sufficient for maximal phosphatase activity. We have identified a highly conserved amino acid motif, WDXNWD, located within the unique N-terminal region, which is required for assembly of PGAM5 into large multimeric complexes. Alanine substitutions within the WDXNWD motif abolish the formation of multimeric complexes and markedly reduce phosphatase activity of PGAM5. A peptide containing the WDXNWD motif dissociates the multimeric complex and reduces but does not fully abolish phosphatase activity. Addition of the WDXNWD-containing peptide in trans to a mutant PGAM5 protein lacking the WDXNWD motif markedly increases phosphatase activity of the mutant protein. Our results are consistent with an intermolecular allosteric regulation mechanism for the phosphatase activity of PGAM5, in which the assembly of PGAM5 into multimeric complexes, mediated by the WDXNWD motif, results in maximal activation of phosphatase activity. Our results suggest the possibility of identifying small molecules that function as allosteric regulators of the phosphatase activity of PGAM5.
Introduction
In humans, the 12 members of the phosphoglycerate mutase (PGAM)2 family are defined by the presence of a catalytic domain of ∼200 amino acids containing a canonical motif of RHGE in which the His residue is critically required for enzymatic activity (1). Although most PGAM members catalyze the dephosphorylation/phosphorylation of small metabolites, three members of the PGAM family, STS1, STS2 and PGAM5, are protein phosphatases (1–3). The tyrosine phosphatase activity of the closely related STS1 and STS2 proteins underlies their involvement in T-cell receptor signaling (2), and the serine/threonine phosphatase activity of PGAM5 has been implicated in mitochondrial fragmentation and cell death (4).
Multiple isoforms of PGAM5 have been reported (5–7). The predominant isoform of PGAM5, denoted PGAM5-L, encodes a protein of 289 amino acids in which amino acids 98–289 compose the PGAM domain. A short isoform (PGAM5-S) has been identified that replaces 50 C-terminal amino acids with 16 amino acids derived from an alternatively spliced mRNA (5). The PGAM5-S isoform has been implicated in mitochondrial fragmentation leading to necrosis (4, 8). Both the long and short isoforms of PGAM5 contain an N-terminal mitochondrial targeting sequence that anchors the protein on the cytosolic face of the outer mitochondrial membrane (8). Proteolytic removal of the mitochondrial targeting sequence, which releases a truncated PGAM5 protein from the mitochondrial membrane, has been implicated in apoptosis and mitophagy (6, 7).
A number of proteins that interact with one or more isoforms of PGAM5 have been identified. The N-terminal region of the PGAM5-L and PGAM5-S isoforms contains an evolutionarily conserved motif, ESGE, residues 79–82, that mediates binding to KEAP1, a substrate adaptor for a Cul3-dependent ubiquitin ligase complex (5, 8). The canonical KEAP1 substrate, the transcription factor NFE2L2 (also termed Nrf2), has a similar KEAP1-binding motif, and formation of a ternary complex between PGAM5, NFE2L2, and KEAP1 has been reported (8). A ternary complex between KEAP1, PGAM5, and BCL-XL has also been reported, in which the PGAM domain of the PGAM5-L isoform mediates binding to BCL-XL (9). This complex may allow the KEAP1-CUL3 ubiquitin ligase complex to target BCL-XL for degradation by the 26 S proteasome (9). PGAM5 isoforms have been identified in other multiprotein complexes as well, including ASK1, a MAPKKK protein implicated in oxidative stress signaling (3); PINK1, a protein kinase implicated in Parkinson disease and mitophagic turnover of damaged mitochondria (10); and RIP3, a protein kinase involved in necrotic cell death (4). The relationship(s) between the phosphatase activity of PGAM5, the participation of PGAM5 in multiple and distinct multimeric complexes, and the role of PGAM5 in mitochondrial dynamics is not understood.
A crystal structure of the isolated PGAM domain of the PGAM5-L isoform has been reported, in which the PGAM domain forms a dimer via contacts involving C-terminal amino acids that are unique to the PGAM5-L isoform (11). However, as demonstrated in this report, the phosphatase activity of the isolated PGAM domain of the PGAM5-L isoform is markedly reduced relative to the full-length PGAM5-L protein. Thus, amino acid sequences within the unique N terminus of PGAM5 are required for maximal enzymatic activity. We have identified a six-amino acid motif, consisting of residues 58–63 in the N terminus of PGAM5, which is required for maximal phosphatase activity of the PGAM5-L protein. This highly conserved motif, containing the sequence WDXNWD, also mediates the formation of large homomultimeric complexes of PGAM5-L. The formation of multimeric complexes can be abolished either in cis, by alanine substitutions within the WDXNWD motif, or in trans, by incubation of purified PGAM5 with a peptide containing the WDXNWD motif. The WDXNWD-containing peptide is a noncompetitive inhibitor of phosphatase activity. The phosphatase activity of mutant PGAM5-L proteins containing alanine substitutions within the WDXNWD motif is less than 1% that of the wild-type protein. However, the phosphatase activity of these mutant PGAM5-L proteins can be markedly increased, in trans, by the WDXNWD-containing peptide. Our results indicate the presence of an allosteric site in the catalytic domain of PGAM5-L that regulates its phosphatase activity. We propose that this allosteric site is occupied by the WDXNWD motif when PGAM5-L assembles into homomultimeric complexes, resulting in maximal activation of phosphatase activity. Furthermore, as mutant PGAM5-L proteins containing alanine substitutions within the WDXNWD motif induce mitochondrial fragmentation, our results provide a link between regulation of the phosphatase activity of PGAM5-L, its ability to form homomultimeric complexes, and its participation in mitochondrial dynamics.
EXPERIMENTAL PROCEDURES
Peptides
Phosphopeptides at 95% or greater purity were obtained from Genscript or Lifetein. The peptides were resuspended in water, stored at −80 °C, and thawed as needed. The peptide sequences used is provided in the tables and in Fig. 3.
FIGURE 3.

Identification of the WDXNWD motif in PGAM5. A, amino acid sequence of the human (Hs) PGAM5 protein, from residues 51 to 80, is aligned with the corresponding sequences of PGAM5 from seven different species, including rat (Rn), mouse (Mm), frog (Xl), zebrafish (Dr), fruit fly (Dm), and two worms species (Ce and Cb). The conserved WDXNWD motif is in bold. The arrows indicate the first amino acid of three successive N-terminal truncations of the human PGAM5 protein used in this study. The alanine substitutions for each WD pair (W58A/D59A and W62A/D62A) and for both WD pairs (A4) are indicated. The alanine substitutions were made in the context of both the full-length PGAM5 and the ΔN25-PGAM5 protein, with similar effects on the activity of both PGAM5 proteins (data not shown). The majority of the experiments reported in this study were performed with the A4 mutation in the context of the ΔN25 deletion. The wild-type and A4 mutant peptides used in this study, comprising amino acids 52–74 of the human PGAM5 protein, are indicated. B, increasing amounts of the BCL-XL Ser-62 phosphopeptide was incubated with 10 nm of the indicated PGAM5 proteins. Three mutant PGAM5 proteins were used in this experiment. These mutant proteins contained alanine substitutions for either of the WD pair (W58A/D59A or W62A/D62A) or for both WD pairs (A4). The amount of free phosphate was measured, and Michaelis-Menten parameters were determined using KaleidaGraph. ΔN25-PGAM5: Vmax = 1232 (± 21) pmol/min/μg, Km = 59 (± 3) μm, and R2 = 0.999. The kinetic parameters for the W58A/D59A-PGAM5 protein, the W62A/D62A-PGAM5 protein, or the A4-PGAM5 protein could not be determined under these conditions due to insufficient phosphatase activity. C, increasing amounts of the ASK1 Ser-1029 phosphopeptide was incubated with 500 nm of the indicated PGAM5 proteins. Note the difference in scale of the y axis between B and C. The amount of free phosphate was measured, and Michaelis-Menten parameters were determined using KaleidaGraph. A4-PGAM5: Vmax = 20 (± 1) pmol/min/μg, Km = 167 (± 28) μm, and R2 = 0.989. The kinetic parameters for the H105A-PGM5 protein could not determined under these conditions due to insufficient phosphatase activity.
Recombinant Expression Vectors for Protein Production
Expression vectors for the truncated PGAM5 proteins were generated by cloning PCR-derived fragments of PGAM5 into the NdeI and BamHI sites of pET15b (Novagen). Site-directed mutations in PGAM5 were generated using standard overlap-extension PCR techniques. Mammalian expression vectors for the full-length and FLAG-tagged wild-type and A4 (where A4 denotes alanine substitutions for both WD pairs in the WDXNWD motif) mutant PGAM5 proteins were constructed using standard techniques. All DNA constructs were confirmed by sequencing. The long isoform of PGAM5 was used in all experiments reported in this study, as sufficient quantities of the short form could not be obtained from our bacterial expression system.
Protein Purification
The pET15b-PGAM5 expression vectors were transformed into Rosetta cells (BL21 DE3 pLysS) and grown in LB at 37 °C to an absorbance at 595 nm of 0.5. Protein induction was performed at 30 °C in the presence of 1 mm isopropyl thio-β-galactoside and 1 mm phenylmethylsulfonyl fluoride for 3 h. Bacterial cultures were collected by centrifugation and suspended in 500 mm NaCl, 20 mm Hepes, pH 7.2, 10 mm imidazole, 10 mm β-mercaptoethanol in the presence of protease inhibitors (Sigma). Bacteria were lysed by three freeze-thaw cycles. Bacterial lysates were centrifuged at 15,500 × g to remove cellular debris. His-tagged PGAM5 proteins were immobilized onto nickel-nitrilotriacetic acid columns, washed, and eluted with 500 mm NaCl, 20 mm Hepes, pH 7.2, and 500 mm imidazole containing 10 mm β-mercaptoethanol.
Fast Protein Liquid Chromatography
Fast protein liquid chromatography (AKTA, 900 series model, Amersham Biosciences) was used for desalting and size exclusion chromatography. To desalt, proteins eluted from nickel-nitrilotriacetic acid columns were exchanged into 150 mm NaCl, 20 mm Hepes, pH 7.4. For size exclusion chromatography, desalted PGAM5 proteins were separated on a HiLoad 26/60 Superdex 200 size exclusion column (GE Healthcare) at 4 °C in 150 mm NaCl, 20 mm Hepes, pH 7.4, with a flow rate of 2.5 ml/min. Protein standards for size exclusion chromatography (Sigma) were used to calibrate the column. The protein standards used included lactalbumin (14 kDa), carbonic anhydrase (29 kDa), ovalbumin (45 kDa), bovine albumin (132 kDa dimer), and jack bean urease (272-kDa trimer and 545-kDa hexamer). Protein was detected using absorbance at 280 nm.
Phosphatase Assays
Phosphopeptide substrates were sequentially diluted into reaction buffer (150 mm NaCl, 50 mm Hepes, pH 7.4, 2.5 mm EDTA, and 0.5 mm DTT). Reactions were started by the addition of PGAM5 protein to a final concentration ranging from 10 to 500 nm, depending on the intrinsic activity of the protein and the presence of inhibitory or activating peptides. All reactions were performed under conditions in which the production of free phosphate was linear with time, typically from 3 to 15 min for the PGAM5 proteins with robust activity. Longer reaction times, up to 2 h, were used for PGAM5 proteins with reduced activity. The release of phosphate from the phosphopeptide substrates was determined by absorbance at 620 nm using the malachite green assay (R&D Systems). Kinetic parameters were determined using KaleidaGraph or GraphPad software. Double-reciprocal plots were prepared in GraphPad.
Analytical Size Exclusion Chromatography and Molar Mass Determination
High performance liquid chromatography was performed on a TSK G3000SWXL (Toso Haas) column (7.5 mm inner diameter × 30 cm) in 10 mm Hepes, 300 mm NaCl, pH 7.0 at 7 °C. The absolute molar mass of proteins was determined directly using static light scatter by passing the eluent through a multiangle laser light scatter detector followed by a differential refractometer (DAWN-HELEOS and OPTILab Rex, respectively; Wyatt Technology Corp.). The molar mass was determined using a specific refractive index increment (dn/dc) of 0.21 ml/g and the Debye plotting formalism of the Astra software supplied with the instrument. The relationship between the weight average molecular weight (M̄w) and the excess Rayleigh ratio R(θ) at the low protein concentrations used here is given in Equation 1,
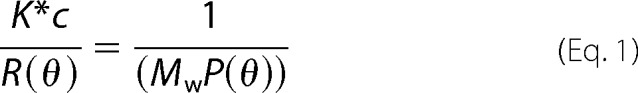 |
where R(θ) is the light scattered by the solution at angle θ in excess of that scattered by pure solvent divided by the incident light intensity; c is the concentration of protein; P(θ) is the form factor that describes the angular dependence of the scatter, and K* is a constant dependent on the parameters of the Wyatt system used.
Cross-linking Experiments
All cross-linking experiments were carried out at 4 °C. In experiments without peptide inhibitors, 15 μm PGAM5 was incubated in the presence of 0–3 mm bis-N-succinimidylpentaethylene glycol ester (BS(PEG)5 (Thermo Scientific) in 50 mm Hepes, pH 7.5, 150 mm NaCl, 2.5 mm EDTA, and 0.5 mm DTT for 1 h. Cross-linking was quenched with 10 mm glycine for 10 min prior to boiling in sample buffer. Samples were resolved by SDS-PAGE and visualized by Coomassie Blue staining. In the presence of peptide, 7 μm PGAM5 and 0–200 μm of various peptides were incubated for 1 h in the buffer listed above prior to the addition of 0 or 1 mm BS(PEG)5 for 10 min. The cross-linking reactions were quenched with 10 mm glycine for 10 min prior to boiling in sample buffer and SDS-PAGE. Protein bands were visualized by silver staining (Thermo Scientific).
Mammalian Cell Culture
A mouse ES cell line, RRZ048, that contains a gene trap disruption of the PGAM5 locus was obtained from Bay Genomics. This cell line was used to generate mice heterozygous for the wild-type and gene trap alleles. Embryos were obtained at E14.5 from a heterozygous mating and used to generate mouse embryo fibroblasts (MEFs). One MEF cell line, 666, was found to be negative for PGAM5 protein expression. A retroviral expression vector was used to introduce the PGAM5-FLAG cDNA into this cell line. The expression level of the FLAG-tagged PGAM5 was equivalent to the level of PGAM5 expression in wild-type MEFs generated in parallel. The MEF cell lines and COS1 cells were cultured in DMEM with 10% fetal bovine serum (FBS) in a humidified atmosphere at 37 °C. Cell lysates for size exclusion chromatography were prepared in 50 mm Hepes, pH 8, 1% Triton X-100, 1% sodium deoxycholate, and 150 mm NaCl. Clarified lysates were loaded onto a HiLoad 26/60 Superdex 200 column with a flow rate of 2.5 ml/min in 300 mm NaCl, 10 mm Hepes, pH 7. 10-ml fractions were collected and precipitated with trichloroacetic acid (TCA). Protein pellets were resuspended in sample buffer and boiled. Samples were resolved by SDS-PAGE and transferred to nitrocellulose membranes, and PGAM5-FLAG was detected by immunoblot analysis using anti-FLAG (Sigma).
COS1 Transfections and Immunofluorescence
For transfections, 1 × 105 COS1 cells were grown overnight on glass coverslips and transfected using Lipofectamine-PLUS reagents according to the manufacturer's guidelines (Invitrogen). At 48 h post-transfection, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100 for 10 min, and rinsed with 1× PBS. Permeabilized cells were incubated with primary antibodies anti-PGAM5 (affinity-purified chicken, 0.2 μg/μl (8)), anti-TOM20 (Santa Cruz Biotechnology; rabbit, 0.2 μg/μl), and anti-FLAG (Sigma; mouse, 0.2 μg/μl) diluted 1:100 in 10% FBS and 1× PBS, pH 7.4, for 1 h at room temperature. Cells were washed twice with 1× PBS and incubated with secondary antibodies (Invitrogen; anti-rabbit 488; anti-mouse 568; and anti-chicken 568) diluted 1:500 in 10% FBS, 1× PBS, pH 7.4, and 1:200 Hoechst (Invitrogen) for 1 h at room temperature. Stained cells were washed with 1× PBS and mounted using ProLong Gold (Invitrogen). Microscopy was performed on a Leica DMI 6000B microscope.
RESULTS
PGAM5 Has a Marked Preference for Peptide Substrates That Contain Negatively Charged Amino Acids
To characterize the phosphatase activity of PGAM5, a full-length PGAM5-L isoform containing an N-terminal hexahistidine tag was purified to homogeneity following expression in bacteria. As the PGAM5-S protein could not be obtained as a soluble protein following expression in bacteria, all subsequent experiments were performed with proteins containing the C terminus of the PGAM5-L isoform. To simplify the description of our proteins, we have dropped the -L designation, simply referring to the protein(s) used in this study as PGAM5.
To provide insight into the substrate specificity of PGAM5, eight model phosphopeptides were tested as substrates (Table 1). As reported previously (3), PGAM5 has no detectable activity against phosphopeptides containing phosphotyrosine. Phosphatase activity was readily detected with phosphopeptides containing serine or threonine. Comparison of kcat/Km values for various peptides revealed that PGAM5 is most active with threonine-containing phosphopeptides that also contain negatively charged residues (Table 1). The published structure of the catalytic domain of PGAM5 reveals a high density of positive charge near the catalytic pocket (Protein Data Bank code 3MXO),3 suggesting an explanation for PGAM5's preference for substrate phosphopeptides containing negatively charged residues (Fig. 1).
TABLE 1.
PGAM5 phosphatase activity against model phosphopeptide substrates
The sequence of phosphopeptides used as model substrates to characterize PGAM5 phosphatase activity is shown, with the phosphorylated residue site in parentheses. Negatively charged residues are in boldface. The indicated kinetic parameters were determined using KaleidaGraph software. ND = not determined due to insufficient phosphatase activity.
| Peptide | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | min−1 | min−1·m−1 | |
| DDA(T)VA | 72 ± 2 | 77.8 ± 1.2 | 1.08 (± 0.03) × 106 |
| END(T)INASL | 113 ± 4 | 72.1 ± 0.9 | 0.64 (± 0.02) × 106 |
| AAA(T)VA | 206 ± 7 | 63.1 ± 1.3 | 0.31 (± 0.01) × 106 |
| RRA(S)VA | 1354 ± 203 | 55.7 ± 2.9 | 0.04 (± 0.01) × 106 |
| RRA(T)VA | 1954 ± 185 | 36.3 ± 5.5 | 0.02 (± 0.01) × 106 |
| RRA(Y)VA | ND | ND | ND |
| END(Y)INASL | ND | ND | ND |
| DADE(Y)LIPQQG | ND | ND | ND |
FIGURE 1.
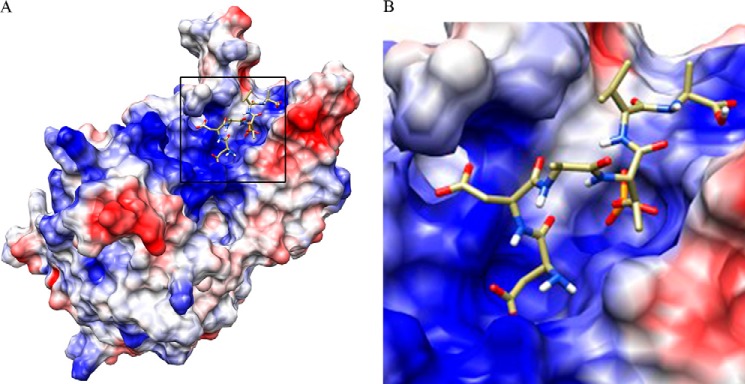
Three-dimensional model of PGAM5 reveals a positively charged substrate-binding pocket. A, three-dimensional model of PGAM5 using coordinates from Protein Data Bank code 3MXO was generated using UCSF Chimera (supported by National Institutes of Health Grant P41-GM103311 from NIGMS (24)). Docking of the phosphopeptide DDA(pT)VA into the catalytic pocket of PGAM5 was performed using AutoDock Vina (11). Positively charged surface areas are indicated in blue and negatively charged surface areas in red. B,a close-up view of the substrate-binding pocket.
To further characterize the substrate specificity of PGAM5, 16 phosphopeptides that correspond to known phosphorylation sites found within candidate protein substrates for PGAM5 were tested (Table 2). These candidate protein substrates of PGAM5 include ASK1, BCL-XL, DRP1, and NRF2, all of which have been reported to be present in PGAM5-containing protein complexes (3–5, 9). Phosphopeptides from BCL-2 were also tested. PGAM5 displayed activity against 13 of the 16 tested phosphopeptides. The four peptides with the highest kcat/Km values, ranging from 3.4 to 1.1 min−1 μm−1, were derived from ASK1 or BCL-XL and contained one or more negatively charged residues. Two of these peptides, containing Ser-1029 from ASK1 or Ser-62 from BCL-XL, with nearly identical Km values of 23 and 28 μm and similar kcat/Km values of 3.4 and 2.2 min−1 μm−1, were used for subsequent experiments.
TABLE 2.
PGAM5 phosphatase activity against phosphopeptides from candidate protein substrates
Candidate protein substrates of PGAM5 are indicated along with the amino acid of specific phosphorylation sites within each protein. The sequences of peptides that contain each phosphorylation site are shown, with the phosphorylation site in parentheses. Negatively charged residues are boldface. The indicated kinetic parameters were determined using KaleidaGraph software. ND = not determined due to insufficient phosphatase activity.
| Protein phosphorylation site | Sequence | Km | kcat | kcat/Km |
|---|---|---|---|---|
| μm | min−1 | min−1·m−1 | ||
| ASK1 Ser-1029 | NFEDH(S)APPSP | 23 ± 2 | 79.9 ± 2.4 | 3.4 (± 0.4) × 106 |
| Bcl-XL Ser-62 | WHLAD(S)PAVNG | 28 ± 2 | 61.6 ± 1.2 | 2.2 (± 0.1) × 106 |
| ASK1 Ser-1033 | HSAPP(S)PEEKD | 26 ± 2 | 51.4 ± 1.2 | 1.9 (± 0.2) × 106 |
| ASK1 Thr-838 | NPCTE(T)FTGTL | 65 ± 5 | 69.8 ± 1.9 | 1.1 (± 0.1) × 106 |
| Bcl-XL Thr-47 | ESEME(T)PSAIN | 50 ± 7 | 23.1 ± 0.8 | 0.5 (± 0.1) × 106 |
| Bcl2 Ser-70 | PVART(S)PLQTP | 98 ± 9 | 38.5 ± 1.1 | 0.4 (± 0.1) × 106 |
| Drp1 Ser-548 | ASQEP(S)PAASA | 114 ± 33 | 36.9 ± 4.9 | 0.3 (± 0.1) × 106 |
| Bcl2 Ser-87 | AGPAL(S)PVPPV | 357 ± 13 | 78.4 ± 1.2 | 0.2 (± 0.1) × 106 |
| Drp1 Ser-637 | VARKL(S)AREQR | 409 ± 69 | 51.7 ± 3.1 | 0.13 (± 0.02) × 106 |
| ASK1 Ser-966 | YLRSI(S)LPVPV | 237 ± 30 | 29.0 ± 2.2 | 0.12 ± 0.02 |
| Bcl-XL Thr-115 | SQLHI(T)PGTAY | 480 ± 29 | 23.7 ± 0.4 | 0.05 ± 0.01 |
| ASK1 Ser-958 | ALSAG(S)NEYLR | 2749 ± 476 | 39.1 ± 3.6 | 0.01 ± 0.01 |
| ASK1 Ser-83 | RGRGS(S)VGGGS | 656 ± 22 | 1.53 ± 0.03 | 0.002 ± 0.001 |
| Bcl2 Thr-69 | DPVAR(T)SPLQT | ND | ND | ND |
| Drp1 Ser-616 | PIMPA(S)PQKGH | ND | ND | ND |
| Nrf2 Ser-40 | EVFCF(S)QRRKE | ND | ND | ND |
N Terminus of PGAM5 Is Required for Phosphatase Activity
The PGAM domain of PGAM5 starts at amino acid 98 in human PGAM5 and continues to amino acid 289. This domain contains a histidine residue, His-105, that is part of the active site and is required for phosphatase activity (3). However, a truncated PGAM5 protein that starts at amino acid 89 has greatly reduced phosphatase activity compared with the full-length PGAM5 protein (data not shown), suggesting that the N terminus of PGAM5, which is not part of the conserved PGAM domain, is required for maximal phosphatase activity of PGAM5.
To define residues in the N terminus of PGAM5 that are required for phosphatase activity, a series of N-terminal truncated PGAM5 proteins were assayed for phosphatase activity (Fig. 2). Deletion of the first 25 amino acids of PGAM5 (ΔN25-PGAM5, where ΔN denotes N-terminal truncations of PGAM5) increased solubility and protein recovery from bacteria, with only a modest reduction in the kinetic parameters of the enzyme (data not shown). Further truncations up to residue 55 (ΔN55-PGAM5) did not significantly alter the phosphatase activity of PGAM5 toward either a phosphopeptide containing Ser-62 from BCL-XL (Fig. 2C) or a phosphopeptide containing Ser-1029 from ASK1 (data not shown). In contrast, the phosphatase activities of PGAM5 proteins truncated to residue 60 and beyond were markedly reduced (Fig. 2C and data not shown).
FIGURE 2.

Deletion mutants in the N terminus of PGAM5 identify an amino acid sequence required for phosphatase activity. A, diagram of the full-length and N-terminal truncations of PGAM5 used in this study. The full-length PGAM5 protein contains five discrete domains, including an N-terminal mitochondrial targeting sequence (8) (A); a multimerization motif (current work) (B); a Keap1-binding motif (C) (5); PGAM motif characteristic of all proteins in the phosphoglycerate family, including His-105 (1) (D); PGAM domain that shares significant identity with other members of the phosphoglycerate family (1) (E). B, proteins used in this study were separated by SDS-PAGE and visualized by Coomassie Blue staining. Molecular size markers are indicated on the left. C, increasing amounts of the BCL-XL Se-r62 phosphopeptide was incubated with 10 nm of each of the indicated PGAM5 proteins. The amount of free phosphate was measured, and Michaelis-Menten parameters were determined using KaleidaGraph. Full-length PGAM5: Vmax = 1224 (± 51) pmol/min/μg, Km = 58 (± 6) μm, R2 = 0.999, and ΔN55-PGAM5: Vmax = 1041 (± 57) pmol/min/μg, Km = 57 (± 7) μm, and R2 = 0.998. The kinetic parameters for ΔN60-PGAM5 could not be determined due to insufficient phosphatase activity.
Conserved WDXNWD Motif Is Required for Phosphatase Activity of PGAM5
Alignment of the amino acid sequence of PGAM5 proteins from several species revealed a highly conserved motif (WDXNWD), located between residues 58 and 63, in the N terminus of human PGAM5 (Fig. 3A). This motif was present in ΔN55, partially deleted in ΔN60, and completely deleted in further truncations. Mutant PGAM5 proteins were generated with alanine substitutions in the WDXNWD motif in place of the tryptophan and aspartate residues, either as individual pairs (W58A/D59A or W62A/D63A) or in combination (W58A/D59A/W62A/D63A; designated A4) (Fig. 3A). The mutations were introduced into both full-length PGAM5 and ΔN25-PGAM5 proteins. Mutant proteins containing alanine substitutions within the WDXNWD motif had phosphatase activity, although markedly reduced in comparison with the wild-type protein (Fig. 3, B and C, and Table 3). Relative to the ΔN25-PGAM5 protein, the Km value of the A4-PGAM5 protein increased about 5-fold and kcat decreased at least 100-fold against the BCL-XL Ser-62 phosphopeptide (Table 3). Nevertheless, residual catalytic activity was observed with the A4-PGAM5 protein (Fig. 3C). In contrast, no phosphatase activity was detected for a mutant PGAM5 protein containing an alanine substitution in place of the catalytically active His-105 residue (Fig. 3C).
TABLE 3.
Regulation of PGAM5 phosphatase activity by WDXNWD-containing peptides
Kinetic parameters of the indicated PGAM5 proteins against the indicated phosphopeptide substrates were determined in the absence or presence of the WT(52–74) or A4(52–74) peptides, as indicated. The kinetic parameters were determined using GraphPad software.
| Protein | Substrate | Peptide | Km | kcat | kcat/Km |
|---|---|---|---|---|---|
| μm | min−1 | min−1·m−1 | |||
| ΔN25-PGAM5 | BCL-XL Ser62 | 0 μm WT(52–74) | 102 (± 15) | 130 (± 9) | 1.3 (± 0.2) × 106 |
| ΔN25-PGAM5 | BCL-XL Ser62 | 40 μm WT(52–74) | 76 (± 14) | 33 (± 3) | 0.4 (± 0.1) × 106 |
| ΔN25-PGAM5 | BCL-XL Ser62 | 100 μm A4(52–74) | 61 (± 11) | 28 (± 3) | 0.4 (± 0.1) × 106 |
| A4-PGAM5 | BCL-XL Ser62 | 0 μm WT(52–74) | 503 (± 57) | 0.44 (± 0.02) | 0.9 (± 0.1) × 103 |
| A4-PGAM5 | ASK1 Ser1029 | 0 μm WT(52–74) | 446 (± 17) | 0.77 (± 0.02) | 1.7 (± 0.1) × 103 |
| A4-PGAM5 | ASK1 Ser1029 | 4 μm WT(52–74) | 818 (± 42) | 11 (± 0.4) | 13.4 (± 0.8) × 103 |
| A4-PGAM5 | ASK1 Ser1029 | 15 μm WT(52–74) | 480 (± 113) | 29 (± 4) | 60 (± 16) × 103 |
| A4-PGAM5 | ASK1 Ser1029 | 15 μm A4(52–74) | 721 (± 49) | 0.93 (± 0.04) | 1.3 (± 0.1) × 103 |
PGAM5 Forms a Multimeric Complex That Requires the WDXNWD Motif
The crystal structures of the PGAM domains of both STS1 and PGAM5 exist as dimers (2, 11). To determine whether amino acid substitutions within the WDXNWD motif of PGAM5 perturbed dimer formation, a series of purified PGAM5 proteins were analyzed by size exclusion chromatography. Greater than 95% of the full-length and ΔN25-PGAM5 proteins eluted in the void volume with an apparent molecular mass greater than 500 kDa but less than 5% of the protein mass entered the column and eluted with an apparent molecular mass of 45 kDa (Fig. 4A and data not shown). The phosphatase activity eluted as a single peak in the void volume. Phosphatase activity was not detected in the 45-kDa peak (data not shown). Analysis by light scattering suggested a molecular mass of ∼600 kDa for the full-length and ΔN25-PGAM5 proteins; however, an accurate molecular mass could not be determined, presumably due to heterogeneity of the multimeric protein complexes (data not shown). The bulk of the ΔN55-PGAM5 protein also eluted in the void volume, and about 25% of ΔN55-PGAM5 protein entered the column and eluted with an apparent molecular mass of about 250 kDa (Fig. 4B). The two peaks had equivalent phosphatase activity per mol of protein (data not shown). The 250-kDa peak was stable, as it remained a single peak following a second round of chromatography (Fig. 4C). As measured by light scattering, the molecular mass of this complex was 334 kDa, consistent with this complex being a dodecamer (Fig. 4F). In contrast, both the ΔN65-PGAM5 and A4-PGAM5 proteins readily entered the size exclusion column and eluted with approximate molecular masses of 29 and 45 kDa, respectively (Fig. 4, D and E). Molecular mass determination by light scattering indicated that the molecular mass of the A4-PGAM5 protein was 66 kDa (Fig. 4F), in close agreement with the expected molecular mass of a dimer. Taken together, these results indicate that highly active PGAM5 proteins form stable multimeric complexes in solution, whereas PGAM5 proteins with low phosphatase activity exist as stable dimers.
FIGURE 4.

Characterization of PGAM5 proteins by size exclusion chromatography. The indicated PGAM5 proteins were subjected to size exclusion chromatography as described under “Experimental Procedures.” The elution profiles of the molecular masses of proteins used to calibrate the size exclusion column are shown for A–E. A, ΔN25-PGAM5. B, ΔN55-PGAM5. The asterisk indicates a peak that eluted with an apparent molecular mass of ∼250 kDa. C, ΔN55-PGAM5. The peak indicated with the asterisk in B was collected, concentrated, and re-chromatographed. D, ΔN65-PGAM5. E, A4-PGAM5. F, peak of the ΔN55-PGAM5 protein indicated with the asterisk in B and the A4-PGAM5 protein peak, as indicated in E, were re-chromatographed on an analytical column coupled to a light scattering detector. The chromatographic traces of each protein were combined together to highlight the differences in elution profiles. The y axis indicates the molar mass of each peak, as determined by light scattering.
PGAM5 Is Present in Multimeric Complexes in Lysates from Mouse Embryo Fibroblasts
To determine whether PGAM5 is present in multimeric complexes in mammalian cells, cell lysates were prepared from MEFs that do not express the endogenous PGAM5 protein but express a C-terminal FLAG-tagged PGAM5 protein at levels equivalent to the endogenous PGAM5 protein present in wild-type MEFs. Cell lysates were fractionated over size exclusion columns. The presence of PGAM5-FLAG in fractions from the size exclusion column was determined by immunoblot analysis. Two distinct fractions of PGAM5-FLAG were detected, one of ∼250–400 and another of ∼40–60 kDa (Fig. 5). A similar distribution of endogenous PGAM5 in size-fractionated cell lysates from COS1 cells was also observed (data not shown). Thus, the ability of PGAM5 to form multimeric complexes is not simply an artifact resulting from bacterial overexpression. Size exclusion chromatography of cell lysates does not differentiate between homomeric or heteromeric complexes. However, PGAM5 has been identified in multiple heteromeric protein complexes that have been purified from mammalian cells (3, 4, 10).
FIGURE 5.
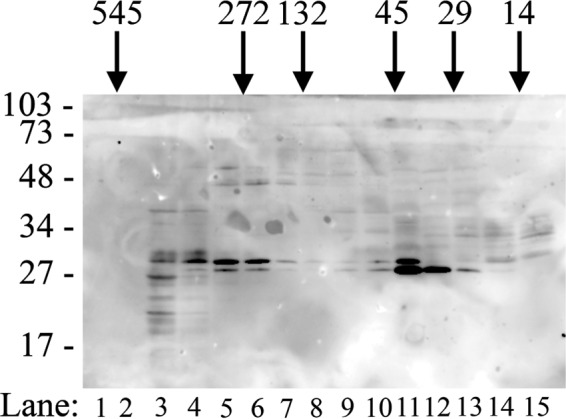
PGAM5 forms multimeric complexes in vivo. A cell lysate from MEFs that express the wild-type FLAG-tagged PGAM5 protein was separated by size exclusion chromatography. Fractions were pooled and analyzed by SDS-PAGE followed by immunoblot analysis using the anti-FLAG antibody (lanes 1–15). The elution positions of molecular mass standards used to calibrate the column are indicated across the top. The migration of molecular size markers during electrophoresis is indicated on the left.
Mutation of the WDXNWD Motif Results in Mitochondrial Fragmentation
Stable expression of the full-length A4-PGAM5 protein could not be achieved in MEFs (data not shown). However, ectopic expression of the full-length A4-PGAM5 protein in COS1 cells resulted in marked fragmentation of mitochondria, in contrast to nuclear clustering of mitochondria induced by ectopic expression of the wild-type PGAM5 protein (Fig. 6). Expression of the catalytically inactive PGAM5-H105A protein resulted in nuclear clustering behavior similar to that induced by expression of the wild-type protein (data not shown). Thus, when a mutant PGAM5 protein that is unable to assemble into multihomomeric complexes is expressed in cells, mitochondrial dynamics are significantly perturbed, resulting in mitochondrial fragmentation.
FIGURE 6.

Ectopic expression of the A4-PGAM5 protein in COS1 cells induces mitochondrial fragmentation. COS1 cells were mock-transfected or transfected with expression vectors for FLAG-tagged wild-type or A4 mutant PGAM5 proteins, as indicated. The cells were processed for indirect immunofluorescence and visualized with epifluorescence with a Leica DMI 6000B microscope. The following antibodies were used: A, chicken anti-PGAM5 antibodies followed by anti-chicken IgY antibodies coupled to Alexa 568; D and G, mouse anti-FLAG antibodies followed by anti-mouse IgG antibodies coupled to Alexa 568; B, E, and H, rabbit anti-TOM20 antibodies followed with anti-rabbit IgG antibodies coupled to Alexa 488. The merged images are shown on the right (C, F, and I).
WDXNWD Peptide Disrupts Multimeric PGAM5 Complexes and Reduces Phosphatase Activity
Cross-linking was used to confirm the presence of multimeric PGAM5 protein complexes. In the absence of cross-linker, the ΔN25-PGAM5 migrated as a monomer during SDS-PAGE with a mass of ∼30 kDa. Following incubation with increasing amounts of cross-linker, higher molecular weight complexes of the ΔN25-PGAM5 protein were readily apparent following SDS-PAGE (Fig. 7A). At the highest levels of cross-linker used, the majority of the ΔN25-PGAM5 protein was captured in high molecular mass complexes, some of which did not enter the resolving gel during electrophoresis (Fig. 7A). In contrast, neither the A4-PGAM5 nor ΔN65-PGAM5 proteins were captured in multimeric complexes (Fig. 7A).
FIGURE 7.
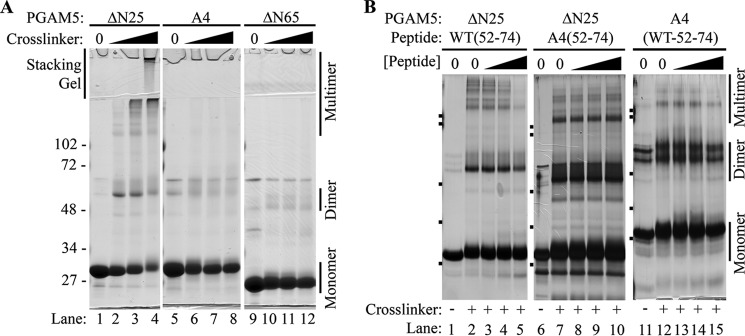
WT(52–74) peptide disrupts multimeric PGAM5 complexes. A, indicated PGAM5 proteins, at 15 μm, were either untreated (lanes 1, 5, and 9) or incubated with increasing amounts of BS(PEG)5 (from 1 to 3 μm; lanes 2–4, 6–8, and 10–12). The protein samples were analyzed by SDS-PAGE and visualized by Coomassie Blue staining. The migration of molecular size markers during electrophoresis is indicated on the left. The position of the stacking gel is indicated on top. The positions of monomeric, dimeric, and multimeric PGAM5 complexes are indicated on the right. B, indicated PGAM5 proteins, at 7 μm, were incubated in buffer only (lanes 1, 6, and 11) or in the presence of the 1 μm BS(PEG)5 without peptides (lanes 2, 7, and 12) or with increasing amounts of the indicated peptides (lanes 3–5, 8–10, and 13–15) prior to the addition of 1 μm BS(PEG)5. The proteins samples were analyzed by SDS-PAGE and visualized by silver staining. The migration of molecular size markers during electrophoresis is indicated on the left side of each panel. The samples in each panel were electrophoresed through different polyacrylamide gels, resulting in slightly different mobilities of the proteins. The positions of monomeric, dimeric, and multimeric PGAM5 complexes are indicated on the right. The mobility of molecular size markers identical to those used in A are indicated on the left side of each panel.
Because mutant PGAM5 proteins containing alanine substitutions within the WDXNWD motif were not able to form multimeric protein complexes, we hypothesized that the WDXNWD motif participates in intermolecular contacts between PGAM5 polypeptides. Therefore, we asked whether a peptide that contains this motif could dissociate the multimeric complexes. A peptide containing residues 52–74 of PGAM5, WT(52–74), disrupted multimeric complex formation of the ΔN25-PGAM5 protein in a concentration-dependent manner (Fig. 7B). In contrast, the corresponding peptide containing four alanine substitutions in place of the two WD pairs, A4(52–74), did not perturb multimer formation (Fig. 7B). Neither the WT(52–74) nor the A4(52–74) peptides induced multimer formation by the mutant A4-PGAM5 protein (Fig. 7B and data not shown).
As the dimer-multimer equilibrium will shift toward the dimeric form at a lower protein concentration, the specific activity of the full-length PGAM5 protein was measured as a function of enzyme concentration (Table 4). A modest decrease in the specific activity of PGAM5, from 0.081 (± 0.002) to 0.032 (± 0.002) μm phosphate/min/nm, was observed as PGAM5 concentration decreased from 10 to 0.6 nm. Within the measured standard error, the specific activity of PGAM5 did not change upon a further decrease in PGAM5 concentration to 0.1 nm (Table 4). The specific activity of PGAM5 could not be reliably determined at concentrations less than 0.1 nm (data not shown). At all concentrations of enzyme tested, the WT(52–74) peptide was able to inhibit phosphatase activity (Table 4). As the multimeric complex is dissociated by the WT(52–74) peptide (Fig. 7B), the data support a hypothesis that the multimeric form, not residual amounts of the dimeric form, are responsible for the measured phosphatase activity of the wild-type enzyme.
TABLE 4.
PGAM5 phosphatase activity as a function of PGAM5 concentration
The full-length PGAM5 protein at the indicated concentrations was assayed for phosphatase activity in the absence or presence of 10 μm WT(52–74) peptide, using 50 μm of the BCL-XL Ser-62 phosphopeptide as substrate. Velocity was determined from the slope of phosphate released (y axis) versus reaction time (x axis), using GraphPad software. ND (not determined) indicates that no measurable phosphate was released.
| No peptide |
With WT(52–74) peptide |
|||||
|---|---|---|---|---|---|---|
| [PGAM5] | Velocity | Velocity/[PGAM5] | R2 | Velocity | Velocity/[PGAM5] | R2 |
| nM | μm phosphate/min | μm phosphate/min/nm | μm phosphate/min | μm phosphate/min/nm | ||
| 10 | 0.807 ± 0.019 | 0.081 ± 0.002 | 0.998 | 0.118 ± 0.014 | 0.012 ± 0.001 | 0.944 |
| 2 | 0.120 ± 0.001 | 0.060 ± 0.001 | 0.999 | 0.025 ± 0.001 | 0.013 ± 0.001 | 0.993 |
| 1 | 0.053 ± 0.005 | 0.053 ± 0.005 | 0.969 | ND | ND | ND |
| 0.6 | 0.019 ± 0.001 | 0.032 ± 0.002 | 0.992 | ND | ND | ND |
| 0.3 | 0.008 ± 0.001 | 0.027 ± 0.003 | 0.998 | ND | ND | ND |
| 0.1 | 0.004 ± 0.001 | 0.040 ± 0.010 | 0.988 | ND | ND | ND |
To further examine the relationship between multimeric complex formation and phosphatase activity of PGAM5, the ability of the WT(52–74) and the A4(52–74) peptides to inhibit the phosphatase activity of PGAM5 was determined. The WT(52–74) peptide inhibited the phosphatase activity of the ΔN25-PGAM5 protein in a concentration-dependent manner (Fig. 8A). Inspection of a double-reciprocal plot of the velocity curves shown in Fig. 8A indicated a noncompetitive mode of inhibition (Fig. 8B). The A4(52–74) peptide was also able to inhibit phosphatase activity of the ΔN25-PGAM5 protein, but a higher concentration of peptide was required to achieve comparable inhibition (Fig. 8, C and D).
FIGURE 8.

WT(52–74) peptide inhibits phosphatase activity of PGAM5. A, ΔN25-PGAM5 protein (10 nm) was incubated with increasing amounts of the WT(52–74) peptide prior to incubation with increasing amounts of the BCL-XL Ser-62 phosphopeptide. The amount of free phosphate was measured, and Michaelis-Menten parameters for the ΔN25-PGAM5 protein in the presence of the indicated concentrations of the WT(52–74) peptide were determined using GraphPad. 0 μm peptide: Vmax = 4349 (± 346) pmol/min/μg, Km = 102 (± 15) μm, and R2 = 0.992; 5 μm peptide: Vmax = 2318 (± 70) pmol/min/μg, Km = 114 (± 7) μm, and R2 = 0.999; 20 μm peptide: Vmax = 1896 (± 273) pmol/min/μg, Km = 126 (± 35) μm, and R2 = 0.981; 30 μm peptide: Vmax = 1155 (± 89) pmol/min/μg, Km = 74(± 13) μm, and R2 = 0.989; 40 μm peptide: Vmax = 1090 (± 95) pmol/min/μg, Km = 76 (± 14) μm, and R2 = 0.987. B, double-reciprocal plots for the velocity curves shown in A. The line slopes are as follows: 0 μm peptide, 0.025 (± 0.001); 5 μm peptide, 0.045 (± 0.001); 20 μm peptide, 0.047 (± 0.007); 30 μm peptide, 0.072 (± 0.001); and 40 μm peptide, 0.089 (± 0.003). C, ΔN25-PGAM5 protein (10 nm) was incubated with increasing amounts of the A4(52–74) peptide prior to incubation with increasing amounts of the BCL-XL Ser-62 phosphopeptide. The amount of free phosphate was measured, and Michaelis-Menten parameters for the ΔN25-PGAM5 protein in the presence of the indicated concentrations of the A4(52–74) peptide were determined using GraphPad. 0 μm peptide: Vmax = 3845 (± 223) pmol/min/μg, Km = 96 (± 9) μm, and R2 = 0.999; 10 μm peptide: Vmax = 2218 (± 154) pmol/min/μg, Km = 56 (± 8) μm, and R2 = 0.995; 50 μm peptide: Vmax = 1064 (± 68) pmol/min/μg, Km = 46 (± 6) μm, and R2 = 0.994; 100 μm peptide: Vmax = 920 (± 95) pmol/min/μg, Km = 61 (± 11) μm, and R2 = 0.990. D, double-reciprocal plots for the velocity curves shown in C. The line slopes are as follows: 0 μm peptide, 0.025 (± 0.001); 10 μm peptide, 0.032 (± 0.001); 50 μm peptide, 0.042 (± 0.002); and 100 μm peptide, 0.066 (± 0.003).
Allosteric Activation of Phosphatase Activity of the Mutant A4-PGAM5 Protein
Neither the WT(52–74) nor the A4(52–74) peptide were able to achieve complete inhibition of phosphatase activity (Fig. 8, A and C, and Table 3). For example, as indicated in Table 3, the WT(52–74) peptide at 40 μm only reduced the kcat/Km value of the ΔN25-PGAM5 protein from 1.3 to 0.4 min−1 μm−1. In contrast, alanine substitutions within the WDXNWD motif abolished multimer formation (Fig. 4E) and markedly reduced the phosphatase activity of PGAM5, as the catalytic efficiency of the A4-PGAM5 protein was less than 0.1% that of either the full-length or ΔN25-PGAM5 proteins (Fig. 3C and Table 3). These results suggested the possibility that the WT(52–74) peptide might increase the catalytic activity of the mutant A4-PGAM5 protein. The WT(52–74) peptide increased the phosphatase activity of the A4-PGAM5 protein in a dose-dependent manner (Fig. 9A). The kcat/Km value of the A4-PGAM5 protein increased from 1.7 to 60 min−1 mm−1 in the presence of 15 μm WT(52–74) peptide (Table 3). In contrast, the A4(52–74) peptide at 15 μm had no effect on the phosphatase activity of the A4-PGAM5 protein (Fig. 9B and Table 3). PGAM5 proteins containing truncations up to amino acid 85 were activated by the WT(52–74) peptide (data not shown). However, the WT(52–74) peptide had no effect on the catalytically inactive PGAM5 protein containing the H105A substitution (data not shown). Taken together, these data indicate that the WDXNWD-containing peptide acts through an allosteric site located within the catalytic PGAM domain to increase the catalytic efficiency of PGAM5.
FIGURE 9.
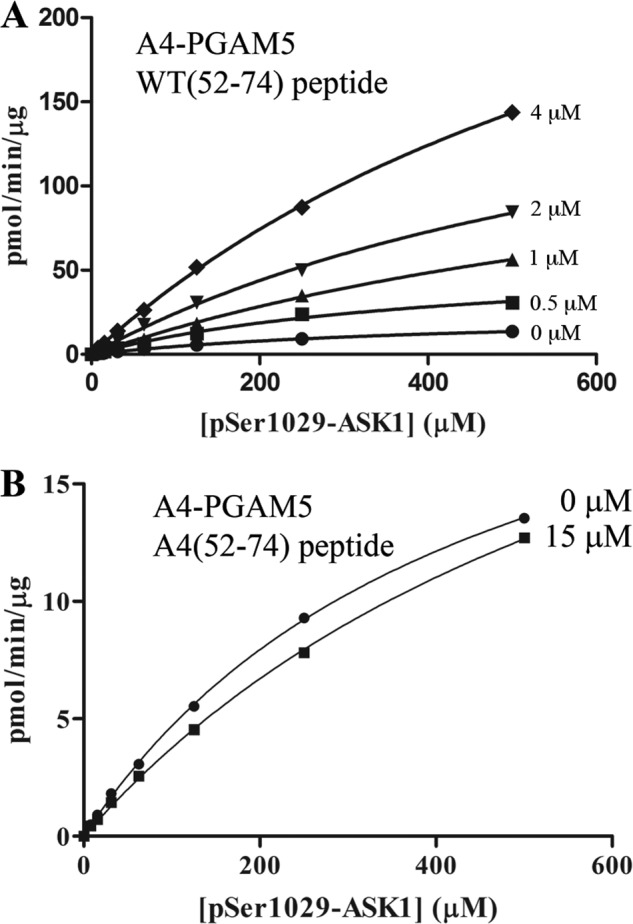
WT(52–74) peptide activates the phosphatase activity of the mutant A4-PGAM5 protein. A, A4-PGAM5 protein (500 nm) was incubated with increasing amounts of the WT(52–74) peptide prior to incubation with increasing amounts of the ASK1 Ser-1029 phosphopeptide. The amount of free phosphate was measured, and Michaelis-Menten parameters for the A4-PGAM5 protein in the presence of the indicated concentrations of the WT(52–74) peptide were determined using GraphPad. 0 μm peptide: Vmax = 26 (± 1) pmol/min/μg, Km = 446 (± 17) μm, and R2 = 0.999; 0.5 μm peptide: Vmax = 58 (± 7) pmol/min/μg, Km = 423 (± 94) μm, R2 = 0.991; 1 μm peptide: Vmax = 163 (± 18) pmol/min/μg, Km = 948 (± 143) μm, and R2 = 0.998; 2 μm peptide: Vmax = 214 (± 17) pmol/min/μg, Km = 777 (± 92) μm, and R2 = 0.999; 4 μm peptide: Vmax = 378 (± 13) pmol/min/μg, Km = 818 (± 42) μm, and R2 = 0.999. B, A4-PGAM5 protein (500 nm) was incubated in the absence or the presence of 15 μm A4(52–74) peptide prior to incubation with increasing amounts of the ASK1 Ser-1029 phosphopeptide. The amount of free phosphate was measured, and Michaelis-Menten parameters for the A4-PGAM5 protein in the absence or presence of the indicated concentration of the A4(52–74) peptide were determined using GraphPad. 0 μm peptide: Vmax = 26 (± 1) pmol/min/μg, Km = 446 (± 17) μm, and R2 = 0.999; 15 μm peptide: Vmax = 31 (± 1) pmol/min/μg; Km = 721 (± 49) μm, and R2 = 0.999.
DISCUSSION
PGAM5 is an atypical Ser/Thr protein phosphatase that contains a unique N-terminal region followed by a catalytic domain that is homologous to the other members of the PGAM family. Among the 12 members of the PGAM family, the catalytic domain of PGAM5 is most similar to that of STS1, a Tyr protein phosphatase. The catalytic domains of both STS1 and PGAM5 crystallize as dimeric complexes (2, 11). In agreement, we have shown that, using both gel filtration and light scattering, the isolated PGAM domain of PGAM5 behaves as a dimeric complex in solution. However, the full-length PGAM5 protein and N-terminal truncated proteins lacking up to 55 N-terminal residues behave as large multimeric complexes in solution. Assembly of PGAM5 into large multimeric complexes was readily apparent during size exclusion chromatography, in which the bulk of the purified PGAM5 protein remained in the void volume of a column capable of resolving proteins or protein complexes up to 500 kDa. Multimeric complexes of PGAM5 proteins were also captured in solution cross-linking experiments. Although the molecular mass of the largest multimeric complexes could not be accurately determined by light scattering, a significant fraction of the ΔN55-PGAM5 protein was resolved on the size exclusion columns used in these experiments, enabling an accurate mass of 344 kDa to be determined for this complex, consistent with a dodecameric composition. Detailed mutational analysis of the unique N terminus of PGAM5 revealed a highly conserved six-amino acid motif, WDXNWD, located between amino acids 58 and 63, that is required for the formation of multimeric complexes. Importantly, a peptide containing this motif was able to dissociate the multimeric complexes, although a peptide containing alanine substitutions for the conserved WD residues did not interfere with complex formation. Our results indicate that the WDXNWD motif mediates protein-protein contacts between PGAM5 dimers, resulting in the assembly of multimeric complexes.
Our results also demonstrate that the WDXNWD motif is required for maximal phosphatase activity of PGAM5. Mutant PGAM5 proteins with deletions of or alanine substitutions within the WDXNWD motif have less than 0.1% of the phosphatase activity of the wild-type PGAM5 protein. Although the A4-PGAM5 protein has higher Km and much lower Vmax values than the wild-type enzyme, the residual catalytic activity of the A4-PGAM5 protein follows Michaelis-Menten kinetics, suggesting that the catalytic site is intact but not functioning very efficiently.
Our data indicate that the phosphatase activity of the PGAM5 protein, when inhibited in trans by the WDXNWD-containing peptide, is significantly greater than the phosphatase activity of the A4-PGAM5 protein, containing a cis-acting mutation within the WDXNWD motif. Yet both the trans-acting peptide and the cis-acting mutation abolish multimer formation. These observations suggested that the trans-acting WDXNWD-containing peptide not only dissociated the multimeric form of the wild-type enzyme but also, when bound, increased the catalytic activity of the dimeric form of the wild-type enzyme. In support of this idea, the WDXNWD-containing peptide increased the catalytic efficiency of the dimeric A4-PGAM5 protein more than 30-fold, whereas an alanine-containing peptide had no effect on the catalytic activity of the A4-PGAM5 protein. Taken together, these results suggest that the WDXNWD peptide binds to an allosteric site within the catalytic PGAM domain. We propose an intermolecular allosteric model, in which the WDXNWD motifs in one dimeric subunit bind into an allosteric activation site in the PGAM domains of an adjacent dimeric subunit, resulting in the formation of multimeric complexes and maximal activation of phosphatase activity.
Although a comprehensive list of PGAM5 substrates has not been defined, the presence of PGAM5 in multiple and distinct multimeric protein complexes suggests that PGAM5 regulates the phosphorylation status of multiple proteins. We characterized multiple phosphopeptides as substrates for PGAM5, including a set of model peptides and a set of peptides derived from five proteins that have been identified in PGAM5-containing complexes. In both sets, the phosphopeptides with the highest kcat/Km values contained one or more negatively charged amino acids located within two amino acids on either side of the phosphorylated Ser or Thr residue. We suggest that the substrate specificity of PGAM5 will be determined, in part, by electrostatic interactions between negatively charged residues in the substrate and positively charged residues on the surface of PGAM5 located near the active site of the enzyme.4
Our results are in agreement with a previous result indicating that PGAM5 can activate ASK1 by dephosphorylating one or more sites in ASK1 (3). We find that three ASK1-derived phosphopeptides, containing Thr-838, Ser-1029, and Ser-1033, are among the best phosphopeptide substrates of PGAM5. Two phosphopeptides from BCL-XL, containing Thr-47 or Ser-62, have Km values comparable with those of the ASK1-derived peptides. As a PGAM5·BCL-XL complex has been demonstrated (5, 9), our results suggest that BCL-XL is likely to be an in vivo substrate of PGAM5. PGAM5 has also been reported to bind to both KEAP1 and BCL-XL, enabling KEAP1 to target BCL-XL for ubiquitin-dependent degradation (9). The relationship between phosphorylation and steady-state levels of BCL-XL is poorly understood and will likely be highly dependent on cell type and physiological status. The mitochondrial fission regulator, DRP1, has also been suggested to be a substrate of PGAM5 (4). We find that two DRP1-derived phosphopeptides, containing Ser-548 or Ser-637, can be dephosphorylated by PGAM5, although kinetic parameters for a third phosphopeptide, containing Ser-616, could not be determined. Phosphorylation of Ser-548 in DRP1 has been reported in a phosphoproteomic study of mitotic phosphorylation (12), although several reports have indicated that phosphorylation of Ser-637 inhibits DRP1-dependent mitochondrial fission (13–15). Although our results are consistent with the possibility that these phosphorylation sites in DRP1 can be dephosphorylated by PGAM5 and possibly lead to increased mitochondrial fragmentation, it is likely that DRP1-independent pathways for mitochondrial fragmentation exist. In particular, neither of these DRP1 phosphopeptides are good substrates for the A4-PGAM5 protein, which induces mitochondrial fragmentation when ectopically expressed in COS1 cells. Furthermore, mitochondrial fragmentation induced by the A4-PGAM5 protein is not inhibited by mdivi-1, a cell-permeable small molecule inhibitor of DRP1-induced mitochondrial fission (data not shown). The short isoform of PGAM5, PGAM5-S, also induces mitochondrial fragmentation in an mdivi-1-independent manner when overexpressed in cells (data not shown). Taken together, these results suggest that PGAM5 can induce mitochondrial fragmentation in a DRP1-independent manner, and further work is needed to define the molecular mechanisms that underlie the induction of mitochondrial fragmentation by PGAM5 proteins.
PGAM5 contains an N-terminal mitochondrial targeting sequence that is responsible for localization to the outer mitochondrial membrane (8). Although our identification of the WDXNWD multimerization motif is based on the use of soluble proteins and in vitro biochemical assays, we believe that WDXNWD-dependent multimerization of PGAM5 is relevant for understanding how PGAM5 is regulated in vivo. First, strict phylogenetic conservation of the WDXNWD motif suggests that the function of this motif has been conserved throughout evolution. Second, the mutant A4-PGAM5 protein, like that of the wild-type PGAM5 protein, is localized to mitochondria, and ectopic expression of the mutant A4-PGAM5 protein, but not wild-type PGAM5 protein, induces mitochondrial fragmentation. Although the mechanisms that underlie this mitochondrial fragmentation phenotype are not fully understood, our results indicate that the inability of a PGAM5 protein to assemble into multihomomeric complexes in vitro correlates with the induction of mitochondrial fragmentation in vivo. Finally, two reports have suggested that, under certain conditions, the mitochondrial targeting sequence of PGAM5 is cleaved (6, 7), releasing soluble PGAM5 into either the cytosol or into the mitochondrial inner membrane space. The release of soluble PGAM5 into the cytosol following cleavage of its mitochondrial targeting sequence has been suggested to promote apoptosis (7). Our results indicate that catalytic activity of these soluble forms of PGAM5 will be regulated by their ability to participate in multimeric complexes.
Mitochondria participate in multiple physiological processes, including metabolic regulation, calcium homeostasis, and both apoptotic and necrotic cell death (16–19). Mitochondria are also highly dynamic organelles, constantly in motion and undergoing complex cycles of fission and fusion (20, 21). It is increasingly clear that the dynamic morphology of mitochondria is tightly linked with mitochondrial function. For example, increased mitochondrial fission has been linked to necrotic cell death (4), although mitochondrial clustering around the nucleus has been reported to occur prior to mitophagic turnover of damaged mitochondria (22, 23). PGAM5 has been implicated in mitochondrial clustering and fragmentation, mitophagy, and both apoptotic and necrotic cell death (3, 4, 8, 10). In this report, we show that PGAM5 is present in both high molecular weight and low molecular weight complexes in mammalian cell extracts. We also show that purified PGAM5 proteins readily assemble into large multihomomeric complexes, although a mutant PGAM5 protein that is incapable of forming homomultimeric complexes is a potent inducer of mitochondrial fragmentation. We propose that the relative distribution of PGAM5 between multimeric and dimeric complexes in cells is regulated by cytosolic and/or mitochondrion-derived signals. Increased accumulation of dimeric PGAM5, with a concomitant reduction in phosphatase activity, may induce fragmentation of mitochondria and thereby sensitize cells to death signals. Conversely, increased accumulation of PGAM5 into either homomultimeric or heteromultimeric complexes, with a concomitant increase in phosphatase activity, may induce nuclear clustering of mitochondria, perhaps leading to mitophagy as a cell-protective mechanism for handling damaged mitochondria. Our results, which have identified the WDXNWD motif as a key mediator of multimeric PGAM5 complexes with robust phosphatase activity, suggest the possibility of developing small molecules that modulate the formation of multimeric complexes containing PGAM5 and, consequently, mitochondrial dynamics and cell death processes.
Acknowledgments
We thank Dr. Stefan Sarafianos, Dr. Lin Randall, and Dr. Steve Alexander for their helpful advice and assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant P50AT006273.
A. Chaikuad, I. Alfano, S. Picaud, P. Filippakopoulos, A. Barr, F. von Delft, C. H. Arrowsmith, A. M. Edwards, J. Weigelt, C. Bountra, K. Takeda, H. Ichijo, and S. Knapp, unpublished data.
A. Chaikuad, I. Alfano, S. Picaud, P. Filippakopoulos, A. Barr, F. von Delft, C. H. Arrowsmith, A. M. Edwards, J. Weigelt, C. Bountra, K. Takeda, H. Ichijo, and S. Knapp, unpublished data.
- PGAM
- phosphoglycerate mutase
- MEFs
- mouse embryonic fibroblasts
- BS(PEG)5
- bis-N-succinimidylpentaethylene glycol ester.
REFERENCES
- 1. Sadatomi D., Tanimura S., Ozaki K., Takeda K. (2013) Atypical protein phosphatases: emerging players in cellular signaling. Int. J. Mol. Sci. 14, 4596–4612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mikhailik A., Ford B., Keller J., Chen Y., Nassar N., Carpino N. (2007) A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol. Cell 27, 486–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takeda K., Komuro Y., Hayakawa T., Oguchi H., Ishida Y., Murakami S., Noguchi T., Kinoshita H., Sekine Y., Iemura S., Natsume T., Ichijo H. (2009) Mitochondrial phosphoglycerate mutase 5 uses alternate catalytic activity as a protein serine/threonine phosphatase to activate ASK1. Proc. Natl. Acad. Sci. U.S.A. 106, 12301–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Z., Jiang H., Chen S., Du F., Wang X. (2012) The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 [DOI] [PubMed] [Google Scholar]
- 5. Lo S. C., Hannink M. (2006) PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J. Biol. Chem. 281, 37893–37903 [DOI] [PubMed] [Google Scholar]
- 6. Sekine S., Kanamaru Y., Koike M., Nishihara A., Okada M., Kinoshita H., Kamiyama M., Maruyama J., Uchiyama Y., Ishihara N., Takeda K., Ichijo H. (2012) Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J. Biol. Chem. 287, 34635–34645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhuang M., Guan S., Wang H., Burlingame A. L., Wells J. A. (2013) Substrates of IAP ubiquitin ligases identified with a designed orthogonal E3 ligase, the NEDDylator. Mol. Cell 49, 273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lo S. C., Hannink M. (2008) PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 314, 1789–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niture S. K., Jaiswal A. K. (2011) Inhibitor of Nrf2 (INrf2 or Keap1) protein degrades Bcl-xL via phosphoglycerate mutase 5 and controls cellular apoptosis. J. Biol. Chem. 286, 44542–44556 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10. Imai Y., Kanao T., Sawada T., Kobayashi Y., Moriwaki Y., Ishida Y., Takeda K., Ichijo H., Lu B., Takahashi R. (2010) The loss of PGAM5 suppresses the mitochondrial degeneration caused by inactivation of PINK1 in Drosophila. PLoS Genet. 6, e1001229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trott O., Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang C. R., Blackstone C. (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587 [DOI] [PubMed] [Google Scholar]
- 14. Gomes L. C., Di Benedetto G., Scorrano L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cribbs J. T., Strack S. (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Reports 8, 939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murgia M., Giorgi C., Pinton P., Rizzuto R. (2009) Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. J. Mol. Cell. Cardiol. 46, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeyaraju D. V., Cisbani G., Pellegrini L. (2009) Calcium regulation of mitochondria motility and morphology. Biochim. Biophys. Acta 1787, 1363–1373 [DOI] [PubMed] [Google Scholar]
- 18. Suen D. F., Norris K. L., Youle R. J. (2008) Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kepp O., Galluzzi L., Lipinski M., Yuan J., Kroemer G. (2011) Cell death assays for drug discovery. Nat. Rev. Drug Discov. 10, 221–237 [DOI] [PubMed] [Google Scholar]
- 20. Archer S. L. (2013) Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251 [DOI] [PubMed] [Google Scholar]
- 21. Chen H., Chan D. C. (2009) Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y. S., Kimura M., Sato S., Hattori N., Komatsu M., Tanaka K., Matsuda N. (2010) p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15, 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Narendra D., Kane L. A., Hauser D. N., Fearnley I. M., Youle R. J. (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]