

Background: Tomosyn-2 is an inhibitor of insulin secretion.
Results: Glucose induces phosphorylation, ubiquitination, and degradation of tomosyn-2. Hrd-1 is an E3-ligase responsible for tomosyn-2 degradation.
Conclusion: We identified a novel mechanism whereby pancreatic β-cells degrade an inhibitor of insulin secretion.
Significance: Inability of β-cells to degrade tomosyn-2 upon activation will lead to reduced insulin secretion and contribute to the pathogenesis of type 2 diabetes.
Keywords: Diabetes, Insulin, Insulin Secretion, SNARE Proteins, Type 2 Diabetes
Abstract
The abundance and functional activity of proteins involved in the formation of the SNARE complex are tightly regulated for efficient exocytosis. Tomosyn proteins are negative regulators of exocytosis. Tomosyn causes an attenuation of insulin secretion by limiting the formation of the SNARE complex. We hypothesized that glucose-dependent stimulation of insulin secretion from β-cells must involve reversing the inhibitory action of tomosyn. Here, we show that glucose increases tomosyn protein turnover. Within 1 h of exposure to 15 mm glucose, ∼50% of tomosyn was degraded. The degradation of tomosyn in response to high glucose was blocked by inhibitors of the proteasomal pathway. Using 32P labeling and mass spectrometry, we showed that tomosyn-2 is phosphorylated in response to high glucose, phorbol esters, and analogs of cAMP, all key insulin secretagogues. We identified 11 phosphorylation sites in tomosyn-2. Site-directed mutagenesis was used to generate phosphomimetic (Ser → Asp) and loss-of-function (Ser → Ala) mutants. The Ser → Asp mutant had enhanced protein turnover compared with the Ser → Ala mutant and wild type tomosyn-2. Additionally, the Ser → Asp tomosyn-2 mutant was ineffective at inhibiting insulin secretion. Using a proteomic screen for tomosyn-2-binding proteins, we identified Hrd-1, an E3-ubiquitin ligase. We showed that tomosyn-2 ubiquitination is increased by Hrd-1, and knockdown of Hrd-1 by short hairpin RNA resulted in increased abundance in tomosyn-2 protein levels. Taken together, our results reveal a mechanism by which enhanced phosphorylation of a negative regulator of secretion, tomosyn-2, in response to insulin secretagogues targets it to degradation by the Hrd-1 E3-ubiquitin ligase.
Introduction
Plasma glucose is the major determinant of insulin secretion from pancreatic β-cells. The secretion of insulin in response to glucose is biphasic (1–3). Glucose enters β-cells via the glucose transporter type 2. Once inside the cell, glucose is metabolized to yield an increase in the ATP:ADP ratio, which leads to closing of K-ATP channels, membrane depolarization, and opening of the voltage-dependent calcium channels. The subsequent increase in cytosolic free calcium causes insulin granules to fuse to the plasma membrane in a soluble N-ethylmaleimide-sensitive attachment protein receptor (SNARE)-dependent manner and release insulin into the bloodstream. This K-ATP channel-dependent first phase of secretion occurs from a relatively small pool of readily releasable granules. Glucose stimulates the second phase of insulin secretion by activating K-ATP channel-independent amplifying pathways (4).
The insulin granule is mobilized from a reserve pool of granules and ultimately fuses with the plasma membrane through the formation of the SNARE complex. The SNARE complex is formed between the plasma membrane proteins, syntaxin and SNAP-25, and the insulin granule protein, VAMP-2/synaptobrevin. Another vesicle-bound protein, synaptotagmin, upon Ca2+ binding, accelerates the last step in the fusion of two membranes by promoting SNARE complex formation (5). Each step in exocytosis is spatially and temporally regulated by several accessory proteins that undergo protein-protein interactions to affect the rate of formation of the SNARE complex and exocytosis. Phosphorylation/dephosphorylation of the SNARE and SNARE-interacting proteins regulates protein-protein interactions and modulates SNARE assembly during exocytosis (6).
Tomosyn-1 and tomosyn-2 are important regulators of the SNARE complex and predominantly function to inhibit exocytosis (7, 8). Overexpression of either tomosyn-1 or tomosyn-2 inhibits insulin secretion from pancreatic β-cells (9–11). Additionally, tomosyn-1 inhibits regulated secretion from other cell types such as neurons and chromaffin cells (11, 12). Knockdown of tomosyn-1 in mouse neurons (13) and Caenorhabditis elegans enhances synaptic transmission (14). In bovine chromaffin cells, tomosyn-1 inhibits exocytosis by decreasing the degree of depletion and replenishment of dense core vesicles (15).
Tomosyn-1 and tomosyn-2 are syntaxin-1A-binding proteins. Their inhibitory effect on exocytosis was attributed to the C-terminal R-SNARE domain. However, this model has undergone several revisions. The N-terminal domain of tomosyn-1, which makes up 90% of the protein, is required for the inhibitory effects of tomosyn-1 on exocytosis (8, 11). An N-terminal region deletion mutant of tomosyn-1 was able to bind syntaxin-1A but lacked the ability to inhibit exocytosis (16). Additionally, a tomosyn-1 fragment lacking the R-SNARE domain was able to attenuate exocytosis (16). The N-terminal domain of tomosyn-1 binds and inhibits synaptotagmin-1-mediated neurotransmitter release (17). Recently, Williams et al. (18) demonstrated that fragments containing loop 1 (537–578 amino acids) or loop 3 (933–955 amino acids) deletions in the N-terminal region of tomosyn-1 were able to bind syntaxin-1A by the R-SNARE domain but failed to inhibit exocytosis. Together, this suggests that the N-terminal domain in tomosyn-1 and, by extension, tomosyn-2 plays a critical role for imparting the inhibitory effects on exocytosis.
The N-terminal domain of tomosyn-1 contains a hypervariable region (HVR),2 between amino acids 702 and 822. This is the least conserved region between isoforms of tomosyn-1 and tomosyn-2 (19). A protein kinase A (PKA) phosphorylation site at serine 724, was identified in the HVR of tomosyn-1 (20). Phosphorylation of this residue decreases the inhibitory function of tomosyn-1, leading to an increase in exocytosis (20). Additionally, tomosyn-1 is SUMOylated at lysine 730 in the HVR (18). SUMOylation of tomosyn-1 also reduces its ability to inhibit exocytosis. Post-translational modifications in the HVR of tomosyn-1 suggest an important regulatory role of this region.
We previously positionally cloned tomosyn-2 as a gene underlying a diabetes susceptibility locus in an F2 intercross of the C57BL/6 and BTBR mouse strains (10). Islets from congenic mice carrying the BTBR allele of tomosyn-2 (harboring an S912L mutation) had an attenuated response to stimulation of insulin secretion compared with islets carrying the C57BL/6 allele. This reduction in secretion was observed in the presence of normal levels of tomosyn-1, suggesting a nonoverlapping role of tomosyn-1 and tomosyn-2 in inhibiting insulin secretion.
Here, we describe a novel mechanism whereby insulin secretagogues enhance turnover of tomosyn-2 as part of a mechanism to stimulate insulin secretion. We identify phosphorylation sites in tomosyn-2 that respond to insulin secretagogues and an E3-ubiquitin ligase that targets tomosyn-2 for degradation.
EXPERIMENTAL PROCEDURES
Antibodies and Chemicals
Insulin was measured with an in-house ELISA using an anti-insulin antibody from Fitzgerald Industries. Antibodies used to perform experiments were as follows: mouse anti-Myc antibody from Millipore; mouse anti-V5 antibody from Invitrogen; rabbit anti-tomosyn from Synaptic Systems; rabbit anti-ubiquitin from Cell Signaling Technology; and rabbit anti-Hrd-1 from ProteaTech. Mouse and rabbit secondary antibodies were purchased from Cell Signaling Technology. MG132 and cycloheximide were purchased from TOCRIS. Glutathione 4B-Sepharose beads were purchased from GE Healthcare.
Expression Constructs
Moloney murine leukemia virus-based retroviral vector (RVV, 3051) (gift from Dr. Bill Sugden, University of Wisconsin, Madison) containing a multiple cloning site-internal ribosomal entry site-GFP was used to generate b-tomosyn-2-V5-RVV construct for expression studies. The pCR-Script-xb, -b, -m, and s-tomosyn-2 constructs were generously provided by Dr. Alexander Groffen, Virije Universitet, Netherlands. The tomosyn-2 cDNA from these vectors was used for subsequent subcloning. The WT-b-tomosyn-2-V5-RVV was generated by using Gibson cloning methodology. Subsequently, Ser → Ala-11mut-b-tomosyn-2-V5-RVV and Ser → Asp-11mut-tomosyn-2-V5-RVV constructs, referred to as Ser → Ala and Ser → Asp, respectively, were generated form WT-b-tomosyn-2-V5-RVV by performing standard site-directed mutagenesis. For phospho-proteomics studies and tomosyn-2 binding studies, the tomosyn-2-pcDNA/TO/myc-His was generated by subcloning a PCR-amplified tomosyn-2 cDNA into 5′-BamHI and 3′-XhoI sites of the pcDNA4/TO/myc-His C vector (Invitrogen). The following primers were used to amplify tomosyn-2 cDNA with the restriction sites for cloning, a partial KOZAK, and a 3′-precision protease cleavage site: forward (5′-TTAAAGGATCCGCCACCATGAAGAAGTTTAATTTCCG) and reverse (5′-ATATCTCGAGGGGCCCCTGGAACAGAACTTCCAGGAACTGGTACCACTTCTTATCCT). Wild type and dominant negative Hrd-1 plasmids were gift from Dr. Deyu Fang. Ubiquitin, sh-Hrd-1, and control sh-Hrd-1 plasmids were gifts from Dr. Alan Weisman.
Cell Culture, Transfection, and Insulin Secretion
The glucose-responsive rat β-cell line, INS1 (832/13), a gift from Dr. Chris Newgard, Duke University, was cultured in RPMI 1640 medium containing 11 mm glucose supplemented with 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 1 mm sodium pyruvate, 10 mm HEPES, 100 units/ml antibiotic/antimycotic, and 50 μm β-mercaptoethanol. Approximately 100,000 cells/well were plated in a 96-well plate. The following day, cells at 80–90% confluency were transfected with 0.4 μg of plasmid DNA using Lipofectamine 2000 (Invitrogen). After 36 h of incubation, cells were washed once with 200 μl and incubated for 2 h in 100 μl of modified Krebs-Ringer bicarbonate buffer (KRB: 118.41 mm NaCl, 4.69 mm KCl, 1.18 mm MgSO4, 1.18 mm KH2PO4, 25 mm NaHCO3, 20 mm HEPES, 2.52 mm CaCl2, pH 7.4, and 0.2% BSA) containing 1.5 mm glucose. After 2 h, cells were stimulated for 2 h in 100 μl of KRB buffer containing 3 mm glucose + 3 mm 8-bromo (Br)-cAMP. The incubation buffer was collected to determine the amount of insulin secreted under varying conditions. The cells were lysed (lysis buffer: 1 m Tris-HCl, pH 8.0, 1 m NaCl, 0.5 m NaF, 200 mm Na3VO4, 2% Nonidet P-40, and protease inhibitor mixture tablet (Roche Applied Science)) to determine insulin content. The percent fractional insulin secretion was calculated as the amount of insulin secreted divided by the total insulin of the cell. Insulin concentration was determined via ELISA.
The human embryonic kidney 293FT cells (HEK293FT) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 25 mm glucose supplemented with 10% fetal bovine serum, 0.1 mm nonessential amino acid, 6 mm l-glutamine, 1 mm sodium pyruvate, 100 units/ml penicillin, 100 units/ml streptomycin, and 500 μg/ml geneticin. HEK293FT cells at 70–80% in 100-mm tissue culture dishes were transfected with plasmid DNA constructs using 40 μl of 1 mg/ml polyethyleneimine. The following day, cells were lysed (lysis buffer: 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm PMSF, and protease inhibitor mixture tablet (Roche Applied Science)), and total protein lysates were prepared, and the immunoblot was performed as described (21).
Protein Turnover Measurement
INS1 (832/13) cells were plated in 35-mm tissue culture dishes and cultured in RPMI 1640 media for 36 h. After 36 h, media were aspirated and replaced with the media containing 3 mm glucose. After overnight incubation, 50 μm cycloheximide was added to the cells for 2 h. Following a 2-h incubation, cells were stimulated with 3 or 15 mm glucose prior to sample collection for Western blot.
Phosphorylation of Tomosyn-2
INS1 (832/13) cells were either transfected with tomosyn-2-V5 or control plasmid for 36 h. The cells were washed once and incubated with phosphate-free KRB for 2 h containing 0.2 mCi/ml of [32P]orthophosphoric acid. After 2 h, cells were stimulated with glucose for 30 min and harvested, and tomosyn-2 was immunoprecipitated via anti-V5 antibody, followed by SDS-PAGE and autoradiography.
Mass Spectrometry
Immunoprecipitated samples were incubated for 15 min in 0.1% TFA followed by 15 min in 8 m urea at ambient temperature and filtered after each incubation step. The filtrate was neutralized with Tris buffer, pH 8.0, reduced, alkylated, digested, desalted, and dried using previously described methods (22). Peptides from each sample were independently resuspended in 100 mm tetraethylammonium bicarbonate and combined with isobaric labels iTRAQ (ABSciex) or TMT (Thermo) labels dissolved in isopropyl alcohol or acetonitrile, respectively. Alternatively, some phosphorylation samples were examined without labeling and quantified by comparing area under the curve. All samples were prepared in at least biological duplicates. The mixture was incubated at ambient temperature for 2 h followed by pooling the samples together and drying in a speed vac. At this point, samples were enriched for phosphorylation using Fe(III) immobilized metal affinity chromatography according to previously described protocols (23). Unbound material was retained for binding partner interaction analyses.
Peptide samples were injected onto a nano-LC reversed phased column in 0.2% formic acid and eluted with increasing acetonitrile. Eluted peptides were analyzed with a Velos Orbitrap (Thermo) or Orbitrap Elite (Thermo). In brief, MS1 survey scans were performed at 30,000 or 60,000 resolving power at 400 m/z in the Orbitrap. Peptides with charge state 2 or higher, selected for MS/MS, were fragmented by HCD (beam type collision), and the fragments were analyzed in the Orbitrap at 7,500 to 15,000 resolving power.
Spectra were converted to text files, searched against a target-decoy Rattus norvegicus database, downloaded from Uniprot, with the sequence of the clones appended, using the OMSSA search algorithm (24). The COMPASS software suite was used to filter peptide search results to 1% false discovery rate based on E-value and mass error, to quantify isobaric tag reporter ion intensities and group proteins, and to further filter protein results to 1% false discovery rate (25, 26). Phosphorylation sites were localized using a modified A-score algorithm with a 95% confidence cutoff (27).
Isolation and Quantitation of Total RNA
RNA from INS1 (832/13) and HEK293FT cells were extracted using Qiagen RNeasy plus kit. Following extraction, RNA was used for cDNA synthesis (Applied Biosystems). The mRNA abundance was determined by quantitative PCR using FastStart SYBR Green (Roche Applied Science), and gene expression was calculated by comparative ΔCT method.
Immunoprecipitation
INS1 (832/13) and HEK293FT cells were lysed in 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm PMSF, and protease inhibitor mixture tablet (Roche Applied Science). Total cell lysate (1 mg) was precleared with 25 μl of equilibrated protein A/G beads for 30 min in lysis buffer with 0.2% BSA, followed by incubation with ∼2 μg of primary antibody for 16 h at 4 °C. After 16 h, 50 μl of equilibrated beads were added to the lysate to mix for 2 h at 4 °C. The beads were spun at 2,000 × g for 3 min and were washed three times with 1× lysis buffer. Immunoprecipitated protein was eluted in 2.5× Western Loading buffer containing 1 mm DTT. The samples were subjected to immunoblot for analysis.
Ubiquitination Experiments
Hrd-1 (WT)-pCMV or Hrd-1(C296A, C294A)-pCMV plasmids were co-transfected with the tomosyn-2-V5-RVV plasmid in INS1 (832/13) cells. After 36 h of transfection, 50 μm MG132 was added to the cells for 2 h, followed by lysis: 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm PMSF, and protease inhibitor mixture tablet (Roche Applied Science)), 2.5 mm iodoacetamide, 1 mm N-ethylmaleimide. Immunoprecipitation and immunoblot were performed.
Statistical Analysis
Data were expressed as means ± S.E. The statistical comparisons were made using Student's t test at p < 0.05.
RESULTS
Glucose Increases Tomosyn Protein Turnover
We tested the ability of tomosyn-2 to inhibit insulin secretion from human islets. Adenovirus encoding either mouse tomosyn-2 or LacZ control was transduced in human islets for 48 h at 8 mm glucose. After viral infection, glucose-stimulated insulin secretion was performed in islets expressing tomosyn-2 or LacZ control at varying glucose concentrations. Tomosyn-2 inhibited fractional insulin secretion by ∼70%, at 11, 16.7, and 20 mm glucose but had no effect on basal insulin secretion (Fig. 1A).
FIGURE 1.

Glucose promotes turnover of tomosyn protein. A, primary human islets were infected with adenovirus containing tomosyn-2 or LacZ at 200 multiplicities of infection in 8 mm glucose for 48 h in standard supplemented RPMI 1640 growth medium. Following incubation, glucose-stimulated insulin secretion was performed as described under “Experimental Procedures.” Fractional insulin secretion in response to varying concentrations of glucose from tomosyn-2-infected cells was normalized to that of cells infected with LacZ control. Values are means ± S.E. of N ≥4. *, p ≤ 0.05 for the fold change in fractional insulin secretion from cells overexpressing tomosyn-2 versus LacZ. NS, not significant. B, INS1 (832/13) cells were cultured overnight in RPMI 1640 growth media containing 3 mm glucose. Following incubation, 50 μm cycloheximide was added to the cells. After 2 h, cells were either treated with 3 or 15 mm glucose, and samples were collected for protein measurement at various time points. The abundance of tomosyn protein was determined using anti-tomosyn antibody. The graph shows relative abundance of tomosyn at 3 and 15 mm glucose. Values are means ± S.E. of N ≥4. *, p ≤ 0.05 for the tomosyn protein abundance in cells treated with 15 mm glucose versus 3 mm. The protein abundance of tomosyn at time = 0 was set to one for both glucose concentrations and was normalized to a nonspecific band. C, INS1 (832/13) cells were cultured overnight in standard supplemental RPMI 1640 growth media containing 3 mm glucose. Following incubation, cells were treated with 15 mm glucose in the presence and absence of 50 μm MG132 for 4 h. Samples were collected for protein measurements. Protein abundance of tomosyn was determined, and data were normalized to β-actin. The graph shows relative abundance of tomosyn. Values are means ± S.E. of n = 4. *, p ≤ 0.05 for the change in tomosyn protein abundance over 4 h in cells treated with 15 mm glucose. NS, not significant.
Because tomosyn-2 inhibits insulin secretion, we hypothesized that cellular signals that activate insulin secretion should block the inhibitory action of tomosyn-2. Thus, we asked whether glucose stimulates degradation of endogenous tomosyn. We treated INS1 (832/13) cells with 15 mm glucose and assessed the abundance of tomosyn protein after treatment with cycloheximide (50 μm). Within 1 h of treatment with cycloheximide, the abundance of tomosyn protein was reduced by 50% (Fig. 1B). No further reduction in tomosyn protein abundance was observed after 1 h of high glucose treatment. In contrast to cells treated with 15 mm glucose, the turnover of tomosyn protein was very slow in cells treated with 3 mm glucose.
We determined whether high glucose increases tomosyn protein turnover by the proteasomal pathway. INS1 (832/13) cells were treated with 15 mm glucose in the presence of either 50 μm proteasome inhibitor MG132 or DMSO for 4 h. Treatment with MG132 reduced tomosyn degradation by ∼40%, suggesting that tomosyn is degraded by the proteasomal pathway.
Identification of Phosphorylation Sites in Tomosyn-2 Protein
Phosphorylation is an established post-translational modification signal for enhancing the turnover of proteins (28). We asked whether tomosyn-2 is phosphorylated in response to the activation of signaling pathways known to promote insulin secretion. We first examined the effect of glucose on the phosphorylation of tomosyn-2 in intact INS1 (832/13) cells. INS1 (832/13) cells overexpressing tomosyn-2-V5 and control plasmid were pre-labeled with 0.2 mCi/ml [32P]orthophosphate for 2 h. After 2 h, the cells were treated with 3, 5, 7, 11, or 15 mm glucose for 30 min. Whole cell lysates were prepared, and the phosphorylation of tomosyn-2-V5 was assessed by quantifying incorporation of 32P by autoradiography. Glucose caused a dose-dependent increase in the phosphorylation of tomosyn-2 (Fig. 2A).
FIGURE 2.

Phosphorylation of tomosyn-2 by insulin secretagogues. INS1 (832/13) cells were transfected with a mammalian expression plasmid containing tomosyn-2-V5. Following 24 h post-transfection, cells were incubated overnight in 3 mm glucose. After 16 h, A, cells were labeled in a phosphate-free buffer with 0.2 mCi/ml of [32P]orthophosphoric acid for 2 h. Glucose was added to the cells for 30 min. Lysates were prepared, and tomosyn-2-V5 was immunoprecipitated by using an anti-V5 antibody. Incorporation of 32P in tomosyn-2 was measured by autoradiography. Abundance of total tomosyn-2 was determined by immunoblot using anti-V5 antibody. The blot is representative of three independent experiments. B, annotated spectra for the peptide bearing the phosphorylation site in tomosyn-2 at Ser-795. C, cells were treated for 30 min in the presence of secretagogues. Three different treatment groups used in the experiments are as follows: (a) 1.5 or 15 mm glucose; (b) 7 mm glucose or 7 mm glucose with 3 mm 8-Br-cAMP; and (c) glucose 1.5 mm or glucose 1.5 mm with 1 μm TPA. Samples harvested for protein were subjected to immunoprecipitation for b-tomosyn-2-myc followed by mass spectrometry. Top, mass spectrometry results for the relative change in phosphorylation measured at each site between the listed treatment and control as log of means ± log of S.D. from two independent experiments. * represents data from one experiment. Bottom, additional regulatory candidate phosphorylation sites that were detected by mass spectrometry but were not confidently localized or quantified or did not exhibit changes in phosphorylation between conditions. D, modeled tomosyn-2 structure showing phosphorylation sites. Tomosyn-2 structural modeling was conducted using the I-TASSER protein structure prediction server. Specifically, homology modeling was carried out based on the yeast Sro7 crystal structure. Site Ser-768 was included to make alanine and aspartate mutations and to generate a potential AMPK site predicted by ExPASy Motif Scan.
In pancreatic islets, activation of protein kinase A (PKA) and protein kinase C (PKC) was involved in the signaling pathways that link nutrient sensing to insulin secretion. We hypothesized that the activation of PKA and PKC by analogs of cyclic adenosine monophosphate (8-Br-cAMP) and phorbol esters (TPA), respectively, results in the phosphorylation of tomosyn-2. We quantitated the extent of tomosyn-2 phosphorylation and identified specific sites phosphorylated in response to TPA, 8-Br-cAMP, and glucose.
Tomosyn-2-myc was transiently transfected in INS1 (832/13) cells for 36 h. After 36 h, cells were treated with either 1 μm TPA at 1.5 mm glucose, 3 mm 8-Br-cAMP at 7 mm glucose, or 15 mm glucose for 30 min. We identified 11 amino acid residues that were phosphorylated in response to TPA, 8-Br-cAMP, or glucose (Fig. 2C). Serine-746, Ser-747, Ser-787, Ser-795, and Ser-592 were phosphorylated in response to 8-Br-cAMP. Ser-738, Ser-747, Ser-775, Ser-795, Ser-797, Ser-798, and Ser-592 were phosphorylated in response to TPA. Ser-795 and Ser-746 were phosphorylated in response to glucose. We also detected several nonquantifiable phosphorylation sites in tomosyn-2 in response to glucose. These sites were present in segments spanning amino acids 588–612, 719–743, and 793–804. The effects of each treatment on specific phosphorylation sites compared with the control are summarized in Fig. 2C and in supplemental files S1–S3. A representative high resolution annotated spectrum localizing a phosphorylation moiety to Ser-795 on the peptide spanning the 793–804 region is displayed in Fig. 2B.
Our studies show that tomosyn-2 is phosphorylated by the activation of major signaling pathways that play an important role in stimulating insulin secretion. Tomosyn-2 is hyper-phosphorylated predominantly in the HVR with multiple signaling pathways phosphorylating tomosyn-2 at residues Ser-795, Ser-562, and Ser-746 (Fig. 2D). Together, this indicates that phosphorylation of tomosyn-2 is potentially critical for its regulation.
Serine → Aspartate Mutant of Tomosyn-2 Is Unable to Repress Insulin Secretion
Several of the serine residues we identified to be phosphorylated are clustered in the HVR of tomosyn-2. We mutated all 11 serine residues in tomosyn-2 to either alanine or aspartate. Alanine blocks phosphorylation, and aspartate acts as a mimic of phosphoserine. We tested the effect of these mutations on the abundance of tomosyn-2 protein. Tomosyn-2 wild type (WT), Ser → Ala-tomosyn-2, and Ser → Asp-tomosyn-2 were transiently expressed in HEK293FT cells. After 36 h, the cells were harvested, and the abundance of the protein was determined by immunoblot. The tomosyn-2 WT and the mutant forms were expressed at similar levels (Fig. 3D).
FIGURE 3.

Effect of Ser → Asp-tomosyn-2 mutant on insulin secretion. INS1 (832/13) cells were transfected with WT, Ser → Ala (S→A), or Ser → Asp (S→D) mutant of tomosyn-2-V5. Post-transfection, cells were cultured in 3 mm glucose culture media. A, cell lysates were prepared, and tomosyn-2-V5 protein abundance was measured by using anti-V5 antibody. The graph shows relative protein abundance of WT, Ser → Ala, and Ser → Asp mutant of tomosyn-2. Protein abundance of β-actin was used as a loading control. *, p ≤ 0.05, and #, p ≤ 0.05 for the protein abundance of Ser → Asp tomosyn-2 mutant compared with WT tomosyn-2 and Ser → Ala tomosyn-2, respectively. ^, p ≤ 0.05 for the protein abundance of Ser → Ala tomosyn-2 mutant compared with WT tomosyn-2. Values are means ± S.E. of n = 5. B, relative mRNA abundance was determined by real time PCR of the cDNA. The ΔCt was calculated by subtracting raw Ct of tomosyn-2 gene from the raw Ct of the β-actin gene. C, fractional insulin secretion in response to 3 mm 8-Br-cAMP at 3 mm glucose from WT, Ser → Ala, or Ser → Asp-tomosyn-2 transfected cells was normalized to that of cells transfected with GFP. Values are means ± S.E. of n = 4. *, p ≤ 0.05 for the fractional insulin secretion from the cells overexpressing WT and Ser → Ala tomosyn-2 versus GFP expressing cells. D, HEK293FT cells were transfected with WT, Ser → Ala, or Ser → Asp-tomosyn-2. Cell lysates were prepared, and tomosyn-2 protein abundance was measured using anti-V5 antibody. Protein abundance of β-actin was used loading control. The graph shows relative protein abundance of WT, Ser → Ala, and Ser → Asp mutant of tomosyn-2. The data are representative of three independent experiments.
To investigate the functional role of the phosphorylation sites that we identified, we assessed the effect of tomosyn-2 mutants on protein expression and insulin secretion. Sixteen or 40 h after transfection with the WT or mutant tomosyn-2 cDNAs, the relative protein abundance of the gain-of-function Ser → Asp mutant was 50% lower compared with WT-tomosyn-2. The relative protein abundance of the Ser → Ala mutant was significantly higher than WT-tomosyn-2 (Fig. 3A) at 16 h time. The differences in the expression of tomosyn-2 mutants were not due to differences in mRNA abundance (Fig. 3B), transfection efficiency (data not shown), or translation (Fig. 3D) of the overexpressed mutants. This suggests that Ser → Asp mutation increases the turnover of tomosyn-2.
We examined the effect of Ser → Ala and Ser → Asp mutations on the ability of tomosyn-2 to inhibit insulin secretion from INS1 (832/13) cells. GFP, WT-tomosyn-2, Ser → Ala, and Ser → Asp tomosyn-2 mutants were transiently expressed in INS1 (832/13) cells. After 36 h, the cells were treated with 3 mm 8-Br-cAMP with 3 mm glucose to stimulate insulin secretion. WT-tomosyn-2 and the Ser → Ala mutant decreased fractional insulin secretion by ∼40% compared with GFP-expressing cells. In contrast, the Ser → Asp tomosyn-2 mutant had no significant inhibitory effect on insulin secretion (Fig. 3C). This loss of inhibitory effect by the Ser → Asp mutant was not due to a reduction in mRNA expression (data not shown, experimental design similar to (Fig. 3B)). These studies support our hypothesis that secretagogue-induced phosphorylation of tomosyn-2 attenuates its ability to inhibit insulin secretion.
Hrd-1 Is an E3-Ubiquitin Ligase That Targets Tomosyn-2 to Proteasomal Degradation
To determine the mechanism by which tomosyn-2 is targeted to proteasomal degradation, we first examined whether tomosyn-2 is subjected to ubiquitination. HEK293FT cells were co-transfected with tomosyn-2-myc and HA-Ub-expressing plasmids in the presence of 50 μm MG132. Tomosyn-2 was immunoprecipitated, and ubiquitination was detected using an anti-Ub antibody. Ubiquitination of tomosyn-2 was significantly increased in cells treated with MG132 (Fig. 4A) compared with DMSO control.
FIGURE 4.
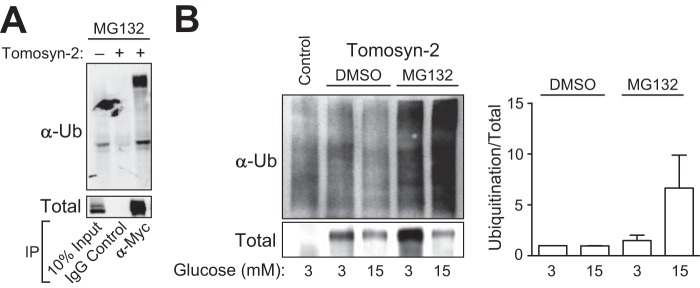
Tomosyn-2 is ubiquitinated by high glucose. A, HEK293FT cells were co-transfected with plasmids expressing HA-Ub and tomosyn-2-myc. After 36 h, 50 μm MG132 was added for 2 h. Cells were harvested for total protein. Immunoprecipitation (IP) was performed by using anti-Myc or control anti-IgG antibody. Ubiquitination and the abundance of tomosyn-2 were determined by Western blot by using anti-Myc and anti-ubiquitin (Ub) antibodies, respectively. B, INS1 (832/13) cells were transfected with plasmid expressing tomosyn-2-V5. After 24 h, cells were cultured in growth media containing 3 mm glucose. Next day, cells were treated with 50 μm MG132 or DMSO at 3 and 15 mm glucose. Cells were harvested for total protein. Immunoprecipitation was performed using anti-V5 antibody. Ubiquitination and the abundance of tomosyn-2 were determined by Western blot by using anti-V5 and anti-ubiquitin antibodies, respectively. The data are representative of three independent experiments in A and B.
Next, we tested the effects of high glucose on the ubiquitination of tomosyn-2. A significant increase in the ubiquitination of tomosyn-2 was observed in cells treated with 15 mm glucose versus 3 mm glucose (Fig. 4B). Together, these results show that tomosyn-2 is subject to ubiquitination, and ubiquitination of tomosyn-2 is progressively increased by high glucose, leading to enhanced turnover of tomosyn-2 protein.
To identify tomosyn-2 binding partners, we performed quantitative mass spectrometry on immunoprecipitated tomosyn-2 from lysates of INS1 (832/13) cells. We detected a significant increase in the binding of Hrd-1, along with its key regulators OS9, Sel1l, and Sel1l2 (Fig. 5A and supplemental files S4–S6). To confirm these results, HEK293FT cells were co-transfected with a plasmid expressing tomosyn-2-V5 and Hrd-1. Co-immunoprecipitation revealed an interaction between tomosyn-2 and Hrd-1. Nonimmune IgG and GFP were used as negative controls and showed that the observed interaction between tomosyn-2 and Hrd-1 is specific (Fig. 5B).
FIGURE 5.
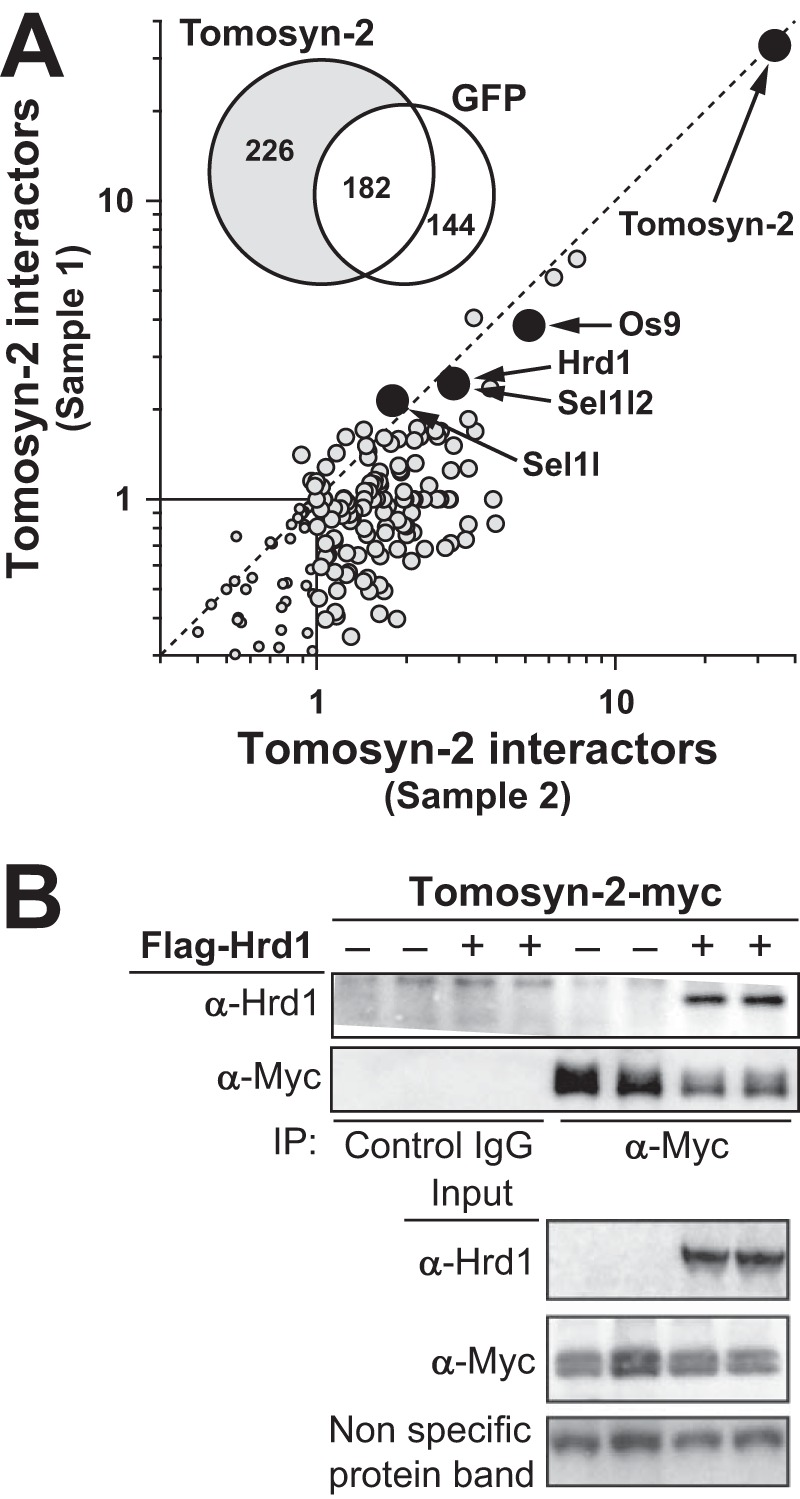
Hrd-1 binds to tomosyn-2. A, INS1 (832/13) cells were transfected with plasmid expressing tomosyn-2-myc. After 36 h, cells were incubated at 1.5 mm glucose for 2 h in KRB. Following a preincubation step, cells were treated with 1 μm phorbol ester (TPA) at 1.5 mm. Cells were harvested for total protein. Immunoprecipitation for tomosyn-2 was performed using protein A-conjugated anti-Myc antibody. Samples were subjected to mass spectrometry. The Venn diagram in the inset of the graph shows the number of proteins identified as tomosyn-2 or GFP-binding partners. The graph shows fold increase in the tomosyn-2 binding partners in response to TPA from two independent experiments. Furthermore, the specificity of binding was measured by comparing the relative abundance of the isobaric reporter ion for a protein in the anti-tomosyn-2 compared with that of the anti-IgG control. B, HEK293FT cells were co-transfected with plasmid expressing Hrd-1 and tomosyn-2-myc. After 36 h, cells were harvested for total protein. Co-immunoprecipitation (IP) was performed. Abundance of Hrd-1 and tomosyn-2 were determined by Western blot by using anti-Hrd-1 and anti-Myc antibodies, respectively. Anti-IgG was used as a control for specificity. The blot is representative of four independent experiments.
To determine whether Hrd-1 can ubiquitinate tomosyn-2, INS1 (832/13) cells were co-transfected with tomosyn-2-V5, along with GFP, Hrd-1, or Hrd-1 (C291A and C294A) (Fig. 6A). Hrd-1 (C291A and C294A) acts as a dominant negative where cysteine at positions 291 and 294 (in the ring domain) were mutated to alanine. Tomosyn-2 was ubiquitinated in cells expressing Hrd-1 compared with the GFP control. No significant ubiquitination was observed in cells expressing a dominant negative form of Hrd-1 (C291A and C294A) above GFP-expressing cells. We then investigated whether the loss of enzymatic activity of Hrd-1 (Cys-291 and Cys-294) affects its ability to bind tomosyn-2. We co-expressed Hrd-1 (Cys-291 and Cys-294) or wild type Hrd-1 along with tomosyn-2 in HEK293FT cells. The dominant negative and wild type Hrd-1 showed comparable binding to tomosyn-2 (Fig. 6A). These results show that tomosyn-2 directly binds and is a substrate of Hrd-1.
FIGURE 6.
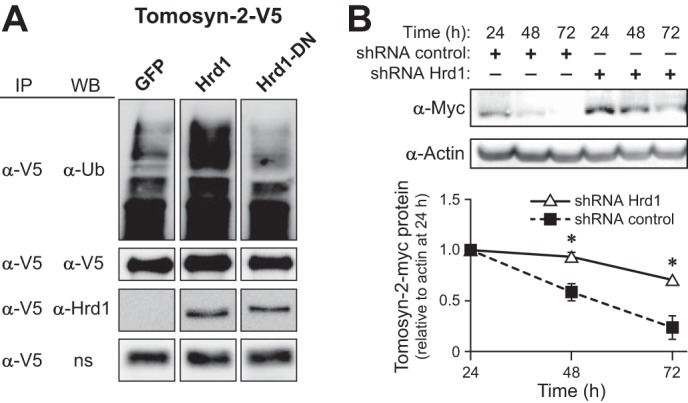
Hrd-1 is an E3-ubiquitin ligase for tomosyn-2. A, INS1 (832/13) cells were co-transfected with plasmid expressing tomosyn-2-V5 along with GFP, Hrd-1 (wild type), or Hrd-1-dominant negative (DN). After 24 h, cells were incubated in growth media containing 3 mm glucose for 16 h. Next day, cells were treated with 11 mm glucose in the presence of 50 μm MG132 for 2 h. Cells were harvested for total protein. Immunoprecipitation (IP) was performed using anti-V5 antibody. The abundance of tomosyn-2, Hrd-1, and ubiquitin was determined by using anti-V5, anti-Hrd-1, and anti-ubiquitin antibodies, respectively. The data are representative of three independent experiments. WB, Western blot. ns, nonspecific band. B, HEK293FT cells were co-transfected with plasmid expressing tomosyn-2-myc with negative control shRNA or shHrd-1. Cells were harvest for total protein at 24, 48, and 72 h post-transfection. The abundance of tomosyn-2 and actin was determined by Western blot by using anti-Myc and anti-actin antibody, respectively. Graph shows relative protein abundance of tomosyn-2 normalized to actin. The abundance of tomosyn-2 after 24 h was set to one. Values are means ± S.E. of N ≥4. *, p ≤ 0.05 for the tomosyn-2 protein abundance in cells treated with shHrd-1 versus control shRNA.
To determine whether Hrd-1 affects the abundance of tomosyn-2, we co-transfected tomosyn-2 along with shHrd-1 or Sh-scramble in HEK293FT cells. We observed an ∼35% reduction in the mRNA abundance of Hrd-1 in cells transfected with shHrd-1 compared with the sh-scrambled control (data not shown). We also observed an ∼40% increase in abundance of tomosyn-2 protein in cells transfected with sh-Hrd-1 versus sh-Scramble (Fig. 6B). These results confirm that Hrd-1 regulates the abundance of tomosyn-2.
DISCUSSION
The role of tomosyn-1/2 as an inhibitor of secretion is well established (8). However, the cellular mechanisms regulating the inhibitory function of tomosyn proteins are not completely understood. The presence of an inhibitor of insulin secretion in β-cells requires a mechanism for insulin secretagogues to block this inhibitory action. We have shown that in response to high glucose, tomosyn protein is degraded. We describe a mechanism by which phosphorylation-mediated degradation of tomosyn-2 de-represses insulin secretion in response to insulin secretagogues. Additionally, we have identified a specific role for Hrd-1, an E3 ubiquitin ligase in the ubiquitin-proteasome pathway that degrades tomosyn-2.
The disappearance of tomosyn upon exposure to glucose followed biphasic kinetics. One possible explanation is that tomosyn exists in two different cellular compartments or pools and that proteasomal degradation accesses just one of these pools. In islets and other cell types, tomosyn fractionates with plasma membrane, cytosol, and dense core granules (8, 11, 29). Tomosyn in the plasma membrane pool likely regulates the formation of the SNARE complex. However, the function of tomosyn in other cellular pools is unknown. Another possibility is that the turnover of tomosyn-1 and tomosyn-2 isoforms is differentially regulated by high glucose. The antibody used in these experiments cannot differentiate between tomosyn-1 and tomosyn-2; therefore, it is possible that one of the two proteins is more rapidly degraded when exposed to high glucose.
The half-life of synaptic proteins ranges between several days and hours (30–33). The half-life of tomosyn-1 in neurons was estimated to be ∼4 days (30). This relatively long half-life could explain why we did not observe a significant change in tomosyn protein abundance after a cycloheximide treatment with low glucose. However, it is possible that the turnover rate of synaptic proteins in β-cells and neurons could be different. In neurons, the site of exocytosis in the pre-synapse, at the distal tip of the axon, is often located far from the site of protein synthesis. This may necessitate the need for increased protein stability.
In pancreatic β-cells, activation of cell signaling pathways through PKA, PKCs, and calcium/calmodulin-dependent protein kinase II (CaMKII) augments insulin secretion by phosphorylating proteins that are involved in the formation of the SNARE complex (34–36). We activated PKA-, PKC-, and CaMK II-mediated signaling pathways by treating INS1 (832/13) cells with 8-Br-cAMP, phorbol esters (TPA), and glucose, respectively.
We identified eleven phosphorylation sites in the N-terminal domain of tomosyn-2. Mutating all phosphoserine sites to aspartate (Ser → Asp) decreased tomosyn-2 protein abundance relative to WT and alanine tomosyn-2 mutant (Ser → Ala). This result suggests an important role of HVR in regulating the abundance of tomosyn-2 protein; 10 of the 11 phosphorylation sites are present in the HVR. The phosphorylated serine residues that we identified in response to glucose, 8-Br-cAMP, and TPA are part of the consensus motif for phosphorylation by kinases such as PKA (Ser-746, Ser-787, and Ser-795), PKC (Ser-738, Ser-741, Ser-775, Ser-795, Ser-798, and Ser-592), CaMKII (Ser-747 and Ser-795), AKT/PKB (Ser-795), AMP-activated kinase (Ser-768), and casein kinase-1 (Ser-797). The kinases listed here represent a diverse network of signaling pathways, and each kinase has been shown to play an important role in regulating SNARE complex formation, SNARE localization, and organelle morphology. Understanding the role of each of the phosphorylation site will provide insight to the mechanism by which tomosyn-2 regulates secretion.
An important facet of protein phosphorylation is the regulation of protein-protein interactions. Here, we focused on the role of phosphorylation sites in regulating the stability of tomosyn-2 protein. Phosphorylation of Ser-724 in the HVR allosterically regulates tomosyn-1's ability to interact with syntaxin-1A in a SNARE complex (20). The N-terminal domain of tomosyn-1 regulates synaptotagmin-1-mediated exocytosis in a Ca2+-dependent manner (17). Therefore, it is possible that one or more phosphorylation sites in the HVR of tomosyn-2 is responsible for allosterically regulating tomosyn-2's ability to interact with its binding partners.
Phosphorylation of Ser-795 in tomosyn-2 was increased in response to glucose, TPA, and 8-Br-cAMP, indicating a potential role of CaMKII, PKCs, and PKA, respectively. This site is also a consensus Akt/PKB kinase phosphorylation site (RXRXXS) and is the only Akt/PKB kinase motif present in tomosyn-2, thus making Ser-795 a top candidate for regulation of the inhibitory activity of tomosyn-2. The Akt/PKB signaling pathway in β-cells is involved in insulin secretion and affects susceptibility to type 2 diabetes (37). There is some evidence that insulin regulates its own secretion (38). Thus, tomosyn-2 might mediate autoregulation of insulin via the Akt/PKB signaling pathway.
The allosteric effects of phosphorylation within the HVR in regulating tomosyn-2 inhibitory function are not known. It is possible that the modes of regulation exerted by the HVR are potentially different between tomosyn-2 and tomosyn-1. Phosphorylation and SUMOylation at Ser-724 and Ser-730, respectively, in the HVR of tomosyn-1 alters its functional activity by allosterically regulating protein-protein interactions (18, 20). Our data show that the phosphorylation in the HVR of tomosyn-2 affects it protein stability. This suggests that even though tomosyn-1 and tomosyn-2 are both inhibitors of secretion, the mechanisms by which cells regulate the functional activity of these two proteins are different.
The ubiquitination-proteasome pathway (UPP) is known to regulate proteins involved in synapse growth and development, synaptic function, and plasticity (39–43), including several of the SNARE proteins, such as syntaxin-1 (44), Munc-13 (45), and RIM-1 (46). In β-cells, the precise role of the UPP is not fully defined. However, it is known to target proteins of the secretory pathway, such as K-ATP channels (47), voltage-dependent calcium channels (48), glucose-dependent insulinotropic polypeptide receptors (GIP-R), and proinsulin (49), and plays a major role in regulating insulin secretion (50–52).
Phosphorylation of proteins sometimes serves as an initiating step for ubiquitination. Protein phosphorylation of K-ATP channels by PKA or SGK1 (serum- and glucocorticoid-sensitive kinase) (53), p35 by SIK2 (AMP-activated protein kinase-related kinase) (54), and GIP-R by PKA (55) promotes degradation by the UPP to regulate insulin secretion. Our results show that high glucose increases ubiquitination and degradation of tomosyn-2 via the UPP. The ubiquitination of a substrate is influenced by proximity of one or multiple phosphorylation sites (28). The ubiquitination sites responsible for regulating tomosyn-2 protein abundance are not known. Sequence analysis of tomosyn-2 by the UbPred program (56) identified four potential ubiquitination sites as follows: Lys-720, Lys-801, Lys-804, and Lys-1131. Of these, Lys-801 and Lys-804 are proximal to several phosphorylation sites that we identified (e.g. Ser-798, Ser-797, and Ser-795). We hypothesize that ubiquitination at sites Lys-801 and Lys-804 in response to phosphorylation is most likely responsible for the degradation of tomosyn-2.
Hrd-1 is an endoplasmic reticulum E3-ligase (57). It is a multispanning membrane protein with a RING domain extending into the cytoplasm (58). Hrd-1 substrates include proteins of the endoplasmic reticulum-associated degradation pathway (58) and several cytoplasmic proteins (59). We show that Hrd-1 binds, ubiquitinates, and regulates the abundance of tomosyn-2, suggesting that Hrd-1 is an E3-ligase for tomosyn-2. Several proteins are involved in the formation of the Hrd-1 E3 ubiquitin complex. Notably, Sel1l is critical for the stability of Hrd-1 protein, and OS9 plays an important role in substrate recognition (60). Sel1l has been shown to play a role in the pathogenesis of type 2 diabetes. Mice expressing just one allele of Sel1l were severely glucose-intolerant and had reduced glucose-stimulated insulin secretion (61). This reduction in insulin secretion was attributed to a defect in the trafficking of insulin granules due to the loss of the β1-integrin (62).
In summary, we show that the induction of phosphorylation of tomosyn-2 by several insulin secretagogues increases turnover of tomosyn-2 protein and thus de-represses insulin secretion. The degradation and/or inactivation of tomosyn-2 appears to be necessary for glucose to fully stimulate insulin secretion. Mutations in tomosyn-2 or in this regulatory pathway contribute to the risk of developing type 2 diabetes (10).
Supplementary Material
Acknowledgments
We gratefully acknowledge Angie T. Oler, Chen-Yu Wang, Donald Stapleton, Kathy Schueler, Matthew Bruss, Melkam Kebede, and Michelle Kimple for their assistance with the studies.
This work was supported, in whole or in part, by National Institutes of Health Grants DK066369 and DK58037 (to A. D. A.).

This article contains supplemental files 1–6.
- HVR
- hypervariable region
- RVV
- retroviral vector
- TPA
- phorbol ester
- CaMKII
- calcium/calmodulin-dependent protein kinase II
- UPP
- ubiquitination-proteasome pathway
- 8-Br-cAMP
- 8-bromo-cyclic AMP.
REFERENCES
- 1. Barbosa R. M., Silva A. M., Tomé A. R., Stamford J. A., Santos R. M., Rosário L. M. (1998) Control of pulsatile 5-HT/insulin secretion from single mouse pancreatic islets by intracellular calcium dynamics. J. Physiol. 510, 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eddlestone G. T., Oldham S. B., Lipson L. G., Premdas F. H., Beigelman P. M. (1985) Electrical activity, cAMP concentration, and insulin release in mouse islets of Langerhans. Am. J. Physiol. 248, C145–C153 [DOI] [PubMed] [Google Scholar]
- 3. Rorsman P., Eliasson L., Renström E., Gromada J., Barg S., Göpel S. (2000) The cell physiology of biphasic insulin secretion. News Physiol. Sci. 15, 72–77 [DOI] [PubMed] [Google Scholar]
- 4. Straub S. G., Sharp G. W. (2002) Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes Metab. Res. Rev. 18, 451–463 [DOI] [PubMed] [Google Scholar]
- 5. Sudhof T. C. (2004) The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547 [DOI] [PubMed] [Google Scholar]
- 6. Nishi H., Hashimoto K., Panchenko A. R. (2011) Phosphorylation in protein-protein binding: effect on stability and function. Structure 19, 1807–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masuda E. S., Huang B. C., Fisher J. M., Luo Y., Scheller R. H. (1998) Tomosyn binds t-SNARE proteins via a VAMP-like coiled coil. Neuron 21, 479–480 [DOI] [PubMed] [Google Scholar]
- 8. Fujita Y., Shirataki H., Sakisaka T., Asakura T., Ohya T., Kotani H., Yokoyama S., Nishioka H., Matsuura Y., Mizoguchi A., Scheller R. H., Takai Y. (1998) Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron 20, 905–915 [DOI] [PubMed] [Google Scholar]
- 9. Cheviet S., Bezzi P., Ivarsson R., Renström E., Viertl D., Kasas S., Catsicas S., Regazzi R. (2006) Tomosyn-1 is involved in a post-docking event required for pancreatic beta-cell exocytosis. J. Cell Sci. 119, 2912–2920 [DOI] [PubMed] [Google Scholar]
- 10. Bhatnagar S., Oler A. T., Rabaglia M. E., Stapleton D. S., Schueler K. L., Truchan N. A., Worzella S. L., Stoehr J. P., Clee S. M., Yandell B. S., Keller M. P., Thurmond D. C., Attie A. D. (2011) Positional cloning of a type 2 diabetes quantitative trait locus; tomosyn-2, a negative regulator of insulin secretion. PLoS Genet. 7, e1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang W., Lilja L., Mandic S. A., Gromada J., Smidt K., Janson J., Takai Y., Bark C., Berggren P. O., Meister B. (2006) Tomosyn is expressed in beta-cells and negatively regulates insulin exocytosis. Diabetes 55, 574–581 [DOI] [PubMed] [Google Scholar]
- 12. Gladycheva S. E., Lam A. D., Liu J., D'Andrea-Merrins M., Yizhar O., Lentz S. I., Ashery U., Ernst S. A., Stuenkel E. L. (2007) Receptor-mediated regulation of tomosyn-syntaxin 1A interactions in bovine adrenal chromaffin cells. J. Biol. Chem. 282, 22887–22899 [DOI] [PubMed] [Google Scholar]
- 13. Sakisaka T., Yamamoto Y., Mochida S., Nakamura M., Nishikawa K., Ishizaki H., Okamoto-Tanaka M., Miyoshi J., Fujiyoshi Y., Manabe T., Takai Y. (2008) Dual inhibition of SNARE complex formation by tomosyn ensures controlled neurotransmitter release. J. Cell Biol. 183, 323–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gracheva E. O., Burdina A. O., Touroutine D., Berthelot-Grosjean M., Parekh H., Richmond J. E. (2007) Tomosyn negatively regulates both synaptic transmitter and neuropeptide release at the C. elegans neuromuscular junction. J. Physiol. 585, 705–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yizhar O., Matti U., Melamed R., Hagalili Y., Bruns D., Rettig J., Ashery U. (2004) Tomosyn inhibits priming of large dense-core vesicles in a calcium-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 101, 2578–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yizhar O., Lipstein N., Gladycheva S. E., Matti U., Ernst S. A., Rettig J., Stuenkel E. L., Ashery U. (2007) Multiple functional domains are involved in tomosyn regulation of exocytosis. J. Neurochem. 103, 604–616 [DOI] [PubMed] [Google Scholar]
- 17. Yamamoto Y., Mochida S., Miyazaki N., Kawai K., Fujikura K., Kurooka T., Iwasaki K., Sakisaka T. (2010) Tomosyn inhibits synaptotagmin-1-mediated step of Ca2+-dependent neurotransmitter release through its N-terminal WD40 repeats. J. Biol. Chem. 285, 40943–40955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Williams A. L., Bielopolski N., Meroz D., Lam A. D., Passmore D. R., Ben-Tal N., Ernst S. A., Ashery U., Stuenkel E. L. (2011) Structural and functional analysis of tomosyn identifies domains important in exocytotic regulation. J. Biol. Chem. 286, 14542–14553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pobbati A. V., Razeto A., Böddener M., Becker S., Fasshauer D. (2004) Structural basis for the inhibitory role of tomosyn in exocytosis. J. Biol. Chem. 279, 47192–47200 [DOI] [PubMed] [Google Scholar]
- 20. Baba T., Sakisaka T., Mochida S., Takai Y. (2005) PKA-catalyzed phosphorylation of tomosyn and its implication in Ca2+-dependent exocytosis of neurotransmitter. J. Cell Biol. 170, 1113–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bhatnagar S., Damron H. A., Hillgartner F. B. (2009) Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J. Biol. Chem. 284, 10023–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hebert A. S., Dittenhafer-Reed K. E., Yu W., Bailey D. J., Selen E. S., Boersma M. D., Carson J. J., Tonelli M., Balloon A. J., Higbee A. J., Westphall M. S., Pagliarini D. J., Prolla T. A., Assadi-Porter F., Roy S., Denu J. M., Coon J. J. (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Phanstiel D. H., Brumbaugh J., Wenger C. D., Tian S., Probasco M. D., Bailey D. J., Swaney D. L., Tervo M. A., Bolin J. M., Ruotti V., Stewart R., Thomson J. A., Coon J. J. (2011) Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods 8, 821–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 25. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 26. Wenger C. D., Phanstiel D. H., Lee M. V., Bailey D. J., Coon J. J. (2011) COMPASS: A suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 28. Swaney D. L., Beltrao P., Starita L., Guo A., Rush J., Fields S., Krogan N. J., Villén J. (2013) Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 10, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Widberg C. H., Bryant N. J., Girotti M., Rea S., James D. E. (2003) Tomosyn interacts with the t-SNAREs syntaxin4 and SNAP23 and plays a role in insulin-stimulated GLUT4 translocation. J. Biol. Chem. 278, 35093–35101 [DOI] [PubMed] [Google Scholar]
- 30. Cohen L. D., Zuchman R., Sorokina O., Müller A., Dieterich D. C., Armstrong J. D., Ziv T., Ziv N. E. (2013) Metabolic turnover of synaptic proteins: kinetics, interdependencies and implications for synaptic maintenance. PLoS One 8, e63191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baitinger C., Willard M. (1987) Axonal transport of synapsin I-like proteins in rabbit retinal ganglion cells. J. Neurosci. 7, 3723–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petrucci T. C., Macioce P., Paggi P. (1991) Axonal transport kinetics and posttranslational modification of synapsin I in mouse retinal ganglion cells. J. Neurosci. 11, 2938–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ehlers M. D. (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242 [DOI] [PubMed] [Google Scholar]
- 34. Gerst J. E. (2003) SNARE regulators: matchmakers and matchbreakers. Biochim. Biophys. Acta 1641, 99–110 [DOI] [PubMed] [Google Scholar]
- 35. Weinberger A., Gerst J. (2004) in Regulatory Mechanisms of Intracellular Membrane Transport (Keränen S., Jäntti J., eds) pp. 145–170, Springer-Verlag, Berlin [Google Scholar]
- 36. Snyder D. A., Kelly M. L., Woodbury D. J. (2006) SNARE complex regulation by phosphorylation. Cell Biochem. Biophys. 45, 111–123 [DOI] [PubMed] [Google Scholar]
- 37. Bernal-Mizrachi E., Fatrai S., Johnson J. D., Ohsugi M., Otani K., Han Z., Polonsky K. S., Permutt M. A. (2004) Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J. Clin. Invest. 114, 928–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bouche C., Lopez X., Fleischman A., Cypess A. M., O'Shea S., Stefanovski D., Bergman R. N., Rogatsky E., Stein D. T., Kahn C. R., Kulkarni R. N., Goldfine A. B. (2010) Insulin enhances glucose-stimulated insulin secretion in healthy humans. Proc. Natl. Acad. Sci. U.S.A. 107, 4770–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yi J. J., Ehlers M. D. (2007) Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol. Rev. 59, 14–39 [DOI] [PubMed] [Google Scholar]
- 40. Hegde A. N., Inokuchi K., Pei W., Casadio A., Ghirardi M., Chain D. G., Martin K. C., Kandel E. R., Schwartz J. H. (1997) Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89, 115–126 [DOI] [PubMed] [Google Scholar]
- 41. Jiang Y. H., Armstrong D., Albrecht U., Atkins C. M., Noebels J. L., Eichele G., Sweatt J. D., Beaudet A. L. (1998) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811 [DOI] [PubMed] [Google Scholar]
- 42. DiAntonio A., Haghighi A. P., Portman S. L., Lee J. D., Amaranto A. M., Goodman C. S. (2001) Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412, 449–452 [DOI] [PubMed] [Google Scholar]
- 43. Hegde A. N., DiAntonio A. (2002) Ubiquitin and the synapse. Nat. Rev. Neurosci. 3, 854–861 [DOI] [PubMed] [Google Scholar]
- 44. Chin L. S., Vavalle J. P., Li L. (2002) Staring, a novel E3 ubiquitin-protein ligase that targets syntaxin 1 for degradation. J. Biol. Chem. 277, 35071–35079 [DOI] [PubMed] [Google Scholar]
- 45. Tada H., Okano H. J., Takagi H., Shibata S., Yao I., Matsumoto M., Saiga T., Nakayama K. I., Kashima H., Takahashi T., Setou M., Okano H. (2010) Fbxo45, a novel ubiquitin ligase, regulates synaptic activity. J. Biol. Chem. 285, 3840–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yao I., Takagi H., Ageta H., Kahyo T., Sato S., Hatanaka K., Fukuda Y., Chiba T., Morone N., Yuasa S., Inokuchi K., Ohtsuka T., Macgregor G. R., Tanaka K., Setou M. (2007) SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell 130, 943–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yan F. F., Lin C. W., Cartier E. A., Shyng S. L. (2005) Role of ubiquitin-proteasome degradation pathway in biogenesis efficiency of β-cell ATP-sensitive potassium channels. Am. J. Physiol. Cell Physiol. 289, C1351–C1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kawaguchi M., Minami K., Nagashima K., Seino S. (2006) Essential role of ubiquitin-proteasome system in normal regulation of insulin secretion. J. Biol. Chem. 281, 13015–13020 [DOI] [PubMed] [Google Scholar]
- 49. Kitiphongspattana K., Mathews C. E., Leiter E. H., Gaskins H. R. (2005) Proteasome inhibition alters glucose-stimulated (pro)insulin secretion and turnover in pancreatic β-cells. J. Biol. Chem. 280, 15727–15734 [DOI] [PubMed] [Google Scholar]
- 50. López-Avalos M. D., Duvivier-Kali V. F., Xu G., Bonner-Weir S., Sharma A., Weir G. C. (2006) Evidence for a role of the ubiquitin-proteasome pathway in pancreatic islets. Diabetes 55, 1223–1231 [DOI] [PubMed] [Google Scholar]
- 51. Costes S., Vandewalle B., Tourrel-Cuzin C., Broca C., Linck N., Bertrand G., Kerr-Conte J., Portha B., Pattou F., Bockaert J., Dalle S. (2009) Degradation of cAMP-responsive element-binding protein by the ubiquitin-proteasome pathway contributes to glucotoxicity in beta-cells and human pancreatic islets. Diabetes 58, 1105–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Broca C., Varin E., Armanet M., Tourrel-Cuzin C., Bosco D., Dalle S., Wojtusciszyn A. (2014) Proteasome dysfunction mediates high glucose-induced apoptosis in rodent beta cells and human islets. PLoS One 9, e92066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Boehmer C., Laufer J., Jeyaraj S., Klaus F., Lindner R., Lang F., Palmada M. (2008) Modulation of the voltage-gated potassium channel Kv1.5 by the SGK1 protein kinase involves inhibition of channel ubiquitination. Cell. Physiol. Biochem. 22, 591–600 [DOI] [PubMed] [Google Scholar]
- 54. Belgardt B. F., Stoffel M. (2014) SIK2 regulates insulin secretion. Nat. Cell Biol. 16, 210–212 [DOI] [PubMed] [Google Scholar]
- 55. Zhou J., Livak M. F., Bernier M., Muller D. C., Carlson O. D., Elahi D., Maudsley S., Egan J. M. (2007) Ubiquitination is involved in glucose-mediated downregulation of GIP receptors in islets. Am. J. Physiol. Endocrinol. Metab. 293, E538–E547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tung C. W., Ho S. Y. (2008) Computational identification of ubiquitylation sites from protein sequences. BMC Bioinformatics 9, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Amano T., Yamasaki S., Yagishita N., Tsuchimochi K., Shin H., Kawahara K., Aratani S., Fujita H., Zhang L., Ikeda R., Fujii R., Miura N., Komiya S., Nishioka K., Maruyama I., Fukamizu A., Nakajima T. (2003) Synoviolin/Hrd-1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev. 17, 2436–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bordallo J., Plemper R. K., Finger A., Wolf D. H. (1998) Der3p/Hrd-1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 9, 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamasaki S., Yagishita N., Nishioka K., Nakajima T. (2007) The roles of synoviolin in crosstalk between endoplasmic reticulum stress-induced apoptosis and p53 pathway. Cell Cycle 6, 1319–1323 [DOI] [PubMed] [Google Scholar]
- 60. Iida Y., Fujimori T., Okawa K., Nagata K., Wada I., Hosokawa N. (2011) SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J. Biol. Chem. 286, 16929–16939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Francisco A. B., Singh R., Sha H., Yan X., Qi L., Lei X., Long Q. (2011) Haploid insufficiency of suppressor enhancer Lin12 1-like (SEL1L) protein predisposes mice to high fat diet-induced hyperglycemia. J. Biol. Chem. 286, 22275–22282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Diaferia G. R., Cirulli V., Biunno I. (2013) SEL1L regulates adhesion, proliferation and secretion of insulin by affecting integrin signaling. PLoS One 8, e79458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.