

Background: The molecular basis for how the PDZ domain of the scaffolding protein PICK1 selectively binds >30 different ligands is unclear.
Results: NMR and fluorescence polarization analyses reveal both conventional and unconventional binding modes.
Conclusion: Three principal binding modes can account for PICK1 PDZ binding specificity.
Significance: Distinct and unconventional binding modes might evolve to rapidly expand the repertoire of functionally important interactions.
Keywords: Nuclear Magnetic Resonance (NMR), Protein Structure, Protein-Protein Interaction, Scaffold Protein, Signal Transduction, Trafficking, PDZ Domains
Abstract
PDZ domain proteins control multiple cellular functions by governing assembly of protein complexes. It remains unknown why individual PDZ domains can bind the extreme C terminus of very diverse binding partners and maintain selectivity. By employing NMR spectroscopy, together with molecular modeling, mutational analysis, and fluorescent polarization binding experiments, we identify here three structural mechanisms explaining why the PDZ domain of PICK1 selectively binds >30 receptors, transporters, and kinases. Class II ligands, including the dopamine transporter, adopt a canonical binding mode with promiscuity obtained via differential packing in the binding groove. Class I ligands, such as protein kinase Cα, depend on residues upstream from the canonical binding sequence that are likely to interact with flexible loop residues of the PDZ domain. Finally, we obtain evidence that the unconventional ligand ASIC1a has a dual binding mode involving a canonical insertion and a noncanonical internal insertion with the two C-terminal residues forming interactions outside the groove. Together with an evolutionary analysis, the data show how unconventional binding modes might evolve for a protein recognition domain to expand the repertoire of functionally important interactions.
Introduction
PDZ domains constitute the largest family of protein interaction domains in the human genome with at least 270 different domains in more than 150 proteins (1–3). The domains are found in multidomain scaffolding proteins, which operate as molecular adaptors that assemble protein complexes and thereby play an essential role in temporal and spatial control of a wide variety of cellular functions (2). PDZ domains are ∼90 residues long and folded to form an elongated groove consisting of mainly two pockets (S0 and S−2) that, with very few exceptions, bind the last 3–4 C-terminal residues of the target proteins. With such a short binding sequence, it has been a central question how sufficient specificity of PDZ interactions is ensured in a cellular environment (3–8). The canonical PDZ binding involves a carboxylate-binding loop coordinating the free C terminus of the target protein ligand and the hydrophobic S0 pocket accommodating the side chain of the C-terminal residue (P0). Earlier studies classified PDZ domains into two main classes based on the specificity for the antepenultimate residue (position −2 from the C terminus, P−2) of the ligand (4, 8, 9). Class I domains, including PSD-95 and other MAGUK-type proteins, preferentially bind ligands with a Thr/Ser at P−2 forming a hydrogen bond to the side chain of a conserved His at the first position in the αB helix (αB1). Class II PDZ domains preferentially bind ligands with a hydrophobic residue at P−2 that docks into the hydrophobic S−2 pocket, formed by a hydrophobic residue at αB1 and other hydrophobic residues in the α-helix (10). A less abundant class of domains (class III) have a Tyr at the αB1 position that forms a hydrogen bond with the carboxylate of the preferred Asp or Glu at the −2 position of the ligand (11).
It is clear that this classical classification is not sufficient to explain the binding specificity of all PDZ domains (3, 5–8). A large study of mouse PDZ domains suggested that PDZ domains do not fall into discrete classes but instead lie on a continuum with the ligand selectivity derived from interactions throughout the binding pocket (5). Another comprehensive study supported that PDZ domains form at least 16 different specificity classes recognizing up to seven C-terminal ligand residues (7). Moreover, it has been suggested that the specificity of PDZ domain interactions rely on physically contiguous networks of co-evolving amino acids, termed protein sectors (6). Nevertheless, these studies do not explain why specific PDZ domains can accommodate an unusual wide range of very different ligands. The protein interacting with C kinase 1 (PICK1), which plays a key role in regulation of synaptic plasticity and endocrine secretions (12–19), has a PDZ domain that is able to bind the C terminus of more than 30 different proteins, including, for example, the protein kinase Cα (PKCα); the GluA2 subunit in AMPA-type ionotropic glutamate receptors; the metabotropic glutamate receptor mGluR7; aquaporins (1, 2, and 9); the dopamine transporter (DAT)4; the glutamate transporter GLT1b; the human epidermal growth factor receptor 2 (HER2); the netrin receptor (UNC5H1); and acid-sensing ion channel (ASIC1a) (20–31). The ligands represent all three canonical classes, as well as ligands outside the classification (32). Previously resolved structures of PICK1 PDZ in complex with EphrinB1 and GluA2 (33, 34) show that class II specificity depends mainly on hydrophobic interactions in the canonical binding groove, but these structures offer no insight into how PICK1 PDZ can bind other types of ligands.
Here, we employ NMR spectroscopy together with molecular modeling, mutational analysis, and fluorescent polarization (FP) binding experiments to dissect the molecular details underlying the ligand promiscuity of PICK1 PDZ. Surprisingly, despite low micromolar affinities for all classes of ligands, only class II ligands formed PICK1 PDZ-ligand complexes suitable for high resolution structure determination. This included PICK1 PDZ bound to a DAT C-terminal peptide, which revealed a typical class II-like binding mode. For the unclassified ligand ASIC1a and the class I ligand PKCα, we were able, despite instability of the complexes, to obtain structural docking models by taking advantage of chemical shifts and saturation transfer difference (STD) NMR data, respectively. The models were subsequently supported by mutational analysis and FP-binding experiments. Notably, the models revealed unique and distinct binding modes with dependence on discrete interactions outside the binding groove. In addition to providing a molecular explanation for the binding specificity of PICK1 PDZ, our data show how distinct and unconventional binding modes of a protein recognition domain might evolve to rapidly expand the repertoire of functionally important cellular interactions.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of PICK1 PDZ Constructs
A PICK1 PDZ-3C-GluA2 construct consisting of the PICK1 residues 18–110, a linker region with a protease 3C cleavage site (LEVLFGGP), and the residues (VYGIESVKI) derived from the C terminus of the AMPA receptor subunit 2 was purchased from Origene, Germany, and subcloned into the pGEX4T2 vector (Novagen) producing an N-terminal glutathione S-transferase (GST) fusion construct (34). The constructs PICK1 PDZ-3C-DAT (QFTLRHWLKV), PICK1 PDZ-3C-ASIC1a (ARGTFEDFTC), PICK1 PDZ-3C-HER2 (EYLGLDVPV), PICK1 PDZ-3C-UNC5H1 (GLFTVSEAEC), PICK1 PDZ-3C-PKCα (FVHPILQSAV), and PICK1 PDZ-3C-GLT1b (FPFLDIETCI) were generated by PCR using oligonucleotides designed to amplify DNA sequences encoding the specific C-terminal ligands. The coding region of rat PICK1 residues 2–416 was amplified from pCINEO-PICK1 vector by PCR and subcloned into the pET41a vector (Novagen) producing an N-terminal GST fusion construct as described (32). The PICK1 K83V, K83H, Y43S, and Y43A mutations were generated in the pET41a vector as described (32). The plasmids were transformed into Escherichia coli BL21 DE3 pLysS cells (Novagen) and purified essentially as described (32). A 50-ml starter culture was grown in LB media for 18 h and used to inoculate 1 liter of culture media. Protein expression for FP was carried out in standard LB media. Protein expression for NMR analysis was performed in M9 minimal media supplemented with [13C]glucose and [15N]ammonium sulfate. Protein expression was induced by adding isopropyl β-d-1-thiogalactopyranoside to a final concentration of 200 μm at an A600 of ∼0.8–1 and grown overnight at 22 °C. The bacteria were spun down, resuspended in lysis buffer (50 mm Tris, pH 7.4, 125 mm NaCl, 1% Triton X-100, 20 μg/ml DNase I, 1 mm DTT (Sigma)), and lysed by freezing and thawing. The lysate was centrifuged at 38,000 × g for 30 min at 4 °C in an SS-34 rotor, and the supernatant was incubated with glutathione-Sepharose beads (Amersham Biosciences) under slow rotation for 90 min at 4 °C. The beads were washed three times in buffer (50 mm Tris, pH 7.4, 125 mm NaCl, 1 mm DTT (with 0.1% Triton X-100 for full-length PICK1 constructs)) and incubated overnight with thrombin protease (Novagen) at 4 °C (cleavage of PICK1 constructs from glutathione-bound GST). The size, purity, and stability of the protein constructs were verified by SDS-PAGE and size exclusion HPLC.
Fluorescence Polarization Assay
The fluorescence polarization assay was performed essentially as described (32). For saturation binding experiments, full-length PICK1 or mutants was diluted in buffer (50 mm Tris, pH 7.4, 125 mm NaCl, 1 mm DTT, 0.1% Triton X-100) to various concentrations and a final volume of 100 μl in black low-binding 96-well microtiter plates (Corning Glass). A volume of 5 μl of Oregon Green-labeled DAT C13 peptide (OG DAT C13) was added to each well to a final concentration of 20 nm. For the FP competition binding assay, increasing concentrations of unlabeled peptide were diluted in the wells, and a fixed ∼75% saturating concentration of PICK1 or mutants was added together with OG DAT C13 as described above. All plates were incubated on ice for 30 min and analyzed on a PolarStar Omega FP reader (BMG, Germany) using a 488-nm excitation filter and a 520-nm emission filter. Ki values for the peptide ligands were determined by fitting the binding curves to the equation, FP = FPf + ((FPb − FPf) × [Rt])/(Kd × (1 + X/Ki) + [Rt]), with FPf and FPb being the FP values of the free and bound peptide, respectively, [Rt] the concentration of PICK1 or mutant, and Kd the apparent dissociation constant for OG DAT C13. The apparent Kd was obtained as described before (32) from the saturation binding experiments. All peptides were purchased from Shafer, Copenhagen, Denmark.
NMR Spectroscopy
All samples were in TBS (50 mm Tris, 125 mm NaCl, 2 mm DTT, 0,01% Triton X-100), 0.01 mm NaN3 and 0.25 mm DSS, pH 7.4, containing either 10 or 100% D2O. NMR spectra were recorded at 15 °C on an 800 MHz Varian INOVA spectrometer with a cryoprobe. Backbone resonances of PICK1 PDZ-DAT and the PICK PDZ-GluA2 were assigned using 1H,15N HSQC, CBCANH, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO, and HN(CA)CO. Side chain resonances for PICK1 PDZ-DAT were assigned from 1H,13C HSQC and HCCH-TOCSY experiments. Distance restraints were obtained from15N NOESY-HSQC and 13C NOESY-HSQC experiments. Backbone resonances of PICK1 PDZ-ASIC1a were assigned from the same set of spectra as above recorded with a nonlinear sampling scheme (35) and reconstructed using compressed sensing (36) implemented in the qMDD mddNMR software (37). NMRpipe was used for Fourier transformation and phasing of all recorded spectra (38) and CCPNmr analysis for assignments (39).
Structural Determination of PICK1-DAT
Backbone ϕ/ψ torsion angle restraints were estimated from backbone 15N, 1H, 13Cα, 1Hα, 13Cβ, and 13C chemical shifts using the program DANGLE (40). The structure was calculated from 1296 NOEs and 66 chemical shift-based backbone dihedral angle restraints. Automated assignment of NOE restraints and structure calculations were carried out using ARIA2. From an ensemble of 250 structures in the final run, the 20 structures with the lowest energy were selected for refinement in explicit water. The 20 refined structures were analyzed with the iCING webserver.
STD-NMR
STD-NMR experiments were carried out at 5 °C in TBS buffer, pH 7.4, using full-length PICK1 and a C12 PKCα peptide. The concentration of full-length PICK1 was 50 μm and the C12 PKCα peptide (PQFVHPILQSAV) concentration was 4 mm yielding an 80-fold ligand excess. Selective saturation of the protein was achieved at −0.2 ppm by a train of 100 gaussian-shaped excitation pulses of 40 ms, each separated by 1 ms. Power of the selective pulses was set to 86 Hz. Off-resonance saturation was achieved at ∼30 ppm. Subtraction of the on- and off-resonance spectra was achieved by phase cycling. Two-dimensional STD homonuclear proton correlation experiments were all acquired with 32 scans and 512 increments in the 1H indirect dimension, and the mixing time was set to 65 ms. The STD amplification factor was calculated as ASTD = ISTD/I0, and normalized to the largest ASTD value.
Docking
High ambiguity driven biomolecular DOCKing (HADDOCK) (41) was used to dock ASIC1a and PKC onto the PICK1 PDZ domain. HADDOCK uses CNS (42) for structural calculations and uses automation derived from ARIA (43) The STD amplification factors were used as restraints for docking the PKC C11 peptide. For docking the ASIC1a C11 peptide, both the changes in backbone chemical shifts of the PICK1 PDZ domain relative to the complex with the DAT ligand and ASIC1a Ala scan (Table 2) were used as restraints. Residues in the PICK1 PDZ ASIC1a complex that exhibited HSQC shifts larger than the mean value (+σ) when compared with the PICK1 PDZ DAT complex (Fig. 2B) were defined as “ACTIVE” residues (Asn-31, Lys-32, Ile-33, Gly-34, Ile-35, Ile-37, Gly-38, Gly-39, Gly-40, Ala-58, Lys-79, Val-84, Ala-87, Met-89, Ile-90, Val-93, and Val-97), and residues Ala-41 to Tyr-48 were set as fully flexible in the first iteration. For the ASIC1a C11 peptide, active residues included Glu at position P−4, Phe at position P−2, and Cys at position P0 and passive residues included Asp at position P−3 and Thr at position P−1. For the PKCα peptide, ambiguous interaction restraints were generated using the STD amplification factor. His at position P−7, Pro at position P−6, Gln at position P−3, Ser at position P−2, and Val at position P0 residues were defined as ACTIVE. The interface for the interaction was defined by residues Leu-32, Ile-33, Gly-34, Ile-35, Ser-36, Ile-37, Gly-38, Leu-47, Tyr-48, Asp-68, Glu-69, Val-86, Ala-87, and Ile-90 in the PICK1 PDZ domain. Default parameters were used for the HADDOCK run. A total of 1000 initial docked structures were generated. The best 200 were refined using simulated annealing followed by water refinement. A minimum of 10 structures with a root mean square deviation below 7.5 Å was used for clustering.
TABLE 2.
Binding affinities of ligand peptides for class I or class II PICK1 mutants
Ki values for the listed C-terminal peptides were calculated from FP competition assays on purified full-length PICK1 WT or indicated mutants in solution using Oregon Green-labeled DAT C13 peptide as tracer (32). Data are average of six determinations (mean ± S.E. interval).
| Ligand peptide | WT Ki (μm), mean ± S.E. | K83H Ki (μm), mean ± S.E. | K83V Ki (μm), mean ± S.E. |
|---|---|---|---|
| PKCα | 13.1 [12.2; 14.1] | 0.22 [0.19; 0.25] | 3.6 [3.1; 4.1] |
| GluA2 | 6.8 [6.4; 7.2] | 69 [53; 89] | 4.9 [4.7; 5.1] |
| HER2 | 3.8 [3.3; 4.5] | >100 | 9.8 [9.0; 10.8] |
| ASIC1a | 7.1 [6.4; 7.9] | >100 | 20 [18; 23] |
| UNC5H1 | 4.2 [4.2; 4.3] | 23 [22; 25] | 12.5 [9.6; 16.3] |
| DAT | 0.5 [0.5; 0.6] | 12.8 [10.5; 15.6] | 0.28 [0.25; 0.31] |
| GLT1b | 5.3 [4.5; 6.4] | 44 [33; 59] | 6.5 [5.9; 7.0] |
FIGURE 2.
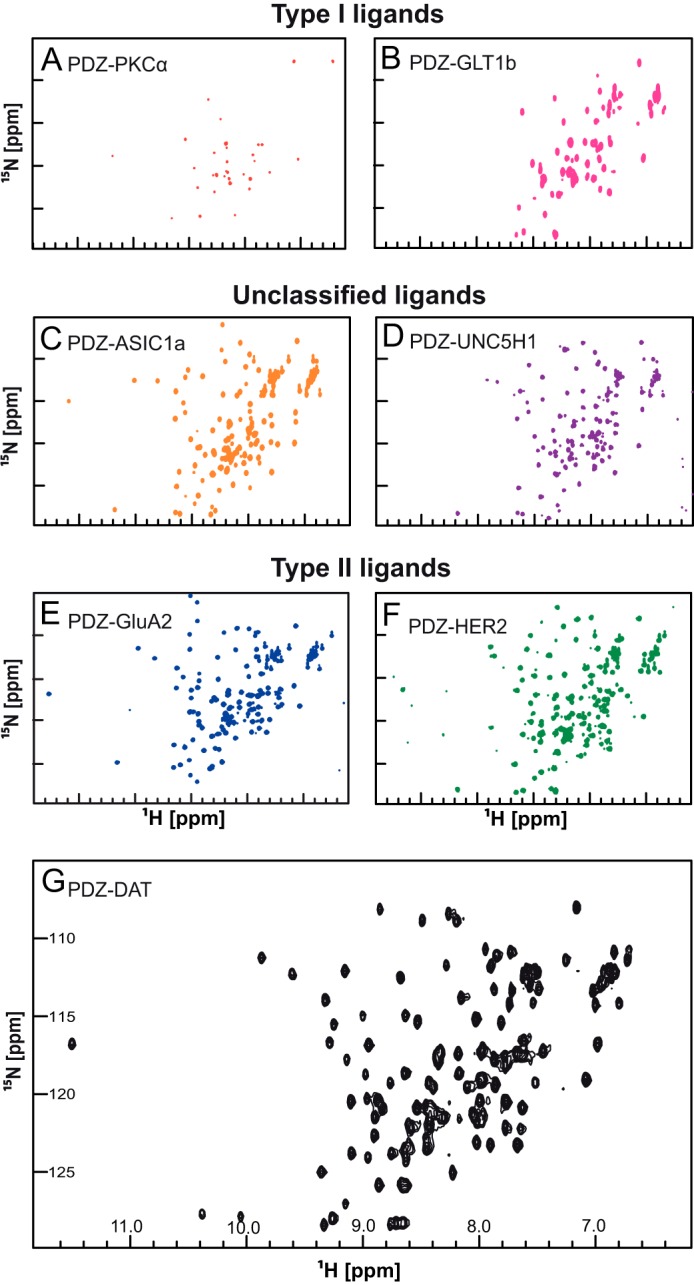
1H-15N HSQC NMR spectra of PICK1 PDZ ligand constructs. The 1H-15N-HSQC NMR experiment shows signals from all the backbone and side chain N-H correlations with 1H along the x axis and 15N along the y axis. Changes in the local environment surrounding a particular residue cause chemical shifts to deviate. Residues in a disordered protein have similar local environments causing the HSQC spectrum to exhibit low dispersion of chemical shifts as observed in this figure. Two class I ligand PDZ domain constructs, PKCα (A) and Glt1b (B) were bacterially expressed and purified. Both constructs showed low dispersion 1H-15N HSQC spectra indicating a low fraction of correctly folded PDZ domain protein. Aggregation was observed in both NMR samples. Fusion constructs with the C terminus of unclassified ligands, ASIC1a (C) and UNC5H1 (D), showed well dispersed HSQC spectra, although the ASIC1a HSQC spectrum was better resolved than the UNC5H1 HSQC spectrum. Three class II ligand, GluA2 (E), HER2 (F), and DAT (G), constructs showed well resolved HSQC spectra. All experiments were performed using 16 scans and 128 increments at 15 °C with an 800 MHz spectrometer.
Accession Numbers
The atomic coordinates for PICK1-DAT have been deposited at the Protein Data Bank (code 2LUI). Chemical shift assignments for PICK1 PDZ-DAT, PICK1 PDZ-GluA2, PICK1 PDZ-ASIC1a, and PKCα C-terminal have been deposited at the BioMagResBank (accessions numbers 18522, 19418, 19466, and 19467).
RESULTS
PICK1 PDZ Binds Peptides Representing Four Different Classes of PDZ Interactions
The list of PICK1 PDZ interaction partners is based on observations from yeast two-hybrid screens, GST pulldown assays, and co-immunoprecipitation studies (32, 44). Computational approaches have recently been used to predict binding energies (45), but otherwise there is little information about mechanism and affinity. We used our previously described FP binding assay (32) to determine the in vitro affinities of peptides corresponding to the C termini (11 last residues) of 15 selected interaction partners (Table 1). We also tested the putative class III ligand, protein kinase Mζ, (PKMζ). PICK1 bound all but one of the tested peptides with affinities in the lower micromolar range, supporting the remarkable promiscuity of PICK1 PDZ.
TABLE 1.
Binding affinities of PICK1 ligand peptide
The Ki values for the listed C-terminal peptides were calculated from FP competition assays on purified full-length PICK1 in solution using Oregon Green-labeled DAT C13 peptide as tracer (32). Data are average of 6–12 determinations (means ± S.E. interval).
| Ligand peptide | Sequence | Type | Ki (μm), mean ± S.E. |
|---|---|---|---|
| PKCα | QFVHPILQSAV | I | 14.1 [13.4; 14.9] |
| Aquaporin 2 | SPQSLPRGSKA | I | 90 [71; 115] |
| GLT1b | PFPFLDIETCI | I | 6.7 [6.3; 7.3] |
| GluA2 | YNVYGIESVKI | II | 7.2 [6.6; 7.7] |
| DAT | RQFTLRHWLKV | II | 0.8 [0.7; 0.9] |
| Ephrin B1 | PQSPANIYYKV | II | 9.4 [7.3; 12.2] |
| HER2 | NPEYLGLDVPV | II | 4.6 [4.1; 5.2] |
| Aquaporin 9 | NLEKHELSVIM | II | 12.2 [11.4; 13.1] |
| PKMζ | NPLLLSAEESV | III | 39 [30; 52] |
| mGluR7B | SVTWYTIPPTV | ? | 23 [19; 27] |
| AE1 | RDEYDEVAMPV | ? | 6.1 [5.3; 7.0] |
| Arf1 GTPase | DWLSNQLRNQK | ? | No affinity |
| ASIC1a | PARGTFEDFTC | ? | 6.3 [5.5; 7.2] |
| UNC5H1 | AGLFTVSEAEC | ? | 2.8 [2.5; 3.1] |
| NCS-1 | VQALSLYDGLV | ? | 21 [17; 25] |
We characterized further the seven ligands with highest affinity, representing class I (PKCα and GLT1b), II (GluA2, DAT, and HER2), and unclassified interactions (ASIC1a and UNC5H1). PICK1 is the only PDZ domain protein with a Lys (Lys-83) at the class-directing αB1 position. To assess whether this residue is important for the promiscuous PICK1 binding, we measured peptide affinities for two PICK1 mutants in which Lys-83 was mutated to either His or Val, the typical residues found in this position in class I and class II PDZ domains, respectively. As shown before (32), the affinity for the class I PKCα peptide was dramatically increased for K83H. All other peptides showed markedly decreased affinities for this mutant (Table 2 and Fig. 1). Thus, the PICK1 PDZ domain loses binding promiscuity when changed to a class I-like PDZ domain. Interestingly, K83V showed binding specificity similar to wild type (WT), although the observed affinities for the class II and unclassified ligand were more dispersed with the lowest affinities for the unclassified ligands (Table 2 and Fig. 1), demonstrating that Lys-83 allows greater flexibility for hydrophobic insertion in the S−2 pocket than Val. Surprisingly, the class I GLT1b peptide also showed preference for K83V compared with K83H (Table 2 and Fig. 1), suggesting that GLT1b does not bind as a typical class I ligand but is dependent on hydrophobic interactions in the S−2 pocket. Altogether, the data suggest that, in addition to a classical class II binding mode with promiscuity related to Lys-83 (33, 34), PICK1 PDZ has alternative binding modes not directly related to the αB1 residue.
FIGURE 1.
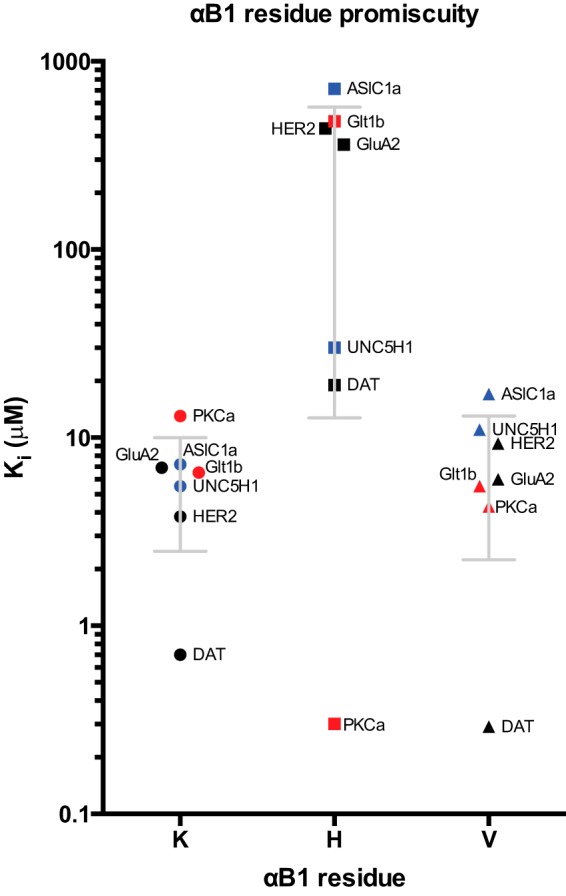
Affinity scatter profile (Ki values) of class-specific mutations in the αB1 position of the PICK1 PDZ domain. The effect of different residue in the αB1 position (x axis) was probed in a competition FP assay. Calculated Ki values are shown on the y- axis. For WT and K83V, OG DAT C13 is used as tracer. For K83H mutant, OG PKCα C13 is used as tracer. The K83H mutant disrupted the ligand binding promiscuity and switched the preference to class I ligands. The class II K83V mutant showed an almost preserved binding pattern, although the observed affinities for the class II and unclassified ligand were more dispersed with the lowest affinities for the unclassified ligands. Class I, II, and unclassified ligand peptides are indicated by red, black, and blue, respectively. See also Table 1.
PICK1 PDZ Forms Stable Structures Only with Class II Ligands
To understand the binding modes in atomic detail, we analyzed by NMR spectroscopy seven constructs of PICK1 PDZ with C-terminal extensions corresponding to the 10 C-terminal residues of the ligands PKCα, GluA2, HER2, ASIC1a, UNC5H1, DAT, and GLT1b. Without a ligand fused to the C terminus, PICK1 PDZ is unstable and rapidly degrades, as noted by others (34). Monomeric and homogeneous 15N-labeled samples of all seven constructs were prepared, and 1H,15N HSQC spectra of all the constructs were recorded. Although the peptides bind PICK1 PDZ with affinities in the same range (Table 1), the quality of the spectra varied considerably (Fig. 2). The class I PKCα and GLTb1 constructs showed spectra of low quality with very few peaks. The absence of peaks suggests that PICK1 PDZ, when bound to these ligands, is structurally dynamic and therefore not suited for NMR structure determination. The remaining constructs with class II and unclassified ligands all resulted in well resolved 1H,15N HSQC NMR spectra characteristic of folded proteins, including those with ligands showing affinities similar to that of GLT1b (e.g. HER2 and GluA2) (Fig. 2). Thus, class II and also to some extent the unclassified ligands form more rigid complexes with the PDZ domain than the class I ligands despite similar affinities.
Structural Flexibility of the Hydrophobic Pocket S−2 Facilitates Optimal Packing of Leu in Position P−2
The C terminus of DAT is the ligand with highest affinity for PICK1, i.e. ∼10-fold higher than for the other class II ligands, EphrinB1, and GluA2, for which high resolution structures in complex with PICK1 PDZ exist (33, 34). To understand the molecular details underlying the high affinity of DAT, we used NMR to determine the solution structure of the PICK1 PDZ-DAT complex (for details see “Experimental Procedures” and Table 3). Like the structures with the EphrinB1 and GluA2 ligands, the structure of PICK1 PDZ-DAT overall resembles a canonical PDZ domain, forming a partially opened β-barrel capped by two α-helices (Fig. 3, A and B). The peptide-binding groove of the domain consists of the two connected hydrophobic pockets, S0 and S−2, between the αB helix and the βB strand. The positions of the residues that form hydrophobic pocket S0 are identical in all three structures (Fig. 3C) (33, 34). However, the packing of the DAT ligand in the binding groove is more compact than for GluA2 and EphrinB1 (33, 34). Val at position P0 (as in DAT and EphrinB1) allows the ligand to dock deeper into the binding groove. Binding of Tyr at P−2 (as in EphrinB1), which is large and bulky, is only allowed because the relatively small Val at P0 residue does not restrict the S−2 pocket as much as Ile or Leu would. In agreement, we have previously demonstrated that the size of the hydrophobic residue in P0 is important for the affinity with the highest affinity seen for Val as compared with Ile or Leu (32). Comparison with the two previous structures shows that the hydrophobic pocket S−2 varies both in size and shape when interacting with the different ligands, indicating that the binding pocket allows binding of a variety of hydrophobic residues at position P−2. The long straight hydrophobic side chain of the Leu at P−2 in DAT is thus positioned deeply into pocket S−2 by slightly displacing Ile-37 relative to its position in the structures with GluA2 and EphrinB1. At the same time, the side chain of Lys-83, of which the aliphatic part stabilizes hydrophobic pocket S−2, is displaced ∼3.5 Å out of the binding groove in the DAT structure while maintaining the hydrogen bond from the amino group to the carbonyl of P−4. This structural rearrangement of the hydrophobic pocket S−2, and in particular Lys-83, allows a tight packing of the DAT ligand in a more elongated conformation relative to the published class II ligands where the residues at P−2 are not intercalating with the side chains from PICK1 (Fig. 3, D–G). Of note, HSQC spectra before and after proteolytic cleavage of the linker between PICK1 and the DAT sequence only showed minor differences for residues next to the cleavage site, showing that the linker between PICK1 PDZ domain and the DAT peptide did not change the structure (data not shown). In summary, the data suggest that the DAT peptide has a near optimal sequence for binding PICK1 via a class II binding mode and that PICK1 PDZ promiscuity among class II ligands is primarily accounted for by Ile-37 and Lys-83, which modulate the hydrophobic pocket S−2.
TABLE 3.
Structural statistics for the family of 20 NMR structures of the PICK1 PDZ domain in complex with the DAT C terminus
None of the structures exhibits distance violations >0.3 Å or dihedral angle violations >4°.
| Statistics | |
|---|---|
| Distance restraints | |
| Intraresidual (|i − j| = 0) | 614 |
| Sequential (|i − j| = 1) | 316 |
| Medium range (2 ≤|i − j| ≤ 4) | 158 |
| Long (|i − j |>5) | 198 |
| Hydrogen bonds | 38 |
| Total | 1324 |
| Dihedral angle restraints | |
| PICK1 PDZ domain | 61 |
| DAT (-HWLKV) | 5 |
| Total | 66 |
| Mean r.m.s.d. from the experimental restraints | |
| Distance, Å | 0.020 ± 0.002 |
| PICK1 PDZ domain dihedral, ° | 0.284 ± 0.099 |
| DAT (-HWLKV) dihedral, ° | 0.511 ± 1.000 |
| Ramachandran plot (ranges 19–37, 49–103, 126–128)a | |
| Core | 75.8% |
| Allowed | 21.5% |
| Generous | 1.3% |
| Disallowed | 1.4% |
| Atomic root mean square differences, Åb | |
| Backbone heavy atoms (N, Ca, and C′) | 0.58 ± 0.11 |
| Heavy atoms | 1.12 ± 0.18 |
a The iCING webserver program was used to assess the overall quality of the structures.
b Residues 19–37,49–103 of the PICK1 PDZ domain were used for superposition analysis.
FIGURE 3.
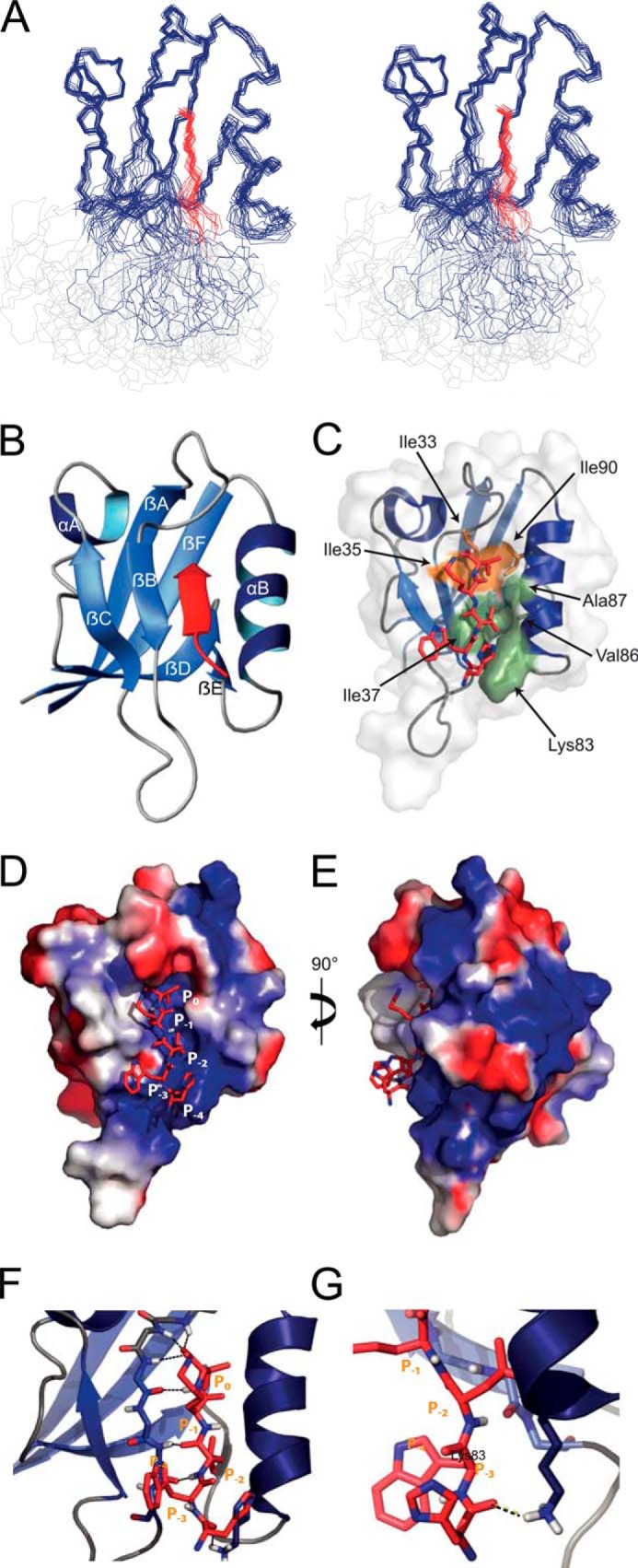
NMR solution structure of the PICK1 PDZ domain in complex with the DAT C terminus. A, stereo view of the 20 lowest energy structures. The PDZ domain is in blue, the DAT peptide in red, and the linker region in light gray. B, closest-to-mean structure of the domain without the linker region. The DAT C5 peptide lining the PDZ-binding groove as an antiparallel β-strand is displayed in red. Secondary structure was predicted using the program DSSP (Define Secondary Structure of Proteins), and secondary structure elements are numbered α(A–B) and β(A–F). C, solvent accessible surface of hydrophobic pocket S0 involving the residues Leu-32, Ile-35, Ile-33, and Ile-90 in the PDZ domain is in olive and solvent-accessible surface of hydrophobic pocket S−2 involving the residues Ile-37, Lys-83, Val-86, and Ala-87 is in orange. C–E, electrostatic surface view of the PICK1 PDZ domain bound to the DAT C5 (shown as red sticks). The PDZ domain is colored with the electrostatic potential from −2 kT/e (blue) to 2 kT/e (red). F, βB strand lines the binding groove and makes backbone interactions with the DAT C terminus. The hydrogen bonds are indicated with dashed black lines. The βA-βB loop binds tightly to the carboxyl of the ligand Val P0 and forms the head of the binding groove. G, interaction between the Lys-83 side chain of the PDZ domain and the carbonyl group of the P-4 His of the DAT ligand stabilizes the interaction.
Evidence for a Dual Noncanonical Binding Mode for ASIC1a
The ASIC1a peptide has an affinity comparable with the canonical class II ligands (e.g. GluA2 and EphrinB1), although it has noncanonical residues at P−0 (Cys) and at P−2 (Phe). Based on this notion and the high quality HSQC spectra obtained from the PICK1 PDZ-ASIC1a, we attempted to resolve its solution structure. Unfortunately, the NMR sample of PICK1 PDZ-ASIC1a was not stable long enough to allow NOE-based structure determination. The spectrum shown in Fig. 2 was based on a freshly prepared sample; however, already after a few hours the spectrum deteriorated making it impossible to obtain high resolution structural information (data not shown). However, using nonlinear sampling of the NMR data (37), the experiment time was significantly reduced, and a set of spectra could be recorded that allowed full backbone resonance assignments of PICK1 PDZ-ASIC1a.
The structure of PICK1 PDZ-ASIC1a, relative to the structures of PICK1 PDZ-DAT and PICK1 PDZ-GluA2, was assessed from the changes in the backbone chemical shifts. The changes in 1HN and 15NH chemical shifts between PICK1 PDZ-DAT and PICK1 PDZ-GluA2 are small and below 0.17 ppm (Fig. 4A). The largest differences were observed for residues in the PDZ groove interacting with the ligand or for adjacent residues (Fig. 4A). This observation is consistent with the different ligand peptide sequences and the subtle differences in the resolved structures of PICK1 PDZ with the two ligands (Fig. 3) (34). In contrast, much larger differences were observed in the amide chemical shifts between PICK1 PDZ-DAT and PICK1 PDZ-ASIC1a (Fig. 4B). Notably, residues Ile-33 and Gly-34, which are situated in the carboxylate-binding loop and constitute part of hydrophobic pocket S0, as well as Ile-37, Gly-40, and Ala-87 in pocket S−2, displayed significant chemical shift changes. The largest difference was 0.76 ppm observed for Ile-37 that forms a backbone hydrogen bond to the ligand residue P−2 in both PICK1 PDZ-DAT and PICK1 PDZ-GluA2. The results suggest that the interactions in both hydrophobic pockets of PICK1 PDZ-ASIC1a are altered compared with the interactions of PICK1 PDZ with class II ligands. In addition, the residues Asn-31, Leu-32, Ala-58, Val-93, Lys-94, Gly-95, and Val-97, which are outside the binding groove and thus not in direct contact with DAT and GluA2, have nearly the same chemical shifts in PICK1 PDZ-DAT and PICK1 PDZ-GluA2. In PICK1 PDZ-ASIC1a, however, they have markedly changed chemical shifts (Fig. 4B). These residues are all clustered adjacent to the loop that binds the C-terminal carboxylate from the ligands in the structures of PICK1 PDZ-DAT and PICK1 PDZ-GluA2.
FIGURE 4.

Ligand-dependent differences in chemical shifts for the PICK1 PDZ domain. The histograms show weighted differences in backbone NH chemical shifts between the DAT construct and the GluA2 construct (A), and the DAT construct and the ASIC1a construct (B), and ASIC1a major and minor state (C). All spectra were recorded at 15 °C using the same experimental settings. The differences in backbone NH chemical shifts between the five C-terminal ASIC residues in the major and the minor state are shown in D, as well as a secondary chemical shift analysis of each state in E and F. 15N-filtered NOEs obtained for the Cys at P0 in ASIC1a in both states are compared with corresponding NOEs obtained for Val in the DAT C terminus used for structural determination (G). Docking models of the ASIC1a C11 peptide onto the PICK1-PDZ domain based on the chemical shift differences in B and C yielded two clusters using the HADDOCK protocol suggesting that the ASIC1a peptide adopts two different binding conformations as follows: a class II-like insertion (H) and an insertion where the ligand is shifted two residues upward (I). ASIC C11 is shown as red sticks, and the PDZ domain is colored with the electrostatic potential from −2 kT/e (blue) to 2 kT/e (red).
Of further interest, double peaks were observed for ligand residues at P−0, P−1, P−2, and P−4 and for several PDZ domain residues involved in the binding of ASIC1a, indicating more than one conformation of the complex. We compared the differences in backbone amide chemical shifts for the two distinct states observed for the residues constituting the PDZ domain (Fig. 4C). In addition, we compared the ligand backbone amide chemical shifts for the two states, denoted major and minor state (Fig. 4D). Furthermore, we compared both the major and the minor state chemical shifts to random coil chemical shifts (Fig. 4, E and F). From these analyses, it was clear that the major state resembled a typical class II binding mode with the C terminus being more extended than in the observed minor state. Equally important, the analyses show that the secondary structure of the minor state is different from that of the major state but still nonrandom. Finally, we were able to get decent NOEs from the Cys P0 of each state using nonlinear sampling (Fig. 4G). Again, it was clear that the NOE pattern for the major state resembled what is observed for Val P0 in the DAT C terminus, whereas the minor state seems to have a completely different NOE pattern. Unfortunately, because of the instability of the complex, we were unable to obtain a decent NOE signal from Phe at P−2. Taken together, our data strongly support the existence of two different binding modes of the ASIC1a ligand.
To characterize the molecular details of the interactions between the PICK1 PDZ and ASIC1a, we modeled the PICK1 PDZ-ASIC1a complex by using the NMR structure of PICK1 PDZ-DAT as template and the HADDOCK docking protocol, which allows docking of ligands on a resolved protein structure using perturbations of the amide chemical shifts from the HSQC spectrum as ambiguous restraints (41). Two different plausible insertion modes for the ligand peptide were observed. This agreed with the additional peaks observed for the ligand and the involved domain residues surrounding P−0, corresponding to two exchanging states that are 60 and 40% populated, respectively, based on their relative intensities in the HSQC. We should note that this distribution could be different for the free ligand as compared with the construct used where the ligand is covalently fused to the C terminus of the PDZ domain. In one of the binding modes, ASIC1a is inserted like a class II ligand with Phe P−2 docked in hydrophobic binding pocket S−2 (Fig. 4H). The other model adopts a structure with Phe P−2 docked in pocket S−0 and the residues in P−1 and P0 interacting with the domain outside the canonical PDZ pocket (Fig. 4I). In addition to the chemical shift changes observed for residues outside the binding groove, this alternative binding mode is supported by the observation that extending the ASIC1a C5 peptide with a single C-terminal Ala only weakened the affinity ∼2-fold (Table 4). This is in contrast to the DAT and GLT1b ligands where a C-terminal extension abolishes binding (26, 28). An Ala scan of the ASIC1a C5 peptide further showed that substitution of Cys in P0 reduced the affinity around 4-fold, supporting the importance of this residue for ASIC1a binding. Substituting P−1 and P−4 did not alter the affinity of the peptide, whereas a 4-fold increase in the affinity for PICK1 was seen when Phe in P−2 was substituted with Ala (Table 4).
TABLE 4.
Binding affinities for ASIC1a Ala scan
Ki values for the listed pentameric peptides were calculated from FP competition assays on purified full-length PICK1 WT in solution using Oregon Green-labeled DAT C13 peptide as tracer (32). Data are average of six determinations (means ± S.E. interval). A indicates where an amino acid residue has been substituted with Ala relative to the wild-type (control) sequence.
| ASIC1a C5 Ala scan | Ki (μm), mean ± S.E. |
|---|---|
| EDFTC (control) | 21 [20; 23] |
| ADFTC | 27 [21; 33] |
| EAFTC | 11 [9.0; 13] |
| EDATC | 6.6 [5.6; 7.9] |
| EDFAC | 20 [17; 22] |
| EDFTA | 93 [82; 107] |
| EDFTC+A | 42 [32; 57] |
Upstream Binding Motifs Are Necessary for the Binding of the Class I Ligand PKCα
We showed previously that residues upstream of the canonical PDZ-binding sequence in GLT1b contribute to the affinity for PICK1 PDZ (28). To test whether this is also the case for other PICK1 PDZ ligands, we investigated the binding affinities of short C5 peptides derived from the termini of the interaction partners described above. All seven C5 peptides displayed decreased affinity compared with the corresponding C11 peptide, although the decrease was smaller for the class II ligands GluA2 and DAT and for ASIC1a and UNC5H1 (Table 5). In contrast, the class I PKCα peptide had dramatically decreased affinity (Table 5), indicating that this ligand has an additional upstream binding motif. None of the tested PICK1 PDZ class I complexes resulted in NMR spectra of suitable quality for chemical shift assignments (Fig. 2). Consequently, we used STD-NMR spectroscopy. The concept of STD-NMR is to saturate all spins in a large protein and measure how this saturation is transferred to a small ligand present in great excess. We characterized the atoms on a C12 PKCα peptide that interacts with the PDZ domain of purified full-length WT PICK1 protein. From two-dimensional TOCSY spectra, we resolved all the 1H resonances from the PKCα peptide and calculated the STD amplification factors (Fig. 5A). High STD amplification factors were observed for residues at P−0 and P−2, which indicates that these residues contribute significantly to peptide binding. We also observed strong saturation of the side chain amide of Gln at P−3 demonstrating that this residue could also interact with the PDZ domain. Moreover, our STD results show that the Hβ protons of Pro P−6, Hβ, Hδ, and Hϵ protons of His P−7, as well as the Hβ protons of Phe P−9 contribute to the binding affinity of PKCα. This suggests binding outside the actual binding cleft consistent with the FP binding data (Table 5). To validate that these two residues do in fact constitute an upstream binding motif, we generated C-terminal PKCα C11 peptides where Pro P−6 or His P−7 was substituted with alanines. In full agreement with these residues being important for PKCα binding to PICK1 PDZ, the FP binding data revealed a marked decrease in binding affinity for both peptides with the largest effect seen for substitution of His P−7 (Table 6).
TABLE 5.
Binding affinities for C5 ligand peptides
Ki values for the listed pentameric peptides were calculated from FP competition assays on purified full-length PICK1 WT in solution using Oregon Green-labeled DAT C13 peptide as tracer (32). Data are average of nine determinations (means ± S.E. interval).
| Ligand peptide | C5 sequence | Ki (μm), mean ± S.E. | Difference from C11 |
|---|---|---|---|
| PKCα | LQSAV | >100 | − >10 |
| GluA2 | ESVKI | 13.3 [12.4; 14.2] | − 1.8 |
| HER2 | LDVPV | 28 [27; 29] | − 6.1 |
| ASIC1a | EDFTC | 11.7 [11.0; 12.4] | − 1.9 |
| UNC5H1 | SEAEC | 6.5 [6.1; 7.1] | − 2.3 |
| DAT | HWLKV | 1.8 [1.4; 2.3] | − 2.3 |
| GLT1b | IETCI | 28 [26; 29] | − 4.2 |
FIGURE 5.

Binding of PKCα to PICK1. A, normalized STD amplification factor (ASTD) measured for the 1H resonances in the PKCα C12 peptide. B, HADDOCK docking model of the PKCα C12 peptide based on the amplification factors in the STD experiment. In the single cluster of structures obtained from the calculation, the C terminus of PKCα binds PICK1 in the canonical binding mode, which is further stabilized by an upstream binding site involving residues located in the Cys-44–Pro-45–Cys-46 βB-βC loop and ligand residues His P−7 and Pro P−6. The PDZ domain is colored with the electrostatic potential from −2 kT/e (blue) to 2 kT/e (red). C, highlight of upstream binding contacts.
TABLE 6.
Binding affinities for PKCα mutated peptides
Ki values for the listed peptides were calculated from FP competition assays on purified full-length PICK1 WT in solution using Oregon Green-labeled DAT C13 peptide as tracer (32). Data are averages of six determinations (means ± S.E. interval).
| Ligand peptide | Sequence | Ki (μm), mean ± S.E. |
|---|---|---|
| PKCα | QFVHPILQSAV | 14.1 [13.4; 14.9]a |
| PKCα Pro P−6 → Ala | QFVHAILQSAV | 243 [186; 317] |
| PKCα His P−7 → Ala | QFVAPILQSAV | 842 [594; 1194] |
a These are the same values as in Table 1.
We then used the STD-derived restraints to model the PKCα peptide insertion into PICK1 PDZ. The calculation returned one single low energy cluster, in which the PKCα peptide binds the PICK1 PDZ using both the C-terminal residues forming interactions within the PDZ-binding groove and the residues Pro P−6 and His P−7 forming interactions with Gln-42 and Tyr-43 located immediately before the Cys-44–Pro-45–Cys-46 motif in the βB-βC loop (Fig. 5, B and C). To obtain support that Tyr-43 is involved in this upstream binding, we mutated Tyr-43 in PICK1 to alanine and serine (Y43A and Y43S). Unfortunately, we were unable to detect any binding using the FP assay to Y43A, suggesting an overall perturbation of the domain by this mutation (data not shown). However, high affinity DAT binding was preserved in Y43S according to an FP saturation assay using OG DAT C13 (Kd = 0.34 (0.26; 0.44) μm, mean (S.E. interval), n = 6). In contrast and in support of the importance of Tyr-43 for PKCα binding, we observed no evidence for binding of the PKCα peptide to the Y43S mutation as it was unable to compete for binding of OG DAT C13 (Ki >100 μm, n = 6).
DISCUSSION
PDZ domain-mediated protein-protein interactions are critical for multiple cellular functions and represent an attractive but largely unexplored class of targets for putative pharmacological intervention (46–48). Yet the molecular details that govern specificity of PDZ domain interactions are not fully understood. Here, we provide a likely structural explanation for how a promiscuous PDZ domain, the PDZ domain of the scaffolding protein PICK1, is capable of recognizing structurally very diverse ligands while at the same time efficiently excluding binding partners for other PDZ domains.
Our data suggest three discriminating binding modes that can account for the PICK1 PDZ promiscuity (Fig. 6). One mode is unique to PICK1 PDZ and depends on flexibility of the Lys-83 in position αB1 that permits promiscuity for canonical class II ligands. Overall, the binding mode of canonical ligands relies on the hydrophobic interactions constituted by residues in the two hydrophobic binding pockets (Fig. 6A). Whereas the hydrophobic S0 pocket is required for all classes of ligands, the hydrophobic pocket S−2 is especially important for canonical class II ligands. Hydrophobic packing within the two pockets is required for formation of hydrogen bonds involving the Ile-33–Gly-34–Ile-35 loop and the βB3 Ile-37, of which the latter binds the P−2 backbone amine of class II ligands. Notably, class II peptides showed preserved or increased affinity for the K83V mutant consistent with the involvement of the aliphatic region of Lys-83 at position αB1 in this binding mode. This indicates that Lys-83 predominantly mimics the binding pattern of a class II domain; however, Lys-83 also adds greater tolerance for different hydrophobic residues in P−2 through its ability to move the aliphatic chain out of the binding pocket. Furthermore, Lys-83 creates an electrostatically charged environment, which allows both the interaction with P−2 but also the P−4 backbone carbonyl of high affinity ligands.
FIGURE 6.

Binding modes of the PICK1 PDZ domain. A, the PICK1 PDZ domain has the highest affinity for class II ligands that is primarily mediated by hydrophobic pocket S0 and S−2 (orange regions). Both patches contribute equally to the affinity. B, class II-like ligands containing a C-terminal Cys may bind similarly to regular class II ligands with the Cys binding in hydrophobic pocket S0. However, we also observed an alternative binding mode in which the hydrophobic P−2 residue binds in hydrophobic pocket S0 and with either hydrophobic or negatively charged residues (shaded red/orange) mimicking in the hydrophobic pocket S−2. The additional upper binding site, situated outside the defined PDZ binding groove and binding the residues in P1 and P0 of the ligand, is slightly hydrophobic (shaded orange). C, class I ligands make hydrophobic contacts in the hydrophobic pocket S0 (orange); Lys-83 contributes to an electrostatic favorable environment for Ser/Thr residues at P−2 of class I ligands (shaded red). Furthermore, an additional upstream binding site preferring positively charged and/or bulky hydrophobic residues resides in the flexible loop region (shaded blue/orange).
A second discriminating binding mode was strongly supported by backbone chemical shift analysis, comparing the ASIC1a construct and the DAT construct. Our data suggested that the noncanonical ASIC1a peptide, in addition to a class II-like insertion mode, was likely to have a different noncanonical, internal insertion mode, which relies on interactions outside the defined PDZ-binding groove. Our models indicated that in this mode, the hydrophobic Phe residue at P−2 is packed into hydrophobic pocket S0 (Fig. 6B). This large residue is presumably not very favorable in hydrophobic pocket S−2 when optimal packing in hydrophobic pocket S0 is not achieved. Non-C-terminal internal interactions have only been observed in few cases, including the PDZ domain of the cell polarity protein Par-6 that binds an internal sequence of Pals1, the C-terminal extension of neuronal NOS PDZ forming a β-hairpin when bound to syntrophin PDZ, and Dishevelled that was suggested to bind an internal sequence of Frizzled (49–51). Interestingly, a phenylalanine is also situated in hydrophobic pocket S0 in the neuronal NOS-syntrophin complex (50).
The existence of the noncanonical internal insertion mode for ASIC1a was supported by FP binding analysis of ASIC1a C5 mutant peptides. Importantly, extending the peptide with a C-terminal Ala was well tolerated in contrast to our findings for the class II ligands such as DAT (32), indicating that an upward-shifted binding mode for ASIC1a is plausible. We find it unlikely that Cys P0 in the extended peptide still occupy the S0 pocket because this would prevent the C-terminal carboxyl group from binding the carboxyl-binding loop, which predictably would be required to maintain affinity in the classical class II ligand binding mode (32). Our analysis of ASIC1a C5 mutant peptides also showed that substituting Phe P−2 for Ala increased the binding affinity 4-fold (Table 4). We speculate that this Ala- substitution shifts the binding mode to a canonical class II interaction with restored backbone interaction between P−2 and Ile-37 in PICK1 PDZ, resulting in an affinity similar to that of UNC5H1 (terminating in -AEC) (Table 1). It is interesting to note that the small ubiquitin-like modifier SUMO-1 was shown to bind the C terminus of the scaffolding protein DAXX in both a parallel and an antiparallel conformation (52); nevertheless, our results for ASIC1a binding to the PDZ domain of PICK1 are, to our knowledge, the first evidence for a possible dual ligand binding mode for a PDZ domain target protein.
It is interesting to compare our ASIC1a data with a recent study suggesting that residues not directly involved in binding contribute to the ligand affinity through an evolutionary conserved sector of 20 residues, which are situated both inside and outside the binding pocket (6). When comparing ASIC1a and DAT binding to PICK1 PDZ, we found that 9 out of the 17 residues with the largest chemical shift changes (top quartile) were included in this 20-residue sector defined for PSD-95 PDZ3. Thus, our results are consistent with the existence of such sectors. Our data also suggest the presence of a second sector of residues, including Asn-31, Leu-32, Val-93, and Val-97, that all are involved in ASIC1a binding. In summary, 14 out of 17 residues in the top quartile can be assigned to two distinct sectors.
The existence of two such networks is also observed when comparing rotameric states and dynamics in the free and bound state of the second PDZ domain of tyrosine phosphatase 1E (53). Comparing these structural networks to residues found in PICK1 PDZ upon binding of two different ligands, the residues in the binding pocket are completely overlapping, and it is clear that a highly conserved structural network can also be identified from our backbone chemical shift differences. However, the second sector that we identify still seems to involve residues that are not considered part of this proposed network, which might be due to the fact that the dual binding mode is not a feature of the tyrosine phosphatase PDZ domain. Finally, we should note that it recently was suggested that several energetic pathways exist in any given PDZ domain and that these pathways may be specifically selected by distinct peptide ligands; hence, selectivity might result from energetic pathway sampling (54).
A third discriminating binding mode for PICK1 PDZ was supported by STD NMR together with FP binding data and mutational analyses. The data suggested that PICK1 PDZ is able to bind PKCα via an upstream binding motif. This mechanism might be particularly important for class I interactions (Fig. 5C). The binding appeared to involve the residues Pro P−6 and His P−7, which showed significant saturation in the STD spectrum. Indeed, previous studies have shown that some class I PDZ domains interact with up to seven residues of the ligand C terminus, which suggests that additional residues outside the defined PDZ-binding groove participate in ligand binding (8, 55). A recent study of the class I PDZ domain of microtubule-associated serine/threonine-protein kinase 2 (MAST2) in complex with the C terminus of phosphatase and tensin homolog also identified an upstream PDZ-binding motif at P−11 (56). Similar to PICK1, MAST2 has a large flexible βB-βC loop region in the PDZ domain, where a highly conserved Trp interacts with the upstream motifs of the ligand. Other studies have also reported upstream binding motifs in the βB-βC loop. For PSD-95 PDZ3, DLG PDZ2, and Par3 PDZ3, it was found that upstream peptide binding is mediated primarily by ionic interactions, although the specific residues interacting seem mostly dependent on the size and composition of the βB-βC loop (57, 58).
Thus, upstream binding facilitated by hydrophobic and/or charged residues might be a common feature for class I binding. Importantly, we note that the three ligands, which lose more than 5-fold affinity upon truncation of the upstream part of the peptides, all have bulky hydrophobic (His, Trp, Tyr, and Phe) residues in the P−7 position, and 8 of the 11 ligands with affinities better than 20 μm have bulky hydrophobic residues in the P−7 position (Table 1), suggesting that these residues should be considered part of the consensus motif for binding to PICK1 PDZ. Furthermore, we note that PICK1 PDZ domain extensions can participate directly in the binding or favor a particular energetic pathway, as it has previously been found for several other PDZ domains (3, 59–61).
We finally considered the binding specificity of PICK1 PDZ in an evolutionary context. The evolution of residues critical for the three binding modes, as well as the evolution of the ligands with affinities >20 μm, are summarized in Tables 7 and 8. In PICK1 PDZ, Lys-83 αB1, which plays a key role in pocket S−2 flexibility, is fully conserved in all investigated species. Correspondingly, the canonical class II ligands (DAT (-LKV), ephrin B (-YKV), and GluA2 (-LVI)) are conserved in all vertebrates (Tables 7 and 8). The extreme C-terminal PDZ-binding sequence of DAT also seems to be fully conserved suggesting that canonical class II ligands might be “old” ligands obtaining their binding affinity for PICK1 PDZ early in evolution. The “second sector,” sensitive to the dual insertion mode seen for ASICa1 and encompassing Asn-31, Leu-32, Val-93, and Val-97, is conserved only in vertebrates, and correspondingly, the binding sequences of ASIC1a and UNC5H1, which presumably use a similar mode of insertion, appear in amphibians and birds, respectively, indicating that these interactions have appeared later in evolution than class II interactions. Finally, the upstream binding motif, involving Gln-42 and Tyr-43, is likewise only conserved in vertebrates. This is in accordance with absence of the bulky P−7 hydrophobic residue in DAT, the only ligand preserved in invertebrates. In contrast, all class II-like ligands with prolines in their binding sequence (HER, -VPV, AE1, -MPV, and -PTV), as well as the class I ligands GLT1b and PKCα, all of which appear in vertebrates from amphibians and so forth, have bulky hydrophobic residues in the P−7 position (Tables 7 and 8).
TABLE 7.
Evolutionary conservation of residues involved in PICK1 promiscuity
The evolution of the residues/sequences were traced using UniGene. αB1 refers to the first position in the αB helix; second sector refers to residues Asn-31, Leu-32, Val-93, and Val-97 involved in ASIC1a binding, and upstream binding motif refers to the motif that binds residues situated upstream from the canonical C-terminal PDZ-binding sequence in the ligand. Boldface letters indicate residues of key importance for ligand binding and specificity.
| PICK1 PDZ |
|||
|---|---|---|---|
| αB1 | Second sector | Upstream binding motif | |
| Vertebrates | |||
| Homo sapiens | Lys | NL-IGI -VKGEV | QY-CPC |
| Rattus norvegicus | Lys | NL-IGI -VKGEV | QY-CPC |
| Gallus gallus | Lys | NL-IGI -VKGEV | QY-CPC |
| Xenopus laevis | Lys | NV-IGI -VKGEV | QH-CPC |
| Danio rerio | Lys | NL-IGI -VQGEV | QF-CPC |
| Arthropods | |||
| Drosophila melanogaster | Lys | NL-IGI -ATDEV | PM-CPC |
| Nematodes | |||
| Caenorhabditis elegans | Lys | GV-IGI -SLNPV | PY-CPC |
| Molluscs | |||
| Aplysia california | Lys | NL-IGI -IKAEV | PL-CPC |
TABLE 8.
C-terminal sequences interacting with PICK1
Sequences were traced using UniGene. In some cases the splice variants or related subunits can be traced further back than indicated but without homology in the C terminus. Bold black indicates class II PDZ-binding sequence; bold red indicates class I PDZ-binding sequence; bold blue indicates atypical binding sequence involved in dual binding mode in PICK1; bold green indicates bulky hydrophobic residue in the P−7 position; gray shading indicates predicted binding to mammalian PICK1.
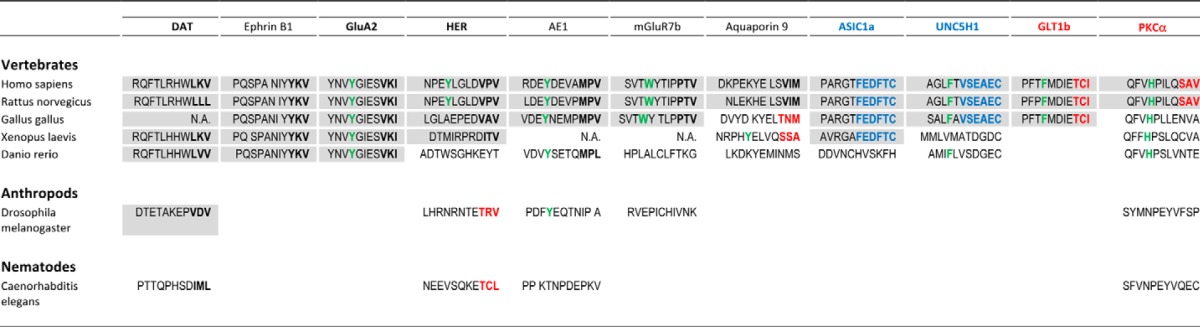
The appearance of the bulky hydrophobic residue in P−7 of AE1 and PKCα precedes the appearance of the PDZ-binding sequence, and in all other cases the appearance coincides. Conversely, a bulky hydrophobic residue is introduced in P−7 of HER in mammals, conceivably to compensate for the Pro in P−1 (−VPV), which is likely to compromise the PDZ-binding sequence compared with the sequence in birds (−VAV) and in amphibians (−ITV) (Tables 7 and 8). This suggests that the appearance of the upstream binding motif in PICK1 in vertebrates has allowed the protein to achieve physiologically relevant interactions with proteins having suboptimal PDZ-binding motifs. Altogether, we speculate that PICK1 PDZ originally evolved to bind canonical class II sequences and that later in evolution other ligands have adapted to bind PICK1 PDZ for specialized functions as well in vertebrates, either by acquiring alternative noncanonical binding modes, as seen for ASIC1a, or by acquiring novel interactions involving residues upstream from the PDZ-binding sequence, as seen for PKCα.
In summary, we provide evidence for three distinct molecular binding modes that can account for the complex binding specificity of the PICK1 PDZ domain. The findings therefore support a remarkably versatile mechanistic basis for PDZ domain binding promiscuity, involving not only binding groove flexibility but also key interactions outside the canonical groove. Our findings might have importance for deciphering composite specificity of protein-protein interaction domains. Moreover, mapping of unconventional interactions should be highly important in putative drug discovery efforts aimed at identifying novel inhibitors targeting PDZ interactions.
Acknowledgment
We thank Jytte Rasmussen for technical support.
This work was supported, in whole or in part, by National Institutes of Health Grant P01 DA 12408 (to U. G.). This work was also supported by the Danish Medical Research Council (to U. G., K. T., and K. L. M.), University of Copenhagen BioScaRT Program of Excellence (to U. G.), Lundbeck Foundation Center for Biomembranes in Nanomedicine (to U. G.), the Novo Nordisk Foundation (to U. G.), and Fabrikant Vilhelm Pedersen og Hustrus Mindelegat (to U. G.).
The atomic coordinates and NMR restraints (code 2LUI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
NMR chemical shift data have been deposited at the BioMagResBank Databank with accession numbers 18522, 19418, 19466, and 19467.
- DAT
- dopamine transporter
- OG
- Oregon Green
- FP
- fluorescence polarization
- STD
- saturation transfer difference
- HSQC
- heteronuclear single quantum coherence
- PDZ domain
- PSD-95/Disc-Large(ZO-1 Homology domain
- ASIC1a
- acid sensing iona channel 1a
- PICK2
- protein interacting with C kinase 1
- HEER2
- human epidermal growth factor receptor 2
- GLT1b
- glutamate transporter 1b.
REFERENCES
- 1. Schultz J., Milpetz F., Bork P., Ponting C. P. (1998) SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. U.S.A. 95, 5857–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Feng W., Zhang M. (2009) Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. 10, 87–99 [DOI] [PubMed] [Google Scholar]
- 3. Ye F., Zhang M. (2013) Structures and target recognition modes of PDZ domains: recurring themes and emerging pictures. Biochem. J. 455, 1–14 [DOI] [PubMed] [Google Scholar]
- 4. Sheng M., Sala C. (2001) PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 24, 1–29 [DOI] [PubMed] [Google Scholar]
- 5. Stiffler M. A., Chen J. R., Grantcharova V. P., Lei Y., Fuchs D., Allen J. E., Zaslavskaia L. A., MacBeath G. (2007) PDZ domain binding selectivity is optimized across the mouse proteome. Science 317, 364–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McLaughlin R. N., Jr., Poelwijk F. J., Raman A., Gosal W. S., Ranganathan R. (2012) The spatial architecture of protein function and adaptation. Nature 491, 138–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tonikian R., Zhang Y., Sazinsky S. L., Currell B., Yeh J. H., Reva B., Held H. A., Appleton B. A., Evangelista M., Wu Y., Xin X., Chan A. C., Seshagiri S., Lasky L. A., Sander C., Boone C., Bader G. D., Sidhu S. S. (2008) A specificity map for the PDZ domain family. PLoS Biol. 6, e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Songyang Z., Fanning A. S., Fu C., Xu J., Marfatia S. M., Chishti A. H., Crompton A., Chan A. C., Anderson J. M., Cantley L. C. (1997) Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275, 73–77 [DOI] [PubMed] [Google Scholar]
- 9. Bezprozvanny I., Maximov A. (2001) Classification of PDZ domains. FEBS Lett. 509, 457–462 [DOI] [PubMed] [Google Scholar]
- 10. Daniels D. L., Cohen A. R., Anderson J. M., Brünger A. T. (1998) Crystal structure of the hCASK PDZ domain reveals the structural basis of class II PDZ domain target recognition. Nat. Struct. Biol. 5, 317–325 [DOI] [PubMed] [Google Scholar]
- 11. Stricker N. L., Christopherson K. S., Yi B. A., Schatz P. J., Raab R. W., Dawes G., Bassett D. E., Jr., Bredt D. S., Li M. (1997) PDZ domain of neuronal nitric oxide synthase recognizes novel C-terminal peptide sequences. Nat. Biotechnol. 15, 336–342 [DOI] [PubMed] [Google Scholar]
- 12. Cao M., Mao Z., Kam C., Xiao N., Cao X., Shen C., Cheng K. K., Xu A., Lee K. M., Jiang L., Xia J. (2013) PICK1 and ICA69 control insulin granule trafficking and their deficiencies lead to impaired glucose tolerance. PLoS Biol. 11, e1001541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holst B., Madsen K. L., Jansen A. M., Jin C., Rickhag M., Lund V. K., Jensen M., Bhatia V., Sørensen G., Madsen A. N., Xue Z., Møller S. K., Woldbye D., Qvortrup K., Huganir R., Stamou D., Kjærulff O., Gether U. (2013) PICK1 deficiency impairs secretory vesicle biogenesis and leads to growth retardation and decreased glucose tolerance. PLoS Biol. 11, e1001542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu W., Ziff E. B. (2005) PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron 47, 407–421 [DOI] [PubMed] [Google Scholar]
- 15. Steinberg J. P., Takamiya K., Shen Y., Xia J., Rubio M. E., Yu S., Jin W., Thomas G. M., Linden D. J., Huganir R. L. (2006) Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron 49, 845–860 [DOI] [PubMed] [Google Scholar]
- 16. Cao M., Xu J., Shen C., Kam C., Huganir R. L., Xia J. (2007) PICK1-ICA69 heteromeric BAR domain complex regulates synaptic targeting and surface expression of AMPA receptors. J. Neurosci. 27, 12945–12956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hanley J. G. (2008) PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol. Ther. 118, 152–160 [DOI] [PubMed] [Google Scholar]
- 18. Thorsen T. S., Madsen K. L., Rebola N., Rathje M., Anggono V., Bach A., Moreira I. S., Stuhr-Hansen N., Dyhring T., Peters D., Beuming T., Huganir R., Weinstein H., Mulle C., Strømgaard K., Rønn L. C., Gether U. (2010) Identification of a small-molecule inhibitor of the PICK1 PDZ domain that inhibits hippocampal LTP and LTD. Proc. Natl. Acad. Sci. U.S.A. 107, 413–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Terashima A., Pelkey K. A., Rah J. C., Suh Y. H., Roche K. W., Collingridge G. L., McBain C. J., Isaac J. T. (2008) An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron 57, 872–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Staudinger J., Lu J., Olson E. N. (1997) Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-α. J. Biol. Chem. 272, 32019–32024 [DOI] [PubMed] [Google Scholar]
- 21. Chung H. J., Xia J., Scannevin R. H., Zhang X., Huganir R. L. (2000) Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J. Neurosci. 20, 7258–7267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dev K. K., Nishimune A., Henley J. M., Nakanishi S. (1999) The protein kinase Cα binding protein PICK1 interacts with short but not long form alternative splice variants of AMPA receptor subunits. Neuropharmacology 38, 635–644 [DOI] [PubMed] [Google Scholar]
- 23. Dev K. K., Nakajima Y., Kitano J., Braithwaite S. P., Henley J. M., Nakanishi S. (2000) PICK1 interacts with and regulates PKC phosphorylation of mGLUR7. J. Neurosci. 20, 7252–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cowan C. A., Yokoyama N., Bianchi L. M., Henkemeyer M., Fritzsch B. (2000) EphB2 guides axons at the midline and is necessary for normal vestibular function. Neuron 26, 417–430 [DOI] [PubMed] [Google Scholar]
- 25. Torres G. E., Yao W. D., Mohn A. R., Quan H., Kim K. M., Levey A. I., Staudinger J., Caron M. G. (2001) Functional interaction between monoamine plasma membrane transporters and the synaptic PDZ domain-containing protein PICK1. Neuron 30, 121–134 [DOI] [PubMed] [Google Scholar]
- 26. Bjerggaard C., Fog J. U., Hastrup H., Madsen K., Loland C. J., Javitch J. A., Gether U. (2004) Surface targeting of the dopamine transporter involves discrete epitopes in the distal C terminus but does not require canonical PDZ domain interactions. J. Neurosci. 24, 7024–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jaulin-Bastard F., Saito H., Le Bivic A., Ollendorff V., Marchetto S., Birnbaum D., Borg J. P. (2001) The ERBB2/HER2 receptor differentially interacts with ERBIN and PICK1 PSD-95/DLG/ZO-1 domain proteins. J. Biol. Chem. 276, 15256–15263 [DOI] [PubMed] [Google Scholar]
- 28. Bassan M., Liu H., Madsen K. L., Armsen W., Zhou J., Desilva T., Chen W., Paradise A., Brasch M. A., Staudinger J., Gether U., Irwin N., Rosenberg P. A. (2008) Interaction between the glutamate transporter GLT1b and the synaptic PDZ domain protein PICK1. Eur. J. Neurosci. 27, 66–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williams M. E., Wu S. C., McKenna W. L., Hinck L. (2003) Surface expression of the netrin receptor UNC5H1 is regulated through a protein kinase C-interacting protein/protein kinase-dependent mechanism. J. Neurosci. 23, 11279–11288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hruska-Hageman A. M., Wemmie J. A., Price M. P., Welsh M. J. (2002) Interaction of the synaptic protein PICK1 (protein interacting with C kinase 1) with the non-voltage gated sodium channels BNC1 (brain Na+ channel 1) and ASIC (acid-sensing ion channel). Biochem. J. 361, 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baron A., Deval E., Salinas M., Lingueglia E., Voilley N., Lazdunski M. (2002) Protein kinase C stimulates the acid-sensing ion channel ASIC2a via the PDZ domain-containing protein PICK1. J. Biol. Chem. 277, 50463–50468 [DOI] [PubMed] [Google Scholar]
- 32. Madsen K. L., Beuming T., Niv M. Y., Chang C. W., Dev K. K., Weinstein H., Gether U. (2005) Molecular determinants for the complex binding specificity of the PDZ domain in PICK1. J. Biol. Chem. 280, 20539–20548 [DOI] [PubMed] [Google Scholar]
- 33. Elkins J. M., Papagrigoriou E., Berridge G., Yang X., Phillips C., Gileadi C., Savitsky P., Doyle D. A. (2007) Structure of PICK1 and other PDZ domains obtained with the help of self-binding C-terminal extensions. Protein Sci. 16, 683–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pan L., Wu H., Shen C., Shi Y., Jin W., Xia J., Zhang M. (2007) Clustering and synaptic targeting of PICK1 requires direct interaction between the PDZ domain and lipid membranes. EMBO J. 26, 4576–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jaravine V. A., Zhuravleva A. V., Permi P., Ibraghimov I., Orekhov V. Y. (2008) Hyperdimensional NMR spectroscopy with nonlinear sampling. J. Am. Chem. Soc. 130, 3927–3936 [DOI] [PubMed] [Google Scholar]
- 36. Kazimierczuk K., Orekhov V. Y. (2011) Accelerated NMR spectroscopy by using compressed sensing. Angew. Chem. Int. Ed. Engl. 50, 5556–5559 [DOI] [PubMed] [Google Scholar]
- 37. Orekhov V. Y., Jaravine V. A. (2011) Analysis of non-uniformly sampled spectra with multi-dimensional decomposition. Prog. Nucl. Magn. Reson Spectrosc. 59, 271–292 [DOI] [PubMed] [Google Scholar]
- 38. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 39. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 40. Cheung M. S., Maguire M. L., Stevens T. J., Broadhurst R. W. (2010) DANGLE: a Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J. Magn. Reson. 202, 223–233 [DOI] [PubMed] [Google Scholar]
- 41. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 42. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 43. Linge J. P., Habeck M., Rieping W., Nilges M. (2003) ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics 19, 315–316 [DOI] [PubMed] [Google Scholar]
- 44. Xu J., Xia J. (2006) Structure and function of PICK1. Neurosignals 15, 190–201 [DOI] [PubMed] [Google Scholar]
- 45. Bolia A., Gerek Z. N., Keskin O., Banu Ozkan S., Dev K. K. (2012) The binding affinities of proteins interacting with the PDZ domain of PICK1. Proteins 80, 1393–1408 [DOI] [PubMed] [Google Scholar]
- 46. Doucet M. V., Harkin A., Dev K. K. (2012) The PSD-95/nNOS complex: new drugs for depression? Pharmacol. Ther. 133, 218–229 [DOI] [PubMed] [Google Scholar]
- 47. Dev K. K., Henley J. M. (2006) The schizophrenic faces of PICK1. Trends Pharmacol. Sci. 27, 574–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dev K. K. (2004) Making protein interactions druggable: targeting PDZ domains. Nat. Rev. Drug Discov. 3, 1047–1056 [DOI] [PubMed] [Google Scholar]
- 49. Penkert R. R., DiVittorio H. M., Prehoda K. E. (2004) Internal recognition through PDZ domain plasticity in the Par-6-Pals1 complex. Nat. Struct. Mol. Biol. 11, 1122–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hillier B. J., Christopherson K. S., Prehoda K. E., Bredt D. S., Lim W. A. (1999) Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 284, 812–815 [PubMed] [Google Scholar]
- 51. Wong H. C., Bourdelas A., Krauss A., Lee H. J., Shao Y., Wu D., Mlodzik M., Shi D. L., Zheng J. (2003) Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol. Cell 12, 1251–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Escobar-Cabrera E., Okon M., Lau D. K., Dart C. F., Bonvin A. M., McIntosh L. P. (2011) Characterizing the N- and C-terminal small ubiquitin-like modifier (SUMO)-interacting motifs of the scaffold protein DAXX. J. Biol. Chem. 286, 19816–19829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dhulesia A., Gsponer J., Vendruscolo M. (2008) Mapping of two networks of residues that exhibit structural and dynamical changes upon binding in a PDZ domain protein. J. Am. Chem. Soc. 130, 8931–8939 [DOI] [PubMed] [Google Scholar]
- 54. Hultqvist G., Haq S. R., Punekar A. S., Chi C. N., Engström Å., Bach A., Strømgaard K., Selmer M., Gianni S., Jemth P. (2013) Energetic pathway sampling in a protein interaction domain. Structure 21, 1193–1202 [DOI] [PubMed] [Google Scholar]
- 55. Zhang Y., Yeh S., Appleton B. A., Held H. A., Kausalya P. J., Phua D. C., Wong W. L., Lasky L. A., Wiesmann C., Hunziker W., Sidhu S. S. (2006) Convergent and divergent ligand specificity among PDZ domains of the LAP and zonula occludens (ZO) families. J. Biol. Chem. 281, 22299–22311 [DOI] [PubMed] [Google Scholar]
- 56. Terrien E., Chaffotte A., Lafage M., Khan Z., Préhaud C., Cordier F., Simenel C., Delepierre M., Buc H., Lafon M., Wolff N. (2012) Interference with the PTEN-MAST2 interaction by a viral protein leads to cellular relocalization of PTEN. Sci. Signal. 5, ra58. [DOI] [PubMed] [Google Scholar]
- 57. Mostarda S., Gfeller D., Rao F. (2012) Beyond the binding site: the role of the β(2)-β(3) loop and extra-domain structures in PDZ domains. PLoS Comput. Biol. 8, e1002429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tyler R. C., Peterson F. C., Volkman B. F. (2010) Distal interactions within the par3-VE-cadherin complex. Biochemistry 49, 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bhattacharya S., Ju J. H., Orlova N., Khajeh J. A., Cowburn D., Bu Z. (2013) Ligand-induced dynamic changes in extended PDZ domains from NHERF1. J. Mol. Biol. 425, 2509–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chi C. N., Haq S. R., Rinaldo S., Dogan J., Cutruzzolà F., Engström Å., Gianni S., Lundström P., Jemth P. (2012) Interactions outside the boundaries of the canonical binding groove of a PDZ domain influence ligand binding. Biochemistry 51, 8971–8979 [DOI] [PubMed] [Google Scholar]
- 61. Wang C. K., Pan L., Chen J., Zhang M. (2010) Extensions of PDZ domains as important structural and functional elements. Protein Cell 1, 737–751 [DOI] [PMC free article] [PubMed] [Google Scholar]