

Background: Sorting nexin 17 (SNX17) binds cytoplasmic proteins in addition to cell surface receptors.
Results: A direct interaction between SNX17 and the cytoplasmic adaptor protein KRIT1 (Krev interaction trapped 1) is characterized by biochemistry and crystallography.
Conclusion: KRIT1 is a SNX17 binding partner.
Significance: Understanding the binding activities of SNX17 may suggest function beyond endosomal sorting.
Keywords: Crystal Structure, Peptide Interaction, Protein Domain, Protein Motif, Protein-Protein Interaction, FERM Domain, KRIT1, NPXY Motif, Cerebral Cavernous Malformations, Sorting Nexin
Abstract
Sorting nexin 17 (SNX17) is a member of the family of cytoplasmic sorting nexin adaptor proteins that regulate endosomal trafficking of cell surface proteins. SNX17 localizes to early endosomes where it directly binds NPX(Y/F) motifs in the cytoplasmic tails of its target receptors to mediate their rates of endocytic internalization, recycling, and/or degradation. SNX17 has also been implicated in mediating cell signaling and can interact with cytoplasmic proteins. KRIT1 (Krev interaction trapped 1), a cytoplasmic adaptor protein associated with cerebral cavernous malformations, has previously been shown to interact with SNX17. Here, we demonstrate that SNX17 indeed binds directly to KRIT1 and map the binding to the second Asn-Pro-Xaa-Tyr/Phe (NPX(Y/F)) motif in KRIT1. We further characterize the interaction as being mediated by the FERM domain of SNX17. We present the co-crystal structure of SNX17-FERM with the KRIT1-NPXF2 peptide to 3.0 Å resolution and demonstrate that the interaction is highly similar in structure and binding affinity to that between SNX17 and P-selectin. We verify the molecular details of the interaction by site-directed mutagenesis and pulldown assay and thereby confirm that the major binding site for SNX17 is confined to the NPXF2 motif in KRIT1. Taken together, our results verify a direct interaction between SNX17 and KRIT1 and classify KRIT1 as a SNX17 binding partner.
Introduction
The sorting nexins (SNX)4 are a family of adaptor proteins involved in trafficking of proteins through the endosomal pathway (1, 2). One member, SNX17, was first identified as a binding partner of the cytoplasmic tail of the cell adhesion protein P-selectin (3). SNX17 has since been shown to mediate internalization, recycling, and/or protection from degradation of multiple cell surface proteins including P-selectin (4), amyloid precursor protein (APP) (5), integrin β1 (6, 7), low density lipoprotein receptor (LDLR) (8), LDLR related protein (Lrp1) (9), ApoER2 (10), and FEEL1 (11) among others. All SNX proteins contain a conserved phox homology (PX) domain that binds specific phosphoinositides, and various scaffolding modules including SH3, BAR, and PDZ domains (2). The SNX17 PX domain displays specificity for phosphatidylinositol 3-monophosphate, which is enriched at the cytoplasmic face of the early endosome. Phosphatidylinositol 3-monophosphate binding drives localization of SNX17 to the early endosomes in addition to its broad cytoplasmic localization (3).
In addition to its N-terminal PX domain, SNX17 contains a C-terminal band 4.1/ezrin/radixin/moesin (FERM) domain (see Fig. 1A) (12). Classical FERM domains, which are lipid and protein interaction domains, contain ∼300 residues and are comprised of three lobes termed F1, F2, and F3. The F1 lobe adopts a ubiquitin-like fold, F2 is a helical structure similar to acyl-CoA binding protein, and F3 is similar to the phosphotyrosine binding (PTB)/pleckstrin homology domain (13). Many FERM domains provide a link from the cytoskeleton to the cell membrane via interaction with phosphatidylinositol phosphates or transmembrane proteins (14). The FERM domain of SNX17 binds directly to a common motif in the cytoplasmic region of its target proteins: a conserved Asn-Pro-Xaa-(Tyr/Phe) (NPX(Y/F), where X is any amino acid) (12). The NPX(Y/F) motif has been shown to serve as an internalization signal for cell surface proteins (15, 16), in addition to its role as a signaling motif that binds adaptor proteins containing PTB (or FERM) domains to trigger signaling events (17), such as the cytoplasmic protein talin binding the NPXY motif in the cytoplasmic tail of integrin β3 leading to integrin activation (18). Recently, the co-crystal structure of the SNX17 FERM domain with the cytoplasmic tail of P-selectin confirmed that the binding mode involves PTB-like recognition of NPX(Y/F) motifs by the F3 lobe of SNX17 FERM (19).
FIGURE 1.
SNX17 binds directly to KRIT1 NPXF2. A, schematic diagram of SNX17 and KRIT1 proteins. In this study, we demonstrate that the F3 lobe of the FERM domain of SNX17 (green) directly binds KRIT1 NPXF2 (orange). B, sequence alignment of human KRIT1 NPX(Y/F) motifs. The position and numbering of the NPX(Y/F) residues is shown. C, endogenous SNX17 from HEK293T cells binds KRIT1-NPXF2 in GST pulldown assays. The cytoplasmic tail of P-selectin (p-sel) is included as a positive control, and GST alone is included as a negative control. Top panel, bound SNX17 is visualized by anti-SNX17 immunoblot (IB). Bottom panel, equal GST loading is shown by Ponceau staining. D, purified recombinant SNX17 PX-FERM binds directly to KRIT1 NPXF2 in a similar GST pulldown experiment from C. Proteins are visualized by Coomassie Blue staining.
Although the binding partners of SNX17 are mostly transmembrane proteins, SNX17 has also been shown to bind several cytoplasmic proteins including Ras GTPases (12), the kinesin-like protein Kif1B (20), and SH3 domain-containing proteins including PLCγ (21). Another reported cytoplasmic binding partner for SNX17 is Krev interaction trapped 1 (KRIT1) (22). The gene encoding KRIT1 is associated with cerebral cavernous malformations (CCM), a class of vascular dysplasias that cause mulberry-shaped lesions leading to increased risk of stroke (23); accordingly, KRIT1 is also commonly referred to as CCM1 (24). The basis of CCM disease is the heterozygous loss of one allele of either KRIT1, CCM2, or CCM3, along with the secondary loss of one of the protein products by a second-hit mutation (25).
KRIT1 is an adaptor protein with numerous protein binding partners and no enzymatic activity reported to date. It contains an N-terminal Nudix fold domain and three NPX(Y/F) motifs: NPXY1, NPXF2, and NPXF3, a central ankyrin repeat domain, and a C-terminal FERM domain (see Fig. 1, A and B). KRIT was first discovered by its interaction with the small GTPase Rap1/Krev, which binds the KRIT1 FERM domain and helps localize KRIT1 to the cell membrane (26, 27). There, KRIT1 acts as a positive regulator of integrin signaling by binding via its Nudix/NPXY1 region to the PTB domain of ICAP1 (integrin cytoplasmic domain associated protein-1) to relieve the suppressive effect of ICAP1 on integrin activation (27–29). KRIT1 is also found in complex with another CCM protein; the NPXF2-NPXF3 region of KRIT1 binds the predicted PTB domain of CCM2 (30–32). Furthermore, the orphan transmembrane protein HEG (heart of glass) binds KRIT1 via the FERM domain (33–35). Despite some knowledge about its structure and binding partners, the cellular pathways in which KRIT1 functions and how its loss contributes to CCM are not well understood (36).
Since the first report of an interaction between SNX17 and KRIT1 (22), no further studies have been presented. To gain a more complete understanding of how SNX17 interacts with KRIT1, we characterize the complex using biochemical, crystallographic, and biophysical approaches. Our results show for the first time with purified proteins that SNX17 binds KRIT1 directly with binding affinity similar to other SNX17 binding proteins. We map the binding site for SNX17 on KRIT1 and reveal the molecular basis for the interaction by solving the co-crystal structure of the FERM-like domain of SNX17 and KRIT1 NPXF2 peptide. We further validate the molecular basis of the interaction by biochemistry and point mutagenesis. Our studies provide a comprehensive description of the SNX17-KRIT1 interaction.
EXPERIMENTAL PROCEDURES
Expression Constructs
cDNA encoding full-length human SNX17 (UniProt Q15036, residues 1–470) was subcloned into a modified pET vector containing an N-terminal GST tag. cDNA encoding SNX17 PX-FERM domains (residues 1–391) and FERM domain alone (residues 108–391) were subcloned in a modified pET vector containing an N-terminal His6 tag, which is removable with tobacco etch virus and thrombin proteases. Full-length human KRIT1 cDNA (UniProt O00522, residues 1–736) was subcloned into the XhoI/EcoRI sites of pEGFP-C1 (Clontech) for expression in a mammalian system as an N-terminally eGFP-tagged protein. Expression constructs for KRIT1 NPX(Y/F) motifs (NPXY1, residues 186–198; NPXF2, residues 225–237; NPXF3, residues 243–255) and the C-terminal NPXY motif of mouse P-selectin (UniProt Q01102, residues 755–768) were generated by insertion of synthetic oligonucleotides by QuikChange mutagenesis (Agilent) 3′ to the GST and PreScission protease recognition sequence of pGEX-6p-1 (GE Healthcare). cDNA encoding the cytoplasmic tail of human HEG1 (UniProt Q9ULI3, residues 1276–1381) was PCR-amplified and subcloned into pGEX-6p-1 (GE Healthcare). Single-point mutations were generated by a QuikChange Lightning mutagenesis kit (Agilent).
Protein Expression and Purification
Proteins were expressed in BL21(DE3) or Rosetta(DE3) cells by induction with 0.2 mm isopropyl β-d-thiogalactopyranoside overnight at 16 °C. For crystallization, His6-SNX17-FERM and GST-KRIT1-NPXF2 were co-expressed in Rosetta(DE3) cells by co-transformation and co-purified. The complex was isolated from lysates by nickel affinity chromatography (HisTrap; GE Healthcare) and eluted with imidazole, and the His6 tag on SNX17-FERM was removed by tobacco etch virus protease cleavage. The complex was then bound to glutathione-Sepharose 4B affinity resin (GE Healthcare) and washed to remove tobacco etch virus and other impurities. Lastly, PreScission protease was added to cleave the KRIT1-NPXF2 peptide from GST and liberate the SNX17-FERM/KRIT1-NPXF2 complex from the glutathione beads. The complex was then run over size exclusion chromatography (Superdex 75; GE Healthcare), concentrated to 10 mg/ml, and used in crystallization trials. GST-tagged full-length SNX17, KRIT1, and P-selectin NPXY motifs, and the cytoplasmic tail of HEG1 were purified on glutathione-Sepharose 4B (GE Healthcare), eluted with 10 mm reduced glutathione in 50 mm Tris, pH 8.0, and 150 mm NaCl, and further purified by gel filtration chromatography (Superdex 200; GE Healthcare) or desalted into gel filtration buffer to remove glutathione. His-tagged SNX17-PX-FERM protein was purified by nickel affinity (HisTrap; GE Healthcare), eluted with imidazole, and desalted into binding buffer (see below) or further purified by ion exchange (Resource Q; GE Healthcare) and size exclusion (Superdex 200; GE Healthcare) chromatography.
Pulldown Assays
GST-tagged proteins or GST alone were reapplied to glutathione-agarose beads at an approximate concentration of 0.5 μg of protein per μl of bead bed. For in vitro pulldown assays, His-tagged SNX17 PX-FERM wild-type or mutant proteins were diluted to 0.05 μg/μl in binding buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 0.01% (w/v) Triton X-100) and incubated with GST coated beads at 4 °C for 2–4 h rocking. The beads were washed three times with binding buffer, and bound proteins eluted by addition of SDS-PAGE Sample buffer. Samples were resolved by SDS-PAGE, and pulldowns were either visualized by staining with Coomassie Blue or probed for bound His-tagged SNX17 with anti-His antibody (Sigma).
For pulldowns from Human embryonic kidney (HEK) 293T lysates, cells were grown to 80–90% confluence in DMEM with 10% (w/v) FBS, 1 mm sodium pyruvate, 10 units/ml penicillin/streptomycin at 37 °C. Cells were transfected using Lipofectamine (Invitrogen) with plasmid encoding KRIT1-eGFP or eGFP alone and harvested 24–48 h later. For binding of endogenous SNX17, cells were harvested at 100% confluence without transfection. Cell lysates were prepared in buffer X (1 mm Na3VO4, 50 mm NaF, 40 mm sodium pyrophosphate, 50 mm NaCl, 150 mm sucrose, 10 mm Pipes, pH 6.8) plus 0.5% Triton X-100, 0.1% deoxycholate, and EDTA-free protease inhibitor (Roche Applied Science) and incubated with glutathione-agarose beads precoated with GST proteins at a final Triton X-100 concentration of 0.1%. After incubation at 4 °C (4 h to overnight), beads were washed three times in buffer X with 0.05% Triton X-100. Bound proteins were eluted by SDS-PAGE sample buffer, resolved by SDS-PAGE, and transferred to PVDF. The presence of bound SNX17 was probed by immunoblot with anti-SNX17 antibody (H-10; Santa Cruz) or bound KRIT1-eGFP with anti-GFP antibody (B-2; Santa Cruz). Immunoblots were imaged on an Odyssey Infrared Imaging System (Li-Cor) and analyzed using ImageJ or Image Studio Lite (Li-Cor). Similar amounts of GST fusion proteins on beads were verified by Coomassie Blue or Ponceau S staining. Quantitation of binding is presented as a percentage of maximum binding and/or a percentage of wild-type binding.
Crystallization and Structure Solution
Sparse matrix screening was performed manually. Initial crystals were obtained by vapor diffusion in sitting drops at room temperature with a 1:1 (v/v) ratio of purified protein complex to reservoir solution containing 2.0 m ammonium sulfate, 0.1 m HEPES, pH 7.5, and 2% PEG 400. Optimization of crystal singularity and size was achieved by preparing a serially diluted seed stock of microcrystals in mother liquor similar to the method described in (37) and adjusting the concentration of ammonium sulfate to 1.9 m and PEG 400 to 5%. The best crystals were obtained by adding seed stock at 1:10 (v/v) ratio to a drop containing 1:1 ratio of protein complex stock and mother liquor. Single crystals were quickly incubated in cryoprotectant solution containing mother liquor supplemented with up to 5% (w/v) ethylene glycol, mounted in cryoloops, and frozen in liquid nitrogen.
Diffraction data were collected at Northeastern Collaborative Access Team (NECAT) Beamline 24-ID-E at Argonne National Laboratory Advanced Photon Source and processed in HKL2000 (38). Data from five individual crystals were indexed, integrated separately, and then scaled together to improve completeness, redundancy, and resolution. A molecular replacement solution was found by Phaser (39) using the previously determined crystal structure of the SNX17 FERM domain (PDB accession number 4GXB chain A (19)) as a search model. Three copies of SNX17 per asymmetric unit were correctly placed by Phaser. Incorrect segments of the initial molecular replacement solution model (as assessed by difference map analysis) were deleted and manual building was performed in Coot (40). Refinement was carried out in Phenix (41) using TLS, NCS, and external model restraints to the high resolution SNX17 structure (PDB accession number 4GXB (19)). The KRIT1 NPXF2 peptides were modeled into unbiased positive Fobs − Fcalc electron density after the SNX17-FERM molecules were built and refined to near convergence. The register of the peptide sequence was guided by the expected position of the NPXF motif and the large positive side chain density of Phe-234 and Tyr-230. Peptide modeling was also guided by NCS maps and by minimizing clashes. Crystallographic software is compiled by SBGrid (42).
Affinity Measurements
Dip and Read Biosensors covalently coupled with anti-GST antibody (ForteBio) were loaded with GST KRIT1-NPXF2 or GST alone at 0.05 μg/μl on a BLItz instrument (ForteBio) in assay buffer (20 mm HEPES, pH 7.3, 150 mm NaCl, 5 mm Tris(2-carboxyethyl)phosphine (TCEP)). SNX17 PX-FERM protein was prepared as dilution series in assay buffer at room temperature. Binding response to GST-NPXF2 or GST was measured as biolayer interferometry during a 30-s association period. The signal from binding GST-NPXF2 was referenced against binding to GST alone, and equilibrium binding responses were plotted against concentration. The dissociation constant was calculated in GraphPad Prism in the nonlinear regression equilibrium binding/specific binding mode. The final KD measurement is the result of curve fitting 18 independently measured SNX17 concentration points.
RESULTS
SNX17 Directly Binds KRIT1 NPXF2 Motif
SNX17 has been shown to bind the N-terminal region of KRIT1 (22), which includes the Nudix domain and three NPXY/NPXF motifs: NPXY1, NPXF2, and NPXF3 (Fig. 1, A and B) (29, 43). Because SNX17 binds directly to NPX(Y/F) motifs in target proteins (4, 5, 19), we hypothesized that SNX17 binds KRIT1 via one or more of these motifs. To test this hypothesis and to determine which motif is responsible for interaction with SNX17, we utilized a GST pulldown assay using peptides derived from individual NPX(Y/F) peptides of KRIT1 immobilized as GST fusion proteins on Sepharose beads, similar to those performed for SNX17 binding to APP (5), LRP1 (9), and others (44). To assess binding of SNX17 to KRIT1, we incubated GST-NPX(Y/F)-coated beads with HEK293T cell lysates and probed for bound endogenous SNX17 by immunoblot. We find that SNX17 binds specifically to KRIT1 NPXF2, similar to its interaction with the P-selectin cytoplasmic NPXY motif (Fig. 1C). In contrast, no binding is detected to KRIT1 NPXY1 or NPXF3 or to GST alone. Thus, we conclude that SNX17 binds KRIT1 via NPXF2. Using the same KRIT1 NPX(Y/F)-coated beads, we next asked whether SNX17 directly binds KRIT1 in a purified protein system. Recombinant SNX17 protein encompassing the PX-FERM domains (Fig. 1A) was expressed in bacteria, purified, and tested for its ability to directly bind KRIT1 GST-NPX(Y/F) motif peptides. We find that similar to endogenous SNX17 from HEK293T cells, purified SNX17-PX-FERM binds directly and specifically to KRIT1 NPXF2, with little binding detected to NPXY1 or NPXF3 (Fig. 1D). Taken together, these results support that SNX17 binds directly and preferentially to the KRIT1 NPXF2 motif.
Co-crystal Structure of SNX17-FERM/KRIT1-NPXF2
The binding of SNX17 to target NPX(Y/F) motifs is mediated by its FERM domain (19). Because the NPXF2 motif in KRIT1 represents the SNX17 binding site (Fig. 1), we hypothesized that SNX17 uses its FERM domain to bind KRIT1. This notion is supported by the previous report that the C-terminal portion of SNX17 containing the FERM-like domain (SNX17-FERM) mediates the interaction with KRIT1 (22). To investigate the molecular details of the interaction between SNX17 and KRIT1, we determined the co-crystal structure of the SNX17 FERM domain in complex with KRIT1 NPXF2 peptide. In our hands, recombinant SNX17-FERM protein expressed alone in bacteria is largely insoluble and difficult to purify via standard purification protocols. However, co-expression of SNX17-FERM with GST-KRIT1-NPXF2 partially rescues SNX17-FERM solubility, and the complex is stable throughout co-purification (data not shown). The SNX17 FERM/KRIT1-NPXF2 complex was co-purified, co-crystals were obtained, and a 3.0 Å co-crystal structure was determined (Table 1 and Fig. 2A).
TABLE 1.
Data collection and refinement statistics
| SNX17-FERM/KRIT1-NPxY2 | |
|---|---|
| Data Collection | |
| X-ray source | APS NECAT (E) |
| No. crystals | 5 |
| Wavelength (Å) | 0.97926 |
| Space Group | C2 |
| Cell dimensions a, b, c (Å) | 117.9, 191.2, 46.6 |
| Cell dimensions α, β, γ (°) | 90, 110.3, 90 |
| Resolution range (Å)a | 50–3.0 (3.12–3.00) |
| Unique reflections | 18,042 |
| Multiplicitya | 15.0 (14.4) |
| Completeness (%)a | 100 (100) |
| Rsym (%)a | 24.1 |
| Rpim (%)a | 7.6 (74.3) |
| <I>/<σI>a | 10.8 (2.3) |
| Wilson B-factor | 86.8 |
| Refinement | |
| Resolution rangea | 47.88–3.00 |
| (3.08–3.00) | |
| No. atoms | |
| Total | 6,422 |
| Non-hydrogen | 6,422 |
| Protein (all) | 6,374 |
| Water | 48 |
| Reflections | |
| Totala | 17,901 (1,032) |
| Free (no.)a | 1,794 (116) |
| Free (%) | 10 |
| R factors | |
| Rwork (%)a | 21.8 (34.4) |
| Rfree (%)a | 26.8 (39.2) |
| Average B factors | |
| Overall | 126.1 |
| Protein | 126.4 |
| Water | 87.0 |
| Chain A/D | 121.2/124.2 |
| Chain B/E | 122.5/121.6 |
| Chain C/F | 134.6/159.0 |
| Model statistics | |
| Ramachandran plot, favored/allowed/outliers (%) | 98.8/1.2/0 |
| MolProbity | |
| Score | 1.42 |
| Percentile | 100th |
| r.m.s.d. | |
| Lengths (Å) | 0.005 |
| Angles (°) | 1.2 |
a Values in parentheses denote highest resolution shell.
FIGURE 2.

Overall crystal structure of SNX17-FERM bound to KRIT1 NPXF2. A, stereo view electron density map for SNX17-FERM/KRIT1-NPXF2 crystal structure. Final refined 2Fobs − Fcalc map is contoured to 1 σ (blue). All-atom view of the final SNX17 model is illustrated, and representative residues are labeled. B, ribbon diagram of a representative chain of SNX17-FERM with F1 (dark green), F2 (green), and F3 (light green) lobes labeled. The KRIT1 NPXF2 peptide (orange) bound to F3 is included. The secondary structure elements for each lobe are shown and lettered according to lobe: F1 (A), F2 (B), and F3 (C). The N and C termini of SNX17-FERM are also labeled. Unstructured loops are connected by dashed lines. C, backbone trace of SNX17-FERM/KRIT1-NPXF2 (green) with SNX17/P-selectin co-crystal structure (gray; PDB accession code 4GXB). The zoomed-in view shows the change in angle between the F2 helices α2B and α4C between the two structures. D, sequence conservation of SNX17 through evolution (across 30 species) mapped onto the SNX17-FERM surface (by CONSURF). The two views are related by a 180° rotation about a vertical axis as shown. Conservation degree is shown by color (dark is most conserved, and white is least conserved). The KRIT1-NPXF2 peptide and lobe numbering are included.
Overall Structure of SNX17-FERM Domain
The SNX17-FERM domain is a three-lobed structure consisting of the F1, F2, and F3 lobes arranged in a compact trefoil (Fig. 2B) similar to the FERM domains of moesin and radixin (13, 45) and superimposes with its closest structural homolog, myosin-X, with r.m.s.d. of 3.0 Å over 232 residues (Dali Server (46)). The F1 lobe (residues 113–205) folds as a prototypical ubiquitin/Ras association (RA) domain with a five-stranded β-sheet (β1A–β5A; numbering according to radixin nomenclature (45) where “A” denotes the F1 lobe, “B” denotes the F2 lobe, and “C” denotes the F3 lobe), a helix α1A between β2A and β3A, and a short α2A between β4A and β5A. The F2 lobe (residues 206–269) folds as a helical bundle and is unique among FERM domains in that it lacks the standard helix α3B. The F3 lobe (residues 270–388) is homologous to a PTB/pleckstrin homology domain and contains two antiparallel β-sheets that form a sandwich and a long helix α1C (Fig. 2B). The linker sequences connecting the three lobes are mostly buried within the core of the tightly packed FERM domain trefoil structure. There are three copies of the SNX17-FERM/KRIT1-NPXF2 complex per asymmetric unit, which are highly similar in structure and superpose well with r.m.s.d. values ranging from 0.4 to 0.6 Å over 276 equivalent Cα positions. Our crystal structure, along with that previously reported (19), confirm that the FERM domain of SNX17 extends to residue 388, in contrast to early predictions that the FERM domain ends at residue 274. This helps to explain why these early studies concluded that an extra C-terminal region of SNX17 (beyond residue 274) along with its predicted FERM domain is required for binding P-selectin and KRIT1 (4, 22).
Overall, the structure of the SNX17 FERM domain is highly similar to that previously reported (19) with r.m.s.d. of 1.2 Å over 254 residues (46) (Fig. 2C). Superposition of individual lobes results in r.m.s.d. of 0.5 Å (92 residues) for F1, 1.5 Å (63 residues) for F2, and 1.0 Å (96 residues) for F3. The larger r.m.s.d. in the F2 lobe is largely due to a noticeable shift of α2B which results in a 7° change in the interhelical angle between the α2B and α4B helices (Fig. 2C). This difference in conformation is likely stabilized by crystal packing in our structure, because the α2B-α4B unit packs against the equivalent region as part of a pseudosymmetric dimer (not shown), whereas in the previous crystal structure it is exposed to a solvent channel and has elevated average B factors compared with the remainder of the protein.
We mapped surface conservation of SNX17-FERM through evolution over 30 species (CONSURF server (47)), which reveals several regions of very high conservation (Fig. 2D). The largest area of conservation is the NPX(Y/F) binding site in F3 (discussed below).
KRIT1 NPXF2 Adopts an Extended Conformation with β-Turn
The co-crystal structure confirms that KRIT1-NPXF2 is bound by the F3 lobe of the SNX17 FERM domain, because there is clear electron density for the bound NPXF2 peptide in all three molecules in the asymmetric unit (Fig. 3A and data not shown). The peptide adopts a largely extended conformation of ∼20 Å in length. It forms a β-turn at the NPXF sequence (Fig. 3B) that is stabilized by hydrogen bonds involving the −3 residue Asn-231: one hydrogen bond is formed between the Asn-231 carbonyl and the backbone amide of Phe-234 at the 0 position, and a second hydrogen bond between the carboxyamide oxygen of the Asn-231 side chain and the backbone amide of Leu-233 at the −1 position (see Figs. 1B and 3B for NPX(Y/F) motif numbering). The β-turn conformation, which is commonly observed in NPX(Y/F) motifs, serves to position the side chain of the 0 Tyr/Phe for binding (48). Thus, the NPXF2 sequence in KRIT1 bound to SNX17 adopts structural characteristics common to NPX(Y/F) motifs. Alignment of KRIT1 NPXF2 with other SNX17 target NPX(Y/F) motifs reveals some sequence similarities including a hydrophobic residue at the −5 position (discussed below; Fig. 3C).
FIGURE 3.

Co-crystal structure of SNX17-FERM bound to KRIT1 NPXF2. A, unbiased positive difference Fo − Fc electron density for KRIT1-NPXF2 peptide in complex with SNX17-FERM calculated before peptide model building (final peptide model is depicted in orange). Map is contoured at +3 σ (green) and −3 σ (red). Map box size was 40 Å (no mask used). B, structural details of KRIT1-NPXF2 peptide alone in the same view as A. The residues are numbered, and intramolecular hydrogen bonds forming the β-turn are depicted as black dashed lines. The NPX(Y/F) motif numbering is included. C, sequence alignment of NPX(Y/F) motifs in SNX17 binding partners. All sequences are human (except P-selectin, mouse). UniProt access numbers are KRIT-1 (O00522), P-selectin (mouse, Q01102), APP (P05067), integrin β1 (P05556), LDLR (P01130), Lrp1 (Q07954), VLDLR (P98155), FEEL1 (Q9NY15), ApoER2 (Q14114), L2 HPV-11 (P04013), and L2 HPV-16 (P03107). D, KRIT1-NPXF2 (in orange) with the final refined 2Fobs − Fcalc electron density map for the peptide contoured to 2 σ (cyan) and 1 σ (blue) bound to SNX17-FERM, depicted as Cα trace (green). The zoomed-in view shows the SNX17-FERM F3 lobe as a ribbon diagram (green) bound to KRIT1 NPXF2 peptide (orange). Secondary structure elements are labeled. Residues involved in the interface are labeled (green for SNX17 and orange for KRIT1). E, surface electrostatic potential of SNX17 F3 lobe with KRIT1 peptide bound. F, surface conservation of SNX17 F3 lobe.
Molecular Recognition of KRIT1-NPXF2 by SNX17
The co-crystal structure reveals that the SNX17 F3 lobe employs canonical PTB-like recognition of the KRIT1 NPXY2 peptide (Fig. 3, A and D). The KRIT1 binding site on the surface of the F3 lobe is a groove made up of residues from α1C, the β4C-β5C loop, and β5C (Fig. 3, D and E). There are numerous features of the contiguous interface that facilitate KRIT1 binding. First, a conserved hydrophobic surface comprised of the side chains of Arg-319, Leu-353, and Leu-359, and the main chains of Thr-316, Arg-317, Met-318, and Arg-319 accommodate binding of KRIT1 Phe-234 at the 0 position of the NPXF motif (Fig. 3, D and E). Over 70% of the available surface area of Phe-234 is buried upon complex formation. As discussed above, the β-turn in the peptide positions the side chain of Phe-234 to fit into this hydrophobic pocket. A second predominant feature is the binding of KRIT1 Asn-231 at the −3 position. In addition to the intramolecular interactions that stabilize the peptide β-turn, Asn-231 makes three critical hydrogen bonds to SNX17: two side chain hydrogen bonds from its carboxyamide nitrogen with the carbonyl groups of Val-315 and Met-318 and a main chain nitrogen H-bond with the main chain carbonyl of Arg-319 (Fig. 3D). Another contribution to binding is the interaction between Leu-233 at the −1 position and a hydrophobic pocket created by Val-380, Leu-383, and Met-384 (Fig. 3D). Pro-232 is also engaged in hydrophobic interactions in this area. Next, the KRIT1 NPXF2 peptide forms a short β-strand at Ile-229 and Tyr-230, which hydrogen bond to Trp-321 in β5C. Lastly, Trp-321 lies at the base of the binding groove and provides a hydrophobic surface for binding of the −5 residue Ile-229 (Fig. 3, D and E), which corresponds to a large hydrophobic residue in other SNX17 binding partners including APP (Tyr) (5), P-selectin (Phe) (4), and Lrp1 (Ile) (9) (Fig. 3C). There are also minor contributions to the interface made by Cys-320, Arg-322, and Gln-377. Taken together, the binding site encompasses a surface area of ∼1073 Å2 (average of 605 Å2 from KRIT1 and 468 Å2 from SNX17) with a shape complementarity score of 0.77 (49). There is considerable surface conservation at the NPX(Y/F) binding site (Fig. 3F).
Structural Comparison of SNX17 Binding KRIT1-NPXF2 and P-selectin
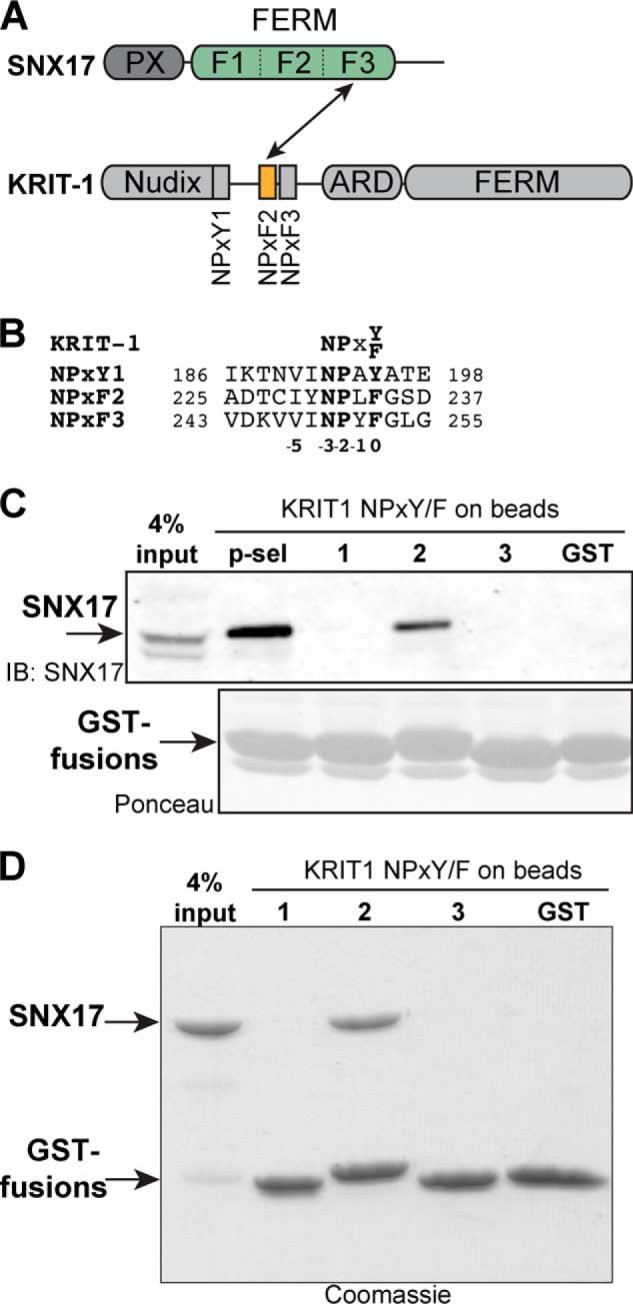
Our co-crystal structure reveals that SNX17 binds KRIT1-NPXF2 via a structurally similar mode to that previously observed for SNX17 binding to the P-selectin cytoplasmic tail (19) (Fig. 4). Superposition of the SNX17 F3 lobes from the two crystal structures (r.m.s.d. 1.0 Å over 96 residues) results in close structural alignment of the KRIT1 and P-selectin peptides, which bind to an identical binding site on the surface of F3 (Fig. 4A). The KRIT1 and P-selectin peptides bind in the same direction from their N to C termini, adopt a similar extended conformation with a β-turn at the NPXY motif, and bind with Phe/Tyr at the 0 position (KRIT1 Phe-234 and mouse P-selectin Tyr-763) and a large hydrophobic residue at the −5 position (Phe-758 in P-selectin and Ile-229 in KRIT1) to facilitate binding by the F3 lobe (Fig. 4, A and B). Despite these similarities, there are several differences observed in the binding site. First, the KRIT1 NPXF2 peptide backbone is translated by an average of 1.4 Å (distance between corresponding Cα positions) toward β5C compared with P-selectin (Fig. 4B). Because of this shift, the aromatic ring of Phe-234 is buried more deeply than its P-selectin Tyr-763 counterpart, whose extra hydroxyl group has to be accommodated in the space of the hydrophobic pocket (Fig. 4B, zoom-in view; notably, the human P-selectin sequence contains a Phe at the 0 position). The shift also allows for accommodation of the larger Leu-233 in KRIT1 (Fig. 4B, zoom-in view). P-selectin adopts a longer β-strand consisting of four residues (Gly-756 to Thr-759) compared with only two residues (Ile-229 and Tyr-230) in KRIT1 NPXF2 (Fig. 4A). Overall, the P-selectin peptide displays a larger contact area than KRIT1 NPXF2 (over 1300 Å2 versus 1073 Å2, respectively) because of additional contacts between the N-terminal −9 through −6 residues of P-selectin and α1C/β5C of SNX17. In contrast, KRIT1-NPXF2 extends away from SNX17 starting at the −7 residue, although the peptide used in crystallization contains residues through the −9 position. Nevertheless, comparison of the two crystal structures confirms that SNX17 binds KRIT1 and P-selectin by very similar F3 lobe-NPX(Y/F) binding modes.
FIGURE 4.
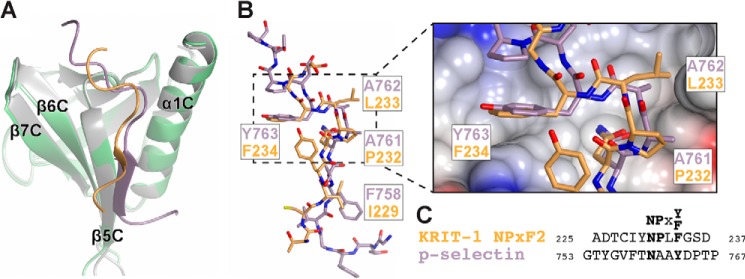
Comparison of KRIT1 and P-selectin binding to SNX17. A, superposition of the SNX17 F3 lobes (green and gray) reveals a common binding site for KRIT1 and P-selectin (orange and violet, respectively). B, all-atom view of the KRIT1 and P-selectin peptides. Superposition in A results in close alignment of the KRIT1 and P-selectin peptides. SNX17 has been removed for clarity. Corresponding residues in the peptides are labeled. The zoomed-in view shows the electrostatic surface potential of SNX17. C, sequence alignment of KRIT1 NPXF2 and P-selectin peptides.
Binding Affinity of SNX17 for KRIT1-NPXF2
We next set out to measure the binding affinity of SNX17 for KRIT1-NPXF2. We used biolayer interferometry to measure the binding of recombinant purified SNX17 PX-FERM protein to GST-NPXF2 protein captured on anti-GST biosensors. By biolayer interferometry, we find that the KD value is 1.3 ± 0.3 μm (95% confidence interval of 0.8–1.9 μm), which is similar to the values reported for SNX17 binding to P-selectin (2.7 μm) and APP (22–33 μm) (12, 19). Therefore, affinity measurements support that KRIT1 is a SNX17 binding partner.
Point Mutations Disrupt Binding of SNX17 to KRIT1 NPXF2
We next set out to confirm the molecular details of the interaction between SNX17 and KRIT1 by point mutagenesis. It has previously been shown that mutations in the F3 lobe of SNX17-FERM negatively impact binding of target NPX(Y/F) sequences in P-selectin and APP (19). To test whether these SNX17 mutations also affect KRIT1 binding, we generated the same W321A, V380D, and M384E mutations in SNX17, which are located in the NPX(Y/F) binding site (Fig. 3). We expressed and purified the wild-type and mutant SNX17 PX-FERM protein from Escherichia coli and assessed binding to GST-KRIT1 NPXF2 peptide or GST alone by pulldown assay. We find that mutations of W321A, V380D, and M384E dramatically diminish SNX17 binding to the GST-KRIT1 NPXF2 peptide (Fig. 5, A and B).
FIGURE 5.

Evaluation of SNX17 interaction with KRIT1 NPXF2 by mutagenesis. A, WT or mutant SNX17 PX-FERM protein (His-tagged) is assessed for binding to GST-KRIT1 NPXF2 peptide immobilized on beads by pulldown assay and anti-His immunoblot. Mutation of SNX17 W321A, V380D, and M384E in the NPX(Y/F) binding site of SNX17 greatly disrupts interaction with KRIT1 NPXF2. Shown are blots/gels from one representative experiment. Equal GST loading is confirmed by Coomassie Blue staining. B, quantitation of binding data (n = 3). Binding is corrected for GST alone and presented as percentage of the wild-type binding. Significance (p value) of the mutants compared with wild type is indicated by asterisks: **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, no significant difference. C, binding of wild-type SNX17 PX-FERM (His-tagged) to GST-KRIT1 NPXF2 WT or mutant protein immobilized on beads is assessed by pulldown assay and anti-His immunoblot. Mutation of KRIT1 NPXF2 I229A and F234A leads to a significant loss in SNX17 binding. Equal GST loading is confirmed by Coomassie Blue staining. D, quantitation and significance (p values) of binding data (n = 3). C.B, Coomassie Blue; IB, immunoblot.
We next tested the effect of mutation of KRIT1 NPXF2 on SNX17 binding. It has been shown that a Tyr/Phe at the 0 position in other SNX17 target proteins is critical for interaction; for example, SNX17 binding is greatly diminished upon mutation of this residue to alanine in P-selectin (4), APP (5), and LDLR (8). Thus, we tested whether Phe-234 in KRIT1 NPXF-2 is required for SNX17 binding. We generated the mutant KRIT1 F234A peptide as a GST fusion protein, assayed SNX17 binding in vitro, and found that KRIT1 F234A mutant does not bind SNX17 (Fig. 5, C and D). Therefore, similar to other SNX17 binding proteins, KRIT1 Phe-234 at the 0 position of NPXF2 is critical for SNX17 binding. In addition, it has been shown that a hydrophobic residue at the −5 position is important for binding: mutation of this residue to alanine in APP (5), P-selectin (4), and Lrp1 (9) leads to a loss of SNX17 binding. To assess the importance of the −5 Ile-229 in KRIT1, we generated I229A mutant peptide as a GST fusion protein and find that binding to SNX17 is also significantly reduced (Fig. 5, C and D). Taken together with data from SNX17 mutagenesis, these results support the conclusion that SNX17 binds the KRIT1 NPXF2 motif via a canonical F3 PTB-like interaction similar to other SNX17 binding partners and confirms the molecular details of the interaction as revealed by the co-crystal structure.
Binding of Full-length KRIT1 to SNX17
The binding assays performed thus far have utilized GST fusion proteins derived from individual NPX(Y/F) motifs in KRIT1 (Figs. 1 and 5). We therefore tested whether the molecular details of the interaction hold in the context of full-length SNX17 and KRIT1 proteins. Toward this end, we expressed and purified full-length SNX17 protein from E. coli, immobilized the protein on glutathione beads, and performed pulldown experiments of full-length KRIT1-eGFP fusion protein heterologously expressed in HEK 293T cells. We find that wild-type KRIT1-eGFP is efficiently pulled down by full-length SNX17 but not GST alone, confirming that an interaction can be detected using intact proteins (Fig. 6A). We next asked whether mutation of the NPX(Y/F) binding site in the SNX17 F3 lobe affects KRIT1 binding and found that single point mutation of W321A, V380D, or M384E in full-length SNX17 leads to a loss of binding of full-length KRIT1-eGFP (Fig. 6, A and B). This result corroborates our finding that mutant SNX17 is unable to bind KRIT1-NPXF2 peptide in GST pulldown assays (Fig. 5, A and B).
FIGURE 6.

Binding of SNX17 to full-length KRIT1 is mediated by NPXF2 motif. A, full-length KRIT1-eGFP fusion protein is heterologously expressed in HEK293T cells and tested for pulldown by recombinant full-length SNX17 WT or mutants immobilized on beads as GST fusion protein. The presence of bound KRIT1-eGFP is visualized by anti-GFP immunoblot, and equal GST loading is verified by Coomassie Blue staining. B, quantitation of binding data (n = 3) in A calculated as the percentage of wild-type binding. Significance (p value) of the mutants compared with wild type is indicated by asterisks. There is no statistical significant difference in binding among the SNX17 mutants. C, mutation of F234A (in the NPXF motif) in full-length KRIT1-eGFP greatly reduces interaction with GST-SNX17 in GST pulldown assays. However, the mutant KRIT1-eGFP retains interaction with GST-HEG1 cytoplasmic tail. D, quantitation and significance (p values) of binding data (n = 5). Binding of KRIT1-eGFP F234A mutant is presented as percentage of wild-type binding to GST-SNX17 and to GST-HEG1. C.B, Coomassie Blue; IB, immunoblot.
We subsequently asked whether the NPXF2 motif in KRIT1 represents the major SNX17 binding site by generating KRIT1-eGFP F234A mutant in HEK293T cells. We find that the KRIT1-eGFP F234A mutant is deficient in SNX17 binding (Fig. 6, C and D); as predicted, this mutation has a similar effect on SNX17 binding when made in the context of GST-peptide fusion (Fig. 5, C and D). However, mutation of F234A in KRIT1 has no effect on the interaction of KRIT1 with another binding partner, HEG1 (which binds to the FERM domain of KRIT1), confirming that the mutant protein is otherwise functional and properly folded (Fig. 6, C and D). Because KRIT1 contains three NPX(Y/F) motifs, our finding that a single point mutation in the NPXF2 motif of intact KRIT1 is sufficient to dramatically disrupt binding to SNX17 supports the conclusion that KRIT1 contains a single SNX17 binding site.
DISCUSSION
An interaction between SNX17 and KRIT1 was preliminarily identified by yeast two-hybrid screening (22). In the current study, we employ biochemical and biophysical methods to conclusively demonstrate that this interaction between SNX17 and KRIT1 is direct. The KRIT1 N-terminus contains three NPX(Y/F) motifs (Fig. 1), and several binding partners have been identified for these sequences. ICAP1 binds NPXY1 (29), whereas CCM2 reportedly binds the NPXF2-NPXF3 region of KRIT1 (30, 31). Additionally, NPXF2-NPXF3 potentially engages the KRIT1 C-terminal FERM domain in an intramolecular interaction (27, 28, 50). In the current study, we map the binding site for SNX17 to KRIT1 NPXF2, raising the question of whether SNX17, CCM2, and/or the KRIT1 FERM domain compete for binding to KRIT1. We also establish that a single point mutation in the NPXF2 region is sufficient to greatly disturb binding of SNX17 to KRIT1. Taken together, our results allow for a more comprehensive understanding of the molecular properties of the interaction between SNX17 and KRIT1 and classify KRIT1 as a SNX17 binding partner.
The experimentally observed preference of SNX17 for KRIT1 NPXF2 is difficult to predict based solely on sequence analysis, because there is sequence diversity in many of the NPX(Y/F) motifs in SNX17 binding partners, and a comprehensive consensus sequence has not yet been established (Fig. 3C). It has been proposed that a large hydrophobic residue at the −5 position helps define a SNX17 binding motif (4, 5, 9, 19); however, KRIT1 NPXY1 and NPXF3 both contain a valine at this position (Fig. 1), which is also present in the SNX17 binding sites in integrin β1 (6, 7) and FEEL1 (11) (Fig. 3C). Other studies have proposed that sequences distal to the −5 xNPX(Y/F) residues of SNX17 partners confer binding specificity; for example, it has been suggested that a region as large as 32 residues defines the SNX17 binding site in the cytoplasmic tail of Lrp1 (51) and that the proline at the 7 position in P-selectin is important for binding (4). However, the GST pulldown assays performed here and elsewhere (for example, in Ref. 11) use minimal KRIT1 NPX(Y/F) peptide fragments that retain SNX17 binding specificity, suggesting that a larger recognition motif is not a universal feature of all SNX17 binding partners.
We demonstrate that the SNX17 FERM F3 lobe domain binds the KRIT1 NPXF2 using the same binding site previously described for other SNX17 binding partners like P-selectin and APP (19) and that there is little structural difference in the F3 lobe when SNX17 binds KRIT1 or P-selectin (Fig. 4). Sequence analysis shows that this binding site possesses a high degree of conservation throughout evolution and that the highly conserved surface extends beyond the direct peptide-contacting residues (Fig. 3F), which may account for the sequence diversity among NPX(Y/F) motifs that bind SNX17 (Fig. 3C). We also find that the binding affinity of the interaction between KRIT1 and SNX17 is similar to other SNX17 binding partners (low micromolar range). Taken together, these results raise the possibility that competition among SNX17 target proteins exists in the cell.
Sequence conservation of the SNX17 FERM domain also raises important questions about potential GTPase binding. SNX17 reportedly binds the small GTPase H-Ras via its FERM domain (12). Interestingly, a GTPase-FERM interaction has been demonstrated for another protein pair, Rap1A and KRIT1 (50). Another SNX protein, SNX27b, lacks a FERM domain but contains a RA domain, which has been shown to bind H-Ras (52). Because a common ubiquitin-like fold is shared between the F1 lobe of FERM domains and RA domains, it is not surprising that the modes of binding to FERM F1 and RA domains exhibited by Ras-like GTPases are highly similar (50). However, the co-crystal structure of KRIT1 FERM bound to Rap1 revealed that the interaction also involves a region of the F2 lobe, raising the possibility that GTPase specificity among FERM domains is determined by a multisite interaction surface (50). Therefore, it is an attractive hypothesis that SNX17 binds H-Ras via an extended interaction surface on its F1-F2 lobes, as has been suggested (19). However, in our alignment of SNX17 from 30 species, there is substantial surface conservation in this predicted GTPase-binding region (Fig. 7), albeit significantly less conservation than the surface involved in NPX(Y/F) binding (Figs. 2D and 3F). It is important to note that the region in the FERM F2 lobe responsible for mediating GTPase binding in KRIT1 is absent in the short F2 lobe of SNX17 (Fig. 7A). Thus, further studies will be needed to help address the mode by which SNX17 binds H-Ras and/or other small GTPases.
FIGURE 7.
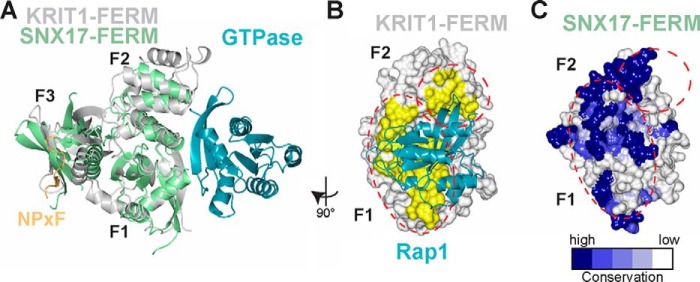
Mapping the potential GTPase binding site on SNX17-FERM. A, superposition of the FERM domains (by F1 lobes) of SNX17 (green) and KRIT1 (gray) reveals the potential binding site for a small GTPase (teal), based on the binding of Rap1 to KRIT1-FERM (PDB accession code 4DXA). The FERM domain lobes are labeled. The location of KRIT1-NPXF2 peptide (orange) bound to SNX17 F3 lobe is included to reflect that GTPase binding is expected to be unaffected by NPX(Y/F) binding. The views in B and C are related to A by a 90° rotation viewed from the right. B, surface representation of KRIT1-FERM domain (white) with Rap1 interface residues highlighted in yellow and indicated with red dotted lines. Rap1 is included in ribbon format (teal). C, SNX17-FERM structure is shown and colored according to surface conservation. The potential GTPase binding site (from superposition of the FERM domains) is indicated with red dotted lines.
SNX17 is part of a small subset of the larger SNX family that also contains SNX27 and SNX31. The distinctive feature of this subfamily is the inclusion of a FERM domain in addition to the PX domain (12). There is significant sequence similarity in the FERM domains of these proteins, specifically at the NPX(Y/F) binding site, suggesting that SNX27 and SNX31 may share binding partners with SNX17 (19). Along these lines, it has been demonstrated that SNX27 binds the intracellular NPXY-containing tail of the SNX17 binding protein APP with 28 μm affinity (19). Therefore, it is intriguing to postulate that SNX27 and/or SNX31 also bind KRIT1.
Molecular dissection of the interactions between KRIT1 and its binding partners has begun to establish roles for KRIT1 in cell signaling (36). For example, KRIT1 binding to ICAP1 promotes integrin activation (29). Additionally, binding to HEG1 and/or Rap1 localizes KRIT1 to cell-cell junctions where KRIT1 is implicated in cardiovascular development in zebrafish (35). Our study, which supports that KRIT1 is a direct binding partner of SNX17, raises new questions about the potential function of KRIT1 in the cell and what role SNX17 may play in KRIT1 signaling. SNX17 typically binds cell surface receptors to help regulate their transport through the endosomal pathway (1, 2). Therefore, the binding of SNX17 to KRIT1, a nonmembrane protein, is a deviation from the typical model. It is an intriguing possibility that KRIT1 serves as a molecular scaffold to link SNX17 to another cell surface protein to aid in its trafficking through the endosomal pathway and control its recycling and/or degradation. One possible candidate is the orphan receptor HEG1, whose cytoplasmic tail binds directly to the FERM domain of KRIT1 (35).
Alternatively, it is possible that binding to SNX17 affects KRIT1 localization, because KRIT1 binding partners are implicated in driving localization of KRIT1 in the cell. Although the overexpression of both KRIT1 and SNX17 leads to localization of a fraction of KRIT1 to SNX17 containing vesicles, presumably the early endosome (22), it remains to be tested whether endogenous KRIT1 can be found in endosomes. Most of the focus on SNX17 function is on its role in endocytic trafficking; however, a cytoplasmic pool of endogenous SNX17 has been detected, whose function has gone largely untested (8). Therefore, it remains possible that cytosolic SNX17 stabilizes a cytoplasmic fraction of KRIT1 or vice versa. In fact, the observation that many SNXs contain protein-protein interaction domains has suggested that SNX localization may be broadly controlled by its binding partners (1); along these lines, binding of SNX17 to P-selectin is potentially involved in SNX17 endosomal localization (19). Additionally, the discovery that SNX17 binds to small GTPases like H-Ras has led to the suggestion that SNX17 serves as signaling platforms in addition to its role in endosomal protein trafficking (53). Therefore, the SNX17/KRIT1 complex could play a role in KRIT1 signaling, either in the cytoplasm or associated with endosomal membranes.
Acknowledgments
We thank O. Fisher, X. Li, D. Calderwood, C. Huet-Calderwood, K. Draheim, and D. Iwamoto. Some data were collected at Advanced Proton Source Northeastern Collaborative Access Team (NECAT) Beamline.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 NS085078 (to T. J. B.).
The atomic coordinates and structure factors (code 4TKN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- SNX
- sorting nexin
- CCM
- cerebral cavernous malformation
- r.m.s.d.
- root mean square deviation
- APP
- amyloid precursor protein
- LDLR
- low density lipoprotein receptor
- PX
- phox homology
- FERM
- 4.1/ezrin/radixin/moesin
- PTB
- phosphotyrosine binding
- RA
- Ras association.
REFERENCES
- 1. Carlton J., Bujny M., Rutherford A., Cullen P. (2005) Sorting nexins: unifying trends and new perspectives. Traffic 6, 75–82 [DOI] [PubMed] [Google Scholar]
- 2. Cullen P. J. (2008) Endosomal sorting and signalling: an emerging role for sorting nexins. Nat. Rev. Mol. Cell Biol. 9, 574–582 [DOI] [PubMed] [Google Scholar]
- 3. Florian V., Schlüter T., Bohnensack R. (2001) A new member of the sorting nexin family interacts with the C-terminus of P-selectin. Biochem. Biophys. Res. Commun. 281, 1045–1050 [DOI] [PubMed] [Google Scholar]
- 4. Knauth P., Schlüter T., Czubayko M., Kirsch C., Florian V., Schreckenberger S., Hahn H., Bohnensack R. (2005) Functions of sorting nexin 17 domains and recognition motif for P-selectin trafficking. J. Mol. Biol. 347, 813–825 [DOI] [PubMed] [Google Scholar]
- 5. Lee J., Retamal C., Cuitiño L., Caruano-Yzermans A., Shin J. E., van Kerkhof P., Marzolo M. P., Bu G. (2008) Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 283, 11501–11508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Böttcher R. T., Stremmel C., Meves A., Meyer H., Widmaier M., Tseng H. Y., Fässler R. (2012) Sorting nexin 17 prevents lysosomal degradation of beta1 integrins by binding to the beta1-integrin tail. Nat. Cell Biol. 14, 584–592 [DOI] [PubMed] [Google Scholar]
- 7. Steinberg F., Heesom K. J., Bass M. D., Cullen P. J. (2012) SNX17 protects integrins from degradation by sorting between lysosomal and recycling pathways. J. Cell Biol. 197, 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burden J. J., Sun X. M., García A. B., Soutar A. K. (2004) Sorting motifs in the intracellular domain of the low density lipoprotein receptor interact with a novel domain of sorting nexin-17. J. Biol. Chem. 279, 16237–16245 [DOI] [PubMed] [Google Scholar]
- 9. van Kerkhof P., Lee J., McCormick L., Tetrault E., Lu W., Schoenfish M., Oorschot V., Strous G. J., Klumperman J., Bu G. (2005) Sorting nexin 17 facilitates LRP recycling in the early endosome. EMBO J. 24, 2851–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sotelo P., Farfán P., Benitez M. L., Bu G., Marzolo M. P. (2014) Sorting nexin 17 regulates ApoER2 recycling and reelin signaling. PLoS One 9, e93672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adachi H., Tsujimoto M. (2010) Adaptor protein sorting nexin 17 interacts with the scavenger receptor FEEL-1/stabilin-1 and modulates its expression on the cell surface. Biochim. Biophys. acta 1803, 553–563 [DOI] [PubMed] [Google Scholar]
- 12. Ghai R., Mobli M., Norwood S. J., Bugarcic A., Teasdale R. D., King G. F., Collins B. M. (2011) Phox homology band 4.1/ezrin/radixin/moesin-like proteins function as molecular scaffolds that interact with cargo receptors and Ras GTPases. Proc. Natl. Acad. Sci. U.S.A. 108, 7763–7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pearson M. A., Reczek D., Bretscher A., Karplus P. A. (2000) Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 101, 259–270 [DOI] [PubMed] [Google Scholar]
- 14. Chishti A. H., Kim A. C., Marfatia S. M., Lutchman M., Hanspal M., Jindal H., Liu S. C., Low P. S., Rouleau G. A., Mohandas N., Chasis J. A., Conboy J. G., Gascard P., Takakuwa Y., Huang S. C., Benz E. J., Jr., Bretscher A., Fehon R. G., Gusella J. F., Ramesh V., Solomon F., Marchesi V. T., Tsukita S., Tsukita S., Hoover K. B. (1998) The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem. Sci. 23, 281–282 [DOI] [PubMed] [Google Scholar]
- 15. Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 16. Chen W. J., Goldstein J. L., Brown M. S. (1990) NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J. Biol. Chem. 265, 3116–3123 [PubMed] [Google Scholar]
- 17. Uhlik M. T., Temple B., Bencharit S., Kimple A. J., Siderovski D. P., Johnson G. L. (2005) Structural and evolutionary division of phosphotyrosine binding (PTB) domains. J. Mol. Biol. 345, 1–20 [DOI] [PubMed] [Google Scholar]
- 18. García-Alvarez B., de Pereda J. M., Calderwood D. A., Ulmer T. S., Critchley D., Campbell I. D., Ginsberg M. H., Liddington R. C. (2003) Structural determinants of integrin recognition by talin. Mol. Cell 11, 49–58 [DOI] [PubMed] [Google Scholar]
- 19. Ghai R., Bugarcic A., Liu H., Norwood S. J., Skeldal S., Coulson E. J., Li S. S., Teasdale R. D., Collins B. M. (2013) Structural basis for endosomal trafficking of diverse transmembrane cargos by PX-FERM proteins. Proc. Natl. Acad. Sci. U.S.A. 110, E643–E652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seog D. H., Han J. (2008) Sorting nexin 17 interacts directly with kinesin superfamily KIF1Bβ protein. Korean J. Physiol. Pharmacol. 12, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu C., Ma M. H., Brown K. R., Geisler M., Li L., Tzeng E., Jia C. Y., Jurisica I., Li S. S. (2007) Systematic identification of SH3 domain-mediated human protein-protein interactions by peptide array target screening. Proteomics 7, 1775–1785 [DOI] [PubMed] [Google Scholar]
- 22. Czubayko M., Knauth P., Schlüter T., Florian V., Bohnensack R. (2006) Sorting nexin 17, a non-self-assembling and a PtdIns3P high class affinity protein, interacts with the cerebral cavernous malformation related protein KRIT1. Biochem. Biophys. Res. Commun. 345, 1264–1272 [DOI] [PubMed] [Google Scholar]
- 23. Yadla S., Jabbour P. M., Shenkar R., Shi C., Campbell P. G., Awad I. A. (2010) Cerebral cavernous malformations as a disease of vascular permeability: from bench to bedside with caution. Neurosurg. Focus 29, E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laberge-le Couteulx S., Jung H. H., Labauge P., Houtteville J. P., Lescoat C., Cecillon M., Marechal E., Joutel A., Bach J. F., Tournier-Lasserve E. (1999) Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat. Genet. 23, 189–193 [DOI] [PubMed] [Google Scholar]
- 25. Fisher O. S., Boggon T. J. (2014) Signaling pathways and the cerebral cavernous malformations proteins: lessons from structural biology. Cell Mol. Life Sci. 71, 1881–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Serebriiskii I., Estojak J., Sonoda G., Testa J. R., Golemis E. A. (1997) Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21–22. Oncogene 15, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 27. Béraud-Dufour S., Gautier R., Albiges-Rizo C., Chardin P., Faurobert E. (2007) Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. FEBS J. 274, 5518–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Francalanci F., Avolio M., De Luca E., Longo D., Menchise V., Guazzi P., Sgrò F., Marino M., Goitre L., Balzac F., Trabalzini L., Retta S. F. (2009) Structural and functional differences between KRIT1A and KRIT1B isoforms: a framework for understanding CCM pathogenesis. Exp. Cell Res. 315, 285–303 [DOI] [PubMed] [Google Scholar]
- 29. Liu W., Draheim K. M., Zhang R., Calderwood D. A., Boggon T. J. (2013) Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol. Cell 49, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zawistowski J. S., Stalheim L., Uhlik M. T., Abell A. N., Ancrile B. B., Johnson G. L., Marchuk D. A. (2005) CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 14, 2521–2531 [DOI] [PubMed] [Google Scholar]
- 31. Zhang J., Rigamonti D., Dietz H. C., Clatterbuck R. E. (2007) Interaction between krit1 and malcavernin: implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery 60, 353–359 [DOI] [PubMed] [Google Scholar]
- 32. Faurobert E., Albiges-Rizo C. (2010) Recent insights into cerebral cavernous malformations: a complex jigsaw puzzle under construction. FEBS J. 277, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gingras A. R., Puzon-McLaughlin W., Ginsberg M. H. (2013) The structure of the ternary complex of Krev interaction trapped 1 (KRIT1) bound to both the Rap1 GTPase and the heart of glass (HEG1) cytoplasmic tail. J. Biol. Chem. 288, 23639–23649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kleaveland B., Zheng X., Liu J. J., Blum Y., Tung J. J., Zou Z., Sweeney S. M., Chen M., Guo L., Lu M. M., Zhou D., Kitajewski J., Affolter M., Ginsberg M. H., Kahn M. L. (2009) Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat. Med. 15, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gingras A. R., Liu J. J., Ginsberg M. H. (2012) Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J. Cell Biol. 199, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Draheim K. M., Fisher O. S., Boggon T. J., Calderwood D. A. (2014) Cerebral cavernous malformation proteins at a glance. J. Cell Sci. 127, 701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luft J. R., DeTitta G. T. (1999) A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Crystallogr. D 55, 988–993 [DOI] [PubMed] [Google Scholar]
- 38. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 39. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., Sliz P. (2013) Collaboration gets the most out of software. Elife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang J., Clatterbuck R. E., Rigamonti D., Chang D. D., Dietz H. C. (2001) Interaction between krit1 and icap1 α infers perturbation of integrin β1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum. Mol. Genet. 10, 2953–2960 [DOI] [PubMed] [Google Scholar]
- 44. Stockinger W., Sailler B., Strasser V., Recheis B., Fasching D., Kahr L., Schneider W. J., Nimpf J. (2002) The PX-domain protein SNX17 interacts with members of the LDL receptor family and modulates endocytosis of the LDL receptor. EMBO J. 21, 4259–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamada K., Shimizu T., Matsui T., Tsukita S., Hakoshima T. (2000) Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 19, 4449–4462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bansal A., Gierasch L. M. (1991) The NPXY internalization signal of the LDL receptor adopts a reverse-turn conformation. Cell 67, 1195–1201 [DOI] [PubMed] [Google Scholar]
- 49. Lawrence M. C., Colman P. M. (1993) Shape complementarity at protein-protein interfaces. J. Mol. Biol. 234, 946–950 [DOI] [PubMed] [Google Scholar]
- 50. Li X., Zhang R., Draheim K. M., Liu W., Calderwood D. A., Boggon T. J. (2012) Structural basis for small G protein effector interaction of Ras-related protein 1 (Rap1) and adaptor protein Krev interaction trapped 1 (KRIT1). J. Biol. Chem. 287, 22317–22327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Farfán P., Lee J., Larios J., Sotelo P., Bu G., Marzolo M. P. (2013) A sorting nexin 17-binding domain within the LRP1 cytoplasmic tail mediates receptor recycling through the basolateral sorting endosome. Traffic 14, 823–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Balana B., Bahima L., Bodhinathan K., Taura J. J., Taylor N. M., Nettleton M. Y., Ciruela F., Slesinger P. A. (2013) Ras-association domain of sorting nexin 27 is critical for regulating expression of GIRK potassium channels. PLoS One 8, e59800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ghai R., Collins B. M. (2011) PX-FERM proteins: A link between endosomal trafficking and signaling? Small GTPases 2, 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]