

Abstract
The pharmacokinetics of an extended-release (XR) formulation of ciprofloxacin has been compared to that of the immediate-release (IR) product in healthy volunteers. The only significant difference in pharmacokinetic parameters between the two formulations was seen in the rate constant of absorption, which was approximately 50% greater with the IR formulation. The geometric mean plasma ciprofloxacin concentrations were applied to an in vitro pharmacokinetic-pharmacodynamic model exposing three different clinical strains of Escherichia coli (MICs, 0.03, 0.5, and 2.0 mg/liter) to 24 h of simulated concentrations in plasma. A novel mathematical model was derived to describe the time course of bacterial CFU, including capacity-limited replication and first-order rate of bacterial clearance, and to model the effects of ciprofloxacin concentrations on these processes. A “mixture model” was employed which allowed as many as three bacterial subpopulations to describe the total bacterial load at any moment. Comparing the two formulations at equivalent daily doses, the rates and extents of bacterial killing were similar with the IR and XR formulations at MICs of 0.03 and 2.0 mg/liter. At an MIC of 0.5 mg/liter, however, the 1,000-mg/day XR formulation showed a moderate advantage in antibacterial effect: the area under the CFU-time curve was 45% higher for the IR regimen; the nadir log CFU and 24-h log CFU values for the IR regimen were 3.75 and 2.49, respectively; and those for XR were 4.54 and 3.13, respectively. The mathematical model explained the differences in bacterial killing rate for two regimens with identical AUC/MIC ratios.
Ciprofloxacin (Cipro), the first broad-spectrum oral fluoroquinolone, was approved by the Food and Drug Administration in 1987. An intravenous formulation was approved in 1991, and an oral suspension formulation followed in 1997. Recently, an extended-release (XR) formulation of ciprofloxacin has been developed by Bayer Healthcare (Cipro XR). Pharmacokinetic studies of XR ciprofloxacin in healthy volunteers have demonstrated a significant difference in peak concentrations in plasma between the XR and immediate-release (IR) formulations: peak concentrations were 40 to 50% higher with the XR formulation, while areas under the concentration-time curve (AUCs) were comparable to those observed with the IR formulation (H. Stass, J. Nagelschmitz, E. Brandel, et al., Abstr. Am. Fed. Med. Res. Cong., abstr. 24 and 25, 2002).
The objectives of this analysis were threefold. The first objective was to characterize the pharmacokinetics of multiple oral doses of ciprofloxacin when given as an IR and an XR formulation in daily doses of either 500 mg or 1,000 mg in healthy male volunteers. The second objective was to apply the geometric mean plasma concentration profiles from the healthy-volunteer study to an in vitro pharmacokinetic-pharmacodynamic (PK-PD) model exposing three different clinical strains of Escherichia coli to 24 h of simulated concentrations in plasma. Finally, the third objective was to develop a mathematical model to describe bacterial replication and the intrinsic rate of bacterial clearance and to model the effects of ciprofloxacin concentrations on these processes.
MATERIALS AND METHODS
PK study design.
Two randomized, single-center, two-way-crossover, multiple-dose studies were conducted by Bayer Healthcare in which ciprofloxacin was given to Caucasian male volunteers. Both of the two studies evaluated a total of 19 healthy volunteers. In a randomized sequence, volunteers received either 250 mg of IR ciprofloxacin twice daily and 500 mg of XR ciprofloxacin once daily or 500 mg of IR ciprofloxacin twice daily and 1,000 mg of XR ciprofloxacin once daily. All doses were given for five consecutive study days under fasting conditions. Urine and plasma samples were taken on the first and on the last day of treatment. Twenty-nine plasma samples were collected: 14 samples on day 1 and 15 samples on day 5 of the study (predose and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 16, 24, 96, 96.5, 97, 97.5, 98, 98.5, 99, 99.5, 100, 102, 104, 108, 112, 120, and 124 h post-first dose). Urine was collected over two 24-h periods, with four collection periods during day 1 (0 to 4, 4 to 8, 8 to 12, and 12 to 24 h postdose) and five collection periods during day 5 of treatment (96 to 100, 100 to 104, 104 to 108, 108 to 120, and 120 to 124 h postdose). Heparinized plasma and urine were stored in plastic vials at −18°C until analysis. Analysis of specimens was performed using validated high-performance liquid chromatography procedures with fluorescence detection (lower limit of quantitation, 10 mg/liter; interday coefficient of variation, <9%) (3).Ofloxacin was used as the internal standard. Quality control samples produced out of blank matrix spiked with a known concentration of the analyte were assessed together with the study samples to control validity of the data. The interday precision and accuracy for the high-performance liquid chromatography assay were 2.4 and 2.1%, respectively, as determined from the quality control samples.
In vitro PD study.
An in vitro PK-PD model was conducted by Bayer Healthcare using three different clinical strains of E. coli for which the MICs were 0.03, 0.5, and 2.0 mg/liter. MICs were determined according to NCCLS broth microdilution methods at an inoculum of 105 CFU/ml. None of the isolates were extended-spectrum β-lactamase producers, as they were susceptible to the cephalosporins. Bacterial inocula were exposed to ciprofloxacin (Bayer Healthcare, Wuppertal, Germany) in concentrations similar to those achieved in the central compartment in the human body. Mathematical corrections for dilution were not made based on our previous work showing that demonstrated dilution did not impact antibacterial effect (A. Dalhoff, A. MacGowan, O. Carrs, et al., Abstr. 43rd Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-1147, 2003). The geometric mean concentrations in plasma from the first 24 h of the four-dosage regimens in the healthy-volunteer trial (ciprofloxacin, 250 mg IR twice daily, 500 mg XR once daily, 500 mg IR twice daily, and 1,000 mg XR once daily) were simulated in the in vitro model. The highest MIC, 2.0 mg/liter, was only tested against the two higher-dosage regimens (ciprofloxacin total daily dose of 1,000 mg).
For the experimental evaluation of the PK-PD relationship of ciprofloxacin against E. coli, a slightly modified in vitro method, that of Grasso et al. (9), was used. The experimental setting consisted of a central compartment without a separating membrane (calibrated Erlenmeyer flasks with a total volume of 300 ml and containing 100 ml of medium) into which the antibiotic is pumped via programmable pumps until the maximum serum concentration to be simulated is reached. Thereafter, antibiotic-free medium is pumped into the central compartment and is continuously eliminated in parallel to mimic half-life values. The variability between the resulting in vitro profiles compared to the desired human target profiles were extremely low, <4% deviation at any given time. Flasks were maintained at 37°C and agitated on a rotary shaker. Control growth in the absence of antibiotic was studied in the same model.
E. coli was grown in brain heart infusion broth (Oxoid, Wesel, Germany). The initial inoculum into the model for the ciprofloxacin 500-mg total daily dose regimens was 108.2 CFU, and the inoculum for the ciprofloxacin 1,000 mg total daily dose regimens was 107.58 CFU. Viable bacterial counts (CFU) were determined on agar plates containing charcoal, which completely adsorbs fluoroquinolones. The lower limit of detectability was 100 CFU/ml. CFU counts were obtained at 16 time points: 0, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 18, 20, 22, and 24 h. At the same time points as those for quantification of CFU, the antibiotic concentrations were measured by a conventional cup-agar diffusion test with Bacillus subtilis spore suspension as the indicator organism (14). Postexposure MIC testing was performed, and changes in MICs were not observed.
PK and PD modeling methods.
The concentrations of ciprofloxacin in plasma and urine and the log10-transformed counts of CFU were characterized by fitting candidate PK-PD models to the data, using a maximum a posteriori-Bayesian parameter value estimator available in Adapt II (User's guide for release 4, Biomedical Simulations Resource, University of Southern California, Los Angeles). A PK-PD model was derived by fitting smooth curves through the experimental data, resulting in the computation of model parameter values which summarize and characterize the observed data and which are more amenable to hypothesis testing. Two PK-PD models were explored: one in which drug effect was hypothesized to inhibit bacterial replication (A. Meagher, A. Forrest, A. Dalhoff, et al. Abstr. 42nd Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-1257, 2002) and one in which drug effect enhanced the rate of bacterial killing. Model discrimination was accomplished using the rule of parsimony (10) and Akaike's information criterion (1).
Experimental measures that were BLQ.
A number of drug plasma samples or counts of number of CFU, for which the signal (e.g., peak area)-to-noise (random assay error) ratio was low, were judged to be below the limit of quantitation (BLQ). These plasma concentration values or CFU counts were originally reported as <0.01 mg/liter or <100 CFU/ml, respectively. The true values for these samples were actually somewhere between zero and the lower limit of quantitation of the assay. It is inappropriate to assign these samples a value of zero, which is biased low, and it is also suboptimal to discard these data as they contain usable information. Our approach for concentrations in plasma was to use 0.0 mg/liter for predose samples. Postdose plasma samples that were BLQ were entered as either 0.0 or 0.005 mg/liter (the midpoint between 0.0 and 0.01 mg/liter), whichever was closer to the values predicted by extrapolation of the other detectable terminal concentrations in plasma. CFU counts that were below 100 were entered as missing.
Procedures for declaring outliers.
Individual plasma or urine drug concentrations, suspected to be outliers, were tested as follows. The data set was fit with and without the suspect value. If the residual of the observation (the difference between fitted and observed values) was at least 3 standard deviations (SDs) of the measurement, and if the trajectory of the fitted line changed when the value was removed, the point was declared an outlier. No CFU values were excluded as outliers.
Residual (“error”) variance models.
The empirical variance model assumed that the random errors in measurements of concentrations of ciprofloxacin in plasma and urine were similar for all of the subjects in the study and that the residual (error) SDs of the observations (σ) were linearly related to the true values (Y): σ = SDslope Y + SDintercept, in which SDslope and SDintercept are the variance parameters. SDintercept is the asymptotic minimum σ (the value as Y approaches zero) and is mainly a measure of sensitivity, and the SDslope is the asymptotic minimum coefficient of variation (the value as Y approaches infinity) and is mainly a measure of precision. In cases where the intercept approaches zero, this relationship collapses to a constant coefficient of variance (CV) model (with SDslope equaling the CV). Even with a nonzero intercept, at values of Y much greater than the SDintercept, the SDslope approximates the CV. In cases where SDslope approaches zero, the relationship collapses to a homoscedastic variance model (with SDintercept equaling the observation SD). The initial empirical estimates for the variance parameters were based on the assay performance. Later in the process, the values for the variance parameters were fitted (determined from the data). The choice was made to model the CFU counts as base 10 log values. This transforms weights data like assuming a constant CV does and no further weighting was used.
SHAM analysis.
A number of data sets were not amenable to mathematical modeling. This was caused by apparent multiphasic absorption. These data were analyzed using slope, height, area, and moment (SHAM) analysis, which is sometimes referred to as noncompartmental analysis (4, 7, 10). The AUC from time zero to 24 h (AUC0→24) for plasma concentration data was determined using the linear-trapezoidal rule. The terminal rate constant of elimination (β) was determined by linear least-squares regression of the last three detectable concentrations in plasma. The AUC from 24 h to infinity (AUC24→∞) was computed as C24/β, where C24 is the drug concentration in plasma at 24 h. The AUC0→∞ was computed as the sum of AUC0→24 and AUC24→∞. The percentage of the AUC0→∞ that was extrapolated was computed as 100 times AUC24→∞ divided by AUC0→∞. Oral plasma clearance (CLT/F) was computed as dose/AUC0→∞. Renal clearance (CLR) was computed as amount of drug (in milligrams) excreted in 24 h divided by AUC0→24.
Summary statistics, including mean, median, SD, and CV, were determined using Systat computer software (16).
RESULTS
PK study subjects and in vitro PD model.
Data for concentrations in plasma and urine were obtained from PK studies of healthy volunteers comparing ciprofloxacin in 500-mg once-daily XR and 1,000-mg once-daily XR formulations with two corresponding twice-daily IR formulation regimens (H. Stass, J. Nagelschmitz, E. Brandel, et al., Abstr. Am. Fed. Med. Res. Cong., abstr. 24, 2002; H. Stass, J. Nagelschmitz, E. Brandel, et al., Abstr. Am. Fed. Med. Res. Cong., abstr. 25, 2002). Volunteer demographics are shown in Table 1. No significant differences were noted between the two groups. Serial determinations of CFU counts of three strains of E. coli following exposure to the geometric mean plasma ciprofloxacin concentrations to the four dosage regimen profiles were obtained. These data were used to determine the PK-PD relationship between concentrations of ciprofloxacin in plasma and net bacterial growth and eradication.
TABLE 1.
Volunteer demographics in this study
| Study group and parametera | Mean | CV (%) | Median | Range |
|---|---|---|---|---|
| 250 mg IR BID and 500 mg XR QD (n = 19) | ||||
| Age (yr) | 31.9 | 25.6 | 31.0 | 19-53 |
| Weight (kg) | 78.9 | 14.7 | 80 | 60-98 |
| Height (cm) | 181 | 4.90 | 180 | 170-199 |
| Dose (mg/kg) | 3.24 | 14.8 | 3.13 | 2.55-4.17 |
| 500 mg IR BID and 1,000 mg XR QD (n = 19) | ||||
| Age (yr) | 32.5 | 21.2 | 33.0 | 23-49 |
| Weight (kg) | 82.0 | 11.1 | 80.0 | 61-100 |
| Height (cm) | 180 | 1.90 | 181 | 172-187 |
| Dose (mg/kg) | 6.17 | 11.9 | 6.25 | 5.00-8.20 |
BID, twice a day; QD, once a day.
Final PK structural model.
A two-compartment PK model was employed for this analysis, and the model fit the data very well (median r2 = 0.996). In this model, drug is administered into an absorptive compartment with the amount of drug available systemically dependent on F, the oral bioavailability of the drug. After a lag time (TLag), ciprofloxacin is absorbed per first-order rate constant (ka) into the central compartment (of apparent volume, V1). Drug in the central compartment equilibrates via a distributional clearance (CLd) with drug in the peripheral compartment (of apparent volume, Vp), and is eliminated from the central compartment by both renal and nonrenal clearance (CLR and CLNR, respectively). Because ciprofloxacin was administered as an oral formulation, the fitted volumes and clearances are conditioned on F, which could not be estimated in a study of this design. Other PK parameter values were derived from the fitted parameters. For example, the oral volume of distribution at steady state, VSS/F = V1/F+Vp/F, was calculated.
Fitted PK parameter values.
Studied doses within subject-regimen were often unable to be comodeled due to apparent within-subject interoccasion variability. For this reason, each dosing event on days 1 and 5 of the healthy-volunteer study for each of the four dosing regimens was modeled as a separate data set, and each subject had two or four data sets, depending upon dosage regimen. Therefore, a total of 218 data sets were created and modeled. Data were not available for two subjects in the XR-ciprofloxacin 500-mg group and for one subject in the IR-ciprofloxacin 500-mg group. For two separate volunteers, one of the four data sets was considered an outlier and was censored. In these cases plasma drug concentrations were suspiciously low and it appeared as though study drug was not administered. Ten (10) of the 218 subject data sets (two in the 250-mg IR-ciprofloxacin twice-daily group, one in the 500-mg XR-ciprofloxacin once-daily group, four in the 500-mg IR-ciprofloxacin twice-daily group, and three in the 1,000-mg XR-ciprofloxacin once-daily group) could not be fitted adequately by the PK model due to apparent multiphasic absorption. For these cases, SHAM analysis was used to calculate the total and renal clearance of ciprofloxacin. Table 2 summarizes the fitted PK parameter values. Although there appears to be an inverse relationship in values of V1 and Vp between the IR and XR formulations, this difference is most likely to be a modeling artifact. Supportive evidence is the opposing trend in CLd between formulations and the relative constancy of VSS across all four regimens. Even if these differences were real, they would be of no clinical significance. The CLR and total clearances and TLag values do not differ between dosage regimens. However, as expected, the ka is approximately 50% greater in the IR than in the XR formulation. Figure 1 depicts the PK profiles for the four ciprofloxacin dosage regimens.
TABLE 2.
Fitted PK parameters
| Parametera | 250 mg IR BID
|
500 mg XR QD
|
500 mg IR BID
|
1,000 mg XR QD
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Median | CV (%) | Mean | Median | CV (%) | Mean | Median | CV (%) | Mean | Median | CV (%) | |
| V1/F (liters) | 47.4 | 41.7 | 64.5 | 62.7 | 66.8 | 45.6 | 56.4 | 55.4 | 66.8 | 80.9 | 77.5 | 43.3 |
| Vp/F (liters) | 203 | 197 | 25.6 | 174 | 166 | 31.8 | 213 | 216 | 33.1 | 198 | 191 | 25.9 |
| Vss/F (liters) | 250 | 242 | 21.1 | 237 | 217 | 25.9 | 269 | 277 | 26.6 | 279 | 277 | 18.4 |
| CLd/F (liters/h) | 59.8 | 58.5 | 39.5 | 34.6 | 31.1 | 46.8 | 58.0 | 55.0 | 48.6 | 32.7 | 29.7 | 39.2 |
| CLt/F (liters/h) | 63.2 | 61.1 | 26.1 | 68.3 | 64.6 | 29.5 | 63.0 | 62.1 | 27.2 | 68.8 | 60.8 | 38.0 |
| CLR (liters/h) | 21.5 | 21.5 | 22.9 | 22.9 | 22.2 | 17.7 | 23.2 | 23.8 | 22.4 | 23.8 | 24.1 | 16.4 |
| TLag (h) | 0.551 | 0.386 | 172 | 0.379 | 0.380 | 69.5 | 0.708 | 0.471 | 146 | 0.556 | 0.464 | 66.8 |
| ka (h−1) | 0.963 | 0.689 | 89.0 | 0.476 | 0.444 | 41.9 | 0.718 | 0.598 | 77.2 | 0.425 | 0.405 | 31.1 |
V1/F, volume of distribution of the central compartment; F, fraction of administered dose systemically available (unidentifiable); Vp/F, volume of distribution of the peripheral compartment; Vss/F, V1/F plus Vp/F; CLd/F, distributional clearance; CLt/F, total clearance; CLR, renal clearance; TLag, lag time before onset of absorption; ka, absorption rate constant.
FIG. 1.

Ciprofloxacin PK profiles for four different dosage regimens. The solid symbols represent the geometric mean concentrations in plasma surrounded by a ±1 SD envelope (dashed lines). The solid line represents the empirical connection from point to point.
Final PD model.
The PD model derived for this analysis is depicted in Fig. 2. The model fit the data very well (see r2 values in Table 3). The total concentration of bacteria in the body was the net result of a saturable capacity-limited mechanism for bacterial replication and a first-order process characterizing the rate of death in a drug-free environment (natural death). The modeled drug effect was to increase the rate constant for bacterial death (Kd) according to a Hill-type model. The other model considered, which assumed drug effect to decrease the rate of replication, yielded similar goodness of fit but was inferior by Akaike's information criterion and generated greater variability in final PD parameter values within strains of bacteria and across dosing regimens. Results from this model are not reported here.
FIG. 2.

Final PD model, where the bacterial load is represented as a pool within the body in which replication is saturable and driven by the concentration of viable bacteria in the pool. Clearance from the body is by a first-order process where the drug effect is to enhance the natural death rate.
TABLE 3.
Fitted PD parameters for four ciprofloxacin dosage regimens against E. coli strains for which the MICs differ
| Parametera | Median (range) for strains for which the MIC (mg/liter) is:
|
||
|---|---|---|---|
| 0.03 (n = 4) | 0.5 (n = 4) | 2.0 (n = 2) | |
| r2 | 0.985 (0.975-0.997) | 0.946 (0.896-0.984) | 0.763 (0.663-0.863) |
| VGmax (CFU/ml/h) | 2.45 × 106 (2.00 × 106-2.87 × 106) | 4.56 × 107 (3.39 × 107-5.71 × 107) | 3.09 × 107 (2.75 × 107-3.43 × 107) |
| CFUm (CFU/ml) | 3.40 × 105 (2.79 × 105-4.34 × 105) | 3.55 × 107 (2.78 × 107-4.36 × 107) | 5.66 × 107 (4.27 × 107-7.04 × 107) |
| Kd (h−1) | 0.297 (0.279-0.320) | 0.274 (0.222-0.308) | 0.199 (0.106-0.293) |
| SITms | 0.040 (0.034-0.040) | 0.099 (0.096-0.106) | 1.89 (1.88-1.91) |
| SITmi | 1.79 (1.17-2.26) | ||
| H | 2.47 (2.44-2.47) | 2.31 (1.00-3.21) | 1.80 (1.46-2.13) |
| Emax | 28.7 (23.9-36.8) | 27.7 (8.39-49.7) | 43.2 (39.5-46.8) |
| IC4 (CFU/ml) | 1.02 × 108 (7.36 × 107-1.27 × 108) | 5.88 × 107 (9.08 × 106-1.39 × 108) | 5.57 × 107 (5.04 × 107-6.09 × 107) |
| IC5 (CFU/ml) | 3.86 × 106 (9.51 × 105-5.11 × 107) | ||
SITms, median effect serum inhibitory titer for a relatively sensitive subpopulation; SITmi, median effect serum inhibitory titer for a relatively intermediately sensitive subpopulation; H, Hill’s constant; Emax, maximum fractional increase in Kd; IC4, initial condition of the sensitive-subpopulation compartment; IC5, initial condition of the intermediate-subpopulation compartment.
The mathematical model for a bacterial inoculum of homogenous sensitivity is shown below.
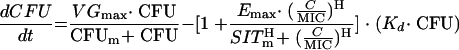 |
This differential equation describes the time course of the total bacterial load (CFU in CFU per milliliter). The rate of replication is parameterized by the maximal velocity of bacterial growth (VGmax) (CFU per milliliter per hour) and the median effect CFU (CFUm) (in CFU per milliliter). The CFUm is the CFU at which the rate of replication is half maximal. The rate of bacterial death due to natural elimination processes (i.e., non-drug-related elimination) is characterized by Kd (where units are hour−1), the first-order bacterial elimination rate constant. In any instant in time, these rates are driven by the current total bacterial load. The effect of ciprofloxacin, which is to increase Kd by a Hill-type function, is contained within the parenthetical phrase. The time course of concentrations of drug (C) (in milligrams per liter), scaled to the nominal MIC, is the forcing function for drug effect. Under certain assumptions, C/MIC is a reasonable estimate of the inverse serum inhibitory titer (SIT). The SITm is the median effect value for C/MIC, the value at which the drug effect is half maximal. Emax is the maximal fractional increase in Kd (i.e., if Emax is 0.3, Kd is increased by 30%). Hill's constant (H) affects the shape of the curve. As H increases, the slope of the PD function also increases.
Neither this equation nor any other considered that assumed a homogenous drug sensitivity throughout the inoculum was able to fit the experimental data. Therefore, we employed a “mixture model” in which the total bacterial load at any moment is characterized as a mixture of as many as three bacterial subpopulations. These populations were allowed to differ only in their initial concentrations (CFU at time zero in the experiment) and in their susceptibility to drug (SITm, the value of C/MIC at which the replication rate is reduced by half). Thus, the final model included three values for SITm: SITms is for the most sensitive subpopulation, SITmi is for the intermediately sensitive subpopulation, and SITmr is for the relatively resistant subpopulation. These SITm values are approximately equal to the MIC for the subpopulation divided by the nominal MIC of the initial inoculum.
Fitted PD parameter values.
The final fitted PD parameter values are summarized in Table 3. IC4 and IC5 are the fitted baseline initial conditions (in CFU per milliliter) for the sensitive and intermediate subpopulations, respectively. Ten experiments were performed: the three bacterial strains were exposed to the geometric mean plasma concentration profiles for each of the four dosage regimens, except for the E. coli for which the MIC was highest, 2.0 mg/liter, which was exposed only to the 1,000-mg total daily dose regimens. None of the experiments required three bacterial subpopulations to fully describe the PD profile of ciprofloxacin. Two bacterial subpopulations were required to describe the dynamics for all the regimens against the E. coli strain for which the MIC was 0.5 mg/liter, and one bacterial population was adequate for all other regimens at all tested MICs.
The PD profiles for the four ciprofloxacin dosage regimens versus E. coli at three different ciprofloxacin MICs (0.03, 0.5, and 2.0 mg/liter) are shown in Fig. 3 through 5. As can be seen graphically, the model fit the data extremely well. In general, comparing the two formulations at equivalent daily doses, the once-daily XR formulation provided a net bacterial kill that was at least as rapid and extensive. At the lowest MIC, 0.03 mg/liter (Fig. 3), near-maximal activity was exhibited with all four dosage regimens against E. coli. Bacterial CFU were at or below the limit of detection (100 CFU/ml) at approximately 5 h with all studied regimens. At the next highest MIC of 0.5 mg/liter (Fig. 4), for the two regimens with 500 mg/day, the shape of the curves were different but the overall effects were similar. For the two regimens with the total daily dose of 1,000 mg, however, the XR formulation showed a moderate advantage in antibacterial effect. For these two regimens, the area under the CFU-time curve was 45% higher for the IR regimen; the nadir log CFU values, for IR versus XR formulations, respectively, were 3.75 and 2.49; and the 24-h log CFU values were 4.54 and 3.13, respectively. If the experiment is extended to 48 h through simulation, only the regimen with 1,000 mg of XR ciprofloxacin once-daily would be predicted to drive the CFU to nondetectable concentrations. Neither the IR nor XR formulation at the highest daily dose, 1,000 mg of ciprofloxacin, was able to achieve more than a 1-log kill during the 24-h experiment with the E. coli strain for which the MIC was 2.0 mg/liter (Fig. 5).
FIG. 3.

PD profiles with E. coli for which the MIC was 0.03 mg/liter. The symbols represent the observations, and the solid lines depict the fitted function for each of the four dosage regimens (median r2 = 0.985).
FIG. 5.

PD profiles with E. coli for which the MIC was 2.0 mg/liter. The symbols represent the observations, and the solid lines depict the fitted function for each of the four dosage regimens (median r2 = 0.763).
DISCUSSION
This data analysis represents our first publication using a new model we have developed for PK and PD of bacteria in vitro and in animal models and should prove useful for modeling human trials. The main characteristic of this approach is describing the initial inoculum of bacteria as a mixture of subpopulations, each with different susceptibilities to a drug. Bacterial replication is modeled as a capacity-limited, saturable process, and rate of clearance of bacteria from the system is modeled as a first-order process. Drug effects can be hypothesized to either inhibit replication or enhance the rate of kill. One major difficulty with these models is their complexity and that no single experiment within a group of experiments is informative about all the parameters in the model. One of our observations is that a large range of drug exposures, including a drug-free control and a large dose that will elicit near-maximal effect, must be considered, and all the experiments with a given bacterial strain should be comodeled and analyzed simultaneously. This is an exceedingly difficult analysis to perform, and we are currently exploring two general approaches to accomplish this task. The first approach is to treat each separate experiment as if it were observed in a separate “subject” and fit the sample of subjects using a population PK-PD tool, such as iterative two-stage analysis, IT2S (15). The other approach could be thought of as a pooled approach in which all of the experiments using a given strain are comodeled as if they had been studied in the same subject multiple times.
These guidelines would have been beneficial in this study in two cases in particular. The first is the experiment performed with the bacterial strain for which the MIC was 0.03 mg/liter. All the drug concentrations for all four tested regimens were far enough above the MIC throughout the study period that the SITms was poorly estimated. The median value of 0.04 mg/liter should simply be taken to be a low value, as there is no way to determine the value satisfactorily from this experiment. The other case is the experiment with the bacterial strain for which the MIC was 2.0 mg/liter. Drug concentrations for the two tested regimens were never high enough above the nominal MIC to allow a precise estimation of Emax, and the fitted value of 43 (i.e., a maximal Kd value that is 44-fold higher than the initial Kd) should again be considered with caution.
Fluoroquinolones are considered to work primarily in a concentration-dependent manner (2, 5, 11, 13). Both AUC/MIC ratios and peak/MIC ratios have been used to describe fluoroquinolone PDs in human and animal models (8, 12). Optimizing these ratios and providing more-rapid bacterial eradication has the potential to improve outcomes as well as prevent the emergence of resistance. There are no clinically significant differences in either PK parameters or AUC between the XR and IR ciprofloxacin formulations (H. Stass et al., Abstr. Am. Fed. Med. Res. Cong., abstr. 24 and 25). As expected, the rate constant of absorption was found to be lower with the XR formulation. What may be clinically significant, however, are the peak concentrations achieved with the newly developed XR formulations. Overall, peak ciprofloxacin concentrations in plasma are approximately 40% higher with the 500-mg once-daily XR formulation and approximately 50% higher with the 1,000-mg once-daily XR formulation than with IR regimens with the same total daily dose given in two separate doses (H. Stass et al., Abstr. Am. Fed. Med. Res. Cong., abstr. 24 and 25). Despite comparable AUC values when the total daily doses are the same, the XR once-daily formulation was generally associated with greater in vitro bacterial killing in this model.
At this time, Cipro XR is approved by the U.S. Food and Drug Administration only for use in urinary tract infections. The concentration of ciprofloxacin achieved in urine is much higher than that in plasma. The present study exposed inocula of bacteria to exposure profiles consistent with concentrations in plasma. We would expect this model to be reasonably predictive of outcomes for infection sites that achieve similar concentrations to that seen in plasma. This probably would include complicated urinary tract infections, where there is a significant tissue component to the infection. If you envision a simple urinary tract infection as being an infection of bacteria in urine only, then all of the regimens considered in this analysis would be predicted to provide maximum rates of kill and rapid eradication of susceptible bacteria in urine.
This model predicts more rapid bacterial killing for the once-daily XR formulation than for the IR product given in two divided doses despite similar AUC values. The two main hypotheses for these findings cannot be discriminated by this study data. One of these hypotheses is that the once-daily XR formulation yields a higher AUC during a portion of the 24-h dosing period and would have increased activity against resistant subpopulations. The second mechanism would be a relationship between rate of kill and peak drug concentration. We believe the first of these hypotheses to be more likely. That is, the once-daily XR formulation, compared to the equivalent twice-daily regimen, provided maximal AUC values during the period when the CFU counts were highest. The mathematical model suggests higher AUCs early in the dosing interval results in greater bacterial killing of both the sensitive and the more resistant bacterial subpopulations.
FIG. 4.

PD profiles with E. coli for which the MIC was 0.5 mg/liter. The symbols represent the observations, and the solid lines depict the fitted function for each of the four dosage regimens (median r2 = 0.946).
REFERENCES
- 1.Akaike, H. 1979. A Bayesian extension of the minimum AIC procedure of autoregressive model fitting. Biometrika 66:237-242. [Google Scholar]
- 2.Andes, D., and W. A. Craig. 2002. Animal model pharmacokinetics and pharmacodynamics: a critical review. Int. J. Antimicrob. Agents 19:261-268. [DOI] [PubMed] [Google Scholar]
- 3.Bannefeld, K. H., H. Stass, and G. Blaschke. 1997. Capillary electrophoresis with laser-induced fluorescence detection, an adequate alternative to high-performance liquid chromatography, for the determination of ciprofloxacin and its metabolite desethyleneciprofloxacin in human plasma. J. Chromatogr. B 692:453-459. [DOI] [PubMed] [Google Scholar]
- 4.Caprani, O., E. Sveinsdottir, and N. Lassen. 1975. SHAM, a method for biexponential curve resolution using initial slope, height, area and moment of the experimental decay type curve. J. Theor. Biol. 52:299-315. [DOI] [PubMed] [Google Scholar]
- 5.Craig, W. 1993. Pharmacodynamics of antimicrobial agents as a basis for determining dosage regimens. Eur. J. Clin. Microbiol. Infect. Dis. 12(Suppl. 1):S6-S8. [DOI] [PubMed] [Google Scholar]
- 6.D'Argenio, D. Z., and A. Schumitzky. 1979. A program package for simulation and parameter estimation in pharmacokinetic systems. Comput. Programs Biomed. 9:115-134. [DOI] [PubMed] [Google Scholar]
- 7.DiStefano, J. J., III. 1982. Noncompartmental vs. compartmental analysis: some basis for choice. Am. J. Physiol 243:R1-R6. [DOI] [PubMed] [Google Scholar]
- 8.Forrest, A., D. E. Nix, C. H. Ballow, T. F. Goss, M. C. Birmingham, and J. J. Schentag. 1993. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob. Agents Chemother. 37:1073-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grasso, S., G. Meinardi, I. de Carneri, and V. Tamassia. 1978. New in vitro model to study the effect of antibiotic concentration and rate of elimination on antibacterial activity. Antimicrob. Agents Chemother. 13:570-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jusko, W. J. 1992. Guidelines for collection and analysis of pharmacokinetic data, p. 1-43. In W. E. Evans, J. J. Schentag, and W. J. Jusko (ed.), Applied pharmacokinetics: principles of therapeutic drug monitoring, 3rd ed. Applied Therapeutics, Inc., Vancouver, Canada.
- 11.MacGowan, A., C. Rogers, and K. Bowker. 2000. The use of in vitro pharmacodynamic models of infection to optimize fluoroquinolone dosing regimens. J. Antimicrob. Chemother. 46:163-170. [DOI] [PubMed] [Google Scholar]
- 12.Preston, S. L., G. L. Drusano, A. L. Berman, C. L. Fowler, A. T. Chow, B. Dornseif, V. Reichl, J. Natarajan, and M. Corrado. 1998. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA 279:125-129. [DOI] [PubMed] [Google Scholar]
- 13.Roosendaal, R., I. A. Bakker-Woudenberg, M. van den Berghe-van Raffe, J. C. Vink-van den Berg, and M. F. Michel. 1987. Comparative activities of ciprofloxacin and ceftazidime against Klebsiella pneumoniae in vitro and in experimental pneumonia in leukopenic rats. Antimicrob. Agents Chemother. 31:1809-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stass, H., and A. Dalhoff. 1997. Determination of BAY 12-8039, a new 8-methoxyquinolone, in human body fluids by high-performance liquid chromatography with fluorescence detection using on-column focusing. J. Chromatogr. B 702:163-174. [DOI] [PubMed] [Google Scholar]
- 15.Steimer, J. L., A. Mallet, J. L. Golmard, and J. F. Boisvieux. 1984. Alternative approaches to estimation of population pharmacokinetic parameters: comparison with the nonlinear mixed effect model. Drug Metab. Rev. 15:265-292. [DOI] [PubMed] [Google Scholar]
- 16.Wilkinson, L. 1999. SYSTAT: the system for statistics. SYSTAT, Inc., Evanston, Ill.