

Abstract
Clostridium perfringens uses its arsenal of >16 toxins to cause histotoxic and intestinal infections in humans and animals. It has been unclear why this bacterium produces so many different toxins, especially since many target the plasma membrane of host cells. However, it is now established that C. perfringens uses chromosomally encoded alpha toxin (a phospholipase C) and perfringolysin O (a pore-forming toxin) during histotoxic infections. In contrast, this bacterium causes intestinal disease by employing toxins encoded by mobile genetic elements, including C. perfringens enterotoxin, necrotic enteritis toxin B-like, epsilon toxin and beta toxin. Like perfringolysin O, the toxins with established roles in intestinal disease form membrane pores. However, the intestinal disease-associated toxins vary in their target specificity, when they are produced (sporulation vs vegetative growth), and in their sensitivity to intestinal proteases. Producing many toxins with diverse characteristics likely imparts virulence flexibility to C. perfringens so it can cause an array of diseases.
Keywords: animal disease, avian necrotic enteritis, Clostridium perfringens, enterocolitis, enterotoxemia, food poisoning, gas gangrene, human disease, toxins
Background
Clostridium perfringens ranks among the most widespread bacteria, with an ubiquitous environmental distribution in soil, sewage, food, feces, and the normal intestinal flora of humans and animals [1]. However, this Gram-positive, anaerobic spore former is also one of the most common and important human and animal pathogens, causing a spectrum of diseases (Table 1). For example, C. perfringens is the leading cause of traumatic gas gangrene [2]. It is also a major cause of foodborne illness, ranking as the second most common bacterial cause of food poisoning in the USA [3,4]. In addition, this bacterium is responsible for approximately 5–15% of all cases of antibiotic-associated diarrhea [5], which develops in 5–40% of all patients receiving antibiotic therapy [6]. It also causes an often-fatal human disease, enteritis necroticans [1]. As an animal pathogen, C. perfringens is responsible for several serious diseases, including avian necrotic enteritis, which drains approximately US$2 billion/year from the global agricultural system [7]. In addition, widespread vaccination is practiced to protect livestock from C. perfringens-induced enteritis and enterotoxemias, the latter characterized by intestinally produced toxins that are absorbed into the circulation and then affect other organs such as the brain [8].
Table 1.
Main diseases caused by the major toxins of Clostridium perfringens.
| Type of Clostridium perfringens† | Toxins produced | Most significant diseases‡ |
|---|---|---|
| A | CPA§ CPA, CPE§ CPA, NetB§ |
Human and animal myonecrosis (gas
gangrene) Human food poisoning and non-foodborne gastrointestinal disease; canine gastrointestinal disease Necrotic enteritis of poultry |
| B | CPA, CPB, ETX | Necrohemorrhagic enteritis of sheep (lamb dysentery) |
| C | CPA, CPB§ | Human necrotic enteritis (enteritis necroticans, pigbel); necrotic enteritis of neonatal individuals of several animal species (horse, cattle, sheep, pigs) |
| D | CPA, ETX§ | Enterotoxemia of sheep and goats |
| E | CPE, ITX | No known association with human disease; suspected, but no confirmed association with gastrointestinal disease of cattle, sheep and rabbits. |
All types of Clostridium perfringens may also produce several other toxins, including, but not limited to, CPB2, perfringolysin O and toxin perfringens large.
Only diseases that have been confirmed to be associated with each type of C. perfringens, and that are significant in terms of prevalence, are included in this table.
Critical toxin for virulence. PFO also contributes to virulence during myonecrosis (see text).
CPA: C. perfringens alpha toxin; CPB: C. perfringens beta toxin; CPE: C. perfringens enterotoxin; ETX: C. perfringens epsilon toxin; ITX: C. perfringens iota toxin; NetB: Necrotic enteritis toxin B-like.
Reproduced with permission from [94].
The virulence of C. perfringens is mediated in large part by its intimidating toxin arsenal (Table 2). Toxin production varies from strain to strain, permitting classification of C. perfringens isolates into five toxinotypes (Table 3) based upon the production of four typing toxins; that is, alpha (CPA), beta (CPB), epsilon (ETX) and iota (ITX). Although historic and somewhat dated, this typing system is still useful since different toxinotypes, and often subtypes, of C. perfringens are associated with certain diseases (Table 1).
Table 2.
Properties of the most relevant toxins produced by Clostridium perfringens.
| Toxin | Location | Molecular mass (kDa) | LD50 (mice†) | Action |
|---|---|---|---|---|
| CPA | C | 43 | 3 μg | Phospholipase C and sphingomyelinase activity; affects host signaling |
| CPB | P | 35 | <400 ng | Pore-forming toxin |
| ETX | P | 33 | 100 ng | Pore-forming toxin |
| ITX | P | Ia 48; Ib 72 | 40 μg | Actin-specific ADP-ribosyltransferase |
| PFO | C | 54 | 15 μg | Pore-forming toxin |
| CPE | C/P | 35 | 81 μg | Pore-forming toxin |
| CPB2 | P | 28 | 160 μg | Pore-forming toxin |
| TpeL | P | 191 | 600 μg?‡ | Ras-specific monoglucosyltransferase |
| NetB | P | 33 | Unknown | Pore-forming toxin |
Per kg of mouse after intravenous injection.
Determined using a much less active TpeL variant.
C: Chromosomal; CPA: C. perfringens alpha toxin; CPB: C. perfringens beta toxin; CPE: C. perfringens enterotoxin; ETX: C. perfringens epsilon toxin; ITX: C. perfringens iota toxin; NetB: Necrotic enteritis toxin B-like; P: Plasmid; PFO: Perfringolysin O; TpeL: Toxin perfringens large.
Reproduced with modification from [8].
Table 3.
Classification of Clostridium perfringens based on the production of the four major toxins.
| Type | Toxin produced | |||
|---|---|---|---|---|
| Alpha | Beta | Epsilon | Iota | |
| A | + | − | − | − |
| B | + | + | + | − |
| C | + | + | − | − |
| D | + | − | + | − |
| E | + | − | − | + |
For many years it remained unclear why C. perfringens produced so many different toxins, particularly since most of these toxins share a common target, that is, the plasma membrane of host cells. However, recent advances in molecular techniques for studying this bacterium, coupled with the development of appropriate animal models for C. perfringens diseases, are now providing insights into this question. Given this progress, it is timely to review our current understanding of the contributions of different C. perfringens toxins to disease.
Histotoxic infections
Clostridial myonecrosis, also known as gas gangrene, is a devastating histotoxic infection of humans and animals caused by C. perfringens type A. Although the incidence of gas gangrene in humans is relatively low, lethality rates remain generally high [9]. The successful invasion of traumatic wounds by vegetative cells or spores leads to rapid and extensive tissue damage and necrosis in the human host. If untreated, infection proceeds to shock, organ failure and ultimately death [9].
CPA and perfringolysin O (PFO) are quintessential to the pathogenesis of gas gangrene. The structural genes encoding these toxins, cpa (or plc) and pfoA, respectively, are located on the chromosome [10,11]. Production of both toxins is regulated by the Agr-like quorum sensing (QS) [12,13] system and also by the VirS/VirR two-component regulatory system [14–16]. However, PFO expression is directly regulated by the VirR response regulator, whereas CPA production is controlled by the VirR-regulated VR-RNA molecule [17].
CPA is a single polypeptide with two domains (Figure 1): an N-terminal zinc-binding domain that has both phospholipase C and sphingomyelinase activity, and a C-terminal calcium-binding domain that has structural similarity to eukaryotic C2-lipid binding proteins and is responsible for the binding of the toxin to cell membranes, making it essential for toxic activity [18]. PFO is a member of the cholesterol-dependent cytolysin (CDC) family of pore-forming toxins [19], which have been shown in other organisms to play a role in virulence [20]. PFO has four structural domains (Figure 1). Contact of domain 4 with the host cell membrane leads to two α-helical domains in domain 3 changing their conformation to form two amphipathic β-hairpins that insert into and span the membrane [21]. Up to 50 PFO monomers oligomerize to form a 15-nm membrane pore, which is much larger than the 1.5–1.7-nm pores formed by beta-pore-forming toxins such as NetB [22].
Figure 1. Crystal structures of Clostridium perfringens toxins.
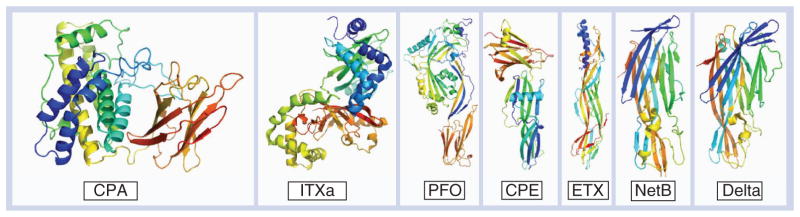
Graphic representations of all known toxin structures were prepared using Pymol [88]. Color coding is shown as a transition from N-terminal (blue) to C-terminal [35] residues. Note that delta toxin is a pore-forming toxin, but its role in disease has not yet been established.
CPA: C. perfringens alpha toxin (1CA1) [89]; CPE: C. perfringens enterotoxin (3AM2) [40,41]; Delta: Delta toxin (2YGT) [93]; ETX: C. perfringens epsilon toxin (1UYJ) [92]; ITXa: C. perfringens iota toxin a subunit (1GIR) [90]; NetB: Necrotic enteritis toxin B (4ION) [50]; PFO: Perfringolysin O (1PFO) [91].
For color images please see www.futuremedicine.com/doi/full/10.2217/fmb.13.168
Early studies suggested a correlation between CPA production and disease [23]. For example, immunization with purified CPA or the C-terminal CPA domain protected mice from experimental gas gangrene [24]. Given its hemolytic nature and similarity to other CDCs, PFO also was proposed to contribute to the pathogenesis of gas gangrene. Definitive evidence for the essential role of CPA, and for an involvement of PFO in virulence only came about when an isogenic set of C. perfringens type A CPA and PFO null mutants was constructed by homologous recombination in type A strain 13 [23]. When the null mutants and complemented strains were tested in a mouse model, the results showed that CPA is essential for the development of myonecrosis, that is, the CPA mutants were avirulent in mice and virulence was restored upon complementation in trans by the wild-type plc gene [23]. Furthermore, these studies revealed that several features of gas gangrene, including tissue necrosis, thrombosis and the absence of a polymorphonuclear (PMN) leukocytic influx at the site of infection (Figure 2), could be attributed to CPA activity [23].
Figure 2. Microscopic lesions in mice inoculated with Clostridium perfringens type A (mouse gas gangrene model).

The inner tight muscle shows severe necrosis of myofibers characterized by flocculation and fragmentation of the sarcolemma, hypereosinophilia and nuclear pyknosis. A mid-size venule (*) shows marked margination of neutrophils, which are mostly absent in the extravascular tissue. Sections were stained with hematoxylin and eosin and photographed at 100× magnification. Modified with permission from [26].
Similar studies using an isogenic PFO null mutant of strain 13 and its complemented derivatives showed that PFO is not essential for disease, but that it does affect the host inflammatory response by contributing to the lack of PMN influx into the lesion and to the vascular accumulation of PMNs bordering the site of infection [25]. Furthermore, PFO appears to be the primary toxin responsible for killing mouse peritoneal macrophages in vitro and, together with CPA, enables C. perfringens to escape from the phagosomes of macrophages [25].
Subsequent studies involving virulence testing of a defined mutant deficient in both CPA and PFO production, and its complemented derivatives, showed that both toxins work synergistically to produce the overall clinical and pathological effects observed in C. perfringens-mediated gas gangrene [26]. It has been suggested that the mechanism of synergy may involve the initial membrane action of CPA facilitating the interaction of PFO with cholesterol in the cell membrane [27]. Both toxins contribute to the leukostasis observed in the peripheral blood vessels and the extracellular matrix of infected host tissues (Figure 2) [28].
The genetic studies outlined above provided insight into the pathogenesis of C. perfringens-mediated gas gangrene, but C. perfringens type A produces >ten extracellular toxins and/or enzymes. Studies involving the construction of other isogenic mutants have shown that collagenase [29], the NanI and NanJ sialidases [30], and alpha-clostripain [31], are not essential for disease in the mouse myonecrosis model. However, these studies are of necessity limited given that the mouse gas gangrene model does not enable conclusions to be drawn about the early stages of a histotoxic infection.
Diseases originating in the intestine
Infections caused by C. perfringens enterotoxin-positive strains of C. perfringens type A
As mentioned in the Introduction, Type A strains producing C. perfringens enterotoxin (CPE) are the second most common cause of bacterial food-borne illness in the USA, where they cause nearly 1,000,000 cases per annum, resulting in a net economic burden of US$382 million [3,4]. Symptoms of C. perfringens type A food poisoning include abdominal cramping, nausea and diarrhea, which usually begin 8–18 h after ingestion of contaminated food and then persist for 12–24 h [1]. Classic CPE-mediated C. perfringens food poisoning occurs when contaminated meat and/or poultry products are undercooked and/or held at improper temperature, allowing for spore germination and growth of CPE-producing type A strains in foods [1]. When that contaminated food is ingested, the bacteria sporulate in the small intestine, at which time CPE is produced. At the completion of sporulation, the toxin is released into the intestinal lumen when the mother cell lyses to release the mature spore [1].
In addition to food poisoning, type A strains producing CPE cause 5–15% of all cases of non-food-borne human gastrointestinal (GI) diseases, including antibiotic-associated diarrhea and sporadic non-foodborne diarrhea [5]. These non-food-borne GI illnesses tend to be more severe and longer lasting than most C. perfringens type A food poisoning cases [5].
Only approximately 1–5% of C. perfringens type A strains carry the cpe gene, which is associated with insertion sequences [1]. The cpe gene can have a chromosomal or plasmid location in type A strains [32], but most type A food poisoning isolates carry a chromosomal cpe gene, whereas non-food poisoning type A isolates typically carry a plasmid-encoded cpe gene [32]. Type A strains carrying a chromosomal cpe gene comprise a distinct sublineage of C. perfringens that appears to be well-adapted for food-borne transmission [33], due in part to their production of spores with exceptional resistance against heating, low temperatures and certain food preservatives [34]. This resistance phenomenon involves, in large part, production of a variant small acid-soluble protein (Ssp4) that provides enhanced protection to the spores of chromosomal cpe type A strains [34,35].
CPE, an approximately 35-kDa protein belonging to the aerolysin family of β-pore-forming toxins, is comprised of a C-terminal receptor binding domain (Domain I) and an N-terminal domain (Domain II) that mediates toxin oligomerization and membrane insertion/pore formation (Figure 1). The action of CPE starts with binding to claudin receptors on enterocytes [36–38]. Claudins are a 24-member protein family that play critical structural and functional roles in the mammalian tight junction [39]. Only certain claudins (e.g., claudins- 3, −4, −8 and −14) can function as CPE receptors [36]. CPE binding to claudins results in the formation of an approximately 90-kDa CPE ‘small complex’ that contains CPE, a receptor claudin, and nonreceptor claudins, likely due to interactions of those claudins with the claudin receptors [1]. Following small complex formation, CPE hexamerizes on the membrane surface to form large (>450 kDa), heterologous prepore complexes that contain CPE, claudins and sometimes occludin, another tight junction protein. Once formed, the CPE prepore inserts amphipathic, transmembrane β-hairpins from Domain II of each toxin monomer into the host membrane to form an active pore [40,41]. The resultant plasma membrane permeability alterations then allow a strong influx of calcium, causing downstream cell death due to apoptosis or oncosis [1]. Enterocyte death disrupts villus integrity, resulting in intestinal damage and electrolyte disregulation [1,42].
Animal model studies and human disease observations suggest that, when diarrhea is impaired in C. perfringens food poisoning victims due to medication side effects, CPE can enter the circulation and damage internal organs such as the kidney or liver, leading to excessive serum potassium. In animal models, and presumably people, this can result in hyperkalemia-associated heart failure and death [43].
A wealth of data [1] supports a critical role for CPE in the pathogenesis of food poisoning caused by CPE-positive type A strains. First, while absent from the feces of healthy individuals, CPE is found in the feces of 80–100% of individuals with C. perfringens type A food poisoning. Second, peroral administration of purified CPE to human volunteers caused the development of classical C. perfringens type A food poisoning symptoms. Third, CPE is present in fecal specimens from patients of C. perfringens type A food poisoning outbreaks at levels known to cause pathogenic effects in animal models. Fourth, CPE-specific antisera can neutralize the intestinal pathology caused by CPE-positive type A isolates in animal models. Fifth, purified CPE was shown to be sufficient to cause morphological damage in human ileal tissue ex vivo. Finally, isogenic cpe null mutants and complemented strains have been used to fulfill molecular Koch’s postulates by demonstrating that CPE is essential for the type A food poisoning strain SM101 to cause fluid accumulation and histological damage in rabbit small intestinal loops (Figure 3). Similar analyses indicated that CPE is also essential for the non-foodborne human GI disease isolate F4969 to cause pathology in rabbit intestinal loops [44].
Figure 3. Histological damage in rabbit ileum treated with lysates from Clostridium perfringens enterotoxin-positive C. perfringens type A strain SM101.
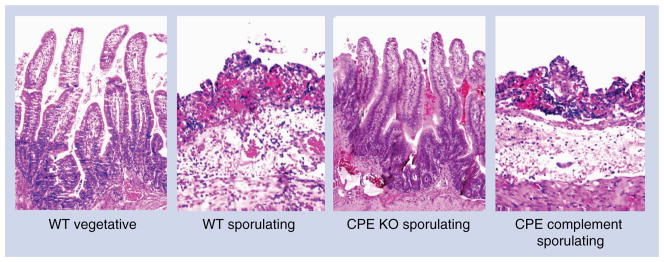
Loops inoculated with WT SM101 vegetative culture (WT vegetative) or an isogenic CPE KO sporulating culture lysate (CPE KO sporulating) show normal, full-length intestinal villi with a well-preserved epithelium and lamina propria. Loops inoculated with sporulating culture lysate of WT SM101 (WT sporulating) or the isogenic CPE complement (CPE complement sporulating) show histological damage consisting of necrosis and loss of epithelium, necrosis of lamina propria, villous blunting, and hemorrhage and edema of the mucosa and submucosa. Sections were stained with hematoxylin and eosin and photographed at 250× magnification.
CPE: C. perfringens enterotoxin; KO: Knockout; WT: Wild-type.
Figure and legend modified with permission from [44].
Infections caused by NetB-positive strains of C. perfringens type A
NetB-positive type A strains of C. perfringens cause necrotic enteritis in chickens [45]. This debilitating disease of the gastrointestinal tract is found in chickens in all poultry-producing regions of the world and, as indicated in the Introduction, imposes a significant economic burden because of reduced production efficiency and the cost of control measures [46]. The disease is most common in broilers, but layer hens and turkeys can also be affected. Liver lesions and erosive lesions of the gizzard have also been associated with C. perfringens infection, but little work has been reported on these syndromes and the involvement of toxins remains unknown [47]. Subclinical disease, which adversely affects feed conversion ratios without causing significant mortality, is more of a problem because it is less obvious and may remain untreated, thus leading to longer grow-out periods and negatively impacting productivity and profitability [46].
The intestinal lesions of avian necrotic enteritis are characterized by a massive infiltration of granulocytes, a finding that was at odds with the previously held theory that CPA was the key virulence factor in necrotic enteritis [48,49]. Furthermore, CPA null mutants retained full virulence in a chicken disease induction model. These findings provided a strong indication that another toxin was involved, which led to the discovery of a new C. perfringens toxin, NetB [49]. Bioinformatic analyses of the NetB sequence suggested it is a β-pore-forming toxin, which was confirmed when NetB was shown to form a heptameric pore that affects membrane ion permeability [50,51]. In addition, structural studies showed that NetB is similar in structure to several other pore-forming toxins, including C. perfringens delta toxin (Figure 1) and α-hemolysin from Staphylococcus aureus [50,51].
Expression of NetB is under the control of the VirSR two-component signal transduction system; virR mutants produce reduced levels of NetB and complementation restores NetB production [52]. Strain surveys have generally found that most C. perfringens isolates from lesions of diseased birds carry the netB gene, whereas carriage of the gene is less frequent in isolates from healthy birds [45,53]. The isolation of NetB-negative strains from diseased birds has called into question the importance of the NetB toxin. However, in most studies in which the virulence of disease isolates has been directly studied, only NetB-positive strains reliably reproduce disease [45,54]. One report did find some disease from NetB-negative strains, but since the disease-causing strains were not re-isolated from diseased birds the exact etiology of the disease in these birds is not clear [47]. The netB gene is carried on a large conjugative plasmid [55–57], which can be lost during culturing, which appears to be an issue particularly when initially culturing from clinical material. Loss of plasmids when the strains are under severe stress, such as moving from the in vivo environment to in vitro culture conditions, may explain why many NetB-negative strains have been isolated from diseased birds. Alternatively, mixed colonization with pathogenic and nonpathogenic strains could give rise to the isolation of NetB-negative strains from diseased birds.
Studies using netB null mutants have provided convincing evidence for the essential role of NetB in the pathogenesis of avian necrotic enteritis (Figure 4). Wild-type Culture supernatant from a netB-positive strain is toxic to chicken leghorn male hepatoma (LMH) cells, but the supernatant from netB null mutants is not toxic. When a functional wild-type netB gene was reintroduced to complement the mutation, wild-type virulence was restored. This cellular toxicity can be neutralized by anti-NetB polyclonal antibodies [49]. These netB null mutants are also completely avirulent in the chicken disease induction model (Figure 4). Finally, recent studies have shown that NetB is efficacious as a vaccine antigen [58–61].
Figure 4. Virulence of Clostridium perfringens type A strains in 24-day-old broiler chickens challenged with different C. perfringens strains are shown.

Solid horizontal bars depict the mean small intestinal lesion score in each group (n = 10). Lesion scores correspond to 0: no gross lesions; 1: thin and/or friable walls; 2: focal necrosis or ulceration (1–5 foci); 3: focal necrosis or ulceration (6 to 15 foci); 4: focal necrosis or ulceration (>16); 5: patches of necrosis 2–3 cm long; 6: diffuse necrosis typical of field cases. The strains tested included: WT; netB KO, netB KO complemented (netB KO complement); netB knockout + shuttle vector (netB KO + shuttle vector). One tailed, nonparametric t-test analysis of the wild-type and complemented mutant derivatives against netB KO showed a statistical difference (*p = 0.05 and **p = 0.01), but no statistical significance was observed between the netB KO and the netB KO + shuttle vector.
KO: Knockout; WT: Wild-type.
Figure and legend modified from [49].
Infections caused by C. perfringens type C strains
C. perfringens type C strains are capable of causing fulminant disease in both humans and animals. In humans, type C infections cause enteritis necroticans (also known regionally as Darmbrand or Pigbel), an intestinal infection resulting in vomiting, bloody stool, abdominal pain and, in severe cases, toxemia leading to rapid death [62]. These infections are endemic in Southeast Asia, where poor diet and consumption of trypsin inhibitor-rich foods, such as sweet potatoes, provide ideal conditions for disease, suggesting that trypsin is a key host defense against disease. Disease is also occasionally observed in developed countries, where it is usually confined to individuals with reduced pancreatic function, such as diabetics [63,64].
Type C infections also occur in most livestock species, producing a hemorrhagic necrotic enteritis and enterotoxemia similar to human infection. Although adult animals can be infected and killed rapidly by this organism, most disease is observed in neonates. The propensity for type C strains to cause disease in young animals is likely due to trypsin inhibition by colostrum. Colonization of animals occurs within hours of birth, most likely originating from contact with contaminated fecal material. Infection in animals typically involves outbreaks in unvaccinated herds, with lethality rates >50% [62].
C. perfringens type C disease begins with multiplication of the bacterium in the host intestine, followed by CPB production. CPB then affects the epithelium of the jejunum, ileum and, to a lesser extent, the duodenum and colon, where it causes fluid accumulation and necrosis of the intestine with variable hemorrhage [65]. Mucosal damage is followed by absorption of CPB into the circulation, from which it acts at distal sites, including the brain [62,66]. At the cellular level, the action of CPB remains incompletely defined. However, CPB is a pore-forming toxin that uses an unidentified receptor for binding to host cells [8]. Once bound, this toxin oligomerizes into hexamers or heptamers on host cells, creating transmembrane pores in susceptible cells [67]. These pores cause K+ efflux and Ca2+, Na+ and Cl− influx, leading to cellular swelling and necrosis of intoxicated cells [68]. Interestingly, several studies using cell culture and tissue samples from infected humans and piglets have shown that CPB binds to endothelial cells, although the contribution of those CPB interactions to disease remains to be delineated [68,69].
The cpb gene is carried on large virulence plasmids of approximately 65–110 kb that can contain additional toxin genes encoding CPE or TpeL [70,71]. CPB is expressed as a 336 amino acid protein containing a 27 amino acid leader sequence that is cleaved to yield an approximately 35-kDa functional protein sharing 20–28% identity with some staphylococcal pore-forming toxins and 38% identity with C. perfringens NetB toxin [8]. Investigations into the regulatory mechanism of CPB production by type C strains have demonstrated the involvement of the Agr-like QS system in regulating the production of CPB during intestinal disease. An AgrB (membrane-bound endopeptidase) null mutant of strain CN3685 [72] exhibited sharply reduced in vivo CPB production, resulting in the mutant being unable to induce necrotizing enteritis in rabbit ileal loops or lethality in mice. In addition to the Agr-like QS system, the VirS/VirR system is also involved in regulating in vivo CPB production and type C virulence [73].
Type C strains must carry genes encoding CPA and CPB, and may also possess genes for additional accessory toxins. However, several recent studies provided direct evidence identifying CPB as the primary toxin responsible for development of disease by type C strains. Initial studies utilizing monoclonal antibodies indicated that neutralization of CPB, but not CPA, protects mice from a lethal IV challenge with type C culture supernatants [62]. More recently, purified CPB was shown to induce necrotizing enteritis in rabbit small intestinal loops (Figures 5 & 6) or lethality in a mouse enterotoxemia assay [65]. The most convincing support for CPB involvement in type C disease was provided by studies using toxin null mutants, where a CPB null mutant of type C strain CN3685 was essentially avirulent in mouse, rabbit (Figures 5 & 6) and goat models of necrotizing enteritis or enterotoxemia [74–76]. Reversing this cpb null mutation restored virulence in all three animal models, ruling out secondary mutation effects. In contrast, isogenic CPA, PFO or double CPA/PFO null mutants retained near full virulence in the rabbit small intestinal loop model (Figures 5 & 6) and the mouse enterotoxemia model [76].
Figure 5. Gross pathology of rabbit-ligated small intestinal loops challenged for 6 h with Clostridium perfringens type C strain CN3685 WT, isogenic single- and double-toxin mutants (PFO KO, CPA KO, CPB KO, CPA/PFO KO), purified CPB (CPB) or sterile tryptic soy broth–glucose–yeast extract broth (control).
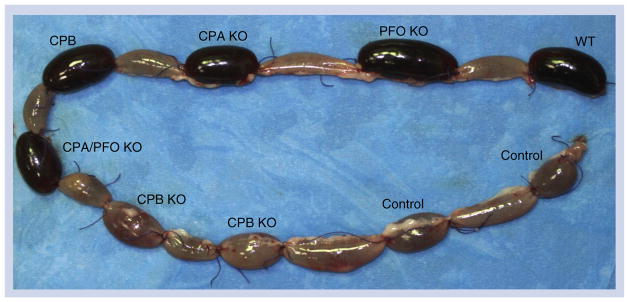
Note that loops inoculated with the WT, PFO KO, CPA KO, CPA/PFO KO or purified CPB are severely hemorrhagic and distended with fluid. No significant gross abnormalities are observed in the loops inoculated with CPB KO or sterile tryptic soy broth–glucose–yeast extract broth.
CPA: C. perfringens alpha toxin; CPB: C. perfringens beta toxin; KO: Knockout; PFO: Perfringolysin O; WT: Wild-type.
Modified with permission from [76].
Figure 6. Histological damage in rabbit small intestine treated for 6 h with an 8-h culture of WT strain CN3685 (WT), single- and double-toxin isogenic mutants (PFO KO, CPA KO, CPB KO, CPA/PFO KO), or sterile tryptic soy broth–glucose–yeast extract broth medium (control).

Control loops and loops inoculated with the CPB KO showed normal, full-length intestinal villi with a well-preserved epithelium and lamina propria. Loops inoculated with WT, CPB reversed mutant, PFO KO, CPA KO, or CPA/PFO KO all showed microscopic changes, including mucosal necrosis, hemorrhage and blunting of the villi, together with neutrophilic infiltration of mucosa and submucosa. Tissue sections were stained with hematoxylin and eosin, and photographed at 200× magnification.
CPA: C. perfringens alpha; CPB: C. perfringens beta; KO: Knockout; PFO: Perfringolysin O; WT: Wild-type.
Figure and legend modified with permission from [76].
Infections due to C. perfringens type D strains
C. perfringens type D produces enterotoxemia in sheep and goats [77]. Less frequently, these strains are responsible for enterotoxemia in cattle and other animal species. ETX increases vascular permeability in the brain and other organs. In the brain, this toxin affects endothelial cells, which is followed by swelling and rupture of the perivascular astrocyte feet. The immediate consequence of this effect is perivascular edema, with an increase of the intracerebral pressure and occasionally parenchymal brain necrosis (focal symmetrical encephalomalacia). Owing to these effects, type D disease is mainly characterized by neurological clinical signs in the most affected animal species, although goats can also suffer intestinal damage [77].
Type D strains by definition must produce ETX, which is encoded by the plasmid-borne etx gene [32]. The regulation of ETX production is more variable than that of many other toxins discussed in this review. For example, while ETX production is regulated by the Agr-like QS system in type D strain CN3718, this is not true for two type B strains [78,79]. In addition, ETX production by type D strain CN3718 is not dependent upon a functional VirS/VirR system [78].
ETX is secreted as a 33-kDa prototoxin that must be proteolytically activated by cleavage of the C-terminal 29 amino acids to achieve full potency [80]; activating ETX with trypsin leads to a 1000-fold increase in toxicity [80]. Structurally, the ETX protein belongs to the same aerolysin pore-forming toxin family that includes CPE (Figure 1). Furthermore, ETX is able to bind to glutamatergic neuronal cells, where it causes release of the neurotransmitter glutamate, which is thought to contribute to the neurological signs of type D disease [80]. ETX is the third most potent clostridial toxin, that is, ETX is 100-times more potent than other members of the aerolysin pore-forming toxin family, suggesting that pore formation may not be the only mechanism of action utilized by ETX [80]. As a result of this potency, ETX has been of interest as a potential bioterrorism agent [80] and was included until recently on the CDC list of select toxins. We have shown that the etx gene is located on large plasmids [32] and, for two type D isolates, the plasmids were shown to be conjugative [81].
Most clinical and pathological changes in type D disease were reproduced by inoculation of ETX in sheep, goats and cattle. Recent studies have confirmed that ETX is necessary for the virulence of type D isolates. Assaying a large number of type D culture supernatants using an intravenous injection mouse model revealed a strong correlation between ETX levels and lethality, suggesting this is the major toxin contributor to mouse lethality [82]. An etx null mutant was later produced by allelic exchange, which resulted in its insertional inactivation by an antibiotic resistance cassette. This process allowed for the tracking of the plasmid encoding the deleted etx gene and led to the discovery that these plasmids could be transferred to other C. perfringens strains at a very high frequency [81]. This etx null mutant was also used to demonstrate the role of ETX in C. perfringens type D virulence. Clinical disease followed by death was observed in sheep, goats and mice challenged intraduodenally with a wild-type isolate of a type D strain (Figure 7). Gross and/or microscopic changes observed in affected sheep included edema of the brain (Figure 8), lungs and heart and hydropericardium. Necrotizing colitis, pulmonary edema and hydropericardium were observed in affected goats. Mice challenged with the wild-type strain died, but presented no significant gross or histological abnormalities [83]. No clinical disease, gross abnormalities, or microscopic damage were observed in any of the sheep, goats and mice treated with the isogenic etx null mutant. Clinical and pathological changes similar to those observed in animals infected with the wild-type strain were observed in most goats, sheep and mice challenged with the complemented mutant (Figure 7). Based on these results, it was concluded that ETX is necessary for type D isolates to cause disease.
Figure 7. Kaplan–Meier survival curves for sheep, goats and mice Clostridium perfringens type D infection models.

Survival over 24 h in (A) sheep and (B) goats or 48 h (C) in mice after intraduodenal treatment with the WT strain CN1020 (WT), the etx mutant JIR4981 (ETX knockout), its complemented derivative JIR12604 (ETX complemented), or TGY. Each inoculum was administered to six sheep, five goats and 15 mice.
etx: C. perfringens epsilon; TGY: Tryptic soy broth–glucose–yeast extract broth; WT: Wild-type.
Figure and portions of the legend used with permission from [83].
Figure 8. Microscopic lesions in sheep after intraduodenal treatment with Clostridium perfringens type D WT strain CN1020 (WT) or isogenic derivatives including an etx null mutant (etx KO), its complemented derivative (etx complement), or sterile, nontoxic culture medium (control) are shown.

Brains from sheep challenged intraduodenally with the WT or complemented strains show proteinaceous perivascular edema (arrow) due to increased vascular permeability. Significant histological lesions were absent from the brains of sheep treated intraduodenally with the etx KO or control animals.
etx: C. perfringens epsilon; KO: Knockout; WT: Wild-type.
Figures and portions of the legend reproduced with permission from [83].
Conclusion
Research is now providing substantial insights as to why C. perfringens produces so many different toxins. The emerging evidence is that some of these toxins (CPA and PFO) are utilized mainly when this bacterium causes histotoxic infection, including gas gangrene (Figure 9) [9]. During gas gangrene, these toxins interfere with the immune response and cause localized necrosis to facilitate the growth of C. perfringens, an anaerobe. These toxins also induce host cell leakage and lysis, which may provide nutrients (particularly many required amino acids) for C. perfringens growth. The combination of regional damage and, especially, distant toxin effects soon kills the host, providing a significant food source for further C. perfringens multiplication.
Figure 9. Summary of toxin involvement in Clostridium perfringens disease.

(A) During histotoxic infections CPA plays a predominant role after C. perfringens (blue rods) are introduced by trauma into muscle tissue. This toxin induces localized necrosis, as well as toxemic effects in other organs when toxins enter the circulation. Both CPA and PFO are important in vascular leukostasis. (B) During intestinal infections, C. perfringens (blue rods) can produce several pore-forming toxins, including CPE during sporulation or (during vegetative growth) NetB, ETX or CPB. Host intestinal protease levels can either destroy (e.g., CPB) or activate (e.g., ETX) these toxins. When present in an active form, these toxins can cause local damage (necrosis) or be absorbed into the circulation to damage internal organs such as the brain, kidney or liver.
CPA: C. perfringens alpha toxin; CPB: C. perfringens beta toxin; CPE: C. perfringens enterotoxin; PFO: Perfringolysin O.
For color images please see www.futuremedicine.com/doi/full/10.2217/fmb.13.168
When causing diseases originating in the intestine, C. perfringens generally relies upon other toxins, namely CPE, NetB, CPB or ETX [8]. Notably, those toxins involved in most intestinal infections are associated with mobile genetic elements, typically a large conjugative plasmid [32]. The major exception is the chromosomal cpe gene that is present in most type A food poisoning strains; however, even this toxin gene is associated with a putative transposon that has integrated onto the chromosome [1].
The progress in dissecting the contributions of individual toxins to C. perfringens virulence is also illuminating why this bacterium produces so many pore-forming toxins. PFO clearly plays a supportive role during gas gangrene [25,26]; however, two lines of evidence suggest this toxin is not a major contributor to most C. perfringens intestinal infections. First, inactivation of the pfoA gene of type C animal disease strain CN3685 did not significantly attenuate that strain for causing necrotizing enteritis or lethal enterotoxemias in animal models [76]. Second, PFO cannot be a major contributor to several important C. perfringens human intestinal infections since the pfoA gene is absent from the chromosomal cpe type A strains causing most cases of C. perfringens type A food poisoning [33] and from the type C strains causing Darmbrand [84].
In contrast, recent studies have clearly demonstrated a key role for a number of other pore-forming toxins in C. perfringens infections originating in the intestine (Figure 9). Why then does this bacterium employ so many different pore-forming toxins to cause intestinal infections? One answer is that these toxins likely provide the species with tremendous versatility in causing these diseases. For example, CPE is only produced during sporulation, which occurs during food poisoning when C. perfringens is ingested in contaminated foods, briefly undergoes a period of intense growth in the anaerobic and nutrient-rich intestinal environment, and then (for still unknown reasons) synchronously sporulates and produces CPE. This pore-forming toxin then induces a strong diarrhea that flushes large numbers of spores back into the environment so they can eventually repeat this cycle [1].
Unlike CPE, the other pore-forming toxins with established roles in intestinal infections are produced by vegetative cells. As for gas gangrene, intestinal infections involving ETX, CPB and NetB often result in rapid death of the host, possibly providing the infecting C. perfringens strains with a food source for their multiplication [8]. Why then might C. perfringens use so many different pore-forming toxins to induce host death during vegetative growth? There could be at least two answers. First, some of these toxins appear to be exquisitely adapted for particular hosts. For example, NetB and CPB are clearly related with respect to sequence, yet NetB-producing strains over-whelmingly cause disease in poultry while CPB-producing strains affect mammals [45,62]. A second potential explanation for producing so many different pore-forming toxins is that it provides C. perfringens with flexibility in causing disease under varying host conditions. For example, ETX is activated by intestinal proteases [80], yet CPB is easily destroyed by those same intestinal proteases [85]. Therefore, ETX-producing strains are adept at causing disease in ‘healthy’ animals, while CPB-producing strains are well-suited for affecting young or compromised hosts (animals or people) with reduced levels of intestinal proteases. Type B strains may illustrate this point particularly well, that is, presumably these strains can use their ETX to cause disease in healthy animals but employ their CPB to cause disease in animals with reduced intestinal protease levels (see next section) [85].
Finally, while it is clear that toxins play a critical role in C. perfringens pathogenicity, it should be appreciated that other factors, such as strain clonality, toxin production regulation, enzyme production and conjugative toxin plasmid transfer [32,33,56,86], are also key contributors to the virulence of this bacterium.
Future perspective
Many gaps still remain in our knowledge of toxin contributions to C. perfringens infections. For example, type B strains cause an often fatal hemorrhagic dysentery in sheep, and possibly in other species. Some earlier studies using IV injection of type B culture supernatants into mice suggested that both CPB and ETX are important for virulence of these strains [85], with host factors (i.e., the presence or absence of trypsin) apparently tipping the scale for the relative importance of these two toxins for lethality. However, there is a clear need to demonstrate molecular Koch’s postulates using a natural infection model to confirm these findings. Similarly, conclusive evidence for a role of type E strains in animal disease remains unclear, although these strains have been associated with enteric disease in rabbits, lambs and calves. These are the only C. perfringens strains producing iota toxin, which is a binary toxin that ADP-ribosylates actin to cause a depolymerization of actin filaments in host cells [8]. There is currently little information available regarding toxin contributions to type E strain virulence. Type E strains are also worthy of study because iota toxin is one of only two toxins (TpeL being the other) made by this bacterium that act intracellularly [8,87]. Therefore, careful virulence dissections of type E strains and TpeL-producing strains now appear warranted to evaluate whether intracellularly active toxins, as well as plasma membrane-active toxins, contribute to virulence.
EXECUTIVE SUMMARY.
Background
Clostridium perfringens causes a spectrum of important human and animal diseases ranging from histotoxic to enteric infections.
These include clostridial myonecrosis (gas gangrene), C. perfringens type A food poisoning, C. perfringens enterotoxin (CPE)-associated nonfoodborne GI diseases, avian necrotic enteritis, human enteritis necroticans, and animal enteritis or enterotoxemia.
These diseases are mediated by different toxins, whose production varies among C. perfringens strains.
Histotoxic infections
Alpha toxin and perfringolysin O are important during histotoxic infections such as gas gangrene.
Diseases originating in the intestines
When produced, enterotoxin (CPE), NetB toxin, epsilon toxin and beta toxin are critical contributors to intestinal infections.
CPE is necessary for C. perfringens type A food poisoning and CPE-associated nonfoodborne human GI diseases.
NetB is required for avian necrotic enteritis.
CPB or ETX, respectively, are important for type C and D infections originating in the intestines.
The toxins contributing to intestinal infections vary in their targets when produced (sporulation vs vegetative growth) and their protease sensitivity, giving C. perfringens the ability to cause disease under varying host intestinal conditions.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Authors’ note
For our colleagues, please note that this review cites many reviews from older work due to reference limitations.
Financial & competing interests disclosure
The National Institute of Allergy and Infectious Diseases grants AI056177 and AI19488 supported this work. The authors also acknowledge the support of grants from the Australian National Health and Research Council, the Australian Research Council and the Poultry Cooperative Research Centre. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
•• of considerable interest
- 1.McClane BA, Robertson SL, Li J. Clostridium perfringens . In: Buchanan RL, Doyle MP, editors. Food Microbiology: Fundamentals and Frontiers. 4. American Society for Microbiology; Washington, DC, USA: 2013. pp. 465–489. [Google Scholar]
- 2.McClane BA, Rood JI. Clostridial toxins involved in human enteric and histotoxic infections. In: Bahl H, Durre P, editors. Clostridia: Biotechnology and Medical Applications. Wiley-VCH; Weinheim, Germany: 2001. pp. 169–209. [Google Scholar]
- 3.Grass JE, Gould LH, Mahon BE. Epidemiology of foodborne disease outbreaks caused by Clostridium perfringens, United States, 1998–2010. Foodborne Pathog Dis. 2013;10(2):131–136. doi: 10.1089/fpd.2012.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scharff RL. Economic burden from health losses due to foodborne illness in the United States. J Food Protect. 2012;75(1):123–131. doi: 10.4315/0362-028X.JFP-11-058. [DOI] [PubMed] [Google Scholar]
- 5.Carman RJ. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev Med Microbiol. 1997;8(Suppl 1):S43–S45. [Google Scholar]
- 6.McFarland LV. Diarrhoea associated with antibiotic use. BMJ. 2007;335(7610):54–55. doi: 10.1136/bmj.39255.829120.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keyburn AL, Bannam TL, Moore RJ, Rood JI. NetB, a pore-forming toxin from necrotic enteritis strains of Clostridium perfringens. Toxins. 2010;2(7):1913–1927. doi: 10.3390/toxins2071913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uzal FA, Vidal JE, McClane BA, Gurjar AA. Clostridium perfringens toxins involved in mammalian veterinary diseases. Open Toxinol J. 2010;3:24–42. [PMC free article] [PubMed] [Google Scholar]
- 9.Stevens DL, Aldape MJ, Bryant AE. Life-threatening clostridial infections. Anaerobe. 2012;18(2):254–259. doi: 10.1016/j.anaerobe.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu T, Ohtani K, Hirakawa H, et al. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci USA. 2002;99(2):996–1001. doi: 10.1073/pnas.022493799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myers GS, Rasko DA, Cheung JK, et al. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 2006;16(8):1031–1040. doi: 10.1101/gr.5238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vidal JE, Chen J, Li J, McClane BA. Use of an EZ-Tn5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS ONE. 2009;4(7):e6232. doi: 10.1371/journal.pone.0006232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohtani K, Yuan Y, Hassan S, Wang R, Wang Y, Shimizu T. Virulence gene regulation by the agr system in Clostridium perfringens. J Bacteriol. 2009;191(12):3919–3927. doi: 10.1128/JB.01455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyristis M, Bryant AE, Sloan J, et al. Identification and molecular analysis of a locus that regulates extracellular toxin production in Clostridium perfringens. Mol Microbiol. 1994;12(5):761–777. doi: 10.1111/j.1365-2958.1994.tb01063.x. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu T, Ba-Thein W, Tamaki M, Hayashi H. The virR gene, a member of a class of two-component response regulators, regulates the production of perfringolysin O, collagenase, and hemagglutinin in Clostridium perfringens. J Bacteriol. 1994;176(6):1616–1623. doi: 10.1128/jb.176.6.1616-1623.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung JK, Low LY, Hiscox TJ, Rood JI. Regulation of extracellular toxin production in C. perfringens. In: Vasil ML, Darwin AJ, editors. Regulation of Bacterial Virulence. American Society for Microbiology; Washington, DC, USA: 2013. pp. 281–294. [Google Scholar]
- 17.Shimizu T, Yaguchi H, Ohtani K, Banu S, Hayashi H. Clostridial VirR/VirS regulon involves a regulatory RNA molecule for expression of toxins. Mol Microbiol. 2002;43(1):257–265. doi: 10.1046/j.1365-2958.2002.02743.x. [DOI] [PubMed] [Google Scholar]
- 18.Oda M, Kabura M, Takagishi T, et al. Clostridium perfringens alpha-toxin recognizes the GM1a-TrkA complex. J Biol Chem. 2012;287(39):33070–33079. doi: 10.1074/jbc.M112.393801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heuck AP, Moe PC, Johnson BB. The cholesterol-dependent cytolysin family of Gram-positive bacterial toxins. Subcell Biochem. 2010;51:551–577. doi: 10.1007/978-90-481-8622-8_20. [DOI] [PubMed] [Google Scholar]
- 20.Chiarot E, Faralla C, Chiappini N, et al. Targeted amino acid substitutions impair streptolysin O toxicity and group A Streptococcus virulence. mBio. 2013;4(1):e00387–12. doi: 10.1128/mBio.00387-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossjohn J, Polekhina G, Feil SC, Morton CJ, Tweten RK, Parker MW. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J Mol Biol. 2007;367(5):1227–1236. doi: 10.1016/j.jmb.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olofsson A, Hebert H, Thelestam M. The projection structure of perfringolysin O (Clostridium perfringens theta-toxin) FEBS Lett. 1993;319(1–2):125–127. doi: 10.1016/0014-5793(93)80050-5. [DOI] [PubMed] [Google Scholar]
- 23••.Awad MM, Bryant AE, Stevens DL, Rood JI. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol Microbiol. 1995;15(2):191–202. doi: 10.1111/j.1365-2958.1995.tb02234.x. Awad et al. constructed the first toxin mutants in Clostridium perfringens and showed a plc null mutant, which is avirulent in mice. Virulence was restored upon complementation, revealing that C. perfringens alpha toxin is essential for the development of myonecrosis. [DOI] [PubMed] [Google Scholar]
- 24.Williamson ED, Titball RW. A genetically engineered vaccine against the alpha-toxin of Clostridium perfringens protects mice against experimental gas gangrene. Vaccine. 1993;11(12):1253–1258. doi: 10.1016/0264-410x(93)90051-x. [DOI] [PubMed] [Google Scholar]
- 25.O’Brien DK, Melville SB. Effects of Clostridium perfringens alpha-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect Immun. 2004;72(9):5204–5215. doi: 10.1128/IAI.72.9.5204-5215.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Awad MM, Ellemor DM, Boyd RL, Emmins JJ, Rood JI. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect Immun. 2001;69(12):7904–7910. doi: 10.1128/IAI.69.12.7904-7910.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moe PC, Heuck AP. Phospholipid hydrolysis caused by Clostridium perfringens alpha-toxin facilitates the targeting of perfringolysin O to membrane bilayers. Biochemistry. 2010;49(44):9498–9507. doi: 10.1021/bi1013886. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy CL, Lyras D, Cheung JK, Hiscox TJ, Emmins JJ, Rood JI. Cross-complementation of Clostridium perfringens PLC and Clostridium septicum alpha-toxin mutants reveals PLC is sufficient to mediate gas gangrene. Microb Infect. 2009;11(3):413–418. doi: 10.1016/j.micinf.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Awad MM, Ellemor DM, Bryant AE, et al. Construction and virulence testing of a collagenase mutant of Clostridium perfringens. Microb Pathog. 2000;28(2):107–117. doi: 10.1006/mpat.1999.0328. [DOI] [PubMed] [Google Scholar]
- 30.Chiarezza M, Lyras D, Pidot SJ, et al. The NanI and NanJ sialidases of Clostridium perfringens are not essential for virulence. Infect Immun. 2009;77(10):4421–4428. doi: 10.1128/IAI.00548-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakravorty A, Awad MM, Hiscox TJ, et al. The cysteine protease alpha-clostripain is not essential for the pathogenesis of Clostridium perfringens-mediated myonecrosis. PLoS ONE. 2011;6(7):e22762. doi: 10.1371/journal.pone.0022762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Adams V, Bannam TL, et al. Toxin plasmids of Clostridium perfringens. Microbiol Mol Biol Rev. 2013;77(2):208–233. doi: 10.1128/MMBR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deguchi A, Miyamoto K, Kuwahara T, et al. Genetic characterization of type A enterotoxigenic Clostridium perfringens strains. PLoS ONE. 2009;4(5):e5598. doi: 10.1371/journal.pone.0005598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, McClane BA. A novel small acid soluble protein variant is important for spore resistance of most Clostridium perfringens food poisoning isolates. PLoS Pathog. 2008;4(5):e1000056. doi: 10.1371/journal.ppat.1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Paredes-Sabja D, Sarker MR, McClane BA. Further characterization of Clostridium perfringens small acid soluble protein-4 (Ssp4) properties and expression. PLoS ONE. 2009;4(7):e6249. doi: 10.1371/journal.pone.0006249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shrestha A, McClane BA. Human claudin-8 and −14 are receptors capable of conveying the cytotoxic effects of Clostridium perfringens enterotoxin. mBio. 2013;4(1) doi: 10.1128/mBio.00594-12. pii:e00594–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robertson SL, Smedley JG, 3rd, McClane BA. Identification of a claudin-4 residue important for mediating the host cell binding and action of Clostridium perfringens enterotoxin. Infect Immun. 2010;78(1):505–517. doi: 10.1128/IAI.00778-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katahira J, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J Cell Biol. 1997;136(6):1239–1247. doi: 10.1083/jcb.136.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angelow S, Ahlstrom R, Yu AS. Biology of claudins. Am J Physiol Renal. 2008;295(4):F867–F876. doi: 10.1152/ajprenal.90264.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Briggs DC, Naylor CE, Smedley JG, 3rd, et al. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J Mol Biol. 2011;413(1):138–149. doi: 10.1016/j.jmb.2011.07.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitadokoro K, Nishimura K, Kamitani S, et al. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J Biol Chem. 2011;286(22):19549–19555. doi: 10.1074/jbc.M111.228478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smedley JG, 3rd, Saputo J, Parker JC, et al. Noncytotoxic Clostridium perfringens enterotoxin (CPE) variants localize CPE intestinal binding and demonstrate a relationship between CPE-induced cytotoxicity and enterotoxicity. Infect Immun. 2008;76(8):3793–3800. doi: 10.1128/IAI.00460-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caserta JA, Robertson SL, Saputo J, Shrestha A, McClane BA, Uzal FA. Development and application of a mouse intestinal loop model to study the in vivo action of Clostridium perfringens enterotoxin. Infect Immun. 2011;79(8):3020–3027. doi: 10.1128/IAI.01342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44••.Sarker MR, Carman RJ, McClane BA. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol Microbiol. 1999;33(5):946–958. doi: 10.1046/j.1365-2958.1999.01534.x. By inactivating and complementing the cpe gene (the first toxin null mutants of C. perfringens in gastrointestinal disease strains), Sarker et al. demonstrated that C. perfringens enterotoxin is essential for C. perfringens type A food-borne and nonfood-borne isolates to cause pathology in rabbit ileal loops. [DOI] [PubMed] [Google Scholar]
- 45.Keyburn AL, Yan XX, Bannam TL, Van Immerseel F, Rood JI, Moore RJ. Association between avian necrotic enteritis and Clostridium perfringens strains expressing NetB toxin. BMC Vet Res. 2010;41(2):21. doi: 10.1051/vetres/2009069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skinner JT, Bauer S, Young V, Pauling G, Wilson J. An economic analysis of the impact of subclinical (mild) necrotic enteritis in broiler chickens. Avian Dis. 2010;54(4):1237–1240. doi: 10.1637/9399-052110-Reg.1. [DOI] [PubMed] [Google Scholar]
- 47.Cooper KK, Songer JG, Uzal FA. Diagnosing clostridial enteric disease in poultry. J Vet Diagn Invest. 2013;25(3):314–327. doi: 10.1177/1040638713483468. [DOI] [PubMed] [Google Scholar]
- 48.Van Immerseel F, Rood JI, Moore RJ, Titball RW. Rethinking our understanding of the pathogenesis of necrotic enteritis in chickens. Trends Microbiol. 2009;17(1):32–36. doi: 10.1016/j.tim.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 49••.Keyburn AL, Boyce JD, Vaz P, et al. NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 2008;4(2):e26. doi: 10.1371/journal.ppat.0040026. In this manusript, Keyburn et al. demonstrated the requirement of NetB for causing necrotic enteritis in broiler chickens using purified native and recombinant NetB toxin, and netB null and complemented strains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan XX, Porter CJ, Hardy SP, et al. Structural and functional analysis of the pore-forming toxin NetB from Clostridium perfringens. mBio. 2013;4(1):e00019–13. doi: 10.1128/mBio.00019-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Savva CG, Fernandes Da Costa SP, Bokori-Brown M, et al. Molecular architecture and functional analysis of NetB, a pore-forming toxin from Clostridium perfringens. J Biol Chem. 2013;288(5):3512–3522. doi: 10.1074/jbc.M112.430223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheung JK, Keyburn AL, Carter GP, et al. The VirSR two-component signal transduction system regulates NetB toxin production in Clostridium perfringens. Infect Immun. 2010;78(7):3064–3072. doi: 10.1128/IAI.00123-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allaart JG, De Bruijn ND, Van Asten AJ, Fabri TH, Grone A. NetB-producing and beta2-producing Clostridium perfringens associated with subclinical necrotic enteritis in laying hens in The Netherlands. Avian Pathol. 2012;41(6):541–546. doi: 10.1080/03079457.2012.729809. [DOI] [PubMed] [Google Scholar]
- 54.Smyth JA, Martin TG. Disease producing capability of netB positive isolates of C. perfringens recovered from normal chickens and a cow, and netB positive and negative isolates from chickens with necrotic enteritis. Vet Microbiol. 2010;146(1–2):76–84. doi: 10.1016/j.vetmic.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 55.Bannam TL, Yan XX, Harrison PF, et al. Necrotic enteritis-derived Clostridium perfringens strain with three closely related independently conjugative toxin and antibiotic resistance plasmids. mBio. 2011;2(5):e00190–11. doi: 10.1128/mBio.00190-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lepp D, Gong J, Songer JG, Boerlin P, Parreira VR, Prescott JF. Identification of accessory genome regions in poultry Clostridium perfringens isolates carrying the netB plasmid. J Bacteriol. 2013;195(6):1152–1166. doi: 10.1128/JB.01032-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lepp D, Roxas B, Parreira VR, et al. Identification of novel pathogenicity loci in Clostridium perfringens strains that cause avian necrotic enteritis. PLoS ONE. 2010;5(5):e10795. doi: 10.1371/journal.pone.0010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jang S, Lillehoj H, Lee S. Vaccination with Clostridium perfringens recombinant proteins in combination with montanide TM ISA 71 adjuvant increases protection against experimental necrotic enteritis in commercial broiler chickens. Vaccine. 2012;30(36):5401–5406. doi: 10.1016/j.vaccine.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 59.Keyburn AL, Portela RW, Sproat K, et al. Vaccination with recombinant NetB toxin partially protects broiler chickens from necrotic enteritis. BMC Vet Res. 2013;44(1):54. doi: 10.1186/1297-9716-44-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fernandes Da Costa SP, Mot D, Bokori-Brown M, et al. Protection against avian necrotic enteritis after immunisation with NetB genetic or formaldehyde toxoids. Vaccine. 2013;31(37):4003–4008. doi: 10.1016/j.vaccine.2013.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keyburn AL, Portela RW, Ford ME, et al. Maternal immunization with vaccines containing recombinant NetB toxin partially protects progeny chickens from necrotic enteritis. BMC Vet Res. 2013;44(1):108. doi: 10.1186/1297-9716-44-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uzal FA, McClane BA. Recent progress in understanding the pathogenesis of Clostridium perfringens type C infections. Vet Microbiol. 2011;153(1–2):37–43. doi: 10.1016/j.vetmic.2011.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petrillo TM, Beck-Sague CM, Songer JG, et al. Enteritis necroticans (pigbel) in a diabetic child. N Engl J Med. 2000;342(17):1250–1253. doi: 10.1056/NEJM200004273421704. [DOI] [PubMed] [Google Scholar]
- 64.Gui L, Subramony C, Fratkin J, Hughson MD. Fatal enteritis necroticans (pigbel) in a diabetic adult. Modern Pathol. 2002;15(1):66–70. doi: 10.1038/modpathol.3880491. [DOI] [PubMed] [Google Scholar]
- 65.Vidal JE, McClane BA, Saputo J, Parker J, Uzal FA. Effects of Clostridium perfringens beta-toxin on the rabbit small intestine and colon. Infect Immun. 2008;76(10):4396–4404. doi: 10.1128/IAI.00547-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fisher DJ, Fernandez-Miyakawa ME, Sayeed S, et al. Dissecting the contributions of Clostridium perfringens type C toxins to lethality in the mouse intravenous injection model. Infect Immun. 2006;74(9):5200–5210. doi: 10.1128/IAI.00534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagahama M, Hayashi S, Morimitsu S, Sakurai J. Biological activities and pore formation of Clostridium perfringens beta toxin in HL 60 cells. J Biol Chem. 2003;278(38):36934–36941. doi: 10.1074/jbc.M306562200. [DOI] [PubMed] [Google Scholar]
- 68.Autheman D, Wyder M, Popoff M, D’herde K, Christen S, Posthaus H. Clostridium perfringens beta-toxin induces necrostatin-inhibitable, calpain-dependent necrosis in primary porcine endothelial cells. PLoS ONE. 2013;8(5):e64644. doi: 10.1371/journal.pone.0064644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miclard J, Jaggi M, Sutter E, Wyder M, Grabscheid B, Posthaus H. Clostridium perfringens beta-toxin targets endothelial cells in necrotizing enteritis in piglets. Vet Microbiol. 2009;137(3–4):320–325. doi: 10.1016/j.vetmic.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 70.Gurjar A, Li J, McClane BA. Characterization of toxin plasmids in Clostridium perfringens type C isolates. Infect Immun. 2010;78(11):4860–4869. doi: 10.1128/IAI.00715-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sayeed S, Li J, McClane BA. Characterization of virulence plasmid diversity among Clostridium perfringens type B isolates. Infect Immun. 2010;78(1):495–504. doi: 10.1128/IAI.00838-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vidal JE, Ma M, Saputo J, Garcia J, Uzal FA, McClane BA. Evidence that the Agr-like quorum sensing system regulates the toxin production, cytotoxicity and pathogenicity of Clostridium perfringens type C isolate CN3685. Mol Microbiol. 2012;83(1):179–194. doi: 10.1111/j.1365-2958.2011.07925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma M, Vidal J, Saputo J, McClane BA, Uzal F. The VirS/VirR two-component system regulates the anaerobic cytotoxicity, intestinal pathogenicity, and enterotoxemic lethality of Clostridium perfringens type C isolate CN3685. mBio. 2011;2(1):e00338–10. doi: 10.1128/mBio.00338-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Uzal FA, Saputo J, Sayeed S, et al. Development and application of new mouse models to study the pathogenesis of Clostridium perfringens type C Enterotoxemias. Infect Immun. 2009;77(12):5291–5299. doi: 10.1128/IAI.00825-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garcia JP, Beingesser J, Fisher DJ, et al. The effect of Clostridium perfringens type C strain CN3685 and its isogenic beta toxin null mutant in goats. Vet Microbiol. 2012;157(3–4):412–419. doi: 10.1016/j.vetmic.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76••.Sayeed S, Uzal FA, Fisher DJ, et al. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol Microbiol. 2008;67(1):15–30. doi: 10.1111/j.1365-2958.2007.06007.x. The critical role of Clostridium perfringens beta (CPB) in C. perfringens type C intestinal disease was demonstrated by Sayeed et al. using isogenic cpb null and reversed mutants in rabbit ileal loops. Neutralizing anti-CPB antibody also blocked intestinal damage by CPB-producing strain CN3685 or by purified native CPB. [DOI] [PubMed] [Google Scholar]
- 77.Uzal FA, Songer JG. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J Vet Diagn Invest. 2008;20(3):253–265. doi: 10.1177/104063870802000301. [DOI] [PubMed] [Google Scholar]
- 78.Chen J, Rood JI, McClane BA. Epsilon-toxin production by Clostridium perfringens type D strain CN3718 is dependent upon the agr operon but not the VirS/VirR two-component regulatory system. mBio. 2011;2(6) doi: 10.1128/mBio.00275-11. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 79.Chen J, McClane BA. Role of the Agr-like quorum-sensing system in regulating toxin production by Clostridium perfringens type B strains CN1793 and CN1795. Infect Immun. 2012;80(9):3008–3017. doi: 10.1128/IAI.00438-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Popoff MR. Epsilon toxin: a fascinating pore-forming toxin. FEBS J. 2011;278(23):4602–4615. doi: 10.1111/j.1742-4658.2011.08145.x. [DOI] [PubMed] [Google Scholar]
- 81.Hughes ML, Poon R, Adams V, et al. Epsilon-toxin plasmids of Clostridium perfringens type D are conjugative. J Bacteriol. 2007;189(21):7531–7538. doi: 10.1128/JB.00767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sayeed S, Fernandez-Miyakawa ME, Fisher DJ, et al. Epsilon-toxin is required for most Clostridium perfringens type D vegetative culture supernatants to cause lethality in the mouse intravenous injection model. Infect Immun. 2005;73(11):7413–7421. doi: 10.1128/IAI.73.11.7413-7421.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83••.Garcia JP, Adams V, Beingesser J, et al. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect Immun. 2013;81(7):2405–2414. doi: 10.1128/IAI.00238-13. Using isogenic etx null mutants and complemented strains of a C. perfringens type D isolate, Garcia et al. demonstrate that ETX is essential for causing intestinal disease and enterotoxemias in sheep, goats and mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ma M, Li J, McClane BA. Genotypic and phenotypic characterization of Clostridium perfringens isolates from Darmbrand cases in post-World War II Germany. Infect Immun. 2012;80(12):4354–4363. doi: 10.1128/IAI.00818-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fernandez-Miyakawa ME, Fisher DJ, Poon R, et al. Both epsilon-toxin and beta-toxin are important for the lethal properties of Clostridium perfringens type B isolates in the mouse intravenous injection model. Infect Immun. 2007;75(3):1443–1452. doi: 10.1128/IAI.01672-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guttenberg G, Hornei S, Jank T, et al. Molecular characteristics of Clostridium perfringens TpeL toxin and consequences of mono-O-GlcNAcylation of Ras in living cells. J Biol Chem. 2012;287(30):24929–24940. doi: 10.1074/jbc.M112.347773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carter GP, Cheung JK, Larcombe S, Lyras D. Regulation of toxin production in the pathogenic clostridia. Mol Microbiol. 2014 doi: 10.1111/mmi.12469. In Press. [DOI] [PubMed] [Google Scholar]
- 88.Schrodinger L. The PyMOL Molecular Graphics System. 2008. [Google Scholar]
- 89.Naylor C, Eaton J, Howells A, et al. Structure of the key toxin in gas gangrene. Nat Struct Biol. 1998;5:738–746. doi: 10.1038/1447. [DOI] [PubMed] [Google Scholar]
- 90.Tsuge H, Nagahama M, Nishimura H, et al. Crystal structure and site-directed mutagenesis of enzymatic components from Clostridium perfringens iota-toxin. J Mol Biol. 2003;325(3):471–483. doi: 10.1016/s0022-2836(02)01247-0. [DOI] [PubMed] [Google Scholar]
- 91.Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell. 1997;88:685–692. doi: 10.1016/s0092-8674(00)80251-2. [DOI] [PubMed] [Google Scholar]
- 92.Cole AR, Gibert M, Popoff M, Moss DS, Titball RW, Basak AK. Clostridium perfringens epsilon-toxin shows structural similarity to the pore-forming toxin aerolysin. Nat Struct Biol. 2004;11(8):797–798. doi: 10.1038/nsmb804. [DOI] [PubMed] [Google Scholar]
- 93.Huyet J, Naylor CE, Savva CG, Gibert M, Popoff MR, Basak AK. Structural Insights into delta toxin pore formation. PLoS ONE. 2013;8(6):e66673. doi: 10.1371/journal.pone.0066673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen J, Ma M, Uzal FA, McClane BA. Host cell-induced signaling causes Clostridium perfringens to upregulate production of toxins important for intestinal infections. Gut Microbes. 2013;5:1. doi: 10.4161/gmic.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]