

Abstract
Estrogen receptor-alpha36 (ER-α36) is a 36-kDa variant of estrogen receptor-alpha (ER-α) firstly identified and cloned by Wang et al in 2005. It lacks both transactivation domains (activation function 1 and activation function 2) and has different biological characteristics compared to traditional ER-α (ER-α66). ER-α36 primarily locates on plasma membrane and cytoplasm and functions as a mediator in the rapid membrane-initiated non-genomic signaling pathway. Additionally, it inhibits the traditional genomic signaling pathway mediated by ER-α66 in a dominant-negative pattern. Accumulating evidence has demonstrated that ER-α36 regulates the physiological function of various tissues. Thus, dysregulation of ER-α36 is closely associated with plenty of diseases including cancers. ER-α36 is recognized as a molecular abnormality which solidly correlates to carcinogenesis, aggressiveness, and therapeutic response of breast cancer. Additionally, special attention has been paid to the role of ER-α36 in endocrine therapy resistance. Therefore, ER-α36 provides a novel biomarker of great value for diagnosis, prognosis, and treatment of breast cancer. It may also be a potential therapeutic target for breast cancer patients, especially for those who are resistant to endocrine therapy. In this review, we will overview and update the biological characteristics, underlying mechanism, and function of ER-α36, focusing on its biological function in breast cancer and endocrine therapy resistance. We will evaluate its application value in clinical practice.
Keywords: ER, ER-α36, breast cancer, endocrine therapy resistance
Introduction
Estrogen is essential to the development and physiology of target organs, such as uterus, ovary, and breast. Dysregulation of estrogen results in pathological processes including osteoporosis, breast cancer, and endometrial cancer.1 These pleiotropic effects of estrogen are related to estrogen receptors (ERs) which are generally regarded as ligand-activated transcription factors belonging to the nuclear receptor superfamily.1–3 Two different forms of ER have been discovered, ER-α and ER-β.3,4 They share similar architecture composed of three independent but interactional function domains.3,5 The N-terminal A/B domains possess a ligand-independent transactivation domain (activation function 1 [AF-1]), interacting with co-activators and activating transcription of target genes.6 The C or DNA-binding domain contains two zinc finger-like structures, allowing dimerization of receptors and then binding to target DNA sequences.6 The C-terminal D/E/F domains or ligand-binding domains mediate ligand binding, receptor dimerization, and nuclear localization and have a ligand-dependent activation domain (activation function 2 [AF-2]) (Figure 1B).6
Figure 1.
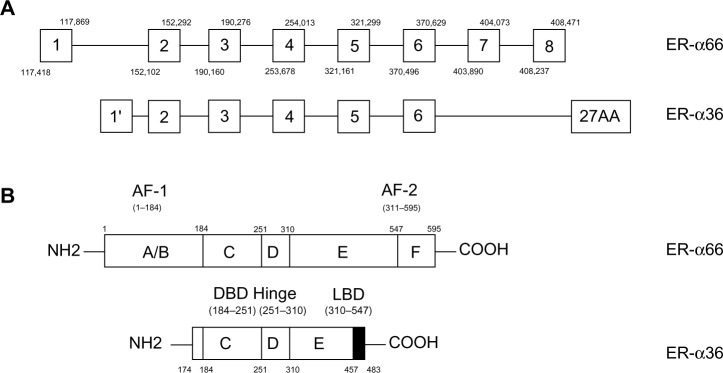
Structure of ER-α66 and ER-α36.
Notes: (A) Transcription of ER-α36 initiates from a previously unidentified promoter in the first intron of ER-α66. We labeled the first exon of ER-α36 as “1” to distinguish it. Besides, ER-α36 has an extra, unique 27-amino-acid sequence at C-terminus which may broaden ligand-binding spectrum of ER-α36. (B) Compared to the protein structure of ER-α66, ER-α36 lacks both transactivation domains of AF-1 and AF-2 and retains DBD, Hinge, and LBD of ER-α66. Therefore, ER-α36 functions as a powerful competitor of ER-α66.
Abbreviations: AF-1, activation function 1; AF-2, activation function 2; DBD, DNA binding domain; ER-α36, estrogen receptor-alpha36; ER-α66, estrogen receptor-alpha66; LBD, ligand binding domain.
ER-α36 is a 36-kDa variant of ER-α generating from full length ER-α by alternative splicing or different usage of promoters.6–9 Compared to ER-α66, ER-α36 retains DNA-binding, partial dimerization, and ligand-binding domains but lacks both transactivation domains (AF-1 and AF-2).3,14,15 ER-α36 mainly locates on plasma membrane and cytoplasm mediating non-genomic estrogen signaling.6,10,11 Additionally, it inhibits traditional genomic estrogen signaling mediated by ER-α66 in a dominant-negative pattern.10 Accumulating evidence has demonstrated that ER-α36 participates in the development and physiological function of many tissues. Therefore, it is related to diseases such as postmenopausal osteoporosis,12 airway hyperresponsiveness,13 and even malignancies including gastric,14–16 hepatic,17 endometrial,18 and breast cancers.19 ER-α36 is crucial in the carcinogenesis and aggressiveness of breast cancer; in addition, it explains why antiestrogen therapy loses efficacy in ER-positive breast cancer patients.20–22 These achievements imply that ER-α36 is a promising biomarker for diagnosis, prognosis, and treatment of various diseases, especially breast cancer.23–25 What’s more, it provides a novel pharmaceutical approach to individualized treatment of breast cancer.
This review will overview and update the biological characteristics, molecular mechanism, and biological role of ER-α36, focusing on its function in carcinogenesis, progression, and endocrine therapy resistance of breast cancer and its potential application value in clinical practice.
Basic characteristics of ER-α36
Transcription of ER-α36 initiates from a previously unidentified promoter in the first intron of ER-α66, which continues from exon 2 to exon 6 and skips the final exon 7 and exon 8 of ER-α66 (Figure 1A).3,6,10,14,15 As a result, ER-α36 lacks both transactivation domains (AF-1 and AF-2) and retains DNA-binding, partial dimerization, and ligand-binding domains of ER-α66 (Figure 1B). The last 138 amino acids encoded by the final exon 7 and exon 8 are replaced by an extra, unique 27-amino-acid sequence at C-terminus. This extra sequence may broaden the ligand-binding spectrum of ER-α36 so that it is able to interact with more factors other than estrogen.10 ER-α36 primarily locates on plasma membrane (50%) and cytoplasm (40%) rather than nucleus.10,19,26–28 Because it has three potential myristoylation sites near N-terminus instead of the nuclear-location signals of ER-α66, ER-α36 may be modified by palmitoylation posttranslationally and then locate on plasma membrane and cytoplasm just like ER-α46, another variant of ER-α66.6,10,11 Therefore, ER-α36 is a potential regulator of membrane-initiated mitogenic signaling pathways responding to E2α, E2β, E3, E4, and even tamoxifen, an estrogen antagonist.6,10,24
Though ER-α36 exists in both ER-α66-positive and ER-α66-negative breast cancer cells,10,28 it is highly expressed in the majority of ER-α66-negative breast cancer.19,29 Furthermore, overexpression of ER-α36 is usually accompanied with a decrease of ER-α66,10,24,28,30 indicating that ER-α66 and ER-α36 are mutually regulated and inversely associated.10,20,24,27–30 Wilms’ tumor suppressor WT1 is one of the coordinators which activates promoters of ER-α66 but suppresses ER-α36 promoter activity.31 Another coordinator is synuclein γ (SNCG), which binds to ER-α66 or ER-α36 depending on E2 concentration.32,33 In addition, ER-α66 can negatively regulate the promoter activity of ER-α36.30 Deng et al found that proteasome inhibitors dramatically increased the steady level of ER-α66, indicating that the proteasome system took part in the expression regulation of ER-α36 and ER-α66.34 With the dynamic regulation of ER-α66 and ER-α36, genomic and non-genomic estrogen signaling pathways are also coordinated to maintain a balance.31 Although the explicit mechanism needs further investigation, it is obvious that an imbalance of ER-α66 and ER-α36 may result in abnormal proliferation and differentiation, finally leading to diseases including breast cancer.1
Molecular mechanism underlying ER-α36 action
Under normal conditions, ERs bind to estrogen and then transfer to the nucleus, subsequently binding to specific estrogen response elements and regulating transcription of downstream DNA (Figure 2).35 Other than the traditional genomic pathway, ERs mediate the non-genomic pathway by membrane- associated receptors.36–39 The non-genomic pathway controls more genes than the genomic pathway, regulating cellular growth, survival, motility, invasion, and apoptosis. What’s more, non-physiologic estrogen also activates the non-genomic pathway, which is important to so-called “nontarget” tissue such as the cardiovascular system.40,41 The unique structure and intracellular location of ER-α36 decide its molecular mechanism. Lacking both transcriptional activation domains, ER-α36 binds to the same target DNA sequence as ER-α66 does, but possesses no intrinsic transcriptional activity. Thus, ER-α36 functions as a powerful competitor of the genomic signaling pathway mediated by ER-α66.6,10 Additionally, ER-α36 mediates rapid membrane-initiated estrogen signaling cascades, which regulate various biological functions and are associated with tumorigenesis, aggressive phenotype, and treatment sensitivity of carcinomas (Figure 2).16,23,24,42–44
Figure 2.

Genomic and non-genomic activities mediated by ERs.
Notes: ER-α66 binds to estrogen and then transfers to the nucleus to interact with specific EREs and induce transcription of target genes. ER-α36 has a broader ligand-binding spectrum than ER-α66 so that it can respond to E2α, E2β, E3, E4, and even tamoxifen. ER-α36 can mediate rapid MIES such as MAPK/ERK, PI3K/Akt, and PKC δ to regulate biological functions of cells.
Abbreviations: Akt, protein kinase B; BCSCs, breast cancer stem cells; cdk 4, cyclin-dependent kinase 4; ER-α36, estrogen receptor-alpha36; ER-α66, estrogen receptor-alpha66; EREs, estrogen response elements; ERK, extracellular signal-regulated kinase; ERs, estrogen receptors; GSK3 β, glycogen synthase kinase 3 beta; JNKs, c-Jun N-terminal kinases; MAPK, mitogen-activated protein kinases; MIES, membrane-initiated estrogen signaling; PKC δ, protein kinase C delta; PI3K, phosphatidylinositide 3-kinase.
ER-α36 and downstream kinases
The mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway is the most popular mitogenic signaling in cancers, which can be rapidly activated by E2.45,46 Research has demonstrated that membrane-initiated signaling pathways mediated by ER-α36 could stimulate the transcription of ELK1 in the nucleus.10,26,42,47,48 Tong et al found that ER-α36 also activated ERK1/2 through the protein kinase C delta signaling pathway and then elevated expression of cyclin D1/cyclin-dependent kinase 4, a transcriptional coregulator that regulates cell cycle progression.48 The MAPK/ERK signaling pathway mediated by ER-α36 contributes to the potential invasion and metastasis of cancer cells.23,24,44 What’s more, it induces paclitaxel resistance through c-Jun N-terminal kinases (JNKs), an important component of the MAPK family mediating paclitaxel-induced apoptosis and death (Figure 2).23,24,44
Phosphatidylinositide 3-kinase/protein kinase B (PI3K/Akt) is another classical pathway associated with malignant cancers.49–51 Rapid phosphorylation of Akt, which is induced by E2, testosterone, and tamoxifen, can be blocked by a PI3K inhibitor or knockdown of ER-α36, suggesting that the PI3K/Akt pathway is one of the non-genomic pathways mediated by ER-α36 (Figure 2).26,47
ER-α36 associated proteins
Several proteins are involved in the stability and function of ER-α36, thus promoting mitogenic estrogen signaling mediated by ER-α36. Glucose-regulated protein 94, an endoplasmic reticulum chaperone, is a potential downstream effector of the ER-α36-mediated pathway in gastric cancer.15 SNCG, previously regarded as breast cancer-specific gene BCSG1,52 binds to ER-α36 without heat shock protein 90 to prevent degradation of ER-α36 and significantly strengthen ER-α36-mediated mitogenic estrogen signaling pathways.33
Many investigators discovered a significant co-expression of ER-α36 and epidermal growth factor receptor (EGFR) in primary breast cancers, indicating that ER-α36 took part in EGFR-related carcinogenesis.19 Further studies elucidated that epidermal growth factor (EGF) induced phosphorylation of ERK1/2 via ER-α36 in a time- and dose-dependent pattern.18 A positive feedback loop was confirmed that EGFR signaling activated transcription of ER-α36 through an activator-protein-1-binding site in the promoter of ER-α36. In turn, ER-α36 interacted with the EGFR/Src/Shc complex to strengthen the EGFR signaling pathway and stabilize EGFR protein (Figure 3).20 A similar feedback loop between ER-α36 and human epidermal growth factor receptor 2 (HER-2), a member of the EGFR family, was reported.53 Interestingly, our previous study found that HER-2 expression didn’t increase in tamoxifen-resistant cells, which overexpressed ER-α36 and EGFR.23 In the Src/EGFR/signal transducer and activator of transcription 5 (STAT5) pathway mediated by ER-α36, Src functions as a switch to adjust phosphorylation of EGFR and then recruits STAT5 as a downstream effector, which regulates activation of the MAPK/ERK signaling pathway and expression of cyclin D1 (Figure 3).16,20,54 Therefore, the positive feedback loops between ER-α36 and growth factor receptors are important for cellular function, although the definite mechanism has not been clarified.
Figure 3.

Positive feedback loop between EGFR and ER-α36.
Notes: A positive feedback loop has been confirmed that EGFR signaling activates transcription of ER-α36 through an AP-1-binding site in the promoter region of ER-α36. In turn, ER-α36 is able to stabilize EGFR protein and mediate MAPK/ERK and Src/EGFR/STAT5 pathways. In the Src/EGFR/STAT5 pathway, Src functions as a switch by phosphorylation to enhance proliferation and malignant properties of cancer.
Abbreviations: AP-1, activator protein 1; EGFR, epidermal growth factor receptor; ER-α36, estrogen receptor-alpha36; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinases; STAT5, signal transducer and activator of transcription 5.
Biological roles of ER-α36
ER-α36 participates in the development and function of target tissues. For example, expression and subcellular localization of ER-α36 are necessary for folliculogenesis, oocyte meiotic maturation, ovulation, and luteinization of the postnatal ovary, thus facilitating maturation of the female reproductive system.55 Additionally, ER-α36 maintains the bone density of postmenopausal women.12 However, dysregulation of ER-α36 leads to various dysfunctions and diseases, such as osteoporosis,12 airway hyperresponsiveness,13 and even cancers.14,17–19 Since the mammary gland is estrogen-dependent, ER-α36 plays a particularly vital role in breast cancer. Therefore, we will focus on its role in breast cancer and endocrine therapy resistance and its potential application value in clinical practice.
Biological function of ER-α36 in breast cancer
In order to investigate the biological function of ER-α36 in breast cancer, Zhang et al established ER-α36-knockdown MDA-MB-231 cell lines in which expression of ER-α36 decreased obviously. They found knockdown of ER-α36 suppressed proliferation, migration, and invasion and increased sensitivity to paclitaxel in MDA-MB-231 cells.24 Chaudhri et al got similar results that ER-α36 was able to enhance cellular proliferation and metastasis and protect against apoptosis.43 Recently, Yu et al studied the relationship between ER-α36 and chemotherapy response in 120 breast cancer patients and found that ER-α36-negative patients had better response to anthracycline and/or paclitaxel than ER-α36-positive patients.25
ER-α36 and breast cancer stem cells (BCSCs)
BCSCs, a subpopulation of stem/progenitor cells that possesses the ability to self-renew, play a key role in occurrence, recurrence, metastasis, and chemotherapy resistance of breast cancer.56,57 CD44+/CD24−/low and aldehyde dehydrogenase 1 (ALDH1) were identified as markers of BCSCs.56,58 Kang et al found that ER-α36 was highly expressed in ALDH1-positive SK-BR-3 cells. Additionally, the depletion of ER-α36 reduced the growth rate and proportion of ALDH1high cells, indicating that ER-α36 contributed to the proliferation and maintenance of stem-like cells.53 Deng et al observed that estrogen significantly increased the population of CD44+/CD24−/low cells in MCF-7 and T47D breast cancer cells, but failed to do so after knocking down ER-α36, suggesting that estrogen positively regulated BCSCs through the ER-α36-mediated signaling pathway.34 Furthermore, they found that overexpression of ER-α36 tended to promote self-renewal and tumor-seeding efficiency of BCSCs through the PI3K/AKT/glycogen synthase kinase 3 beta/β-catenin signaling pathway (Figure 2).34
ER-α36 and endocrine therapy resistance
Endocrine therapy is a dominant anticancer method for ER-positive breast cancer patients. Although aromatase inhibitors are increasingly applied in postmenopausal women,59 antiestrogens are still used as first-line therapy, especially when aromatase inhibitors are ineffective. However, less or no response after long-term treatment is the major obstacle in antiestrogen treatment.60,61
Tamoxifen
Tamoxifen is a competitive antagonist of ER-α, effectively blocking traditional genomic signaling pathway mediated by ER-α66.61,62 However, it can activate different kinase cascades and produce second messengers, thus acting as an agonist that is equal to estrogen.63 For example, tamoxifen strongly activates the MAPK/ERK signaling pathway to stimulate cell growth; additionally, it activates the p38/MAPK and SAPK/JNK pathways to induce apoptosis.10,64 In recent years, a large number of explorations have concentrated on the association between ER-α36 and tamoxifen resistance. Due to the unique 27-amino-acid domain, ER-α36 has a broader ligand-binding spectrum than ER-α66 so that it can respond to tamoxifen.10,24 Usually, ER-α36 is highly expressed in ER-α66-negative breast cancer, which is insensitive to endocrine therapy.10,28,29 Clinical research demonstrated that high expression of ER-α36 was associated with decreased tamoxifen sensitivity and poorer survival in ER-α66-positive breast cancer patients who received tamoxifen as adjuvant therapy.29 Additionally, laboratory studies revealed that the tamoxifen-resistant MCF-7 cell line (MCF-7/TAM) highly expressed ER-α36.22,23,65 However, knocking down ER-α36 in MCF-7/TAM cells could not restore their sensitivity to tamoxifen completely.23 This may be because the expression level of ER-α36 in MCF-7/TAM cells is too high to be completely blocked; besides, biological reprogramming during development of tamoxifen resistance is much more complicated than upregulation of ER-α36. Furthermore, proteins maintaining the stability and function of ER-α36 such as SNCG can strengthen tamoxifen resistance mediated by ER-α36.33 Therefore, ER-α36 is an underlying cause of tamoxifen resistance in breast cancer patients.
Previous explorations have demonstrated that ER-α36 is a potent mediator of membrane-initiated signaling pathways associated with agonist effects of tamoxifen, which leads to tamoxifen resistance.10,24 For example, the MAPK/ERK and PI3K/AKT signaling pathways that are mediated by ER-α36 induce the expression of protooncogene c-Myc, which has profound mitogenic effects (Figure 4).10,44 ER-α36 also mediates the Src/EGFR/STAT5 pathway regulating activity of the MAPK/ERK signaling pathway and expression of the growth-promoting cyclin D1 (Figure 4).66 However, it is still unknown whether other non-genomic pathways, such as protein kinase C , protein kinase A , and calcium channels are involved in tamoxifen resistance.
Figure 4.

ER-α36 and tamoxifen resistance.
Notes: ER-α36 is an important cause of tamoxifen resistance. On the one hand, it activates membrane-initiated signaling pathways, such as MAPK/ERK, PI3K/AKT, and Src/EGFR/STAT5 pathways, causing the agonist effect of tamoxifen. On the other hand, ER-α36 upregulates EGFR and downregulates ER-α66 switching growth status from estrogen-dependent to growth-factor-dependent. Therefore, ER-α36 leads to tamoxifen resistance.
Abbreviations: Akt, protein kinase B; EGFR, epidermal growth factor receptor; ER-α36, estrogen receptor-alpha36; ER-α66, estrogen receptor-alpha66; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinases; MCF-7/TAM, tamoxifen-resistant MCF-7 cell line; PI3K, phosphatidylinositide 3-kinase; STAT5, signal transducer and activator of transcription 5.
Further research has revealed that overexpression of EGFR with downregulation of ER-α66 is responsible for tamoxifen resistance.67–69 Li et al established a tamoxifen-resistant MCF-7 cell line (MCF-7/TAM) that had an accelerated proliferation rate as well as enhanced migratory and invasive ability. In MCF-7/TAM cells, ER-α36 upregulated EGFR expression and downregulated ER-α66 expression, switching growth status from estrogen-dependent to growth-factor-dependent, which was a critical step in the development of tamoxifen resistance (Figure 4).23
ICI 182,780 (Fulvestrant, Faslodex®)
ICI 182,780 (Fulvestrant, Faslodex), a ‘pure’ antiestrogen, not only accelerates degradation but also impairs dimerization and nuclear localization of ER-α66.70,71 However, several studies reported that ICI 182,780 failed to induce degradation of ER-α36,6,72 presumably because ER-α36 has a truncated ligand-binding domain lacking the last four helices (helix 9–12) of ER-α66, which are essential for protein degradation induced by ICI 182,780.6,73 Therefore, failure of ER-α36 degradation is a highly possible reason for ICI 182,780 resistance.
Application value of ER-α36
Metastasis and recurrence are the most intractable issues in the management of cancer; thus, using molecular alteration to assess tumor degree and prognosis is of great significance. A large number of studies have confirmed that ER-α36-positive breast cancers tend to have more lymph node metastasis, advanced degree, less sensitivity to chemotherapy or endocrine therapy, and poorer outcome than ER-α36-negative breast cancer. Therefore, ER-α36 is a promising biomarker for malignant behavior, therapeutic response, and outcome of breast cancer, guiding clinical decisions in early diagnosis, prognosis estimation, and personalized treatment. For example, we can combine the expression of ER-α66 and ER-α36 to predict whether endocrine therapy is suitable and effective for breast cancer patients.
Given its important role in carcinogenesis, progression, and therapeutic response of breast cancer, ER-α36 is emerging as a potential therapeutic target for breast cancer. The surface localization of ER-α36 makes it possible to generate specific surface epitopes for treatment intervention with small molecules or biologics which directly down-modulate the ER-α36 protein or indirectly block its downstream signaling pathways, such as biological antagonists, monoclonal antibodies, and small kinase inhibitors. Inhibition of ER-α36 can suppress tumor growth and progression, benefitting breast cancer patients, especially those who are resistant to endocrine therapy. Though few studies have focused on the therapeutic application of ER-α36 and no clinical trials currently, a targeted agent suitable for clinical testing will be available in the near future. Broussoflavonol B, a flavonoid purified from the bark of Broussonetia papyrifera (Moraceae), exhibits a more potent growth inhibition effect than tamoxifen in ER-negative breast cancer.74,75 It decreases the steady level of ER-α36 and EGFR and restricts growth of stem-like cells in MDA-MB-231 cells.75
Conclusion
ER-α36 is a variant of ER-α with different characteristics and functions than ER-α66. It locates on plasma membrane and cytoplasm. Therefore, ER-α36 mediates the non-genomic signaling pathway and suppresses the traditional genomic signaling pathway. ER-α36 plays a key role in breast cancer, leading to an accelerated proliferation rate together with enhanced migratory and invasive ability. Additionally, it mediates the agonist action of tamoxifen and switches the growth status of breast cancer cells from estrogen-dependent to growth-factor-dependent, both of which can be the molecular mechanisms of tamoxifen resistance. Furthermore, failure of ER-α36 degradation is a possible reason for Fulvestrant resistance. Therefore, ER-α36 is a promising biomarker for tumor behavior and therapeutic response, being a new indicator to select optimal candidates for therapeutic strategies. Additionally, it is a potential therapeutic target to improve survival of breast cancer patients.
These achievements make the heterogeneity of the estrogen effect more clear and put a new insight into the properties of ER-α36. Additionally, studies of ER-α36 deepen our knowledge of the carcinogenesis and progression of breast cancer and provide an alternative explanation for endocrine therapy resistance. However, there are still many challenges in the future. More research is needed to clarify the biological function and deep mechanism of ER-α36. Additionally, developing an effective and specific inhibitor of ER-α36 is a difficult work. Before the inhibitor is widely used, the inhibitory consequences should be validated regarding proliferation, invasion, migration, and survival of tumor cells and clinical trials with large sample sizes and long-term observation are also needed.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No 81272676), the National Science and Technology Major Project of the Ministry of Science and Technology of China (No 2013ZX09506015), and the Medical Science and Technology Project of Zhejiang Province (No 2011ZDA009).
Footnotes
Disclosure
The authors declare no conflicts of interest in this work.
References
- 1.Nilsson S, Mäkelä S, Treuter E, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81(4):1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 2.Weihua Z, Andersson S, Cheng G, Simpson ER, Warner M, Gustafsson JA. Update on estrogen signaling. FEBS Lett. 2003;546(1):17–24. doi: 10.1016/s0014-5793(03)00436-8. [DOI] [PubMed] [Google Scholar]
- 3.Kong EH, Pike AC, Hubbard RE. Structure and mechanism of the oestrogen receptor. Biochem Soc Trans. 2003;31(Pt 1):56–59. doi: 10.1042/bst0310056. [DOI] [PubMed] [Google Scholar]
- 4.Gustafsson JA. Estrogen receptor beta – a new dimension in estrogen mechanism of action. J Endocrinol. 1999;163(3):379–383. doi: 10.1677/joe.0.1630379. [DOI] [PubMed] [Google Scholar]
- 5.Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. 1986;231(4742):1150–1154. doi: 10.1126/science.3753802. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. 2005;336(4):1023–1027. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- 7.Hirata S1, Shoda T, Kato J, Hoshi K. Isoform/variant mRNAs for sex steroid hormone receptors in humans. Trends Endocrinol Metab. 2003;14(3):124–129. doi: 10.1016/s1043-2760(03)00028-6. [DOI] [PubMed] [Google Scholar]
- 8.Murphy LC, Dotzlaw H, Leygue E, Douglas D, Coutts A, Watson PH. Estrogen receptor variants and mutations. J Steroid Biochem Mol Biol. 1997;62(5–6):363–372. doi: 10.1016/s0960-0760(97)00084-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhang QX, Hilsenbeck SG, Fuqua SA, Borg A. Multiple splicing variants of the estrogen receptor are present in individual human breast tumors. J Steroid Biochem Mol Biol. 1996;59(3–4):251–260. doi: 10.1016/s0960-0760(96)00120-3. [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen-and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci U S A. 2006;103(24):9063–9068. doi: 10.1073/pnas.0603339103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci U S A. 2003;100(8):4807–4812. doi: 10.1073/pnas.0831079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie H, Sun M, Liao XB, et al. Estrogen receptor α36 mediates a bone-sparing effect of 17β-estrodiol in postmenopausal women. J Bone Miner Res. 2011;26(1):156–168. doi: 10.1002/jbmr.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia S, Zhang X, He DZ, et al. Expression and function of a novel variant of estrogen receptor-α36 in murine airways. Am J Respir Cell Mol Biol. 2011;45(5):1084–1089. doi: 10.1165/rcmb.2010-0268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng H, Huang X, Fan J, et al. A variant of estrogen receptor-alpha, ER-alpha36 is expressed in human gastric cancer and is highly correlated with lymph node metastasis. Oncol Rep. 2010;24(1):171–176. [PMC free article] [PubMed] [Google Scholar]
- 15.Fu Z, Deng H, Wang X, Yang X, Wang Z, Liu L. Involvement of ER-α36 in the malignant growth of gastric carcinoma cells is associated with GRP94 overexpression. Histopathology. 2013;63(3):325–333. doi: 10.1111/his.12171. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Deng H, Zou F, et al. ER-α36-mediated gastric cancer cell proliferation via the c-Src pathway. Oncol Lett. 2013;6(2):329–335. doi: 10.3892/ol.2013.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miceli V, Cocciadiferro L, Fregapane M, et al. Expression of wild-type and variant estrogen receptor alpha in liver carcinogenesis and tumor progression. OMICS. 2011;15(5):313–317. doi: 10.1089/omi.2010.0108. [DOI] [PubMed] [Google Scholar]
- 18.Tu BB, Lin SL, Yan LY, Wang ZY, Sun QY, Qiao J. ER-α36, a novel variant of estrogen receptor α, is involved in EGFR-related carcinogenesis in endometrial cancer. Am J Obstet Gynecol. 2011;205(3):227.e1–227.e6. doi: 10.1016/j.ajog.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Vranic S, Gatalica Z, Deng H, et al. ER-α36, a novel isoform of ER-α66, is commonly over-expressed in apocrine and adenoid cystic carcinomas of the breast. J Clin Pathol. 2011;64(1):54–57. doi: 10.1136/jcp.2010.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang XT, Kang LG, Ding L, Vranic S, Gatalica Z, Wang ZY. A positive feedback loop of ER-α36/EGFR promotes malignant growth of ER-negative breast cancer cells. Oncogene. 2011;30(7):770–780. doi: 10.1038/onc.2010.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, Zhang J, Xu ZZ, et al. Quantitative profiles of the mRNAs of ER-alpha and its novel variant ER-alpha36 in breast cancers and matched normal tissues. J Zhejiang Univ Sci B. 2010;11(2):144–150. doi: 10.1631/jzus.B0900266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Wang ZY. Estrogen receptor-α variant, ER-α36, is involved in tamoxifen resistance and estrogen hypersensitivity. Endocrinology. 2013;154(6):1990–1998. doi: 10.1210/en.2013-1116. [DOI] [PubMed] [Google Scholar]
- 23.Li G, Zhang J, Jin K, et al. Estrogen receptor-α36 is involved in development of acquired tamoxifen resistance via regulating the growth status switch in breast cancer cells. Mol Oncol. 2013;7(3):611–624. doi: 10.1016/j.molonc.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Li G, Li Z, et al. Estrogen-independent effects of ER-α36 in ER-negative breast cancer. Steroids. 2012;77(6):666–673. doi: 10.1016/j.steroids.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Yu L, Ke W, Wang Y, et al. Predictive and prognostic value of ER-α36 expression in breast cancer patients treated with chemotherapy. Steroids. 2014;84:11–16. doi: 10.1016/j.steroids.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Lin SL, Yan LY, Liang XW, et al. A novel variant of ER-alpha, ER-alpha36 mediates testosterone-stimulated ERK and Akt activation in endometrial cancer Hec1A cells. Reprod Biol Endocrinol. 2009;7:102. doi: 10.1186/1477-7827-7-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelekanou V, Notas G, Kampa M, et al. ERα36, a new variant of the ERα is expressed in triple negative breast carcinomas and has a specific transcriptomic signature in breast cancer cell lines. Steroids. 2012;77(10):928–934. doi: 10.1016/j.steroids.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 28.Lee LM, Cao J, Deng H, Chen P, Gatalica Z, Wang ZY. ER-alpha36, a novel variant of ER-alpha, is expressed in ER-positive and -negative human breast carcinomas. Anticancer Res. 2008;28(1B):479–483. [PMC free article] [PubMed] [Google Scholar]
- 29.Shi L, Dong B, Li Z, et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27(21):3423–3429. doi: 10.1200/JCO.2008.17.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou Y, Ding L, Coleman M, Wang Z. Estrogen receptor-alpha (ER-alpha) suppresses expression of its variant ER-alpha 36. FEBS Lett. 2009;583(8):1368–1374. doi: 10.1016/j.febslet.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang L, Wang L, Wang ZY. Opposite regulation of estrogen receptor-α and its variant ER-α36 by the Wilms’ tumor suppressor WT1. Oncol Lett. 2011;2(2):337–341. doi: 10.3892/ol.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Y, Liu YE, Goldberg ID, Shi YE. Gamma synuclein, a novel heat-shock protein-associated chaperone, stimulates ligand-dependent estrogen receptor alpha signaling and mammary tumorigenesis. Cancer Res. 2004;64(13):4539–4546. doi: 10.1158/0008-5472.CAN-03-3650. [DOI] [PubMed] [Google Scholar]
- 33.Shi YE, Chen Y, Dackour R, et al. Synuclein gamma stimulates membrane-initiated estrogen signaling by chaperoning estrogen receptor (ER)-alpha36, a variant of ER-alpha. Am J Pathol. 2010;177(2):964–973. doi: 10.2353/ajpath.2010.100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng H, Zhang XT, Wang ML, Zheng HY, Liu LJ, Wang ZY. ER-α36-mediated rapid estrogen signaling positively regulates ER-positive breast cancer stem/progenitor cells. PLoS One. 2014;9(2):e88034. doi: 10.1371/journal.pone.0088034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wehling M. Specific, nongenomic actions of steroid hormones. Annu Rev Physiol. 1997;59:365–393. doi: 10.1146/annurev.physiol.59.1.365. [DOI] [PubMed] [Google Scholar]
- 37.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 38.Kelly MJ, Levin ER. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab. 2001;12(4):152–156. doi: 10.1016/s1043-2760(01)00377-0. [DOI] [PubMed] [Google Scholar]
- 39.Driggers PH, Segars JH. Estrogen action and cytoplasmic signaling pathways. Part II: the role of growth factors and phosphorylation in estrogen signaling. Trends Endocrinol Metab. 2002;13(10):422–427. doi: 10.1016/s1043-2760(02)00634-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bulayeva NN, Wozniak AL, Lash LL, Watson CS. Mechanisms of membrane estrogen receptor-alpha-mediated rapid stimulation of Ca2+ levels and prolactin release in a pituitary cell line. Am J Physiol Endocrinol Metab. 2005;288(2):E388–E397. doi: 10.1152/ajpendo.00349.2004. [DOI] [PubMed] [Google Scholar]
- 41.Simoncini T, Mannella P, Genazzani AR. Rapid estrogen actions in the cardiovascular system. Ann N Y Acad Sci. 2006;1089:424–430. doi: 10.1196/annals.1386.001. [DOI] [PubMed] [Google Scholar]
- 42.Kang L, Zhang X, Xie Y, et al. Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Mol Endocrinol. 2010;24(4):709–721. doi: 10.1210/me.2009-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaudhri RA, Olivares-Navarrete R, Cuenca N, Hadadi A, Boyan BD, Schwartz Z. Membrane estrogen signaling enhances tumorigenesis and metastatic potential of breast cancer cells via estrogen receptor-α36 (ERα36) J Biol Chem. 2012;287(10):7169–7181. doi: 10.1074/jbc.M111.292946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiscox S, Morgan L, Barrow D, Dutkowskil C, Wakeling A, Nicholson RI. Tamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype: inhibition by gefitinib (‘Iressa’, ZD1839) Clin Exp Metastasis. 2004;21(3):201–212. doi: 10.1023/b:clin.0000037697.76011.1d. [DOI] [PubMed] [Google Scholar]
- 45.Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM. Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology. 1997;138(9):4030–4033. doi: 10.1210/endo.138.9.5489. [DOI] [PubMed] [Google Scholar]
- 46.Márquez DC, Pietras RJ. Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cancer cells. Oncogene. 2001;20(39):5420–5430. doi: 10.1038/sj.onc.1204729. [DOI] [PubMed] [Google Scholar]
- 47.Lin SL, Yan LY, Zhang XT, et al. ER-alpha36, a variant of ER-alpha, promotes tamoxifen agonist action in endometrial cancer cells via the MAPK/ERK and PI3K/Akt pathways. PLoS One. 2010;5(2):e9013. doi: 10.1371/journal.pone.0009013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tong JS, Zhang QH, Wang ZB, et al. ER-α36, a novel variant of ER-α, mediates estrogen-stimulated proliferation of endometrial carcinoma cells via the PKCδ/ERK pathway. PLoS One. 2010;5(11):e15408. doi: 10.1371/journal.pone.0015408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leevers SJ, Vanhaesebroeck B, Waterfield MD. Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr Opin Cell Biol. 1999;11(2):219–225. doi: 10.1016/s0955-0674(99)80029-5. [DOI] [PubMed] [Google Scholar]
- 50.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 51.Fresno Vara JA1, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30(2):193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 52.Ji H, Liu YE, Jia T, et al. Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res. 1997;57(4):759–764. [PubMed] [Google Scholar]
- 53.Kang L, Guo Y, Zhang X, Meng J, Wang ZY. A positive cross-regulation of HER2 and ER-α36 controls ALDH1 positive breast cancer cells. J Steroid Biochem Mol Biol. 2011;127(3–5):262–268. doi: 10.1016/j.jsbmb.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang XT, Ding L, Kang LG, Wang ZY. Involvement of ER-α36, Src, EGFR and STAT5 in the biphasic estrogen signaling of ER-negative breast cancer cells. Oncol Rep. 2012;27(6):2057–2065. doi: 10.3892/or.2012.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu BZ, Lin SL, Li M, et al. Changes in estrogen receptor-alpha variant (ER-alpha36) expression during mouse ovary development and oocyte meiotic maturation. Histochem Cell Biol. 2009;131(3):347–354. doi: 10.1007/s00418-008-0526-4. [DOI] [PubMed] [Google Scholar]
- 56.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Charafe-Jauffret E, Ginestier C, Iovino F, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69(4):1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buzdar AU, Cuzick J. Anastrozole as an adjuvant endocrine treatment for postmenopausal patients with breast cancer: emerging data. Clin Cancer Res. 2006;12(3 Pt 2):1037s–1048s. doi: 10.1158/1078-0432.CCR-05-2458. [DOI] [PubMed] [Google Scholar]
- 60.Urruticoechea A. The oestrogen-dependent biology of breast cancer. Sensitivity and resistance to aromatase inhibitors revisited: a molecular perspective. Clin Transl Oncol. 2007;9(12):752–759. doi: 10.1007/s12094-007-0136-y. [DOI] [PubMed] [Google Scholar]
- 61.Clarke R, Liu MC, Bouker KB, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22(47):7316–7339. doi: 10.1038/sj.onc.1206937. [DOI] [PubMed] [Google Scholar]
- 62.Dutertre M, Smith CL. Molecular mechanisms of selective estrogen receptor modulator (SERM) action. J Pharmacol Exp Ther. 2000;295(2):431–437. [PubMed] [Google Scholar]
- 63.Vivacqua A, Bonofiglio D, Recchia AG, et al. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol. 2006;20(3):631–646. doi: 10.1210/me.2005-0280. [DOI] [PubMed] [Google Scholar]
- 64.Zhang CC, Shapiro DJ. Activation of the p38 mitogen-activated protein kinase pathway by estrogen or by 4-hydroxytamoxifen is coupled to estrogen receptor-induced apoptosis. J Biol Chem. 2000;275(1):479–486. doi: 10.1074/jbc.275.1.479. [DOI] [PubMed] [Google Scholar]
- 65.Zhao Y, Deng C, Lu W, et al. let-7 microRNAs induce tamoxifen sensitivity by downregulation of estrogen receptor α signaling in breast cancer. Mol Med. 2011;17(11–12):1233–1241. doi: 10.2119/molmed.2010.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang X, Ding L, Kang L, Wang ZY. Estrogen receptor-alpha 36 mediates mitogenic antiestrogen signaling in ER-negative breast cancer cells. PLoS One. 2012;7(1):e30174. doi: 10.1371/journal.pone.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gutierrez MC, Detre S, Johnston S, et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23(11):2469–2476. doi: 10.1200/JCO.2005.01.172. [DOI] [PubMed] [Google Scholar]
- 68.Johnston SR, Saccani-Jotti G, Smith IE, et al. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995;55(15):3331–3338. [PubMed] [Google Scholar]
- 69.Massarweh S, Osborne CK, Creighton CJ, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68(3):826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 70.Fawell SE, White R, Hoare S, Sydenham M, Page M, Parker MG. Inhibition of estrogen receptor-DNA binding by the “pure” antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc Natl Acad Sci U S A. 1990;87(17):6883–6887. doi: 10.1073/pnas.87.17.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dauvois S, Danielian PS, White R, Parker MG. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci U S A. 1992;89(9):4037–4041. doi: 10.1073/pnas.89.9.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang L, Wang ZY. Breast cancer cell growth inhibition by phenethyl isothiocyanate is associated with down-regulation of oestrogen receptor-alpha36. J Cell Mol Med. 2010;14(6B):1485–1493. doi: 10.1111/j.1582-4934.2009.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mahfoudi A, Roulet E, Dauvois S, Parker MG, Wahli W. Specific mutations in the estrogen receptor change the properties of antiestrogens to full agonists. Proc Natl Acad Sci U S A. 1995;92(10):4206–4210. doi: 10.1073/pnas.92.10.4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo M, Wang M, Zhang X, Deng H, Wang ZY. Broussoflavonol B restricts growth of ER-negative breast cancer stem-like cells. Anticancer Res. 2013;33(5):1873–1879. [PubMed] [Google Scholar]
- 75.Guo M, Wang M, Deng H, Zhang X, Wang ZY. A novel anticancer agent Broussoflavonol B downregulates estrogen receptor (ER)-α36 expression and inhibits growth of ER-negative breast cancer MDA-MB-231 cells. Eur J Pharmacol. 2013;714(1–3):56–64. doi: 10.1016/j.ejphar.2013.05.047. [DOI] [PubMed] [Google Scholar]