

Background: The bacteriocin LsbB targets a membrane-bound zinc-dependent peptidase.
Results: The structure of LsbB was resolved by NMR. The C-terminal unstructured domains of LsbB and several other related bacteriocins were responsible for receptor binding.
Conclusion: A subgroup of leaderless bacteriocins has been found to share a similar mechanism in receptor recognition.
Significance: The study highlights the structure-function relationship of LsbB.
Keywords: Antimicrobial Peptide (AMP), Nuclear Magnetic Resonance (NMR), Peptide Interaction, Receptor, Structural Biology, Zn-dependent Protease, Bacteriocin Receptor, Leaderless Bacteriocin
Abstract
LsbB is a class II leaderless lactococcal bacteriocin of 30 amino acids. In the present work, the structure and function relationship of LsbB was assessed. Structure determination by NMR spectroscopy showed that LsbB has an N-terminal α-helix, whereas the C-terminal of the molecule remains unstructured. To define the receptor binding domain of LsbB, a competition assay was performed in which a systematic collection of truncated peptides of various lengths covering different parts of LsbB was used to inhibit the antimicrobial activity of LsbB. The results indicate that the outmost eight-amino acid sequence at the C-terminal end is likely to contain the receptor binding domain because only truncated fragments from this region could antagonize the antimicrobial activity of LsbB. Furthermore, alanine substitution revealed that the tryptophan in position 25 (Trp25) is crucial for the blocking activity of the truncated peptides, as well as for the antimicrobial activity of the full-length bacteriocin. LsbB shares significant sequence homology with five other leaderless bacteriocins, especially at their C-terminal halves where all contain a conserved KXXXGXXPWE motif, suggesting that they might recognize the same receptor as LsbB. This notion was supported by the fact that truncated peptides with sequences derived from the C-terminal regions of two LsbB-related bacteriocins inhibited the activity of LsbB, in the same manner as found with the truncated version of LsbB. Taken together, these structure-function studies provide strong evidence that the receptor-binding parts of LsbB and sequence-related bacteriocins are located in their C-terminal halves.
Introduction
Bacteriocins are ribosomally synthesized antimicrobial peptides produced by bacteria. They are often cationic and/or amphiphilic and contain between 25 and 70 amino acids (aa).2 Although most eukaryotic antimicrobial peptides have broad spectra of antimicrobial activity, most bacteriocins have narrow inhibitory spectra and are normally active against only related species or genera. Bacteriocins show antimicrobial activity at pico- to nanomolar concentrations and are thus ∼100–1000 times more potent than their eukaryotic counterparts (1). It has been suggested that between 30 and 99% of all bacteria produce at least one bacteriocin, but the specific roles of these bacteriocins in nature are not well understood (2). It is, however, generally believed that bacteria may use bacteriocins as a means to compete with other bacteria for common resources and/or that bacteriocins may play an active role in defining the microbial composition in certain niches (3). Although hundreds of bacteriocins and/or their producers have been discovered during the last two or three decades, and many of them are believed to have great potential as preservatives or drugs in diverse applications, nisin and pediocin PA-1 are so far the only bacteriocins that have been authorized as preservatives in the food industry (4–6). One of the main reasons for the limited success rate in the application of bacteriocins is probably the lack of detailed knowledge of their mode of action. Knowledge of the structure and the mode of action is essential for rational design of new bacteriocin variants with properties that make them especially useful for medical and biotechnological applications.
The three-dimensional structures of only a few bacteriocins have presently been determined; these mostly include some modified bacteriocins called lantibiotics and some nonmodified bacteriocins belonging to the pediocin-like and two-peptide bacteriocin subgroups (7–11). Even for these bacteriocins, very little is known about the part of these peptides that is directly involved in receptor binding or pore formation (12).
LsbB is a small (30 aa) and nonmodified bacteriocin produced by Lactococcus lactis BGMN1-5 isolated from cheese. This bacteriocin belongs to the so-called leaderless bacteriocin group whose members are different from most bacteriocins in that they do not involve an N-terminal leader sequence or signal peptide for export (6, 13, 14). Many, if not all, leaderless bacteriocins have formylmethionine at their N termini, a feature that distinguishes these molecules from other processed bacteriocins (15). Most of the leaderless bacteriocins have been identified in recent years, and insight into their biosynthetic pathways, structures, and mode of action is still limited (16). To our knowledge, the only leaderless bacteriocin that has been determined structurally is the two-peptide bacteriocin enterocin 7, which consists of the peptides Ent7A and Ent7B (7).
LsbB has a narrow inhibitory spectrum that includes mostly lactococcal cells (17). Recently a zinc-dependent membrane-bound metallopeptidase has been identified as the receptor for LsbB on target cells (18). In the present work, we determined the three-dimensional structure of LsbB and defined the part of the bacteriocin involved in the receptor binding. The structure of LsbB appeared to be different from Ent7A and Ent7B. In addition, we report the identification of some sequence-related bacteriocins with receptor binding domains that most likely target the same receptor as LsbB.
MATERIALS AND METHODS
CD Spectroscopy
CD spectra were recorded using a Jasco J-810 spectropolarimeter (Jasco International Co.) calibrated with d-camphor-10-sulfonate (Icatayama Chemical). All measurements were done using a quartz cuvette (Starna) with 0.1-cm path length. Samples were scanned five times with a scanning rate of 50 nm/min with a bandwidth of 0.5 nm and response time of 1 s over the wavelength range 190–245 nm. At least two replicates of each measurement were made. Spectra were recorded at different dodecylphosphocholine (DPC) (Avanti Polar Lipids) to peptide ratios, at different trifluoroethanol (TFE) (Aldridge) concentrations, and at temperatures between 20 and 50 °C.
NMR Spectroscopy
The experiments were run on LsbB samples containing ∼1 mm of the peptide. The DPC micelle experiments were run with a molar ratio between peptide and d38-DPC (98% 2H) (Cambridge Isotopes) of ∼1:200. The TFE experiments were run in 50% d3-TFE (99.5% 2H) (Aldrich). All samples contained 0.2 mm of 4,4-dimethyl-4-silapentane-1-sulfonic acid (Larodan) for internal chemical shift referencing. 5% of D2O was added to the samples to provide lock signal.
Two-dimensional NOESY (19), two-dimensional total correlation spectroscopy (20), two-dimensional double-quantum filtered correlation spectroscopy, 15N heteronuclear single quantum coherence (21), and 13C heteronuclear single quantum coherence (22) were recorded for LsbB in TFE and DPC. In addition, a double filtered heteronuclear multiple bond correlation (23) was recorded for the TFE sample. The data were acquired on a 600 MHz Bruker Avance II spectrometer with four channels and a 5-mm triple resonance cryoprobe (Bruker Biospin). NOESY spectra with mixing times between 120 and 300 ms were obtained for both samples. Total correlation spectroscopy mixing times of 80 ms were used. The experiments were run at temperatures of 298.15 and 293.15 K for the DPC and TFE samples, respectively.
Spectra were processed using the Topspin program (Bruker Biospin). 4,4-Dimethyl-4-silapentane-1-sulfonic acid was used as a chemical shift standard, and 15N and 13C data were referenced using frequency ratios as described (24).
For visualization, assignment and integration of the spectra the computer program CARA was used (25). The spectra were assigned using standard methods (26).
Dihedral angle restraints were obtained from the chemical shift values using the program TALOS+ (27). NOE distance restraints were calculated from the peak volumes in the NOESY spectra with NOESY mixing times of 120 and 200 ms for the DPC and TFE samples, respectively.
All structure calculations were made using the structure calculation program CYANA 2.1 (28, 29). All the applied restraints are presented in Table 1. The annealing macro in CYANA calculated 100 structures. The 20 structures with the lowest energy were kept and analyzed further. The root mean square deviation was calculated, and the structures were visualized using MolMol software (30).
TABLE 1.
Structure data and structure characteristics of the obtained NMR structures
| LsbB in 50% TFE | LsbB in DPC micelles | |
|---|---|---|
| Distance constraints | 433 | 400 |
| Intraresidue | 120 | 147 |
| Sequential (ji − jj = 1) | 155 | 156 |
| Medium range (1 < ji − jj < 5) | 158 | 97 |
| Long range (ji − jj > 5) | 0 | 0 |
| Dihedral angle constraints from TALOS+ | 36 | 32 |
| Energies (Å2)a | 1.51 ± 0.00 × 10−2 | 4.3 ± 0.1 × 10−1 |
| Violationsb | 0 | 3 |
| Deviations from idealized geometry | ||
| Bonds (Å) | 5 ± 0 × 10−4 | 1.78 ± 0.02 × 10−1 |
| Angles (°) | 3.6 ± 0.1 × 10−2 | 3.76 ± 0.02 × 10−1 |
| Ramachandran plot | ||
| In favored regions (%) | 78.5 | 74.8 |
| In allowed regions (%) | 21.5 | 25.0 |
| In additionally allowed regions (%) | 0 | 0.2 |
| Mean pairwise RMSD | ||
| Backbone | 3.90 ± 1.13 | 5.14 ± 1.47 |
| Heavy atoms | 4.79 ± 1.23 | 5.99 ± 1.52 |
| Backbone (3–17) | 0.05 ± 0.02 | 0.24 ± 0.10 |
| Heavy atoms (3–17) | 0.71 ± 0.16 | 1.07 ± 0.24 |
a CYANA energy.
b Only violations larger than 0.2 Å or 5°.
Bacterial Growth Conditions, Biological Antimicrobial Assays, and Peptides
The bacterial strain L. lactis IL1403, which was used as indicator for LsbB activity, was grown in M17 medium (Oxoid) supplemented with 0.5% (w/v) glucose (GM17) at 30 °C without shaking. The bacteriocin activity was assayed using a microtiter plate assay (31). The minimum inhibitory concentration (MIC) was defined as the minimal bacteriocin concentration that inhibited the growth of the indicator strain by at least 50% (50% of the turbidity of the control culture without bacteriocin) in 200 μl of culture. MIC value of LsbB against L. lactis IL1403 was shown to be 0.125 ng/ml (36 pm).
To determine the blocking activity of the truncated peptides, a microtiter plate assay was performed as follows. The peptides were first diluted 2-fold in 50 μl of medium on a microtiter plate. To each well, 145 μl of a diluted and fresh culture of L. lactis IL1403 (i.e. 50 times diluted overnight), and 5 μl of LsbB at concentration 50 ng/ml were added so that the final concentration of LsbB in each well was 10 MIC (1.25 ng/ml). Growth assay was measured spectrophotometrically at 600 nm (A600) for 8 h with 15-min intervals using SPECTROstarNano (BMG LABTECH).
In addition to the full-length LsbB molecule, 22 peptide fragments derived from the sequence of LsbB were synthesized (Table 2). For the sequence-related bacteriocins, enterocin EJ97 (EntEJ97) and a peptide with no annotation (National Center for Biotechnology Information Reference Sequence: WP_002339170.1, hereafter referred to as enterocin K1 (EntK1)), their fragments consisting of the last 20 aa were synthesized. All peptides were synthesized by GenScript Company (USA), all with purity of at least 95%. The purity of LsbB used for NMR was 98.4%. Synthesized peptides were solubilized to a concentration of 0.1–10 mg/ml in 0.1% (v/v) trifluoroacetic acid and stored at −20 °C until use.
TABLE 2.
Blocking activity of truncated peptides
| Name | Sequence | Coordinates | MMRBa | Ratio to P4b |
|---|---|---|---|---|
| LsbB | MKTILRFVAGYDIASHKKKTGGYPWERGKA | 1–30 | Not determined | |
| Series 1: each 15 amino acids in length | ||||
| P1 | MKTILRFVAGYDIAS | 1–15 | NBAc | |
| P2 | RFVAGYDIASHKKKT | 6–20 | NBA | |
| P3 | YDIASHKKKTGGYPW | 11–25 | NBA | |
| P4 | HKKKTGGYPWERGKA | 16–30 | 100 (35 nm) | 1 |
| Series 2: each 10 amino acids in length | ||||
| P4–1 | HKKKTGGYPW | 16–25 | NBA | |
| P4–2 | KKKTGGYPWE | 17–26 | NBA | |
| P4–3 | KKTGGYPWER | 18–27 | NBA | |
| P4–4 | KTGGYPWERG | 19–28 | NBA | |
| P4–5 | TGGYPWERGK | 20–29 | NBA | |
| P4–6 | GGYPWERGKA | 21–30 | 19000 (7.0 μm) | 190 |
| Series 3: each shorter than 10 amino acids in length | ||||
| PC-8 | YPWERGKA | 23–30 | 22000 (7.8 μm) | 220 |
| PC-7 | PWERGKA | 24–30 | 100000 (37 μm) | 1000 |
| PC-6 | WERGKA | 25–30 | 470000 (168 μm) | 4700 |
| PC-5 | ERGKA | 26–30 | NBA | |
| PC-4 | RGKA | 27–30 | NBA | |
| Series 4: alanine substitutiond | ||||
| PA-1 | GGAPWERGKA | 21–30 | 21000 (7.6 μm) | 210 |
| PA-2 | GGYAWERGKA | 21–30 | 19700 (7.1 μm) | 200 |
| PA-3 | GGYPAERGKA | 21–30 | NBA | |
| PA-4 | GGYPWARGKA | 21–30 | 11000 (3.8 μm) | 110 |
| PA-5 | GGYPWEAGKA | 21–30 | 21000 (7.6 μm) | 210 |
| PA-6 | GGYPWERGAA | 21–30 | 21000 (7.4 μm) | 210 |
| Series 5: C-terminal part of related bacteriocins, each of 20 amino acids in length | ||||
| LsbB-C20 | YDIASHKKKTGGYPWERGKA | 10–30 | 250 (81.6 nm) | 2.5 |
| EntEJ97-C20 | YEINWYKQQYGRYPWERPVA | 25–44 | 16000 (5.9 μm) | 160 |
| EntK1-C20 | YEIAWFKNKHGYYPWEIPRC | 18–37 | 8300 (3.0 μm) | 83 |
a Minimal molar ratio of blocking (MMRB) is defined as the least molar ratio between the truncated peptide and LsbB required to attain blocking. The number in parentheses denotes the actual concentration of the blocking peptide at the minimum blocking ratio. The concentration of LsbB used in the assay was approximately 0.36 nm, which is ∼10 MIC.
b The molar ratio between the indicated fragment and P4 fragment at their lowest concentrations that cause blocking of LsbB.
c NBA, no blocking activity at highest concentration tested, ∼0.14–0.15 mm for P1 to P3; 0.2 mm for P4–1 to P4–5; 0.4–0.6 mm for PC-4 and PC-5; and 0.25 mm for PA-3.
d Residues replaced with alanine are indicated with bold letters.
Construction of LsbB Mutants
Selected residues at the C-terminal end of the whole LsbB molecule were mutated to alanine by a PCR approach. A forward primer (F-lsbB) was combined with individual primers designed to introduce the appropriate nucleotides (Table 3). PCR was performed in 30 cycles each consisting of a denaturation step at 95 °C for 30 s, an annealing step at 48 °C for 30 s, and a polymerization step at 72 °C for 30 s and then finished up with 7 min at 72 °C. Obtained PCR DNA products were first cloned into pGemT easy vector (Promega) before the inserts were obtained using EcoRI and EcoRV as restriction enzymes and cloned into the expression vector pAZIL predigested with EcoRI and SmaI. Obtained constructs were expressed in L. lactis subsp. cremoris MG7284, and their bacteriocin activity was tested against L. lactisIL1403 on plate assay. The identities of all inserts were confirmed by DNA sequencing.
TABLE 3.
Primers used for construction of LsbB alanine mutants
| Name of primer | Sequence of primera | Name of LsbB with A replacement | Amino acid sequence of proteina |
|---|---|---|---|
| F-lsbB | 5′-CTCCAAGAATTCCTAAAAAAATAGG-3′ | ||
| R-lsbB WT | 5′-TGATATCTTAAGCTTTTCCACGTTCCC-3′ | LsbB-WT | MKTILRFVAGYDIASHKKKTGGYPWERGKA |
| R-lsbB-Y23/A23 | 5′-TGATATCTTAAGCTGCTCCACGTTCCC-3′ | LsbB-A23 | MKTILRFVAGYDIASHKKKTGGAPWERGKA |
| R-lsbB-P24/A24 | 5′-TGATATCTTAAGCTTTTCCAGCTTCCC-3′ | LsbB-A24 | MKTILRFVAGYDIASHKKKTGGYAWERGKA |
| R-lsbB-W25/A25 | 5′-TGATATCTTAAGCTTTTCCACGTGCCCATGG-3′ | LsbB-A25 | MKTILRFVAGYDIASHKKKTGGAPAERGKA |
| R-lsbB-E26/A26 | 5′-TGATATCTTAAGCTTTTCCACGTTCCGCTGGATAGCC-3′ | LsbB-A26 | MKTILRFVAGYDIASHKKKTGGAPWARGKA |
| R-lsbB-R27/A27 | 5′-TGATATCTTAAGCTTTTCCACGTTCCCATGCATAGCC-3′ | LsbB-A27 | MKTILRFVAGYDIASHKKKTGGAPWEAGKA |
| R-lsbB-K29/A29 | 5′-TGATATCTTAAGCTTTTCCACGTTCCCATGGAGCGCCGCC-3′ | LsbB-A29 | MKTILRFVAGYDIASHKKKTGGAPWERGAA |
a Underlined letters indicate the site for the restriction enzyme EcoRV; underlined triplets in italic indicate alanine substitution sites.
b Underlined letters indicate residues replaced with alanine.
RESULTS
Structural Analysis by CD Spectroscopy
CD spectra of LsbB showed that LsbB was unstructured in water but became structured in TFE and DPC solutions (supplemental Fig. S1). Maximum structuring was obtained at the peptide to lipid ratio of 1:200 (12 mm DPC) and in 50% TFE. The conditions were consequently used in the respective NMR experiments.
Structural Analysis of LsbB by NMR Spectroscopy
The structures were assigned using standard methods as described under “Materials and Methods.” Supplemental Fig. S2 shows the assigned 15N heteronuclear single quantum coherence spectrum of LsbB in DPC. Fig. 1 shows connected strips from NOESY, the torsion angle restraints used in structure calculations, and the Cα chemical shift indexes (CSIs). CSIs based on the Cα chemical shifts of LsbB (32) are shown in Fig. 1 (A and B) for TFE and DPC, respectively. Chemical shift indexing indicates that there is an α-helix in LsbB from residues 5–16 in TFE and from residues 2–15 in DPC. TALOS+ torsion angle predictions indicated backbone torsion angles (φ- and ψ-angles) consistent with α-helical regions between and including residues 4–18 and 3–15 in the presence of TFE and DPC, respectively (Fig. 1, C and D)
FIGURE 1.
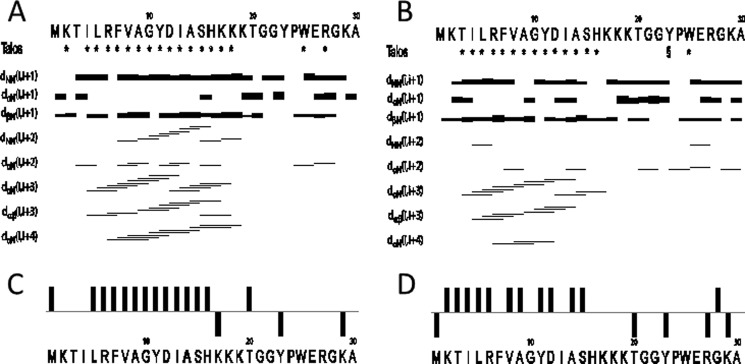
The NMR data obtained of LsbB in TFE (A and C) and DPC (B and D). The restraints used in structure calculation are shown in A and B. The signs * and § indicate that TALOS determined phi and psi torsion angle restraints for this residue were in the α-helical and β-sheet region of the Ramachandran plot, respectively. The lines indicate typical α-helical restraints obtained from analysis of the NOE spectra. The thicknesses of the lines shown are related to NOE cross peak intensities. CSI of C-α carbons in TFE and DPC is shown in C and D.
A total of 433 (14.4 per residue) and 400 (13.3 per residue) unique NOE connectivities were assigned for LsbB in the presence of TFE and DPC, respectively. The total number of distance restrictions and their classes are shown in Table 1. The mid-range NOE connectivities that are especially important for α-helices, NN (i,i+2), αN(i,i+2), αN(i,i+3), αβ(i,i+3), and αN(i,i+4), are shown in Fig. 1 (C and D) for LsbB in TFE and DPC, respectively. For LsbB in the presence of TFE, the observed connectivities indicated an α-helical region stretching from residues 3 to 18, and in DPC the helical region was from residues 3 to 16. Thus, these NOE connectivities are in accordance with what was predicted by CSI and TALOS+.
Structures of LsbB in TFE and DPC were calculated using CYANA. A superimposition over the helical part of the 20 lowest energy structures of LsbB exposed to TFE or DPC is shown in Fig. 2 (A and B, respectively). The peptide structures have been deposited to the Protein Data Bank with access codes 2MLU (LsbB in DPC) and 2MLV (LsbB in TFE), and the NMR data have been deposited to the Biomagnetic Resonance Data Bank with the access codes 19832 (LsbB in DPC) and 19833 (LsbB in TFE). Cartoon drawings of LsbB in the presence of TFE and DPC are shown in Fig. 2 (C and D, respectively). Well defined structures were obtained when superimposition was made over a selection of residues comprising regions where CSI, J-couplings, TALOS, and NOE connectives predicted secondary structure.
FIGURE 2.

LsbB NMR structures in TFE (A and C) and DPC (B and D). The structures ensembles of the 20 lowest energy structures superimposed over residues 3–17 are shown in A and B, and cartoon representations of the lowest energy structures are shown in C and D.
The Receptor Binding Domain of LsbB Is Located in the C-terminal Half
It has recently been shown that LsbB requires the membrane-bound protease YvjB as a receptor on target cells (18). Given that the structure of LsbB revealed distinct domains within the molecule, we wished to examine which part of LsbB is involved in receptor binding. A set of four truncated fragments of the LsbB molecule were synthesized: P1-P4, with coordinates: aa 1–15 for P1, aa 6–20 for P2, aa 11–25 for P3, and aa 16–30 for P4 (Table 2). These peptides were assessed for their antimicrobial activity, as well as their ability to block the activity of the full-length peptide LsbB. None of these truncated peptides, either as individuals or in any combinations, had any antimicrobial activity, as judged from our standard microtiter plate assay (data not shown). However, when assessing their blocking activity, P4 (consisting of the last 15 C-terminal residues) was found to be the only peptide that blocked the antimicrobial activity of LsbB. The minimal molar concentration of P4 was ∼100 times higher than that of LsbB to attain blocking (Table 2). The remaining three fragments, P1–P3 did not protect L. lactis IL 1403 from the killing by LsbB, even at the highest concentrations applied (Table 2).
The aa sequence of P4 is HKKKTGGYPWERGKA. It contains only one acidic residue (E) but six basic residues with four (HKKK) of them being located at the N terminus of this peptide. The high content of basic residues in P4 results in a highly cationic peptide with a pI of 10.68. To further examine the importance of these residues in receptor binding, a series of six shorter peptides, each of ten amino acids in length, was generated (Table 2). The coordinates of these peptides moved along the sequence of P4, one amino acid a time. Surprisingly, only the peptide (P4-6) with the last 10 residues (GGYPWERGKA) but without the preceding four basic residues (HKKK) had blocking activity, whereas the other peptides located N-terminally had not. However, the blocking activity of P4-6 is much less potent compared with that of P4, ∼200-fold lower (Table 2).
To define the shortest truncated peptide with blocking activity, further N-terminal truncation of P4-6 was performed, giving rise to the peptides PC-8 (YPWERGKA), PC-7 (PWERGKA), PC-6 (WERGKA), PC-5 (ERGKA), and PC-4 (RGKA) (Table 2). PC-8 appeared to have almost the same blocking potency as P4-6, whereas PC-7 and PC-6 had ∼5- and 25-fold lower blocking activity than P4-6, respectively. PC-5 and PC-4 did not have any detectable blocking activity at the highest concentrations tested (Table 2).
The Residue Trp25 Is Crucial for Receptor Binding
Given that the short sequence P4-6 (10 amino acids) had a relatively good blocking activity, we performed alanine substitution to most of the residues of this peptide to assess their importance in receptor binding (Table 2). Surprisingly, only one of these changes had a significant impact: the substitution of the tryptophan at position 25 to alanine (W25A) totally abolished the blocking activity of the resulting peptide PA-3. On the contrary, the substitution of glutamic acid (E26A) in PA-4 increased its blocking potency 2-fold. Substitution at the other residues had no apparent impact on the blocking potency (Table 2). We also generated similar alanine substitutions at the same residues on the full-length LsbB. As expected, only the substitution of the tryptophan (W25A) caused total loss of antimicrobial activity, whereas the other substitutions still retained the antimicrobial activity of the molecules, although with various strength (Fig. 3).
FIGURE 3.

Antimicrobial activity of LsbB (WT) and the LsbB mutants: Y23A, P24A, W25A, E26A, R27A, and K29A. Transformants of these mutants were spotted on a lawn of the indicator L. lactis Il1403. Inhibition is seen in the clear zones around the spotted cells.
C-terminal Parts of Homologous Peptides Block the Activity of LsbB
A search for LsbB homologues in public data bases revealed several hits of significance; they all are from Enterococcus species. These are two known enterocins, EntQ and EntEJ97, and three unannotated open reading frames, of which one (termed EntEJ97T) differs from EntEJ97 at only one position (Fig. 4). Notably, all these peptides contain a consensus motif, KXXXGXXPWE (where X is less conserved or any amino acid), at their C-terminal halves. One of the unannotated peptides was termed EntK1 after its antimicrobial activity was confirmed by using synthetic peptide (data not shown).
FIGURE 4.

Sequence alignment of LsbB-like bacteriocins. Asterisks (*) indicate identical amino acids; Colons (:) indicate very similar amino acids, and dots (.) indicate similar amino acids.
Given the blocking activity of the C-terminal half of LsbB, we obtained truncated peptides with sequence derived from the last 20 amino acids of EntEJ97 and Ent K1 (EntEJ97-C20 and EntK1-C20) and assessed for their blocking activity against LsbB. Both EntEJ97-C20 and EntK1-C20 were, as expected, capable to block LsbB activity (Table 2), indicating that these two peptides most probably bind to the same receptor as LsbB.
DISCUSSION
Structural analyses using CD and NMR spectroscopy suggest that the structure of LsbB can be defined into three functional parts (Fig. 2): (i) an N-terminal domain consisting of an amphiphilic α-helix of ∼15 residues; (ii) a small middle region (residues 15–18) consisting of a series of basic amino acids (HKKK motif) that contribute to a relatively high pI, a typical characteristic of most bacteriocins; and lastly (iii), the C-terminal part (residues 19–30), which is unstructured in water and in organic solvents (TFE and DPC). Further analyses revealed that the receptor binding domain of LsbB is likely to be located at the C-terminal half, more specifically within the last C-terminally located 8 aa of LsbB. This notion is based on the fact only truncated peptides derived from this region were able to prevent LsbB from killing of target cells. It is reasonable to believe that these truncated peptides compete with the full-length LsbB for the same receptor on target cells, and at higher concentrations, they efficiently prevent LsbB from gaining access to the receptor and thereby rescuing cells from being killed.
Among the truncated peptides tested for blocking activity, P4 appears to be the most potent one. The minimum molar ratio of P4 in excess to LsbB to prevent killing was ∼100 to 1. This peptide covers the last 15 aa (HKKKTGGYPWERGKA) and includes the series of cationic residues (HKKK) at its N terminus. Surprisingly, removal of these basic residues, such as the resulting P4-6 (GGYPWERGKA), could still retain blocking activity. Notably, the blocking activity of P4-6 is specific because the five flanking and overlapping fragments P4-1 to P4-5 did not have any blocking activity at all. Nevertheless, the blocking activity of P4-6 was ∼200 times less potent than that of P4. Based on these results, our interpretation is that the series of basic residues is not directly required for specific targeting, but it is apparently crucial for the initial and unspecific interaction between the positively charged bacteriocin and the negatively charged phospholipid layer of the membrane. This unspecific electrostatic interaction will then be followed by a specific interaction between the receptor binding domain (the last 8 aa) of LsbB and the receptor (YvjB), which subsequently leads to disruption of membrane integrity and killing of the target cells. This mode of action resembles to some extent the model proposed for plantaricin A, a peptide pheromone that binds to the membrane-located histidine protein kinase involved in a quorum sensing regulation of a bacteriocin locus (33). Like LsbB, plantaricin A is a bimodular and small peptide (26 aa); it is composed of an N-terminal unstructured part and a highly cationic C-terminal helical part (34). The latter part has been proposed to be involved in the initial and unspecific (chirally independent) binding to the membrane by electrostatic interaction, whereas the former (i.e. the unstructured N-terminal part) becomes structured upon specific binding to the kinase protein. Because most bacteriocins are highly cationic and often contain an unstructured part in water or in membrane-like environments, it is tempting to speculate that most of them might apply a two-step receptor targeting strategy as proposed for LsbB and plantaricin A, i.e. an initial unspecific interaction facilitated by electrostatic interaction and a subsequent and specific interaction between the receptor binding domain of the bacteriocin and its membrane-located receptor.
The structure of LsbB is quite different from that of the well characterized pediocin-like bacteriocins (10). It has been shown that this group of bacteriocins exploits the membrane-bound sugar permease, mannose phosphotransferase system (man-PTS) as receptor on target cells (35). These bacteriocins bind directly to the membrane-located components IIC and IID of the man-PTS. Using hybrid bacteriocins (containing parts from different pediocin-like bacteriocins), it has been shown that the receptor binding domain of these bacteriocins is most likely located in their C-terminal halves (36, 37). Interestingly, lactococcins A and B, which are nonmodified and non-pediocin-like bacteriocins, also exploit man-PTS as receptor on target cells (38). These lactococcins, like LsbB, differ greatly from the pediocin-like bacteriocins in that their inhibition spectrum is much narrower, targeting primarily lactococcal cells, whereas most pediocin-like bacteriocins have a much broader inhibition spectrum, and they somehow do not target lactococcal cells (38). This extreme species specificity has been found to rely on sequence differences between the lactococcal man-PTS and the non-lactococcal man-PTS (38).
It has been suggested that most two-peptide bacteriocins probably form a transmembrane helix-helix structure (11). In the case of the two-peptide bacteriocin lactococcin G, one of the peptides (LcnG-α), like LsbB, contains a series of cationic residues (residues 35–39: RKKKH) at the C-terminal part. This feature is believed to function as a means to force the peptide through the target cell membrane by the membrane potential (39). Whether this is the case also for LsbB with regard to this basic feature awaits further investigation.
There are several other features on the C-terminal part of LsbB worthy of note because they appear to be essential in receptor binding. The residues Ala30 (the last residue) and Trp25 are indispensable. The six fragments P4-1 to P4-6 are all of the same length (10 aa), and together they cover the last 15 aa of LsbB. Only P4-6, which contains Ala30, had blocking activity, whereas the remaining peptides that lack Ala30 did not block LsbB at all. Similarly, replacement of several residues in P4-6 with alanine did not have any detrimental effect on their blocking activity, except for the substitution of Trp25, which totally abolished the blocking activity of the resulting peptide (PA-3, Table 2). The latter result is in line with the same substitution (W25A) in the full-length LsbB, which showed that the resulting LsbB-W25A totally lost the antimicrobial activity (Fig. 3). At present there are no clues as to how these two residues interact with the receptor. Because both residues (Trp25 and Ala30) have a hydrophobic nature, it is feasible that they are placed in important hydrophobic environments, either within the receptor or the lipid core of the membrane.
Interestingly, it has been shown that tryptophan residues in similar positions as found with LsbB are important for the activity of some pediocin-like bacteriocins (40, 41). Only further investigation can provide more molecular details on these residues in terms of receptor binding or mode of killing.
The substitution E26A increased blocking potency of the resulting peptide PA-4 (GGYPWARGKA; see Table 2). It is reasonable to think that removal of the negative charge (Gly to Ala) will increase the affinity of the resulting peptide to the negatively charged membrane, thereby strengthening its blocking ability. Based on this result, one would anticipate that the same substitution (E26A) on the full-length peptide would outperform the other peptides in antimicrobial activity. Surprisingly, it did not turn to be the case (Fig. 3). This unexpected result might indicate that the receptor binding domain and the membrane-disruptive domain on LsbB are not physically distinct. To delineate the role of this glutamate residue in these two processes probably requires structural detail from the interaction between the bacteriocin and its receptor; however, this is beyond the scope of the present study.
Trp25 and Glu26 are part of a conserved motif KXXXGXXPWE, which is found in all the LsbB-like peptides, a group presently containing six members: LsbB, EntQ, EntK1, EntEJ97, EntEJ97T-HP, and Duracin 41D-HP (Fig. 4), the last two being hypothetical peptides (HP) as their antimicrobial activity has not been confirmed yet. We suggest that this motif is referred to as the LsbB-like consensus. The high sequence similarity at their C-terminal halves suggests that they all might recognize the same receptor as LsbB. This notion is indeed supported by the fact that truncated versions representing the last 20 aa of EntEJ97 (EntEJ97T-HP) and EntK1 displayed the ability to block the antimicrobial activity of LsbB (Table 2).
Although LsbB shares with the other LsbB-like peptides a high sequence similarity at its receptor binding domain, it differs greatly from the others in terms of inhibition spectra. LsbB targets primarily lactococcal cells (17), whereas the enterocins (EntQ, EntEJ97, and EntK1) have a much wider inhibition spectrum that also includes lactococcal cells (13, 42) (data not shown for enterocin K1). This species specificity resembles, to some extent, to that of Lactococcin A/B and the pediocin-like bacteriocins as described above. Preliminary results indicated that some strains of Enterococcus, Pediococcus, and Lactobacillus that are sensitive to EntQ, EntEJ97, and EntK1 (but not to LsbB) also contain an YvjB homologue in their genomes. These proteins share homology with the lactococcal YvjB (428 aa) over their entire length, with a score varying from 46 to 54% identity. It is of great interest to examine in future work whether these YvjB homologues could serve as receptors for these enterocins and whether there are sequence differences in these proteins that define their observed species specificity.
Most, if not all, leaderless bacteriocins are small peptides produced without posttranslational modification or processing (6, 14). These features make it relatively easy to obtain them by chemical synthesis and to subject them to structural and mutation analyses. Such approaches can provide important insights into the interactions at amino acid levels between the bacteriocins and their receptors and hence their mode of action, a field that is poorly understood for most bacteriocins. Further, delineating the differences behind target specificity is also of great interest because it can not only help explain the biological and evolutionary aspect of these bacteriocins but also can provide leads to design drugs with desired specificity and/or improved potency.
This work was supported by a quota scholarship from Norwegian University of Life Sciences and a grant from the Norwegian Centennial Chair program (to K. V. O.) and by Grant 173019 from the Ministry of Education, Science and Technological Development of the Republic of Serbia (to G. U., L. T., and M. K.).

This article contains supplemental Figs. S1 and S2.
The atomic coordinates and structure factors (codes 2MLU and 2MLV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- aa
- amino acid(s)
- CSI
- chemical shift index
- DPC
- dodecylphosphocholine
- MIC
- minimum inhibitory concentration
- TFE
- trifluoroethanol
- HP
- hypothetical peptide
- man-PTS
- mannose phosphotransferase system.
REFERENCES
- 1. Nes I. F., Diep D. B., Håvarstein L. S., Brurberg M. B., Eijsink V., Holo H. (1996) Biosynthesis of bacteriocins in lactic acid bacteria. Antonie van Leeuwenhoek 70, 113–128 [DOI] [PubMed] [Google Scholar]
- 2. Riley M. A. (1998) Molecular mechanisms of bacteriocin evolution. Annu. Rev. Genet. 32, 255–278 [DOI] [PubMed] [Google Scholar]
- 3. Nes I. F., Diep D. B., Holo H. (2007) Bacteriocin diversity in Streptococcus and Enterococcus. J. Bacteriol. 189, 1189–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benmechernene Z., Fernandez-No I., Kihal M., Böhme K., Calo-Mata P., Barros-Velazquez J. (2013) Recent patents on bacteriocins: food and biomedical applications. Recent Pat. DNA Gene Seq. 7, 66–73 [DOI] [PubMed] [Google Scholar]
- 5. Arthur T. D., Cavera V. L., Chikindas M. L. (2014) On bacteriocin delivery systems and potential applications. Future Microbiol. 9, 235–248 [DOI] [PubMed] [Google Scholar]
- 6. Cotter P. D., Ross R. P., Hill C. (2013) Bacteriocins: a viable alternative to antibiotics? Nat. Rev. Microbiol. 11, 95–105 [DOI] [PubMed] [Google Scholar]
- 7. Lohans C. T., Towle K. M., Miskolzie M., McKay R. T., van Belkum M. J., McMullen L. M., Vederas J. C. (2013) Solution structures of the linear leaderless bacteriocins enterocin 7A and 7B resemble carnocyclin A, a circular antimicrobial peptide. Biochemistry 52, 3987–3994 [DOI] [PubMed] [Google Scholar]
- 8. Lohans C. T., Vederas J. C. (2014) Structural characterization of thioether-bridged bacteriocins. J. Antibiot. (Tokyo) 67, 23–30 [DOI] [PubMed] [Google Scholar]
- 9. Wang Y., Henz M. E., Gallagher N. L., Chai S., Gibbs A. C., Yan L. Z., Stiles M. E., Wishart D. S., Vederas J. C. (1999) Solution structure of carnobacteriocin B2 and implications for structure-activity relationships among type IIa bacteriocins from lactic acid bacteria. Biochemistry 38, 15438–15447 [DOI] [PubMed] [Google Scholar]
- 10. Nissen-Meyer J., Rogne P., Oppegård C., Haugen H. S., Kristiansen P. E. (2009) Structure-function relationships of the non-lanthionine-containing peptide (class II) bacteriocins produced by gram-positive bacteria. Curr. Pharm. Biotechnol. 10, 19–37 [DOI] [PubMed] [Google Scholar]
- 11. Nissen-Meyer J., Oppegård C., Rogne P., Haugen H. S., Kristiansen P. E. (2010) Structure and Mode-of-Action of the Two-Peptide (Class-IIb) Bacteriocins. Probiotics Antimicrob. Proteins 2, 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Snyder A. B., Worobo R. W. (2014) Chemical and genetic characterization of bacteriocins: antimicrobial peptides for food safety. J. Sci. Food Agric. 94, 28–44 [DOI] [PubMed] [Google Scholar]
- 13. Cintas L. M., Casaus P., Herranz C., Hâvarstein L. S., Holo H., Hernández P. E., Nes I. F. (2000) Biochemical and genetic evidence that Enterococcus faecium L50 produces enterocins L50A and L50B, the sec-dependent enterocin P, and a novel bacteriocin secreted without an N-terminal extension termed enterocin Q. J. Bacteriol. 182, 6806–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arnison P. G., Bibb M. J., Bierbaum G., Bowers A. A., Bugni T. S., Bulaj G., Camarero J. A., Campopiano D. J., Challis G. L., Clardy J., Cotter P. D., Craik D. J., Dawson M., Dittmann E., Donadio S., Dorrestein P. C., Entian K. D., Fischbach M. A., Garavelli J. S., Göransson U., Gruber C. W., Haft D. H., Hemscheidt T. K., Hertweck C., Hill C., Horswill A. R., Jaspars M., Kelly W. L., Klinman J. P., Kuipers O. P., Link A. J., Liu W., Marahiel M. A., Mitchell D. A., Moll G. N., Moore B. S., Müller R., Nair S. K., Nes I. F., Norris G. E., Olivera B. M., Onaka H., Patchett M. L., Piel J., Reaney M. J., Rebuffat S., Ross R. P., Sahl H. G., Schmidt E. W., Selsted M. E., Severinov K., Shen B., Sivonen K., Smith L., Stein T., Sussmuth R. D., Tagg J. R., Tang G. L., Truman A. W., Vederas J. C., Walsh C. T., Walton J. D., Wenzel S. C., Willey J. M., van der Donk W. A. (2013) Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Natural product reports 30, 108–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu X., Vederas J. C., Whittal R. M., Zheng J., Stiles M. E., Carlson D., Franz C. M., McMullen L. M., van Belkum M. J. (2011) Identification of an N-terminal formylated, two-peptide bacteriocin from Enterococcus faecalis 710C. J. Agric. Food Chem. 59, 5602–5608 [DOI] [PubMed] [Google Scholar]
- 16. Zendo T. (2013) Screening and characterization of novel bacteriocins from lactic acid bacteria. Biosci. Biotechnol. Biochem. 77, 893–899 [DOI] [PubMed] [Google Scholar]
- 17. Gajic O., Buist G., Kojic M., Topisirovic L., Kuipers O. P., Kok J. (2003) Novel mechanism of bacteriocin secretion and immunity carried out by lactococcal multidrug resistance proteins. J. Biol. Chem. 278, 34291–34298 [DOI] [PubMed] [Google Scholar]
- 18. Uzelac G., Kojic M., Lozo J., Aleksandrzak-Piekarczyk T., Gabrielsen C., Kristensen T., Nes I. F., Diep D. B., Topisirovic L. (2013) A Zn-dependent metallopeptidase is responsible for sensitivity to LsbB, a class II leaderless bacteriocin of Lactococcus lactis subsp. lactis BGMN1-5. J. Bacteriol. 195, 5614–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeener J., Meier B. H., Bachmann P., Ernst R. R. (1979) Investigation of exchange processes by 2-dimensional NMR-spectroscopy. J. Chem. Phys. 71, 4546–4553 [Google Scholar]
- 20. Braunschweiler L., Ernst R. R. (1983) Coherence transfer by isotropic mixing: application to proton correlation spectroscopy. J. Magn. Reson. 53, 521–528 [Google Scholar]
- 21. Davis A. L., Keeler J., Laue E. D., Moskau D. (1992) Experiments for recording pure absorption heteronuclear correlation spectra using pulsed field gradients. J. Magn. Reson. 98, 207–216 [Google Scholar]
- 22. Hurd R. E., Boban K. J. (1991) Gradient enhanced proton-detected heteronuclear multiple quantum coherence spectroscopy. J. Magn. Reson. 91, 648–653 [Google Scholar]
- 23. Keeler J. (2010) Understanding NMR Spectroscopy, Wiley, United Kingdom [Google Scholar]
- 24. Wishart D. S., Bigam C. G., Yao J., Abildgaard F., Dyson H. J., Oldfield E., Markley J. L., Sykes B. D. (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140 [DOI] [PubMed] [Google Scholar]
- 25. Keller R. L. J. (2004) The Computer Aided Resonance Assignment Tutorial, Cantina Verlag, Goldau, Switzerland [Google Scholar]
- 26. Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, John Wiley & Sons, New York [Google Scholar]
- 27. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 29. Herrmann T., Güntert P., Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 30. Koradi R., Billeter M., Wuthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- 31. Holo H., Nilssen O., Nes I. F. (1991) Lactococcin A, a new bacteriocin from Lactococcus lactis subsp. cremoris: isolation and characterization of the protein and its gene. J. Bacteriol. 173, 3879–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wishart D. S., Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 33. Diep D. B., Håvarstein L. S., Nes I. F. (1995) A bacteriocin-like peptide induces bacteriocin synthesis in Lactobacillus plantarum C11. Mol. Microbiol. 18, 631–639 [DOI] [PubMed] [Google Scholar]
- 34. Kristiansen P. E., Fimland G., Mantzilas D., Nissen-Meyer J. (2005) Structure and mode of action of the membrane-permeabilizing antimicrobial peptide pheromone plantaricin A. J. Biol. Chem. 280, 22945–22950 [DOI] [PubMed] [Google Scholar]
- 35. Diep D. B., Skaugen M., Salehian Z., Holo H., Nes I. F. (2007) Common mechanisms of target cell recognition and immunity for class II bacteriocins. Proc. Natl. Acad. Sci. U.S.A. 104, 2384–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haugen H. S., Fimland G., Nissen-Meyer J. (2011) Mutational analysis of residues in the helical region of the class IIa bacteriocin pediocin PA-1. Appl. Environ. Microbiol. 77, 1966–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnsen L., Fimland G., Nissen-Meyer J. (2005) The C-terminal domain of pediocin-like antimicrobial peptides (class IIa bacteriocins) is involved in specific recognition of the C-terminal part of cognate immunity proteins and in determining the antimicrobial spectrum. J. Biol. Chem. 280, 9243–9250 [DOI] [PubMed] [Google Scholar]
- 38. Kjos M., Nes I. F., Diep D. B. (2011) Mechanisms of resistance to bacteriocins targeting the mannose phosphotransferase system. Appl. Environ. Microbiol. 77, 3335–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oppegård C., Rogne P., Kristiansen P. E., Nissen-Meyer J. (2010) Structure analysis of the two-peptide bacteriocin lactococcin G by introducing d-amino acid residues. Microbiology 156, 1883–1889 [DOI] [PubMed] [Google Scholar]
- 40. Fimland G., Eijsink V. G., Nissen-Meyer J. (2002) Mutational analysis of the role of tryptophan residues in an antimicrobial peptide. Biochemistry 41, 9508–9515 [DOI] [PubMed] [Google Scholar]
- 41. Rihakova J., Petit V. W., Demnerova K., Prévost H., Rebuffat S., Drider D. (2009) Insights into structure-activity relationships in the C-terminal region of divercin V41, a class IIa bacteriocin with high-level antilisterial activity. Appl. Environ. Microbiol. 75, 1811–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gálvez A., Valdivia E., Abriouel H., Camafeita E., Mendez E., Martínez-Bueno M., Maqueda M. (1998) Isolation and characterization of enterocin EJ97, a bacteriocin produced by Enterococcus faecalis EJ97. Arch. Microbiol. 171, 59–65 [DOI] [PubMed] [Google Scholar]