

Background: Umbilical cord mesenchymal stem cells (UMSCs) have unique immunosuppressive properties.
Results: UMSCs express a rich glycocalyx, which confers their ability to modulate both macrophages and T-regulatory cells and to lead to inflammatory cell death.
Conclusion: UMSCs actively modulate inflammatory cells by suppressing the immune response and evading rejection.
Significance: Engineering cells to express this rich glycocalyx could increase transplantation success.
Keywords: Chondroitin Sulfate, Glycosaminoglycan, Hyaluronate, Inflammation, Mesenchymal Stem Cells (MSCs), Versican
Abstract
Umbilical cord mesenchymal stem cells (UMSCs) have unique immunosuppressive properties enabling them to evade host rejection and making them valuable tools for cell therapy. We previously showed that human UMSCs survive xenograft transplantation and successfully correct the corneal clouding defects associated with the mouse model for the congenital metabolic disorder mucopolysaccharidosis VII. However, the precise mechanism by which UMSCs suppress the immune system remains elusive. This study aimed to determine the key components involved in the ability of the UMSCs to modulate the inflammatory system and to identify the inflammatory cells that are regulated by the UMSCs. Our results show that human UMSCs transplanted into the mouse stroma 24 h after an alkali burn suppress the severe inflammatory response and enable the recovery of corneal transparency within 2 weeks. Furthermore, we demonstrated in vitro that UMSCs inhibit the adhesion and invasion of inflammatory cells and also the polarization of M1 macrophages. UMSCs also induced the maturation of T-regulatory cells and led to inflammatory cell death. Moreover, UMSCs exposed to inflammatory cells synthesize a rich extracellular glycocalyx composed of the chondroitin sulfate-proteoglycan versican bound to a heavy chain (HC)-modified hyaluronan (HA) matrix (HC-HA). This matrix also contains TNFα-stimulated gene 6 (TSG6), the enzyme that transfers HCs to HA, and pentraxin-3, which further stabilizes the matrix. Our results, both in vivo and in vitro, show that this glycocalyx confers the ability for UMSCs to survive the host immune system and to regulate the inflammatory cells.
Introduction
Corneal transplantation is still the most effective treatment for vision restoration of corneal blindness due to congenital gene mutations and acquired diseases such as trachoma, microbial infections, laceration, and chemical burns. Because of tissue shortage at eye banks as well as the complications entailed by corneal transplantation, alternative treatments are required for the prevention and cure of corneal blindness in lieu of corneal transplantation. Umbilical cord mesenchymal stem cell (UMSC)2 transplantation successfully recovers corneal transparency and increased corneal thickness of lumican null mice (Lum−/−) (1). Interestingly, these cells assume a keratocyte phenotype and promote collagen lamellae re-organization (1). We have also found that UMSCs transplanted into the corneal stroma of mice with the congenital metabolic disorder mucopolysaccharidosis VII impede progression of the stromal disease and improve corneal haze (2). The transplanted UMSCs can differentiate into resident stromal cells, recycle accumulated glycosaminoglycans (GAGs), and secrete exosomes that are taken up by the host keratocytes, which enables them to catabolize GAGs (2). Moreover, treated corneas present reduced corneal haze, and UMSC transplantation is therefore a promising treatment for corneal congenital diseases. Interestingly, these experiments involved the treatment of immunocompetent mouse corneas with human UMSCs, and these cells were able to survive immune rejection. The rejection of transplanted tissue involves the recognition of alloantigens, which activates the host immune system and attacks the transplanted tissue. Xenotransplantation exposes the host to a plethora of antigens and usually triggers acute rejection if immunosuppressive medications are not administered. Previous studies have indicated that mesenchymal stem cells have unique immunosuppressive properties; however, the precise mechanism by which these cells suppress the immune system remains elusive (3).
During inflammation, the tissue microenvironment undergoes major changes. In order for inflammatory cells to invade tissues, they must roll and adhere to the vascular endothelium, extravasate, and migrate through the surrounding extracellular matrix (ECM). The important role of the ECM in the trafficking and function of inflammatory cells has been well established. Monocytes adhere to hyaluronan cables, which become a substrate for active migration (4). Recently, T-cells have also been shown to adhere to and migrate along HA cables (5, 6). Versican, a chondroitin sulfate proteoglycan present in the ECM, is also a substrate for both the adhesion and migration of monocytes (7). This interaction has been shown to involve the hyaluronan receptor CD44 expressed by the monocytes and T-lymphocytes (5, 8).
HA, a nonsulfated GAG, is a major component of the ECM. HA has a fundamental role in maintaining tissue integrity and homeostasis and has the necessary roles in ovulation and fertilization, embryonic development, inflammation, tissue repair and wound healing, and tumorigenesis (9–13). During chronic inflammation, tissues secrete a rich and complex cross-linked HA matrix that actively modulates inflammation. TNF-stimulated gene 6 (TSG6), a member of the family of HA-binding proteins, catalyzes the transfer of heavy chains 1–3 (HC1, HC2, and HC3, respectively) from the CS of inter-α-trypsin inhibitor (IαI) to GlcNAc residues of HA forming an HC-HA ECM that modulates inflammatory responses during inflammatory processes (14–18).
Given the ability of the UMSCs to survive host rejection, we investigated whether UMSCs could regulate mouse immune responses. Our results show that UMSCs transplanted into corneal stroma after alkali burn suppress the severe inflammatory response and enable the recovery of corneal transparency. We also show that UMSCs in vitro actively “turn off” the inflammatory cells by inhibiting their adhesion and invasion and by impeding the polarization of M1 macrophages. UMSCs also induced both the maturation of T-regulatory cells and inflammatory cell death. These results are mediated by the UMSC glycocalyces that contain versican and HA. The knockdown of cell surface-associated CS and HA on UMSCs ablates their ability to modulate the immune responses both in vivo and in vitro. However, removal of cell surface heparan sulfate has no effect on the ability of UMSCs to suppress the immune responses.
EXPERIMENTAL PROCEDURES
Animals
Six-week-old C57/BL6 mice were obtained from The Jackson Laboratory, Bar Harbor, ME. Animal care and use conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All animal protocols were previously approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Cincinnati.
Cell Culture
Umbilical cords were obtained from Christ Hospital, Cincinnati, OH, and kept in Earle's balanced salt solution (EBSS) at 4 °C during transportation. Upon arrival, umbilical cords were washed with 70% ethanol, and subsequently excess blood was removed with sequential washes in EBSS. With the use of a blade, the blood vessels were removed from the umbilical cord, and the tissue was minced into fine pieces. The minced tissue was then incubated with 0.05% trypsin (Invitrogen) and 300 units of collagenase (Stem Cell Technologies) in α-MEM (Invitrogen) for 4 h at 37 °C. Thereafter, the cell suspension was filtered; the filtrate was centrifuged for 10 min at 400 × g, and the cell pellet was cultured in α-MEM supplemented with 10% fetal bovine serum (FBS) (Hyclone, Waltham, MA) in 5% CO2 at 37 °C. After 16 h, the medium was changed to remove nonadherent cells. Thereafter, the medium was changed every 2 to 3 days, and cells were harvested when they reached ∼70% confluence with trypsin/EDTA and subsequently seeded at a density of 3–6 × 103 cells/cm2. The UMSCs were used for experiments between passage 4 and 7. Beyond passage 8, the efficiency of the UMSCs to modulate the inflammatory cells declined (data not shown).
Reducing Surface Glycosaminoglycans
UMSCs were treated with chondroitinase AC (Chase AC) (IBEX), chondroitinase B (Chase B) (IBEX), heparinase II (Hepase II) (IBEX), heparinase III (Hepase III) (IBEX), chondroitinase ABC (Chase ABC) (Sigma), hyaluronidase (Hylase) from testes (Sigma), or hyaluronidase from Streptomyces (HylaseS) (Sigma) for 2 h at 37 °C to digest cell surface and associated ECM GAGs. Subsequently, the cells were washed twice with PBS. For in vivo experiments, the UMSCs were labeled with DiI for 15 min at 4 °C, further washed three times with PBS, and transplanted into the corneal stroma of the mice 24 h after alkali burn. The hepase digest heparan sulfate (HS) chain of HS proteoglycans, Chase AC, is selective for digesting CS and would remove CS from versican; Chase B is selective for dermatan sulfate (DS) and would remove any DS chains from versican; testicular Hylase digests both HA and CS, thereby removing both versican CS and HA from the glycocalyx, and HylaseS, although selective for HA, would also remove the glycocalyx releasing intact versicans.
Agarose Gel Electrophoresis
The efficiency of reducing surface CS and HS was analyzed by 0.6% agarose gel electrophoresis in 0.05 m propanediamine acetate buffer, pH 9, as described previously (19). Following electrophoresis, the gels were submerged in 0.2% CETAVLON (cetyltrimethylammonium bromide, Sigma) for 1 h at room temperature, dried, and stained with 0.1% toluidine blue prepared in a solution of 1% acetic acid, 50% ethanol, and 49% water, destained with the same solution without toluidine blue (for staining CS, DS, and HS), and then restained with 0.1% toluidine blue prepared in 25 mm sodium acetate buffer, pH 5.0, and destained in this solution without toluidine blue (to stain HA).
Alkali Burn
Animals were anesthetized by intraperitoneal injection of ketamine hydrochloride (80 mg/kg) and xylazine (10 mg/kg). The eyes were rinsed with PBS and topically anesthetized with a drop of proparacaine. Ocular surface alkali burns were produced by placing 3MM chromatography paper (Whatman) cut into 1-mm diameter circles previously soaked in 0.1 m NaOH onto the central cornea for exactly 1 min. Subsequently, the eyes were continuously washed with sterile PBS for 1 min. Finally, teramycin ointment was topically administered to the eyes, and the animals were placed on a warming pad. The alkali burn protocol used in our experimental model was designed to avoid damage to the limbal stem cells, thereby allowing us to examine the effects of UMSCs on inflammation suppression and regeneration of a transparent cornea without the complication of limbal deficiency.
Intrastromal Injection
Intrastromal injection was done by first creating a small tunnel through the corneal epithelium into the anterior stroma using a 33-gauge needle with a sharp tip (Hamilton Co., Reno, NV). Thereafter, a blunt 33-gauge needle attached to a 10-μl syringe (Hamilton Co.) was passed through the tunnel into the corneal stroma, and 2 μl containing 10,000 cells were injected into the stroma.
In Vivo Confocal Microscopy
Corneal haze was analyzed as described previously (2) with a Heidelberg Retinal Tomograph-HRTII Rostock Cornea Module (HRT-II, Heidelberg Engineering Inc., Germany) according to the manufacturer's instructions. Briefly, GenTeal® gel (Novartis Pharmaceuticals Corp.) was applied to both the eyeball and the tip of the HRT-II objective as an immersion fluid. Subsequently, a series of 40 images were collected to cover the whole stromal thickness as a continuous z axis scan through the entire corneal stroma at 2-μm increments starting from the basal layer of the corneal epithelium and ending at the corneal endothelium. The lens of HRT-II has a working distance of 77 μm.
Corneal Whole Mount
Eyeballs were excised and fixed for 30 min in 4% paraformaldehyde. After extensive washes in PBS, the cornea button was removed, and subsequently, four small peripheral incisions were made in the cornea to enable a flat mount onto a slide. Corneas were treated for 30 min with 0.2% sodium borohydrate and thereafter extensively washed in PBS containing 0.2% Tween. Corneas were blocked overnight in 4% BSA in PBS containing 0.01 m saponin and incubated overnight with primary antibody rat anti-F4/80 (Abcam, ab6640) or rat anti-CD11b (Invitrogen, RM2800) prepared in 4% BSA in PBS solution at 4 °C. Subsequently, corneas were extensively washed in PBS and further incubated with secondary antibody in block solution for 8 h at room temperature. Corneas were finally incubated with DAPI and mounted in Fluoromount G (SouthernBiotech). Z-stacks were generated in 1.8-μm increments, and three-dimensional reconstructions were made using AxioVision software (Zeiss).
Macrophage Isolation
Macrophages were isolated from murine peritoneum. Naive mouse peritoneal cavities were used in order not to alter the physiologic characteristics of the isolated cells. In short, mice were sacrificed by CO2 inhalation and dipped in 70% ethanol, and excess ethanol was removed with sterile gauze. An incision was made along the midline with sterile scissors, and abdominal skin was retracted with forceps to expose the intact peritoneal wall. A small incision was made in the peritoneal wall, and the peritoneal cavity was washed with 6 ml of Hanks' balanced salt solution using a sterile plastic Pasteur pipette. The lavage was then collected and centrifuged to isolate the inflammatory cells.
Inflammatory Cells and UMSC Bilayer Adhesion Assay
UMSCs were seeded in microplates and left for 24 h to adhere. Cells were washed twice with EBSS and treated with Chase AC+B, Hylase, or Hepase II+III at 37 °C for 2 h to remove specific cell-associated GAGs. Meanwhile, inflammatory cells were isolated by peritoneal lavage using EBSS, labeled with DiI for 30 min on ice, and washed four times with EBSS. Subsequently, UMSCs were washed twice with EBSS, and the inflammatory cells were seeded over the UMSCs in α-MEM (Invitrogen) supplemented with 10% FBS. A control assay was done without UMSCs. The inflammatory cells were left to adhere for 16 h. The adherent cells were fixed with 2% paraformaldehyde, and the numbers of adhered DiI+ inflammatory cells and F4/80+ cells (macrophages) were evaluated. Five images were captured for each experimental point, and the numbers of adhered cells were calculated using CellProfiler (20). The experiment was repeated twice with triplicates.
Macrophage Adhesion Assay
UMSCs were seeded in a transwell insert with 0.44-μm (Millicell, Millipore) pores and left for 24 h to adhere. Cells were washed twice with EBSS and treated with Chase AC+B, Hylase, or Hepase II+III at 37 °C for 2 h to remove specific cell-associated GAGs. Meanwhile, inflammatory cells were isolated by peritoneal lavage using EBSS. Subsequently, UMSCs were washed four times with EBSS, and the transwell insert was placed into a new microplate well. The inflammatory cells were then placed under the UMSCs in α-MEM supplemented with 10% FBS with or without rat anti-pentraxin 3 (Cellsciences, HM2242), goat anti-TSG6 (R&D, AF2104), goat anti-ITI-H1 (sc-21968), goat anti-ITI-H2 (sc-21975), goat anti-ITI-H3 (sc-21979), goat IgG isotype control (Abcam, ab37388), or anti-collagen I (Abcam, ab34710) as a nonrelevant control. A control experiment was done without UMSCs in the transwell insert. The macrophages were left to adhere for 16 h; the nonadherent cells were removed by washing, and adherent inflammatory cells were fixed with 2% paraformaldehyde. The numbers of adhered DiI+ cells (inflammatory cells) and F4/80+ cells (macrophages) were counted using CellProfiler (20). Five images were captured for each experimental point. The experiment was repeated five times with triplicates.
Macrophage Invasion Assay
UMSCs were seeded in a 24-well microplate and left for 24 h to adhere. Cells were washed twice with EBSS and treated with Chase AC, Chase B, Hylase, or Hepase II and III at 37 °C for 2 h to remove specific cell-associated GAGs. Meanwhile, macrophages were isolated by peritoneal lavage using EBSS, labeled with DiI for 30 min on ice, and washed four times with EBSS. Subsequently, UMSCs were washed twice with EBSS, and a transwell insert (3 μm, HTS FluoroBlok Insert, Falcon) was placed into each microplate well over the UMSCs. α-MEM supplemented with 10% FBS was placed in the bottom compartment, and the macrophages in α-MEM were placed in the top compartment. A control experiment was done without UMSCs in the bottom compartment. The macrophages were left to migrate for 4 h, and the transwell inserts were then treated with 2% paraformaldehyde. Transwell membranes were removed, and the bottom side was analyzed under a fluorescent microscope. The numbers of migrated macrophages were counted by two separate individuals using a double blind system. The experiment was repeated three times with triplicates.
Cell Death Detection Assay
To verify whether the UMSCs could induce inflammatory cell death, inflammatory cells and UMSCs were co-cultured using transwell inserts with 0.4-μm pores. UMSCs (2.5 × 104) were seeded inside the culture insert and left to adhere for 24 h. The UMSCs were treated or not with Chase AC+B or Hylase for 2 h and then washed with Hanks' balanced salt solution. Thereafter, inflammatory cells were obtained by peritoneal lavage, and 1 × 105 cells were seeded into each well of a 6-well multiplate. The transwell inserts containing the UMSCs were placed over the inflammatory cells, and antibodies were added to the lower chamber when indicated. The cells were left in co-culture for 16 h, and the nonadherent cells were then removed with the medium for cell death analysis. The 1 ml of culture medium was centrifuged to pellet any nonadherent cells. The medium (necrotic fraction) was stored at 4 °C for cell death analysis. The cell pellet was lysed with the lysis buffer provided with the kit for 20 min and centrifuged, and the supernatant was assayed for cell death. Cell death was assayed utilizing the cell death detection ELISAPLUS kit (Roche Applied Science) according to the manufacturer's instructions. The remainder of the culture medium containing the nonadherent cells was centrifuged, and the cell pellet was suspended in 2% paraformaldehyde and incubated at 4 °C for 30 min. The cells were then centrifuged. The cell pellet was suspended in PBS, and 50 μl was placed on a polylysine-coated microscope slide and then heated on a hotplate at 60 °C. When dry, the cells were stained with hematoxylin and eosin and analyzed under an Eclipse E800 microscope (Nikon) coupled with an Axiocam ICc5 camera (Zeiss).
Analysis of UMSCs after Co-culture with Inflammatory Cells
Macrophages and UMSCs were co-cultured using transwell inserts with 0.4-μm pores, which enable cross-talk through soluble factors between the two cell types. This entailed seeding UMSCs in a 6-well microplate in α-MEM supplemented with 10% FBS and leaving the cells for 24 h to adhere in 5% CO2 at 37 °C. UMSCs were washed three times with EBSS and then treated or not treated with Chase AC+B, Chase ABC, or Hylase in EBSS for 2 h, after which the cells were washed three times with EBSS. Meanwhile, macrophages were isolated from mouse peritoneum and seeded in transwell inserts, which were placed over the previously seeded UMSCs. The cells were left for 16 h in fresh α-MEM supplemented with 10% FBS at 5% CO2 and 37 °C. The cells were then either fixed in 2% paraformaldehyde at 4 °C for 30 min, or total protein was extracted by scraping the cells using a cell scraper in RIPA buffer. The macrophages and UMSCs were analyzed by immunocytochemistry and Western blotting. The immunofluorescence analyses were repeated three times with duplicates, and the Western blotting analyses were repeated twice with pooled triplicates.
Immunocytochemistry and Confocal Microscopy
The cells were fixed in 2% paraformaldehyde for 15 min at 4 °C. Cells were incubated for 1 min in 0.1 m glycine, washed four times with PBS, and subsequently incubated for 1 h in blocking solution (5% FBS) at room temperature. The cells were then incubated with primary antibodies overnight at 4 °C. Primary antibodies used were as follows: rabbit anti-versican (ab19345), rat anti-pentraxin 3 (Cellsciences, HM2242), goat anti-TSG6 (R&D Systems, AF2104), rabbit anti-RHAMM (Abcam, ab124729), and mouse anti-CD44 (Abcam, ab23777). Also, phalloidin 647 or biotinylated HA-binding link protein (Calbiochem, Millipore) were used instead of a primary antibody when stated. Afterward, the cells were washed three times in PBS and then incubated for 1 h at room temperature with appropriate fluorescent secondary donkey antibodies conjugated to Alexa Fluor® 488, Alexa Fluor® 555, Alexa Fluor® 647, or streptavidin Alexa Fluor® 488 conjugate (Molecular Probes/Invitrogen). After incubation with the antibodies or the streptavidin conjugate, the coverslips were mounted on microscope slides with Fluoromount G (2:1 in PBS, Electron Microscopy Sciences) and sealed with nail polish. Cells were examined using a Zeiss Observer Z1 inverted microscope or a Zeiss LSM 710 confocal microscope, and images were analyzed using LSM Image Browser 3.2 software (Zeiss, Germany).
ECM Extraction, Immunoprecipitation (IP), and Western Blotting
Proteins were extracted from the UMSCs by scraping the cells using a cell scraper in RIPA buffer containing an EDTA-free protease inhibitor mixture (Sigma). The cells were lysed using sonication and subsequently heat-inactivated at 95 °C for 15 min. The cell lysates were separated into two fractions, and one fraction was digested with Hylase at 37 °C for 4 h. Loading buffer was added to the samples, which were then incubated at 95 °C for 15 min.
Conditioned medium from UMSCs, or UMSCs co-cultured with inflammatory cells, or solely fresh medium was incubated with 10 μg of anti-HC1 (amino acids 623–672 LifeSpan BioSciences, Seattle, WA), anti-HC2 (amino acids 124–321 LifeSpan BioSciences), or anti-HC3 (ab97758, Abcam) for 2 h at 4 °C, after which 20 μg of Dynabeads® protein G (Invitrogen) were added and further incubated for 1 h at 4 °C. The beads were removed with the use of a magnet, and samples were dislodged from beads with 100 μl of 0.1 m glycine, pH 3. Thereafter, the pH was neutralized, and loading buffer was added to the samples, which were then incubated at 95 °C for 15 min. The samples were then applied to SDS-PAGE, transferred to nitrocellulose membranes, and incubated with goat anti-ITI-H1 (sc-21968), goat anti-ITI-H2 (sc-21975), goat ant-ITI-H3 (sc-21979), and anti-pentraxin 3.
Hylase-digested and -undigested cell extracts and IP products were resolved by SDS-PAGE under reducing conditions and transferred to polyvinylidene fluoride transfer membranes. The membranes were blocked overnight at 4 °C, incubated with the following primary antibodies goat anti-ITI-H1 (sc-21968), goat anti-ITI-H2 (sc-21975), goat anti-ITI-H3 (sc-21979), rabbit anti-versican, anti-pentraxin 3, mouse anti-CD44, or goat anti-TSG6 in block solution for 4 h, followed by four 15-min washes with PBS, and subsequent incubation with the secondary antibodies conjugated to Alexa Fluor® 488, Alexa Fluor® 555, or Alexa Fluor® 647 for 1 h at room temperature.
RNA Extraction and Real Time Reverse Transcription-PCR Analysis
Total RNA was isolated from UMSCs and UMSCs exposed to inflammatory cells 24 h after co-culture using TRIzol® reagent (Invitrogen). The concentration and purity of the RNA in each sample were determined using a spectrophotometer at 260 and 280 nm. First strand cDNA was reverse-transcribed using 2 μg of total RNA and the kit Improm-IITM reverse transcriptase (Promega, Madison, WI), according to the manufacturer's protocol. Quantitative RT-PCR amplification was done with 2 μl of the cDNA (1:5) with specific primers for HC1, HC2, HC3, TSG6, β-tubulin, and GADPH using the kit SYBR Green Master Mix (Applied Biosystems, Foster City, CA) in a CFX96 Real Time System in a C1000 Thermo Cycler (Bio-Rad), with the activation cycle of 95 °C for 10 min, 40 cycles of 95 °C for 20 s, 60 °C for 30 s, and 72 °C for 30 s. The specificities of the amplified products were analyzed through dissociation curves generated by the equipment yielding single peaks. Negative controls were used in parallel to confirm the absence of any form of contamination in the reaction. Analysis of the data were done using the 2−ΔCt method. The primer combination used for HC1 was forward 5′-CCACCCCATCGGTTTTGAAGTGTCT-3′, reverse 3′-TGCCACGGGTCCTTGCTGTAGTCT-5′; HC2 forward 5′-ATGAAAAGACTCACGTGCTTTTTC-3′, reverse 3′-ATTTGCCTGGGGCCAGT-5′; HC3 forward 5′-TGAGGAGGTGGCCAACCCACT-3′, reverse 3′-CGCTTCTCCAGCAGCTGCTC-5′; TSG6 5′-CCAGGCTTCCCAAATGAGTA-3′; β-tubulin forward 5′-TGGAACCCACAGTCATTGATGA-3′, reverse 3′-TGATCTCCTTGCCAATGGTGTA-5′; and GADPH forward 5′-ACCACAGTCCATGCCATCAC-3′, reverse 3′-TCCACCACCCTGTTGCTGTA-5′.
Statistics
All values are presented as means ± S.D. The difference between two groups was compared by unpaired Mann-Whitney test. p < 0.05 was considered to be statistically significant. Statistical analysis was done with the GraphPad Prism version 5 software package (GraphPad Software, San Diego).
RESULTS
UMSCs Suppress in Vivo Inflammatory Response in a Glycocalyx-dependent Manner
UMSCs were transplanted into cornea stromas via intrastromal injection 24 h after the alkali burn. To evaluate the role of UMSC GAG side chains, cells were treated for 2 h with either chondroitinase AC and B (Chase AC+B) or heparinase II and III (Hepase II+III) to remove cell-associated CS and HS, respectively, prior to transplantation. Control animals received solely the vehicle (PBS) through intrastromal injection. Corneas transplanted with UMSCs clearly presented reduced inflammation already at 5 days after alkali burn when compared with PBS controls (Fig. 1A). Interestingly, treating UMSCs with hepase II+III prior to transplantation, to remove cell surface HS, had no effect on the ability of UMSCs to suppress inflammation (Fig. 1A). In contrast, treating UMSCs with Chase AC+B prior to transplantation ablated the ability of UMSCs to suppress inflammation, and the corneal inflammation resembled that of the vehicle controls (Fig. 1A). Two weeks after corneal alkali burn, corneas transplanted with UMSCs or Hepase (II+III)-treated UMSCs were mostly transparent (11/12 and 10/12, respectively), although corneas treated with vehicle control or Chase AC+B-treated UMSCs presented persistent inflammation and corneal clouding (only 2/12 and 3/12 transparent, respectively) (Fig. 1A). The efficiency of the CS and HA removal by the enzymatic digestions was evaluated through immunocytochemistry and agarose gel electrophoresis (Fig. 2) revealing efficient removal of cell-associated CS and HA from the UMSCs prior to transplantation. Both CS and HA were susceptible to Chase AC+B digestion; therefore, reducing cell surface CS and/or HA, but not HS, ablates the anti-inflammatory effect of transplanted UMSCs.
FIGURE 1.

UMSCs suppress the in vivo inflammatory response in a glycocalyx-dependent manner. Corneas were subjected to alkali burn, and UMSCs that were previously treated or not with Hepase or Chase AC+B for 24 h were transplanted into the stroma. A, stereomicroscope images of the corneas were taken 24 h, 5 days, 1 week, and 2 weeks after the alkali burn. Twelve animals were analyzed in each group, and the number of transparent corneas/total at 2 weeks is indicated in the figure. B and C, 2 weeks after the alkali burn, the corneas were subjected to whole mount analysis for inflammatory cell infiltration. The number of F4/80+ (B) and CD11b+ (C) cells/cornea were counted. * is S.D. against PBS-treated, and ** against UMSC-treated, * and **, p < 0.05. D, corneas were analyzed by in vivo confocal microscopy 2 weeks after the alkali burn to evaluate corneal haze. E, UMSCs were stained with DiI prior to transplantation to evaluate their presence initially and in the corneas 2 weeks after transplantation, thereby assessing their ability to survive host rejection.
FIGURE 2.
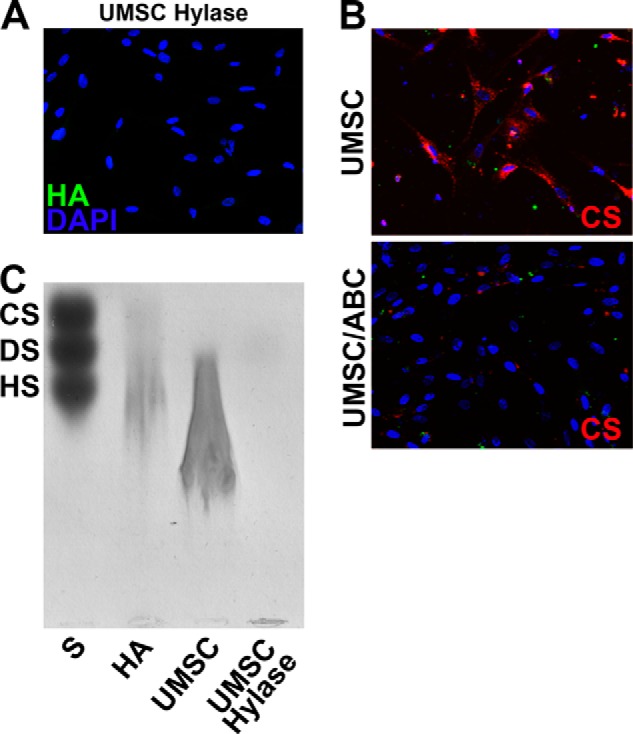
HA and CS shedding after Hylase and Chase ABC treatment. The efficiency of reducing surface CS and HA was analyzed by both immunocytochemistry (A and B) and agarose gel electrophoresis (C) thereby demonstrating successful removal of CS and HA from the UMSCs. S, standard mixture of CS, DS, and HS; HA, standard HA isolated from umbilical cord; UMSC, HA extracted from UMSCs; and UMSC Hylase, HA extracted from UMSCs treated with Hylase.
UMSCs Inhibit Inflammatory Cell Infiltration after Alkali Burn in a CS/HA-dependent Manner
Inflammatory infiltration was analyzed to assess corneal inflammation 2 weeks after UMSC transplantation. Corneal whole mounts were stained with macrophage markers CD11b and F4/80, which recognize leukocytes involved in the innate immune system, including monocytes, granulocytes, macrophages, natural killer cells, and murine macrophages, respectively. Z-stack images were obtained, and the mean number of positive cells present in 10 independent Z-stacks was determined. Corneas transplanted with UMSCs had ∼2 F4/80+ cells and ∼4 CD11b+ cells in each Z-stack analyzed, and UMSCs previously treated with Hepase II+III had slightly higher numbers (∼6 cells and ∼6 cells, respectively) (Fig. 1, B and C). In contrast, corneas transplanted with UMSCs previously treated with Chase AC+B presented an average of ∼18 F4/80+ cells and ∼21 CD11b+ cells in each Z-stack analyzed, closer to the PBS vehicle controls with averages of ∼22 F4/80+ cells and ∼32 CD11b+ cells in each Z-stack analyzed (Fig. 1, B and C). Therefore, treating UMSCs with Chase AC+B prior to transplantation impeded the ability of these cells to suppress inflammatory cell infiltration, whereas treating them with Hepases had little or no effect.
In Vivo Confocal Microscopy
To evaluate corneal inflammation in vivo, confocal microscopy was used to assess corneal clouding. Corneas transplanted with UMSCs or UMSCs treated with Hepase II+III presented clear corneas 2 weeks after alkali burn (Fig. 1D). Animals treated with the vehicle control or with UMSCs treated with Chase AC+B prior to transplantation have significant corneal haze 2 weeks after alkali burn (Fig. 1D).
Survival of Human UMSCs Transplanted in Mouse Corneas after Alkali Burn
Because of the increase in inflammatory response in the corneas transplanted with UMSCs previously treated with Chase AC+B, we investigated the survival of UMSCs and UMSCs treated with Chase AC+B prior to transplantation. UMSCs and Chase AC+B-treated UMSCs were labeled with DiI prior to transplantation, and DiI+ cells were present within the stroma soon after administration (Fig. 1E). Two weeks after administration, DiI-positive UMSCs were evident in the corneal stroma of mice treated with naive UMSCs, whereas corneas transplanted with Chase AC+-treated UMSCs presented no DiI+ cells, indicating that the UMSCs had been rejected by the host. Untreated UMSCs therefore actively modulate inflammatory cells and evade rejection in a CS- and/or HA-dependent manner.
UMSCs Inhibit the Adhesion of Inflammatory Cells
UMSCs actively suppressed the inflammatory response present in mouse corneas 24 h after alkali burn thereby enabling resolution of the inflammatory response within 2 weeks, although the vehicle control-treated mice presented severe inflammation in the same time frame. Therefore, we hypothesize that the UMSCs can actively regulate the inflammatory cells present in the stroma within 24 h after alkali burn. Co-culture assays were used to examine this possibility with inflammatory cells isolated from peritoneal lavages. To investigate the effect of UMSCs on inflammatory cell adhesion, we prepared UMSC 24-h microplate cultures that were digested with/without Chase AC+B or Hylase as described under “Experimental Procedures.” DiI-labeled inflammatory cells, prepared as described under “Experimental Procedures,” were then seeded on the UMSC cultures in the presence or not of anti-pentraxin 3, anti-TSG6, anti-HC1, anti-HC2, anti-HC3, goat IgG, and anti-collagen I. After 16 h, the nonadherent cells were removed by washes, and the adhered cells were fixed and stained with anti-F4/80 to label macrophages. Interestingly, the UMSCs actively inhibited adhesion of the inflammatory DiI+ cells by ∼40%, and previously treating the UMSCs with either Chase AC+B or Hylase disrupted the ability of the UMSCs to inhibit adhesion of the inflammatory cells (Fig. 3A). Similarly, the UMSCs inhibited the adhesion of macrophages (F4/80+ cells) by ∼70%, and this effect was also ablated when the UMSCs were previously digested with Chase AC+B or Hylase (Fig. 3B). To verify whether the ability of the UMSCs to inhibit adhesion of the inflammatory cells was substrate-dependent or required direct cell-cell contact, we performed a co-culture adhesion assay with the UMSCs in transwell inserts with 0.44-μm pores over inflammatory cells in the bottom chamber, which enables cross-talk between the inflammatory cells and UMSCs by soluble factors. Interestingly, the UMSCs inhibited the adhesion of macrophages (F4/80+ cells) by ∼80% (Fig. 3C). Again, previously digesting the UMSCs with Chase AC, Chase AC+B, Chase B, and Hylase impeded the ability of the UMSCs to inhibit adhesion of the macrophages, indicating that soluble factors are involved.
FIGURE 3.

UMSCs inhibit the adhesion and migration/invasion of inflammatory cells. The ability of the UMSCs to modulate immune cells was evaluated using a co-culture assay between UMSCs and inflammatory cells. A and B, inflammatory cells were labeled with DiI and seeded over UMSCs previously treated or not with Chase AC+B or Hylase or over UMSCs in the presence of antibodies. The numbers of adhered DiI+ cells (A) and F4/80+ cells (B) were counted. C, potential requisite of cell-cell contact between the UMSCs and the inflammatory cells was evaluated. UMSCs were seeded in a transwell insert with a 0.44-μm pore and treated or not with Chase ABC, Chase AC, Chase B, or Hylase. Thereafter, inflammatory cells were placed in the bottom chamber, and the numbers of adhered F4/80+ cells were counted. D, UMSCs were treated or not with Chase ABC, Chase AC, Chase B, Hepase, Hylase, or HylaseS. Inflammatory cells labeled with DiI were seeded in a transwell insert with a 3-μm pore. 10% FBS was placed in the bottom chamber as a stimulus to invasion. The numbers of DiI+ cells that invaded into the lower chamber were counted. * < 0.05 compared with UMSCs plus macrophages.
UMSCs Inhibit the Migration/Invasion of Inflammatory Cells
Corneas treated with UMSCs showed a large reduction in the number of inflammatory cells present in the stroma 2 weeks after the alkali burn. This suggests that the UMSCs could be aiding in the resolution of the inflammatory response and/or inhibiting the invasion of more inflammatory cells. Therefore, to further investigate whether UMSCs directly modulate inflammatory cell invasion, an invasion co-culture assay was performed. DiI-labeled inflammatory cells in serum-free medium were seeded in the top compartment of transwell inserts with 3-μm pores over UMSCs previously seeded in medium supplemented with 10% FBS as a stimulus for the inflammatory cells to invade into the bottom compartment. When inflammatory cells were seeded in the top compartment in the absence of UMSCs in the bottom compartment, ∼250 DiI+ cells (inflammatory cells) migrated to the bottom compartment. However, when UMSCs were seeded in the bottom compartment, the number of migrating inflammatory cells decreased by ∼60% (Fig. 3D). When the UMSCs were treated with Chase ABC, Chase AC, Chase B, or Hylase to decrease cell associated CS and HA, the number of macrophages invading the bottom compartment increased by ∼2-fold compared with naive UMSCs (Fig. 3D). Therefore, the absence of cell-associated CS/HA compromised the ability of UMSCs to inhibit inflammatory cell invasion into the bottom compartment. In contrast, the treatment of UMSCs with Hepase had no effect on the ability of UMSCs to suppress the migration of inflammatory cells (Fig. 3D).
UMSCs Affect Macrophage Polarization
In light of the inhibitory effect that UMSCs had on macrophage adhesion and invasion, the role of UMSCs and, more importantly, the role of UMSC CS/HA on macrophage polarization were investigated. Previously adhered and differentiated UMSCs and macrophages were placed in co-culture, using the transwell system with 0.44-μm pores, enabling cross-talk through soluble factors, and macrophage morphology was studied by anti-F4/80 immunostaining. Control macrophages, seeded in the absence of UMSCs, presented a stellate (or dendritic) shape, typical of M1 macrophages, and ∼50% presented this stellate morphology (Fig. 4, A and B). However, when macrophages were exposed to UMSCs, they presented a small rounded cell shape, typical of undifferentiated macrophages, with only ∼15% presenting a stellate shape (Fig. 4, A and B). Moreover, a subpopulation of the macrophages exposed to UMSCs adhered and differentiated into a rounded, flattened morphology (a spread cell body), potentially a M2 macrophage (Fig. 4B, lower panels). When macrophages were exposed to UMSCs that had been previously treated with Chase AC+B or Hylase, ∼55 and 49%, respectively, of the macrophages had a stellate shape (Fig. 4A), essentially the same as macrophages in the absence of UMSCs. The macrophages were stained with antibodies for TNFα and IL10 as markers for M1 and M2 macrophages, respectively. Exposing the inflammatory cells to UMSCs decreased the number of F4/80+TNFα+ (M1) cells by ∼70% relative to macrophages not exposed to UMSCs (Fig. 4C) and increased the number of F4/80+IL10+ cells (M2) ∼3-fold (Fig. 4D), and pretreatment of the UMSCs with Chase AC+B or Hylase abrogated the ability of the UMSCs to inhibit M1 macrophage polarization.
FIGURE 4.

Role of UMSCs on macrophage polarization. The ability of the UMSCs to modulate polarization of macrophages was evaluated. UMSCs were seeded in a transwell insert with a 0.44-μm pore and treated or not with Chase AC+B or Hylase. Inflammatory cells were seeded in a micro-plate and placed in co-culture with the UMSCs seeded in transwell inserts. A and B, morphology of the macrophages (F4/80+ cells) was analyzed (green), and nucleus was stained with DAPI (blue). C, numbers of F4/80+TNFα+ cells were counted to evaluate the M1 phenotype. D, numbers of F4/80+IL10+ cells were counted to evaluate the M2 phenotype. E, IRF5 staining (red) of macrophages is shown, and F4/80+ cells are shown in green. Scale bars, B and E are 20 μm. *, <0.05 compared with macrophages alone.
This was investigated further by staining the inflammatory cells with antibodies for F4/80 and IRF5, a transcription factor that regulates the expression of M1-specific cytokines. There was a significant decrease in F4/80+ cells with IRF5 nuclear staining, which attained a cytoplasmic localization, when the inflammatory cells were co-cultured with UMSCs (Fig. 4E). When the macrophages were seeded in the absence of UMSCs or in the presence of UMSCs previously treated with Chase AC+B or Hylase, the majority of the F4/80+ cells presented IRF5 nuclear staining (Fig. 4E). No nuclear staining was observed for IRF4 (MUM1), a transcription factor that regulates the expression of M2 macrophage-specific cytokines, in any of the experimental conditions (data not shown). Therefore, UMSCs inhibit the polarization of macrophages to the M1 phenotype, and this process is mediated through the associated CS/HA ECM on the UMSCs.
UMSCs Promote the Maturation of T-regulatory Cells
Co-cultures between inflammatory cells and UMSCs using the transwell co-culture system were prepared, and the inflammatory cells were stained for CD44+, F4/80, and IL10. Interestingly, a population of CD44+ cells was present solely in the inflammatory cells exposed to UMSC (Fig. 5A). Moreover, a population of F4/80−IL10+ cells was increased in the inflammatory cells exposed solely to naive UMSC (Fig. 5B). Both populations of CD44+ and F4/80−IL10+ cells were greatly decreased when UMSCs were pretreated with Chase ABC when compared with naive UMSCs. These cells also stained positive for CD4 (results not shown) and CD25 (Fig. 5, C and D) but did not stain positive for Foxp3 (results not shown). Therefore, co-cultivating inflammatory cells with UMSCs led to the maturation of CD4+CD44+CD25+IL10+Foxp3− cells, which have been previously characterized as T-regulatory cells. Previously treating the UMSCs with Chase AC+B or Hylase impeded the maturation of this population of T-regulatory cells.
FIGURE 5.

Role of UMSCs on the maturation of T-regulatory cells. The ability of the UMSCs to activate T-regulatory cells was evaluated. UMSCs were seeded in a transwell insert with a 0.44-μm pore and treated or not with Chase ABC. Inflammatory cells were seeded in a micro-plate and placed in co-culture with the UMSCs. A, numbers of CD44+ cells were counted. B, numbers of F4/80−IL10+ cells were counted. C and D, numbers of CD25+ cells were analyzed and counted, respectively. E, morphology of the macrophages (CD11b+ cells) was analyzed in the presence of UMSCs with neutralizing anti-IL10 or anti-TGFβ antibodies. Scale bar, E is 20 μm. F, numbers of stellate CD11b+ cells were counted to evaluate the M1 phenotype. *, <0.05 compared with control.
T-regulatory cells have previously been shown to suppress inflammatory cells, and TGFβ and IL10 have been characterized as the immunosuppressive cytokines secreted by these cells. Therefore, to verify whether UMSCs can inhibit the polarization of M1 macrophages in a T-regulatory cell-dependent manner, we performed the co-culture assay in the presence of neutralizing antibodies for IL10 and TGFβ. Interestingly, inflammatory cells exposed to UMSCs in the presence of the anti-IL10 neutralizing antibody presented a 5-fold increase in stellate M1 macrophages, and inflammatory cells exposed to UMSCs in the presence of the anti-TGFβ neutralizing antibody presented a 2-fold increase in stellate M1 macrophages (Fig. 5, E and F).
UMSCs Induce Inflammatory Cell Death
UMSCs greatly inhibited adhesion of the inflammatory cells. Therefore, we analyzed whether the nonadherent inflammatory cells after co-culture with UMSCs undergo necrosis or apoptosis using a cell death detection assay and histochemistry. The culture medium (representing the necrotic fraction) and the cell lysate (representing the apoptotic fraction) were analyzed using the cell death detection assay. There was a significant increase in necrosis, ∼40%, when the inflammatory cells were seeded in the presence of UMSCs (Fig. 6A). Therefore, UMSCs induce necrosis in the inflammatory cells. This ability of the UMSCs to induce necrosis was lost when the cells were treated with Chase AC+B or Hylase prior to co-culture (Fig. 6A). Moreover, when the inflammatory cells were co-cultivated with UMSCs in the presence of anti-versican antibody, there was no significant increase in inflammatory cell necrosis. Therefore, when the versican epitopes were masked with the use of a polyclonal antibody, the UMSCs were no longer able to induce necrosis of inflammatory cells. Moreover, when the inflammatory cells were co-cultivated with UMSCs in the combined presence of anti-IL10 and anti-TGFβ antibodies, there was no significant increase in inflammatory cell necrosis. When the cells were co-cultivated in the presence of anti-RHAMM, the ability of UMSCs to induce necrosis in the inflammatory cells was not inhibited (Fig. 6A). When the cell pellets of the nonadherent fraction were analyzed for cell death (apoptosis), again, UMSCs increased cell death of the inflammatory cells ∼15% (Fig. 6B), which was blocked when the UMSCs were previously treated with Chase AC+B. Previous treatment with Hylase and co-culture in the presence of anti-versican had no effect on the ability of the UMSCs to induce cell death (Fig. 6B). Moreover, there was a large increase in inflammatory cell death when the co-culture was done in the presence of anti-RHAMM (Fig. 6B).
FIGURE 6.

UMSCs induce inflammatory cell death. The ability of the UMSCs to induce inflammatory cell death after the indicated treatments was evaluated in the necrotic fraction (A) and apoptotic fraction (B). *, <0.05 compared with inflammatory cells alone. C–F, nonadherent cell fraction was collected 16 h after inflammatory cells were seeded in the presence of UMSCs. Nonadherent cells from solely macrophages (C), macrophages seeded over UMSCs (D), macrophages seeded over UMSCs previously treated with Chase AC+B (E), and macrophages seeded over UMSCs previously treated with Hylase (F) were fixed, attached to polylysine-coated microscope slides, and stained with hematoxylin and eosin to reveal apoptotic cells (arrowheads). Graphs represent arbitrary fluorescence assayed by ELISA (E and F).
The results using the cell death detection ELISA were confirmed by histochemistry of the nonadherent cells. When the macrophages were seeded in the absence of UMSCs, the nonadherent cell fraction contained many cells that were viable. However, when they were seeded in the presence of UMSCs, within 16 h there was a drastic reduction in the overall number of cells in the nonadherent fraction, due to increased necrosis, and the few cells that were detected were apoptotic with characteristic condensed chromatin (arrowhead in Fig. 6D). Therefore, UMSCs induce both necrosis and apoptosis of inflammatory cells, which could account for the reduced number of inflammatory cells that adhered in the adhesion assay. When the inflammatory cells were seeded in the presence of UMSCs previously digested with Chase AC+B, there was an increase in the number of cells in the nonadherent fraction, and again all cells were viable (Fig. 6E). When the inflammatory cells were seeded in the presence of UMSCs previously treated with Hylase, there was an increase in the number of cells in the nonadherent fraction, and ∼5% presented signs of apoptosis (arrowhead in Fig. 6F).
UMSCs Present a Rich Glycocalyx That Is Up-regulated in the Presence of Inflammatory Cells
The co-culture assays between UMSCs and inflammatory cells show that UMSCs can inhibit inflammatory cell adhesion and invasion, inhibit M1 macrophage polarization, and promote inflammatory cell death. We investigated the possible changes that the UMSCs undergo when exposed to inflammatory cells to determine the mechanism by which UMSCs inhibit inflammation. Co-culture experiments were prepared with UMSCs in the bottom chamber, and inflammatory cells were seeded in transwell culture inserts with 0.44-μm pores. Immunocytochemistry revealed that UMSCs have a CS/HA-rich glycocalyx and that this glycocalyx is up-regulated in the presence of inflammatory cells, but is lost upon treatment with Chase AC+B and Hylase. Interestingly, when UMSCs were exposed to inflammatory cells, immunostaining for both CD44 and TSG6 greatly increased, with the formation of thin cellular protrusions extending into the extracellular space (Fig. 7A). These cellular protrusions present positive phalloidin staining (Fig. 8A) demonstrating the presence of F-actin cytoskeleton within these cellular protrusions. These cellular projections also showed some co-localized staining for CD44 and TSG6. Interestingly, when the UMSCs were treated with Chase AC+B prior to co-culture with the inflammatory cells, both CD44 staining and the number of CD44-positive cellular projections increased. In contrast, TSG6 staining decreased to a level similar to that of naive UMSCs (Fig. 7A). The Western blots for CD44 in Fig. 7B verify the increase in CD44 expression in UMSCs exposed to inflammatory cells and the further increase when the UMSCs were treated with Chase AC+B or Hylase prior to co-culture with the inflammatory cells (Fig. 7B).
FIGURE 7.
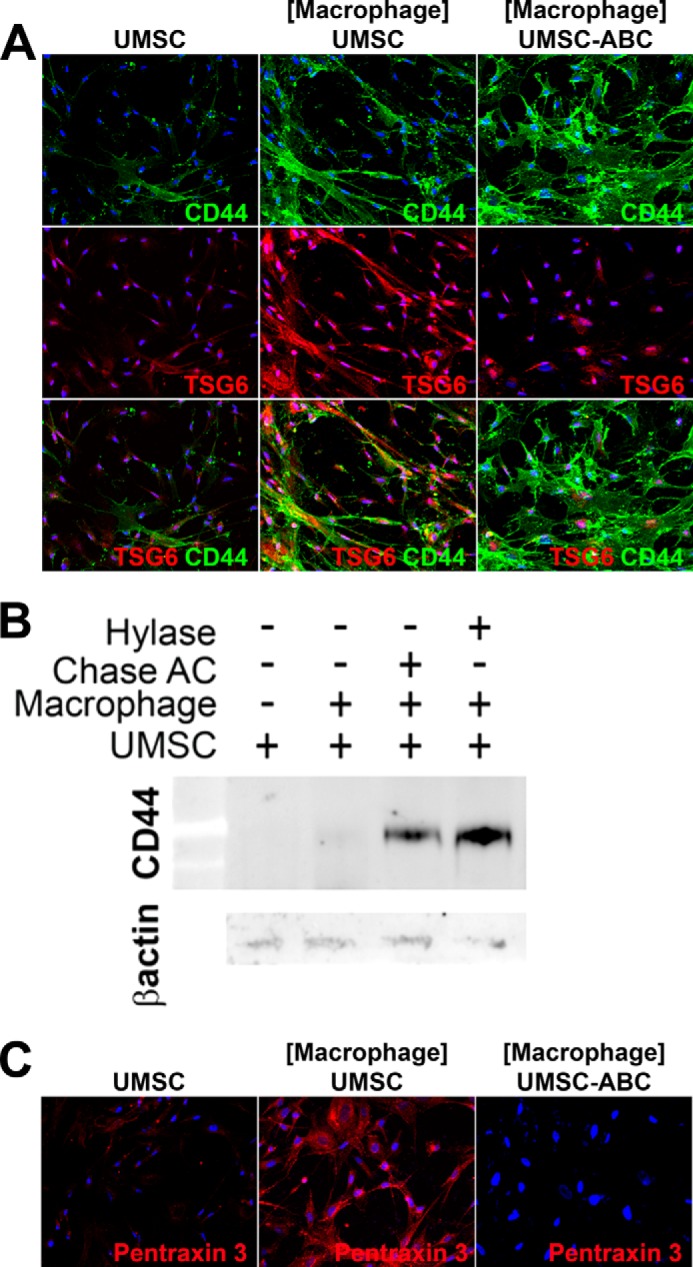
UMSCs have rich glycocalyces. UMSCs were treated or not with Chase ABC or Hylase and placed in co-culture with inflammatory cells seeded in a transwell insert with a 0.44-μm pore. A, expression and localization of TSG6 and CD44 were evaluated by immunocytochemistry. B, expression of CD44 was further evaluated by Western blotting. C, pentraxin 3 expression and localization were analyzed by immunocytochemistry.
FIGURE 8.
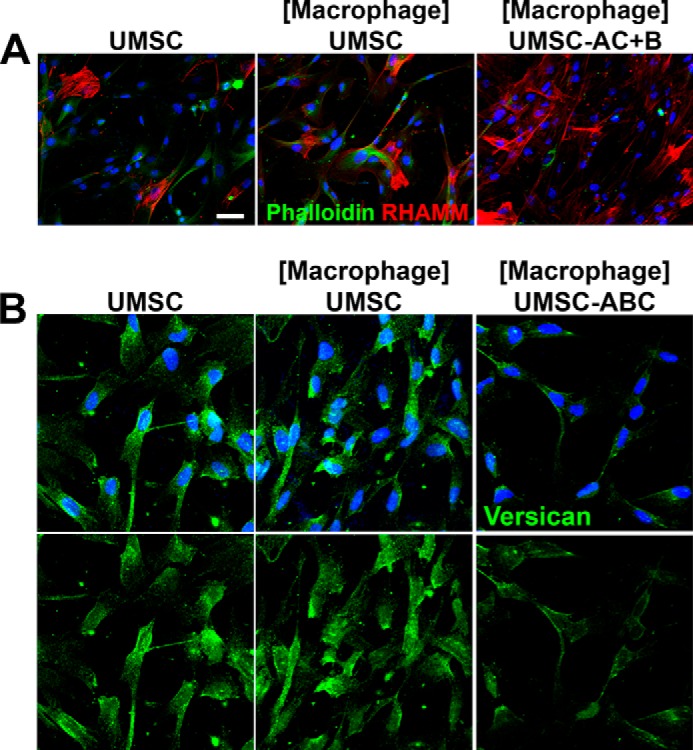
UMSCs form cellular protrusions and “cable-like” structures when exposed to inflammatory cells. UMSCs were treated or not with Chase AC+B or Hylase and placed in co-culture with inflammatory cells seeded in a transwell insert with a 0.44-μm pore. A, expression and localization of RHAMM (red) and F-actin (Phalloidin, green) were evaluated by immunocytochemistry. B, expression and localization of versican (green) were evaluated by immunocytochemistry. Scale bar, 20 μm.
Previous studies have shown that during inflammation, TSG6 catalyzes the transfer of HC from IαI to HA forming an HC-HA ECM that can promote inflammatory processes (21–23). Moreover, pentraxin 3 has been shown to be retained in this HC-HA-rich ECM and has an important role in the immunomodulatory properties of this matrix (24). Therefore, we investigated whether pentraxin 3 was present in the UMSC ECM. Interestingly, pentraxin 3 immunostaining was only present when the UMSCs were exposed to inflammatory cells, and it was also present along the UMSC cellular projections and in cable-like structures present in the ECM (Fig. 7C). Moreover, when the UMSCs were treated with Chase AC+B prior to co-culture, there was no immunostaining for pentraxin 3 (Fig. 7C). An ∼35% increase in pentraxin 3 mRNA expression was also detected in UMSCs when exposed to inflammatory cells by real time PCR (Fig. 10J).
FIGURE 10.

Immunolocalization of HA, HC1, and HC2 in UMSCs exposed to inflammatory cells. UMSCs were treated or not with Hylase or HylaseS and placed in co-culture with inflammatory cells seeded in a transwell insert with a 0.44-μm pore. A, localization of HA, HC1, and HC2 was evaluated by immunocytochemistry. UMSCs were placed in co-culture with inflammatory cells using the transwell system and left for 24 h, after which RNA was extracted from the UMSCs in the bottom chamber. Real time PCR analysis was done to verify the expression levels of HC1 (B), HC2 (C), HC3 (D), TSG6 (E), bikunin (F), RHAMM (H), HAS2 (I), and pentraxin 3 (J). G, inflammatory cells were seeded directly over UMSCs to study the adhesion of the inflammatory cells to UMSC HA-rich cables (green). Nuclei were counterstained with DAPI (blue), and * represents the nuclei of UMSCs. B–J, *, p ≤0.05.
RHAMM, an HA-binding protein that affects cell motility, was also evaluated in the UMSCs exposed to inflammatory cells. Immunocytochemistry with anti-RHAMM antibody revealed RHAMM is also present in UMSCs and is increased when they are exposed to inflammatory cells (Fig. 8A). The expression of RHAMM was also evaluated by real time PCR, which revealed a 20-fold increase in RHAMM expression in UMSCs when exposed to inflammatory cells (Fig. 10F).
Previous studies have also revealed the important role of versican when it is present in the HA ECM (25, 26). Therefore, UMSC cultures were immunostained for versican, which was detected in the ECM. An increase in versican staining was observed when the UMSCs were exposed to inflammatory cells (Fig. 8B). Interestingly, versican was also present in the cellular projections and further confirmed the increase in number of these cable-like projections.
UMSCs Present a Rich Glycocalyx That Is Up-regulated in the Presence of Inflammatory Cells
The components in the UMSC ECM were investigated by Western blot analysis of proteins extracted from UMSCs alone or after co-culture with macrophages. The protein extracts were digested with Hylase for 2 h at 37 °C to release the HA-associated matrix. Fig. 9 shows the results for Western blots analyzed for TSG6, HC1, HC2, HC3, versican, and pentraxin 3. In the absence of Hylase digestion, none of these proteins entered the gel, but they did so after digestion for both UMSCs alone and after co-culture with macrophages. The TSG6 blot showed bands at ∼35 kDa. The HC1, HC2, and HC3 blots show strong bands at ∼75 kDa that are characteristic of HCs released from HA by hyaluronidase and also a band at ∼125 kDa. HC1 and HC3 also present a band at ∼45 kDa, which may represent HC1 and HC3 degradation products that were also present in the Hylase enzyme alone (Fig. 9A, far right panels). No other bands were developed in the Hylase alone (results not shown). Anti-versican primarily revealed an ∼80-kDa species, likely representing one of the versican isoforms (which has undergone CS shedding by Hylase) and also a population at ∼130 kDa. The pentraxin 3 blot revealed primarily a characteristic ∼45-kDa band in the UMSC culture and the co-culture with macrophages. Anti-pentraxin 3 did not reveal any nonspecific band with the Hylase control (Fig. 8A). No differences were observed in the quantities of TSG6, HC1, HC3, versican, and pentraxin 3 between the UMSCs and the UMSCs exposed to the inflammatory cells. The expression and localization of HA in UMSCs were further verified by immunocytochemistry, which demonstrated an up-regulation and change to a cable-like distribution in UMSCs exposed to inflammatory cells (Fig. 8B).
FIGURE 9.

UMSCs express a rich HA/TSG6/HC/pentraxin 3 ECM. UMSCs were placed in co-culture with inflammatory cells seeded in a transwell insert with a 0.44-μm pore. Proteins were extracted and digested or not with Hylase prior to analysis by Western blotting. A, TSG6, HC1, HC2, HC3, versican, and pentraxin 3 were only detected when the protein lysate was digested with Hylase. B, HA staining (green) in UMSCs and UMSCs exposed to inflammatory cells seeded in transwell inserts. C, immunoprecipitation was done with the conditioned medium from UMSCs and UMSCs exposed to inflammatory cells with anti-HC1 and anti-HC2 and subjected to Western blotting with anti-HC1, anti-HC2, and anti-pentraxin 3. The nuclei were stained with DAPI.
The experiments using the transwell inserts require that the UMSCs and inflammatory cells undergo cross-talk through soluble factors. Therefore, to verify whether UMSCs secrete a soluble form of the HA/HC/TSG6, IP was performed from the conditioned medium from UMSCs and UMSCs co-cultured with inflammatory cells with either anti-HC1, anti-HC2, or anti-HC3. Populations of ∼80 and ∼125 kDa were present with anti-HC1, anti-HC2, and anti-HC3, indicating UMSCs secrete a soluble form of the heavy chains (Fig. 9C). An increase in the expression of HC3 was detected in the UMSCs when exposed to inflammatory cells. Moreover, pentraxin 3 was isolated by co-IP with HC1, HC2, and HC3 (Fig. 9C). No HC1, HC2, or HC3 was detected in fresh medium containing 10% FBS (data not shown). Interestingly, when the co-culture adhesion assay was performed in the presence of anti-pentraxin 3, anti-TSG-6, anti-ITI-H1, anti-ITI-H2, and anti-ITI-H3, the ability of the UMSC to inhibit inflammatory cell adhesion was ablated further confirming the involvement of the HA-HC complex (Fig. 3, A and B). The isotype controls, goat IgG and rat IgG (results not shown), and the nonrelevant control, anti-collagen I, had no effect upon the ability of UMSC to inhibit inflammatory cell adhesion (Fig. 3, A and B). The expression and localization of HC1 and HC2 were further verified in UMSCs by immunocytochemistry. Both HC1 and HC2 were detected in UMSCs. However, there was no change in distribution when UMSCs were digested with Hylase or HylaseS prior to co-culture with inflammatory cells (Fig. 10A). HC3 was also detected by immunocytochemistry presenting similar results to HC1 (results not shown). HA staining revealed an increase in both HA and HA cable-like structures in UMSCs exposed to inflammatory cells when compared solely with UMSCs (Fig. 10A). The co-culture of UMSCs and inflammatory cells in the presence of anti-pentraxin 3 had no effect upon the expression and distribution of HCs and HA (Fig. 10A). Interestingly, very weak HA was detected in UMSCs treated with Hylase and HylaseS prior to the co-culture with inflammatory cells with no cable-like HA structures present. This suggests that within the 24 h of co-culture UMSCs are not able to re-synthesize the HA-rich glycocalyx (Fig. 10A).
The expression levels of HC1, HC2, HC3, TSG6, and bikunin were also verified in the UMSCs and UMSCs exposed to inflammatory cells by real time PCR. There was a significant increase of ∼2-fold in the expression of HC1, HC3, TSG6, and bikunin when UMSCs were exposed to inflammatory cells. However, a 30% decrease was observed in HC2 expression in UMSCs exposed to inflammatory cells when compared solely with UMSCs (Fig. 10, B–F).
UMSC HA-Rich Cables Sequester Macrophages
The UMSCs affect both the adhesion and polarization of macrophages and also lead to inflammatory cell death. Here, we have shown that these events are mediated through the cable-like HA-rich matrix formed by the UMSCs when exposed to the inflammatory cells. To verify whether the HA-rich cable-like structures can directly affect macrophages, we performed a bilayer co-culture assay between the UMSCs and inflammatory cells. When inflammatory cells were directly seeded upon UMSCs (asterisks) and left for 24 h, they were detected bound to the HA-rich cables (Fig. 10G), and the HA cables appear to “engulf” the macrophages. All macrophages bound to the HA cables present a small rounded cell shape (Fig. 10G) and presented no MUM1 or IRF5 nuclear staining (results not shown). Finally, to further evaluate the increase in HA, the expression of the hyaluronan synthases were evaluated. The exposure of UMSC to inflammatory cells induced the expression of HAS2 in UMSCs, which was solely expressed below significant levels in naive UMSCs.
DISCUSSION
Recently, transplantation of UMSCs into corneal stroma has been suggested as an alternative to keratoplasty (corneal transplantation) for treating corneal disease. This novel treatment in lieu of current keratoplasty is valuable due to the fact that there is an unlimited supply of UMSCs, although the availability of donor corneas is limited and, recently, the availability is further decreasing due to the increased popularity of laser refractive surgery, which leaves the cornea unsuitable for later organ transplantation. Successful transplantation of human UMSCs has been done in the mouse cornea. However, little is understood about the mechanism by which these cells evade host rejection and differentiate in the mouse cornea. We hereby show that UMSCs can be successfully transplanted into mouse corneas after alkali burn, which is a severe wound to the cornea that leads to severe inflammation, and they survive host rejection. The UMSCs were administered 24 h after the alkali burn into an extensively inflamed cornea, and therefore, a plethora of inflammatory cells is present in the cornea at the time of UMSC administration. In this scenario, the UMSCs are capable of evading host rejection and, more importantly, modulating the inflammatory response. At early time points, such as 5 days after alkali burn, corneas transplanted with UMSCs clearly present less inflammation compared with vehicle controls, which received solely an intrastromal injection of PBS. To elucidate the role that the UMSC glycocalyx has in this host immune modulation, UMSCs were treated with Hepase or Chase AC+B prior to administration to remove cell surface HS and CS/HA, respectively. Interestingly, removal of cell surface HS prior to the administration of UMSCs had no effect on the ability of UMSCs to suppress host inflammation. In contrast, removal of CS/HA prior to administration of UMSCs ablated the ability of these cells to suppress a host inflammatory response. Therefore, UMSC cell surface CS/HA is vital for UMSCs to survive and escape host rejection.
In order for UMSCs to survive host rejection, they either actively modulate the inflammatory response or they present cell surface components that enable them to remain invisible to the host immune system. To further elucidate the mechanism by which UMSCs evade rejection and reduce the inflammatory response, UMSCs were co-cultured with inflammatory cells. To date two subtypes of macrophages have been well established as follows: M1 macrophages and M2 macrophages, which present pro-inflammatory and anti-inflammatory phenotypes, respectively (27, 28). Macrophages present an irregular branched cell shape and are classically detected by the markers CD14, CD40, CD11b, or F4/80 (29, 30). M1 macrophages present the classical macrophage cell shape and may be distinguished from the other macrophage subtypes using markers, such as IL1β, IL6, IL8, IL12, and TNFα, and they can be induced by INFγ. However, M2 macrophages are characterized by IL10 and IL13 expression and can be induced by IL4 (31). Interestingly, a mannose receptor is vital for M2 macrophage polarization (31), which demonstrates a clear role of carbohydrate moieties in macrophage differentiation and polarization. Recently, two more macrophage subtypes have been characterized, referred to as M3 macrophages and M4 macrophages (32). M4 macrophages lack CD163 and can be induced by CXCL4 (33). Macrophages are versatile and may undergo both differentiation and polarization upon subtle variations in the microenvironment (31, 34). The versatility of macrophages to shift between the M1 and M2 phenotype (reversible polarization) has been well established (35). Macrophages isolated from the peritoneal lavage adhered and differentiated into classical M1 macrophages presenting an irregular branched cell shape. When these polarized M1 macrophages were exposed to UMSCs, they assumed a rounded cell shape, and a subpopulation presented markers typical of M2 macrophages. Thus, UMSCs actively changed the polarized state of M1 macrophages into a small rounded phenotype, potentially undifferentiated macrophages (36). Moreover, UMSCs also actively inhibited the adhesion and invasion of inflammatory cells and also induced inflammatory cell death, all in a CS/HA-dependent manner. Therefore, when UMSCs are transplanted into the inflamed cornea 24 h after alkali burn, they have the ability to actively suppress and induce cell death in the inflammatory cells present in the cornea and also to inhibit the recruitment (adhesion and invasion) of more inflammatory cells to the extent that 5 days after alkali burn the corneas transplanted with UMSCs already presented less inflammation than vehicle controls. In accordance with the in vivo data, when the UMSCs were previously treated with Chase ABC, they lost the ability to modulate the inflammatory cells, thus explaining why the UMSCs treated with Chase AC+B prior to transplantation were rejected by the host immune system. Together, these data suggest that UMSCs actively regulate inflammatory cells in a CS/HA-dependent manner.
Our studies also show that UMSCs promote the maturation of T-regulatory cells, which were shown to be CD44+, CD25+, CD4+, and Foxp3−, and to secrete IL10. Previous studies have shown that HA promotes the induction of Foxp3−IL10 producing regulatory T-cells from T-cell precursors in both human and murine systems (37). Moreover, our findings suggest that these cells could have a role in inhibiting the polarization of the macrophages to the M1 phenotype. In order for T-cells to invade the surrounding ECM, they adopt an elongated morphology with a broad lamellipodium at the leading edge and a handle-like protrusion at the rear end (38). Thus, typical T-cell morphology is clear in the CD11b+ inflammatory cells after co-culture with the UMSCs. CD44+ to date is the best characterized receptor of HA, and therefore the dense HA glycocalyx synthesized by the UMSCs could have a role in the maturation and also be a substrate for the T-regulatory cells. Moreover, recent studies have shown that the viscous matrix formed by both HA and versican has an important role in T-cell sequestration and trafficking in inflamed tissues (38).
Recently, studies have shown that bone marrow-derived mesenchymal stem cells modulate inflammatory responses, and when transplanted into animal models of both acute lung injury and corneal injury, they can reduce systemic inflammation, ameliorate lung damage, and improve survival (39–42). Studies have also indicated that UMSCs modulate the immune response by secreting soluble factors creating an immunosuppressive milieu (43, 44).
When the UMSCs are exposed to the inflammatory cells, they proceed to secrete a rich ECM that contains TSG6, pentraxin 3, and versican. Moreover, the formation of “cable-like” structures can be observed when the UMSCs are exposed to inflammatory cells, and these cables become unorganized when the UMSCs are previously treated with Chase AC+B or Hylase. CS/HA are therefore essential for the formation of the UMSC inflammation-specific ECM. The disruption of the inflammation-specific ECM due to the lack of CS and HS could account for the increase in CD44 expression, which is responsible for the bridge between this unorganized ECM and the UMSC cell surface. Therefore, our results provide evidence that exposure of UMSCs to inflammatory cells induces a more organized ECM and up-regulates their synthesis of CD44, which forms the bridge between the UMSC and the HC-HA ECM (glycocalyx). Upon digestion with Chase AC+B, Chase ABC, or Hylase, this ECM is disrupted, and the UMSCs up-regulate CD44 further as a compensatory mechanism. Interestingly, the umbilical cord is considered to be one of the mammalian tissues that has the highest HA content (∼4 mg/ml) (45). Our study shows that the UMSCs produce a rich HA/TSG6/HC ECM that actively modulates the immune response. Previously, the IαI family of proteins was believed to be expressed exclusively by hepatocytes. Recently, however, extra-hepatic expression of IαI has been described (46). The amniotic membrane (AM) was shown to present a rich HA/TSG6/HC ECM, and the HCs come from IαI synthesized by both the AM epithelial and stromal cells (46). This same group recently showed that specifically the HC-HA complex in the AM confers the anti-inflammatory properties to this tissue (24). Moreover, the soluble HC-HA complex purified from the AM contained pentraxin 3, induced apoptosis of neutrophils and macrophages, and polarized the macrophages toward the M2 phenotype. Our findings are in accordance with these studies. We also provide evidence that the umbilical cord is another extra-hepatic tissue that synthesizes IαI. Therefore, the UMSCs synthesize a rich HA/TSG6/HC ECM that protects them from rejection. Moreover, the ability of the UMSCs in the transwell inserts to modulate the inflammatory cells indicates that soluble HA containing pentraxin 3 secreted by the UMSCs could thereby diffuse throughout the cornea stroma enhancing the efficiency of these cells to modulate the severe inflammatory response present after an alkali burn. Oh et al. (3) have shown that solely injecting recombinant human TSG6 into the corneas of mice following chemical burns ameliorated the inflammatory response and improved the corneal opacity. Moreover, this same group was able to reduce the rejection of mouse allogeneic corneal transplants by intravenous administration of mesenchymal stem cells, which were shown to be trapped primarily in the lungs and to increase the levels of TSG6 (47). Dyer et al. (48) have recently shown that free TSG-6 impairs both the binding of CXCL8 to cell surface GAGs and its transport across the endothelium, thereby impeding the CXCL8 gradient formation required for neutrophil chemotaxis and extravasation. Therefore, free TSG6 expressed by the UMSC could also be playing a role in suppressing the inflammatory response. It is of interest to note that fibroblasts and other stem cells, including hematopoietic stem cells and embryonic stem cells, are rejected when transplanted into mouse corneas.3 The reasons for the rejection of these cell types in contrast to UMSCs are not known. Presumably, it may be due to the failure of the cells to synthesize a similar glycocalyx to that synthesized by the UMSCs.
To our knowledge, this is the first study implicating that UMSCs secrete a CS/HA-dense ECM that enables these cells to modulate the host immune response. Taken together, our data show that the xenograft of human UMSCs into mouse corneas suppresses inflammatory cells and thereby regulates the immune response to evade rejection. During inflammatory processes, UMSCs secrete a CS/HA-rich ECM that mediates the mechanism by which UMSCs regulate the inflammatory cells. These data further support that allografting human UMSCs into human corneas can have good potential for successfully treating human corneal clouding. Moreover, this specific CS/HA-rich ECM secreted by UMSCs is likely to be a valuable resource for other therapeutic venues.
Acknowledgments
We thank Shao Hsuan Chang for valuable technical assistance. The RNAseq was supported in part by Center for Environmental Genetics, NIEHS, National Institutes of Health Grant P30-ES006096.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 EY021768 from NEI. This work was also supported by Research to Prevent Blindness, Ohio Lions Eye Research Foundation, and by The Hyaluronan Matrices in Vascular Pathologies funded by National Institutes of Health Grant P01 HL107147 from NHLBI.
V. J. Coulson-Thomas, T. F. Gesteira, V. Hascall, and W. Kao, unpublished observations.
- UMSC
- umbilical cord mesenchymal stem cell
- GAG
- glycosaminoglycan
- HA
- hyaluronan
- HC
- heavy chain
- EBSS
- Earle's balanced salt solution
- α-MEM
- α-minimal essential medium
- ECM
- extracellular matrix
- CS
- chondroitin sulfate
- AM
- amniotic membrane
- DS
- dermatan sulfate
- HS
- heparan sulfate
- IP
- immunoprecipitation
- IαI
- inter-α-trypsin inhibitor.
REFERENCES
- 1. Liu H., Zhang J., Liu C. Y., Wang I. J., Sieber M., Chang J., Jester J. V., Kao W. W. (2010) Cell therapy of congenital corneal diseases with umbilical mesenchymal stem cells: lumican null mice. PLoS One 5, e10707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coulson-Thomas V. J., Caterson B., Kao W. W. (2013) Transplantation of human umbilical mesenchymal stem cells cures the corneal defects of mucopolysaccharidosis VII mice. Stem Cells 31, 2116–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oh J. Y., Roddy G. W., Choi H., Lee R. H., Ylöstalo J. H., Rosa R. H., Jr., Prockop D. J. (2010) Anti-inflammatory protein TSG-6 reduces inflammatory damage to the cornea following chemical and mechanical injury. Proc. Natl. Acad. Sci. U.S.A. 107, 16875–16880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de la Motte C. A., Drazba J. A. (2011) Viewing hyaluronan: imaging contributes to imagining new roles for this amazing matrix polymer. J. Histochem. Cytochem. 59, 252–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bollyky P. L., Lord J. D., Masewicz S. A., Evanko S. P., Buckner J. H., Wight T. N., Nepom G. T. (2007) Cutting edge: high molecular weight hyaluronan promotes the suppressive effects of CD4+CD25+ regulatory T cells. J. Immunol. 179, 744–747 [DOI] [PubMed] [Google Scholar]
- 6. Bollyky P. L., Falk B. A., Wu R. P., Buckner J. H., Wight T. N., Nepom G. T. (2009) Intact extracellular matrix and the maintenance of immune tolerance: high molecular weight hyaluronan promotes persistence of induced CD4+CD25+ regulatory T cells. J. Leukocyte Biol. 86, 567–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Potter-Perigo S., Johnson P. Y., Evanko S. P., Chan C. K., Braun K. R., Wilkinson T. S., Altman L. C., Wight T. N. (2010) Polyinosine-polycytidylic acid stimulates versican accumulation in the extracellular matrix promoting monocyte adhesion. Am. J. Respir. Cell Mol. Biol. 43, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lesley J., Howes N., Perschl A., Hyman R. (1994) Hyaluronan binding function of CD44 is transiently activated on T cells during an in vivo immune response. J. Exp. Med. 180, 383–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang D., Liang J., Noble P. W. (2007) Hyaluronan in tissue injury and repair. Annu. Rev. Cell Dev. Biol. 23, 435–461 [DOI] [PubMed] [Google Scholar]
- 10. Culley F. J., Fadlon E. J., Kirchem A., Williams T. J., Jose P. J., Pease J. E. (2003) Proteoglycans are potent modulators of the biological responses of eosinophils to chemokines. Eur. J. Immunol. 33, 1302–1310 [DOI] [PubMed] [Google Scholar]
- 11. Cantor J. O., Cerreta J. M., Armand G., Osman M., Turino G. M. (1999) The pulmonary matrix, glycosaminoglycans and pulmonary emphysema. Connect. Tissue Res. 40, 97–104 [DOI] [PubMed] [Google Scholar]
- 12. Negrini D., Passi A., Moriondo A. (2008) The role of proteoglycans in pulmonary edema development. Intensive Care Med. 34, 610–618 [DOI] [PubMed] [Google Scholar]
- 13. Jiang D., Liang J., Noble P. W. (2010) Regulation of non-infectious lung injury, inflammation, and repair by the extracellular matrix glycosaminoglycan hyaluronan. Anat. Rec. 293, 982–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoneda M., Suzuki S., Kimata K. (1990) Hyaluronic acid associated with the surfaces of cultured fibroblasts is linked to a serum-derived 85-kDa protein. J. Biol. Chem. 265, 5247–5257 [PubMed] [Google Scholar]
- 15. Zhao M., Yoneda M., Ohashi Y., Kurono S., Iwata H., Ohnuki Y., Kimata K. (1995) Evidence for the covalent binding of SHAP, heavy chains of inter-α-trypsin inhibitor, to hyaluronan. J. Biol. Chem. 270, 26657–26663 [DOI] [PubMed] [Google Scholar]
- 16. Milner C. M., Higman V. A., Day A. J. (2006) TSG-6: a pluripotent inflammatory mediator? Biochem. Soc. Trans. 34, 446–450 [DOI] [PubMed] [Google Scholar]
- 17. Milner C. M., Tongsoongnoen W., Rugg M. S., Day A. J. (2007) The molecular basis of inter-α-inhibitor heavy chain transfer on to hyaluronan. Biochem. Soc. Trans. 35, 672–676 [DOI] [PubMed] [Google Scholar]
- 18. Rugg M. S., Willis A. C., Mukhopadhyay D., Hascall V. C., Fries E., Fülöp C., Milner C. M., Day A. J. (2005) Characterization of complexes formed between TSG-6 and inter-α-inhibitor that act as intermediates in the covalent transfer of heavy chains onto hyaluronan. J. Biol. Chem. 280, 25674–25686 [DOI] [PubMed] [Google Scholar]
- 19. Dietrich C. P., McDuffie N. M., Sampaio L. O. (1977) Identification of acidic mucopolysaccharides by agarose gel electrophoresis. J. Chromatogr. 130, 299–304 [DOI] [PubMed] [Google Scholar]
- 20. Kamentsky L., Jones T. R., Fraser A., Bray M. A., Logan D. J., Madden K. L., Ljosa V., Rueden C., Eliceiri K. W., Carpenter A. E. (2011) Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27, 1179–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanggaard K. W., Sonne-Schmidt C. S., Krogager T. P., Kristensen T., Wisniewski H. G., Thøgersen I. B., Enghild J. J. (2008) TSG-6 transfers proteins between glycosaminoglycans via a Ser28-mediated covalent catalytic mechanism. J. Biol. Chem. 283, 33919–33926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanggaard K. W., Sonne-Schmidt C. S., Krogager T. P., Lorentzen K. A., Wisniewski H. G., Thøgersen I. B., Enghild J. J. (2008) The transfer of heavy chains from bikunin proteins to hyaluronan requires both TSG-6 and HC2. J. Biol. Chem. 283, 18530–18537 [DOI] [PubMed] [Google Scholar]
- 23. Jessen T. E., Ødum L. (2003) Role of tumour necrosis factor stimulated gene 6 (TSG-6) in the coupling of inter-α-trypsin inhibitor to hyaluronan in human follicular fluid. Reproduction 125, 27–31 [DOI] [PubMed] [Google Scholar]
- 24. He H., Zhang S., Tighe S., Son J., Tseng S. C. (2013) Immobilized heavy chain-hyaluronic acid polarizes lipopolysaccharide-activated macrophages toward M2 phenotype. J. Biol. Chem. 288, 25792–25803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang M. Y., Chan C. K., Braun K. R., Green P. S., O'Brien K. D., Chait A., Day A. J., Wight T. N. (2012) Monocyte-to-macrophage differentiation: synthesis and secretion of a complex extracellular matrix. J. Biol. Chem. 287, 14122–14135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keller K. E., Aga M., Bradley J. M., Kelley M. J., Acott T. S. (2009) Extracellular matrix turnover and outflow resistance. Exp. Eye Res. 88, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biswas S. K., Chittezhath M., Shalova I. N., Lim J. Y. (2012) Macrophage polarization and plasticity in health and disease. Immunol. Res. 53, 11–24 [DOI] [PubMed] [Google Scholar]
- 28. Murray P. J., Wynn T. A. (2011) Obstacles and opportunities for understanding macrophage polarization. J. Leukocyte Biol. 89, 557–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamagami S., Usui T., Amano S., Ebihara N. (2005) Bone marrow-derived cells in mouse and human cornea. Cornea 24, S71–SS74 [DOI] [PubMed] [Google Scholar]
- 30. Ji R. C. (2012) Macrophages are important mediators of either tumor- or inflammation-induced lymphangiogenesis. Cell. Mol. Life Sci. 69, 897–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stein M., Keshav S. (1992) The versatility of macrophages. Clin. Exp. Allergy 22, 19–27 [DOI] [PubMed] [Google Scholar]
- 32. Liu G., Yang H. (2013) Modulation of macrophage activation and programming in immunity. J. Cell. Physiol. 228, 502–512 [DOI] [PubMed] [Google Scholar]
- 33. Gleissner C. A. (2012) Macrophage phenotype modulation by CXCL4 in atherosclerosis. Front. Physiol. 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wolfs I. M., Donners M. M., de Winther M. P. (2011) Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb. Haemost. 106, 763–771 [DOI] [PubMed] [Google Scholar]
- 35. Porcheray F., Viaud S., Rimaniol A. C., Léone C., Samah B., Dereuddre-Bosquet N., Dormont D., Gras G. (2005) Macrophage activation switching: an asset for the resolution of inflammation. Clin. Exp. Immunol. 142, 481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim J., Hematti P. (2009) Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp. Hematol. 37, 1445–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bollyky P. L., Evanko S. P., Wu R. P., Potter-Perigo S., Long S. A., Kinsella B., Reijonen H., Guebtner K., Teng B., Chan C. K., Braun K. R., Gebe J. A., Nepom G. T., Wight T. N. (2010) Th1 cytokines promote T-cell binding to antigen-presenting cells via enhanced hyaluronan production and accumulation at the immune synapse. Cell. Mol. Immunol. 7, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evanko S. P., Potter-Perigo S., Bollyky P. L., Nepom G. T., Wight T. N. (2012) Hyaluronan and versican in the control of human T-lymphocyte adhesion and migration. Matrix Biol. 31, 90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gupta N., Su X., Popov B., Lee J. W., Serikov V., Matthay M. A. (2007) Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J. Immunol. 179, 1855–1863 [DOI] [PubMed] [Google Scholar]
- 40. Xu J., Woods C. R., Mora A. L., Joodi R., Brigham K. L., Iyer S., Rojas M. (2007) Prevention of endotoxin-induced systemic response by bone marrow-derived mesenchymal stem cells in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L131–L141 [DOI] [PubMed] [Google Scholar]
- 41. Rojas M., Xu J., Woods C. R., Mora A. L., Spears W., Roman J., Brigham K. L. (2005) Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am. J. Respir. Cell Mol. Biol. 33, 145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu J., Qu J., Cao L., Sai Y., Chen C., He L., Yu L. (2008) Mesenchymal stem cell-based angiopoietin-1 gene therapy for acute lung injury induced by lipopolysaccharide in mice. J. Pathol. 214, 472–481 [DOI] [PubMed] [Google Scholar]
- 43. Fong C. Y., Subramanian A., Gauthaman K., Venugopal J., Biswas A., Ramakrishna S., Bongso A. (2012) Human umbilical cord Wharton's jelly stem cells undergo enhanced chondrogenic differentiation when grown on nanofibrous scaffolds and in a sequential two-stage culture medium environment. Stem Cell Rev. 8, 195–209 [DOI] [PubMed] [Google Scholar]
- 44. Lu L. L., Liu Y. J., Yang S. G., Zhao Q. J., Wang X., Gong W., Han Z. B., Xu Z. S., Lu Y. X., Liu D., Chen Z. Z., Han Z. C. (2006) Isolation and characterization of human umbilical cord mesenchymal stem cells with hematopoiesis-supportive function and other potentials. Haematologica 91, 1017–1026 [PubMed] [Google Scholar]
- 45. Weissmann B., Meyer K., Sampson P., Linker A. (1954) Isolation of oligosaccharides enzymatically produced from hyaluronic acid. J. Biol. Chem. 208, 417–429 [PubMed] [Google Scholar]
- 46. Zhang S., He H., Day A. J., Tseng S. C. (2012) Constitutive expression of inter-α-inhibitor (IαI) family proteins and tumor necrosis factor-stimulated gene-6 (TSG-6) by human amniotic membrane epithelial and stromal cells supporting formation of the heavy chain-hyaluronan (HC-HA) complex. J. Biol. Chem. 287, 12433–12444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Oh J. Y., Lee R. H., Yu J. M., Ko J. H., Lee H. J., Ko A. Y., Roddy G. W., Prockop D. J. (2012) Intravenous mesenchymal stem cells prevented rejection of allogeneic corneal transplants by aborting the early inflammatory response. Mol. Ther. 20, 2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dyer D. P., Thomson J. M., Hermant A., Jowitt T. A., Handel T. M., Proudfoot A. E., Day A. J., Milner C. M. (2014) TSG-6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J. Immunol. 192, 2177–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]