

Background: How the mitotic checkpoint is silenced is not fully understood.
Results: Interference with TRIP13 AAA-ATPase causes delayed activation of the anaphase-promoting complex/cyclosome and stalled metaphase-to-anaphase transition.
Conclusion: TRIP13 is a novel mitotic checkpoint-silencing protein.
Significance: TRIP13 overexpression may lead to premature chromosome segregation and chromosomal instability.
Keywords: ATPases Associated with Diverse Cellular Activities (AAA); Checkpoint Control; E3 Ubiquitin Ligase; Mitosis; Protein Complex; MAD2; Mitotic Checkpoint Complex (MCC); TRIP13; Anaphase-promoting Complex/Cyclosome (APC/C),; p31comet
Abstract
The mitotic checkpoint (or spindle assembly checkpoint) is a fail-safe mechanism to prevent chromosome missegregation by delaying anaphase onset in the presence of defective kinetochore-microtubule attachment. The target of the checkpoint is the E3 ubiquitin ligase anaphase-promoting complex/cyclosome. Once all chromosomes are properly attached and bioriented at the metaphase plate, the checkpoint needs to be silenced. Previously, we and others have reported that TRIP13 AAA-ATPase binds to the mitotic checkpoint-silencing protein p31comet. Here we show that endogenous TRIP13 localizes to kinetochores. TRIP13 knockdown delays metaphase-to-anaphase transition. The delay is caused by prolonged presence of the effector for the checkpoint, the mitotic checkpoint complex, and its association and inhibition of the anaphase-promoting complex/cyclosome. These results suggest that TRIP13 is a novel mitotic checkpoint-silencing protein. The ATPase activity of TRIP13 is essential for its checkpoint function, and interference with TRIP13 abolished p31comet-mediated mitotic checkpoint silencing. TRIP13 overexpression is a hallmark of cancer cells showing chromosomal instability, particularly in certain breast cancers with poor prognosis. We suggest that premature mitotic checkpoint silencing triggered by TRIP13 overexpression may promote cancer development.
Introduction
TRIP13 (thyroid hormone receptor interacting protein 13) was first identified in a yeast two-hybrid screening as a protein fragment that associated with thyroid hormone receptor in a hormone-independent fashion (1). The full-length gene was later renamed 16E1-BP when it was found to interact with HPV16 E1 protein (2). 16E1-BP contains both Walker A and Walker B signature motifs for ATPases, which were missing in the original TRIP13 sequences (Fig. 1A). The functional implication of either interaction is still unclear. Recent work nevertheless found that mouse TRIP13, as well as its orthologs in Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and rice, participates in checkpoint control of defective meiotic recombination (3–9).
FIGURE 1.

TRIP13 is a novel kinetochore protein. A, schematic diagram of the structure of human TRIP13. The AAA-ATPase domain is between residues 171 and 323; the Walker A motif is residues 179–186; and the Walker B motif is residues 248–253. The original TRIP13 sequence lacks residues after residue 173 with a different C terminus and most likely represents a cloning artifact. B, ∼50 μg of HeLa cell lysates prepared either from mock (control) or GFP-TRIP13-transfected cells were loaded together with 5–40 ng of purified recombinant His6-tagged TRIP13. The blot was probed with anti-TRIP13 antibody first and then reprobed with anti-GFP antibody. As can be seen, the antibody recognizes recombinant His-tagged TRIP13, endogenous TRIP13 (at ∼49 kDa), and GFP-TRIP13 fusion protein (∼75 kDa) in HeLa cell lysates. C, control and TRIP13 siRNAs (siRNA#1 and siRNA#2) were transfected at 100 nm final concentrations, and cells were harvested 48 h later. ∼50 μg of cell lysates were loaded and probed with anti-TRIP13 antibody. MAD2 was also probed as a loading control. The specificity of the antibody was confirmed because TRIP13 siRNAs significantly reduced the band intensities corresponding to endogenous TRIP13. D, ∼50 μg of cell lysates from HeLa, MCF10A, and several breast cancer cell lines were probed with anti-TRIP13 and anti-tubulin antibodies followed by IR-800 anti-rabbit and AlexaFluor 680 anti-mouse secondary antibodies in a two-color Odyssey imaging system. The bottom panel shows the ratios of TRIP13/tubulin based on three experiments. E, TRIP13 is enriched at kinetochores in mitotic MCF7 cells. Immunofluorescence of a prometaphase cell was stained with homemade anti-TRIP13 antibody. The centromeres/kinetochores are indicated by anti-MAD2 and anti-centromere autoimmune serum (ACA) staining. DNA is counterstained with DAPI. Shown are maximum projections of z-series confocal images. Bar, 10 μm. Error bars, S.E.
TRIP13 is also expressed in many somatic tissues, and the TRIP13 gene was listed among multiple breast cancer “signatures” due to its overexpression in transcriptional profiling of breast cancer samples (10, 11). The Cancer Genome Atlas project also confirmed TRIP13 overexpression in a large number of breast cancer samples (e.g. 4.4-fold increase in 392 invasive ductal carcinomas versus 61 normal breast tissues) (12). TRIP13 overexpression is associated with poor prognosis in breast cancer patients (10, 11). Interestingly, TRIP13 also ranks as one of the top genes in association with chromosomal instability (CIN)2 in human cancers but has no functional annotations (13).
The mitotic checkpoint is a crucial mechanism to maintain genomic stability that prevents chromosome missegregation when defective kinetochore-microtubule attachment is detected in cells (14). The target of the checkpoint is the E3 ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C), whose activity is essential for anaphase onset through proteasome-mediated destruction of securin and cyclin B (15). When activated, the mitotic checkpoint leads to assembly of the mitotic checkpoint complex (MCC). The MCC, composed of BUBR1, BUB3, CDC20, and closed MAD2 (C-MAD2), directly binds and inhibits APC/C (16–20). C-MAD2 and open MAD2 (O-MAD2) are two distinct native folding states of MAD2. During checkpoint activation, the intracellular concentration of C-MAD2 increases due to O to C conversion catalyzed by MAD1·C-MAD2 heterotetramers localized at unattached kinetochores, promoting MCC assembly (21, 22). Once all sister chromatids have been properly attached with spindle microtubules and bioriented at the metaphase plate, the checkpoint is silenced. The silencing involves events at kinetochores and in the cytoplasm (14, 23, 24). The major events include stopping the generation of C-MAD2, dissociation of MCC from APC/C, and MCC disassembly (14, 23, 24). A likely crucial but long overlooked reaction that underlies all of these events might be the C to O conformational change of MAD2, which must happen during mitotic exit (25, 26). p31comet is a mitotic checkpoint-silencing protein that can block C-MAD2 generation and help MCC disassembly (27–30). MCC disassembly mediated by p31comet employs its structural mimicry to and physical interaction with C-MAD2 but also depends on ATP hydrolysis (28, 29). How ATP hydrolysis is coupled with p31comet activity is not fully understood.
Through data mining, we recently discovered that TRIP13 co-expresses with a group of core centromere/kinetochore components and experimentally demonstrated that GFP-TRIP13 localizes at MAD2-positive kinetochores in HeLa cells (31). Interestingly, TRIP13 also directly interacts with p31comet, confirming and extending previous results obtained in large scale protein-protein interaction analyses in human and mouse cells (31–34). TRIP13 belongs to the family of ATPases associated with diverse cellular activities (AAA-ATPases), and many AAA-ATPases are involved in unfolding and folding protein substrates or assembling and disassembling macromolecular complexes (35). We are intrigued by the hypothesis that TRIP13 coordinates with p31comet to drive mitotic checkpoint silencing by disassembling protein complexes required for the mitotic checkpoint, such as the MCC and the MAD1·C-MAD2 catalyst. Here we report the results that confirmed our hypothesis.
EXPERIMENTAL PROCEDURES
DNA Constructs and Transfection
TRIP13 cDNA (transcript variant 1) was PCR-amplified and cloned into pENTR-TOPO vector (Invitrogen) and then recombined into different Gateway destination vectors for expression in Escherichia coli or mammalian cells. To make the K185T Walker A mutant, site-directed mutagenesis was performed using the QuikChange kit from Stratagene. A TRIP13 shRNA targeting CATTATACCAACTGAGAAA in the 3′-untranslated region of TRIP13 was cloned in pSUPER vector. The shRNA construct was transfected together with pBabe-puromycin in a ratio of 10:1, and puromycin-resistant cells were selected as described previously (36). A doxycycline-inducible Tet-on construct expressing TRIP13-Strep tag fusion protein was prepared in the pLenti-CMVtight Hygro DEST vector (Addgene (37)). DNA transfection was carried out using TransIT-LT1 reagent (Mirus) following the manufacturer's instructions.
RNA Interference
Small interfering RNAs were synthesized by Dharmacon. One sequence (siRNA 1) targeting CATTATACCAACTGAGAAA in the 3′-untranslated region and another (siRNA 2) targeting ACAAGAACGTCAACAGCAA within the open reading frame were found to knock down TRIP13 protein expression efficiently. Mostly results with the first siRNA were shown in this report. siRNA was transfected using Oligofectamine (Invitrogen), following the manufacturer's instructions.
Recombinant Protein and Antibodies
His6-tagged TRIP13 was expressed in E. coli BL21 (DE3) Codon Plus RIPL (Stratagene) and purified using cobalt beads (Clontech). The same protein was used to immunize a rabbit for antiserum. Anti-TRIP13 antibody was purified through an affinity column containing the antigen cross-linked to Affi-Gel 10 gel (Bio-Rad). Custom-made rabbit anti-p31comet, anti-MAD1, and anti-MAD2 antibodies were ordered from Immunosoft China and also affinity-purified. Information about all primary antibodies used in this study can be provided upon request.
Cell Lines and Cell Culture
HeLaM cells were maintained in DMEM with 10% fetal bovine serum (FBS) at 37 °C in 5% CO2 (17). MCF7 cells stably expressing mRFP-H2A were established by transfection with a pcDNA3-mRFP-H2A construct (a gift from Dr. Elena Pugacheva, West Virginia University) and selection against 100 μg/ml Zeocin. The MCF7 Tet-on Advanced cell line was purchased from Clontech and maintained in DMEM with 10% Tet system-approved FBS and 300 μg/ml G418. Cells that inducibly express a TRIP13-Strep fusion protein were established by transfecting MCF7 Tet-on cells with a pLenti-CMVtight Hygro DEST construct (Addgene (37)) and selection against G418 and 150 μg/ml hygromycin. When induced, doxycycline was added to 10 μg/ml final concentration. Cell synchronization was performed as described before (17, 36, 38) and detailed in the figure legends.
Soft Agar Assay
The assay was performed following an online protocol from the Provost and Wallert laboratories (Minnesota State University, Moorhead, MN) with slight modifications. MCF7 cells treated with control or TRIP13 siRNA for 48 h were plated in triplicates in 6-well plates for soft agar assays. For each well, the base contained 1.5 ml of 0.5% agarose in 1× DMEM, and the top contained 104 cells in 1.5 ml of 0.35% agarose in DMEM. The wells were layered with 0.5 ml of DMEM medium every 3 days, and the colonies with a diameter of >100 μm were counted 21 days later after staining with 0.005% Crystal Violet.
Cell Fractionation
Cells were harvested, washed twice with 1× PBS, and resuspended in 10 volumes of RSB (10 mm Tris-HCl, pH 7.4, 1.5 mm MgCl2, 10 mm NaCl). After incubation on ice and examination of the swollen cells under a microscope, cells were subjected to several quick upward strokes of the pestle in a prechilled Dounce homogenizer. The homogenized cell suspension was centrifuged at 1000 × g for 3 min to obtain the nuclear fraction from the pellet.
Cell Lysates, Immunoblotting, and Immunoprecipitation
These procedures were mostly performed as described previously (36, 38). The Western blot in Fig. 1D was probed with fluorescence-labeled secondary antibodies and scanned with the Odyssey infrared imager system (LI-COR Biosciences).
APC/C Activation Assay Using Concentrated HeLa Mitotic Extracts
The extracts were prepared and assayed as described before (38). Immunodepletion of the extracts was carried out by incubation with Affiprep-Protein A beads (Bio-Rad) prebound with either anti-TRIP13 antibody or an equal amount of control rabbit IgG (∼1 μg of antibody/μl of beads). The supernatants were collected after centrifugation. Two rounds of depletion were routinely performed to remove endogenous TRIP13 from the extracts.
Immunofluorescence
Cells grown on number 1.5 coverslips were simultaneously fixed and extracted in freshly prepared 3.5% paraformaldehyde containing 0.5% Triton X-100 for 10 min and then blocked with KB (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mg/ml BSA) for >5 min prior to immunofluorescence. The coverslips were incubated with each primary and secondary antibody sequentially, all at 37 °C for 30 min in a wet chamber. The secondary antibodies were the Alexa Fluor series from Molecular Probes. The stained coverslips were mounted on slides using Vectashield mounting medium (Vector Laboratories) and imaged on a Leica TCS SP8 confocal microscope with a ×63 numerical aperture 1.40 objective. Usually, z-stacks of 0.5 μm were collected, and maximum projections or single focal planes were presented. All images for comparison were acquired and processed in the same manner.
Time Lapse Live Cell Imaging
MCF7 cells stably expressing mRFP-H2A were grown in 6-well plates, transfected with control or TRIP13 siRNA, synchronized by double thymidine block, and imaged ∼8 h after release from thymidine block. Before imaging, cells were changed into HEPES-buffered and phenol red-free DMEM, and OxyFluor (Oxyrase) was added to 0.3 units/ml final concentration. Cells were then covered with mineral oil, and images were collected on an automated Olympus IX-81 microscope using a ×40 numerical aperture 0.60 LWD (long working distance) (WD 2.8) objective, whereas cells were maintained at 37 °C in a heated chamber. Single-plane images were acquired for ∼8 h at 4- or 6-min intervals at multiple positions using a CoolSNAP HQ2 (Photometrics) camera with 2 × 2 binning.
Statistical Analysis
Statistical analyses were performed, and graphs were generated using Prism 5 software (GraphPad Software).
RESULTS
TRIP13 Is a Kinetochore Protein
We previously demonstrated that GFP-TRIP13 co-localizes with MAD2 at kinetochores in HeLa cells (31). To examine subcellular localization of endogenous TRIP13, we prepared an anti-TRIP13 antibody that specifically recognizes TRIP13 (Fig. 1, B and C). In HeLa cells, immunofluorescence using the anti-TRIP13 antibody found that endogenous TRIP13 only appeared at kinetochores that also stained positive for MAD1 in prometaphase cells, and no kinetochore signals could be detected on aligned chromosomes in metaphase cells or in anaphase cells (data not shown). As noted for GFP-TRIP13, the kinetochore localization of TRIP13 does not depend on microtubules because the kinetochore signals persisted in nocodazole-treated cells (31) (data not shown). TRIP13 also had diffuse distribution in the cytoplasm. RNAi treatment profoundly reduced TRIP13 immunofluorescence signals at kinetochores and in the cytoplasm (data not shown). Because TRIP13 overexpression is a “signature” of breast cancers, we also probed TRIP13 protein expression among different breast cancer cell lines. MCF7 showed a significantly elevated TRIP13/tubulin ratio as compared with non-tumorigenic MCF10A cells (1.81 ± 0.36-fold higher, n = 3, p < 0.05, unpaired Student's t test) (Fig. 1D). Immunofluorescence using the anti-TRIP13 antibody clearly demonstrated that endogenous TRIP13 also localizes at kinetochores in prometaphase MCF7 cells (Fig. 1E).
TRIP13 Knockdown Delays Metaphase-to-anaphase Transition
TRIP13 was previously shown to directly interact with the mitotic checkpoint-silencing protein p31comet (31–34). Together with the finding that TRIP13 is a kinetochore protein, this suggests a possible role of TRIP13 in silencing the mitotic checkpoint. To test this hypothesis, mitosis progression of an MCF7-mRFP-H2A stable cell line was compared between control and TRIP13 siRNA-treated cells by time lapse microscopy. TRIP13 knockdown significantly prolonged the metaphase-to-anaphase transition, from ∼23 to ∼51 min (p < 0.05, Student's t test, n = 50), while having little impact on the progression from nuclear envelope breakdown to metaphase (Fig. 2, A and B). Furthermore, co-transfection with either wild type GFP-TRIP13 or its catalytically inactive mutant (lacking ATPase activity due to K185T substitution at the Walker A motif) indicated that only wild type TRIP13 could restore the metaphase-to-anaphase transition duration to that in control cells (Fig. 2C). The results were comparable with those obtained using p31comet siRNA in similar live cell imaging experiments (30, 39). The combined results strongly argued for a role of TRIP13 in mitotic checkpoint silencing and also indicated that TRIP13 ATPase activity is essential for its checkpoint silencing function.
FIGURE 2.
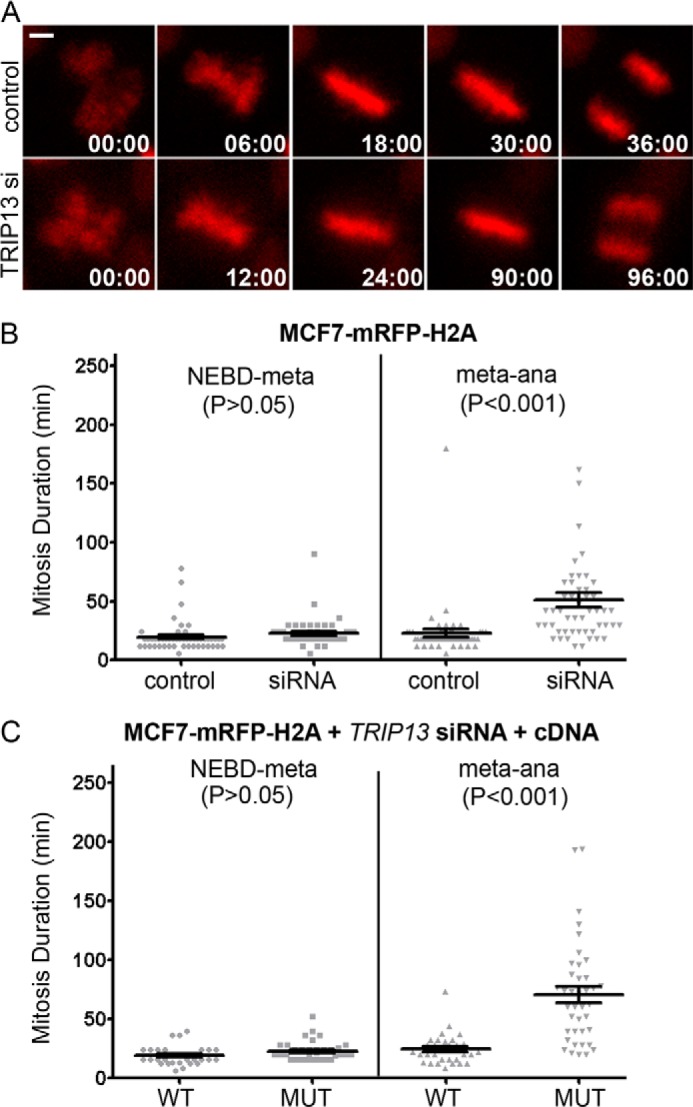
TRIP13 affects mitosis progression in MCF7 cells. A, still images of time lapse movies of MCF7-mRFP-H2A cells going through mitosis 48 h after control or TRIP13 siRNA transfection. The time stamps show minutes:seconds. The frame first showing nuclear envelope breakdown (NEBD) is set as time 0. Bar, 10 μm. B, comparison of mitosis duration (from nuclear envelope breakdown to metaphase and from metaphase to anaphase onset) in control or TRIP13 siRNA-transfected cells (Student's t test, n = 50). Bars, mean ± S.E. (error bars). C, comparison of mitosis duration of cells co-transfected with TRIP13 siRNA and GFP-TRIP13WT or TRIP13K185T.
TRIP13 Is Required for Timely Disassembly of the MCC and MCC·APC/c Complexes
To further analyze the mechanisms underlying delayed mitotic exit in TRIP13-depleted cells, we conducted mechanistic studies using concentrated HeLa mitotic extracts that recapitulated APC/C activation or mitotic checkpoint silencing in vitro when supplemented with ubiquitin, E1, E2, and an ATP regeneration system (17, 38, 40, 41). When TRIP13 was immunodepleted from the extract, degradation of cyclin B was delayed for at least 1 h (Fig. 3A). When recombinant wild type TRIP13 was added back to the immunodepleted extract, the degradation of cyclin B was largely restored, whereas the catalytically inactive K185T mutant added at the same concentration could not rescue the delay in cyclin B degradation (Fig. 3A). Moreover, delayed cyclin B degradation was also observed when endogenous TRIP13 was neutralized by adding the antibody directly into the mitotic extract or when extracts prepared from mitotic cells after TRIP13 siRNA knockdown were used (Fig. 3, B and C). Taken together, the above results indicated that interference with TRIP13 protein level or ATPase activity causes a delay in cyclin B degradation, which may in turn result in slowed mitosis progression, as seen in Fig. 2.
FIGURE 3.

Interference with TRIP13 in HeLa mitotic extracts delays cyclin B degradation. A, mitotic extracts prepared from nocodazole-treated checkpoint-active HeLa cells were subjected to either control IgG (mock) or anti-TRIP13 antibody immunodepletion. The extracts were then incubated with or without adding back recombinant His-tagged TRIP13 proteins. WT, wild type; MUT, K185T Walker A mutant. Samples were taken at the indicated minutes and analyzed by immunoblotting. APC/C activity was evaluated by degradation of endogenous cyclin B1. B, ∼5 μg of either control IgG or anti-TRIP13 antibody were added to the HeLa mitotic extract, and the extracts were then incubated and assayed as in A. The IgG and endogenous TRIP13 bands are indicated by arrows. C, the mitotic extracts were prepared from nocodazole-treated HeLa cells that had been transfected with either control or TRIP13 siRNA and assayed as in A. ATM was probed as a loading control.
Delay in cyclin B degradation was also detected when TRIP13 shRNA-transfected cells were released from nocodazole arrest (Fig. 4A). BUBR1 immunoprecipitation of cell lysates prepared at different times after release indicated prolonged association between BUBR1 and two other MCC subunits, CDC20 and MAD2, and with APC/C subunit CDC27 (Fig. 4B). Furthermore, steady-state levels of MCC, as evidenced by association between BUBR1 and MAD2, also slightly but reproducibly increased in TRIP13 knockdown mitotic cells as compared with control cells (Fig. 4C). The level of the increase was reminiscent of that when p31comet was depleted (42, 43). These results suggested that delayed cyclin B degradation in TRIP13-depleted cells was caused by belated activation of APC/C, most likely due to persistence of the MCC and its association and inhibition of the APC/C. The results therefore support the importance of TRIP13 in disassembling the MCC and the MCC·APC/C protein complexes.
FIGURE 4.

TRIP13 knockdown causes prolonged MCC-APC/C association. A and B, HeLa cells were co-transfected with TRIP13 shRNA or control vector and pBabe-puromycin for 24 h and selected against puromycin while synchronized by single thymidine block for 24 h and then directly released into medium containing nocodazole. Puromycin-resistant mitotic cells were collected 12 h after release, washed, and replated using drug-free medium. Cell lysates were prepared at the indicated hours (hr) after nocodazole release and analyzed either directly by immunoblotting (A) or first subjected to BUBR1 immunoprecipitation (IP) and then probed (B). Note that both unattached mitotic cells and attached early G1 cells were harvested at later release time points (after 1 h). C, HeLa cells transfected with control or TRIP13 siRNA were arrested in mitosis with nocodazole. Mitotic cell lysates and immunoprecipitates using either anti-BUBR1 or anti-MAD2 antibodies were probed by immunoblotting. The smudges marked by asterisks in the TRIP13 blot are IgG signals.
TRIP13 Activity Is Required for p31comet-mediated Mitotic Checkpoint Silencing
As mentioned, ATP was known to be required for p31comet to help disassemble the MCC, but how ATP is utilized is not fully understood (29, 44). As reported before (17, 29), the addition of p31comet into the mitotic HeLa cell extract accelerated degradation of cyclin B, whereas anti-TRIP13 antibody delayed cyclin B degradation (Fig. 3B). Interestingly, the acceleration of cyclin B degradation by p31comet was drastically mitigated if the recombinant protein was added together with anti-TRIP13 antibody (Fig. 5, right). The result suggested that interference with TRIP13 ATPase negatively affected p31comet-mediated checkpoint silencing. The coordination between TRIP13 and p31comet in silencing the mitotic checkpoint was also demonstrated through biochemical assays by Dr. Avram Hershko's laboratory.3
FIGURE 5.
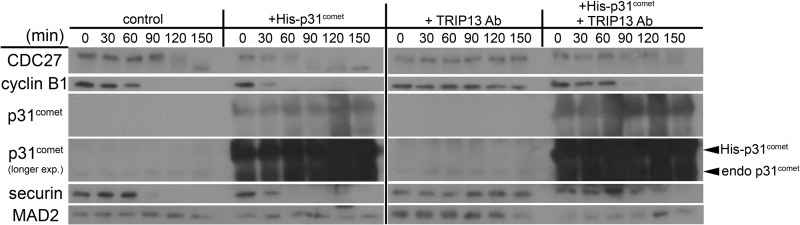
TRIP13 interference abolished p31comet-mediated mitotic checkpoint silencing. Mitotic HeLa cell extracts were incubated alone or with recombinant p31comet and/or anti-TRIP13 antibody and assayed as in Fig. 3. CDC27 and MAD2 served as loading controls, with the mobility shift of CDC27 also indicating loss of Cdk phosphorylation. Both cyclin B1 and securin were probed. A longer exposure (exp.) of p31comet was presented to show endogenous (endo) p31comet.
TRIP13 Disrupts the MAD1·MAD2 Complex
When examining interphase MCF7 cells overexpressing GFP-TRIP13, we found reduced MAD2 signals at the nuclear envelope, as indicated by both immunofluorescence and cell fractionation (Fig. 6, A and B, note the disappearance of MAD2 signals around nuclear rims). The nuclear envelope localization of MAD1 was not notably affected, as well as several other proteins, such as Nup133, Nup103, RanBP2, and RanGAP, indicating no overall structural distortions at the nuclear envelope or nuclear pores (Fig. 6A) (data not shown). More importantly, no MAD2 mislocalization was observed when the catalytically inactive K185T mutant was overexpressed (Fig. 6, A and B). The impact of TRIP13 overexpression on MAD2 localization can be observed in synchronized G1/S or G2 cells and also in MCF10A and HeLa cells (data not shown). Because only C-MAD2 can localize to the nuclear envelope through its association with MAD1 (26), our results suggest that TRIP13 overexpression might disrupt the MAD1·C-MAD2 complex. We derived stable MCF7 Tet-on cell lines that inducibly express TRIP13. Immunoprecipitation with anti-MAD1 antibody using asynchronous cell lysates (primarily interphase cells) confirmed that induced TRIP13 overexpression reduced the level of MAD2 associated with MAD1, without affecting total protein levels in the lysates (Fig. 6C). MAD1 did not co-immunoprecipitate with TRIP13 or p31comet. Because the MAD1-MAD2 interaction is very stable, tolerating up to 1 m urea treatment or other harsh conditions (45), TRIP13 must rely on its ATPase activity to disrupt the complex.
FIGURE 6.

TRIP13 overexpression disrupts MAD1·MAD2 complex in interphase cells. A, MCF7 cells transfected with wild type or catalytically inactive (K185T, Walker A mutant) GFP-TRIP13 were stained with anti-MAD1 and anti-MAD2 antibodies. Images were obtained on a Leica TCS SP8 confocal microscope. Bar, 10 μm. Single focal planes are shown for clarity. The arrowheads point to nuclear rims of transfected cells, whereas the arrows indicate those of untransfected cells. B, GFP-TRIP13- or mock-transfected MCF7 cells were harvested, and nuclei were isolated. Equivalent amounts of nuclear and cytosolic fractions were loaded and probed for different proteins. ATM was probed as a marker for nuclei preparation and loading control. Note the reduction of the nuclei/cytosol ratio of MAD2 in GFP-TRIP13-transfected cells as compared with control. C, cell lysates and anti-MAD1 immunoprecipitates were probed using either uninduced or induced (Doxcycline − or +) asynchronous MCF7 Tet-on cells that inducibly express a TRIP13-Strep fusion protein. Note that the fusion protein cannot be distinguished from endogenous TRIP13 because of the small size of the Strep tag, but the induction can be detected based on the level of TRIP13 in induced samples. The arrowhead indicates protein bands corresponding to TRIP13/TRIP13-Strep. The asterisk marks IgG heavy chain. D, GFP-TRIP13 overexpression does not affect MAD2 kinetochore localization in nocodazole-treated mitotic cells. Transfected MCF7 cells were treated with 3.3 μm nocodazole for 4 h before being fixed for immunofluorescence. Shown are a transfected and an untransfected cell in the same field. Single focal planes are shown for clarity. Bar, 10 μm. Right, quantitation of the intensity ratio of kinetochore MAD2 to anti-centromere autoimmune serum (ACA) in untransfected and GFP-TRIP13WT- or TRIP13K185T-transfected MCF7 cells. E, cell lysates and anti-MAD1 immunoprecipitates were probed as in C except using either uninduced or induced (Doxcycline − or +) and nocodazole-arrested mitotic MCF7 Tet-on cells that inducibly express TRIP13-Strep fusion protein. Error bars, S.E.
Disrupting the MAD1·C-MAD2 complex would have significant functional consequence of compromising O- to C-MAD2 conversion and mitotic checkpoint strength. However, no reduction in MAD2 intensity was observed at kinetochores in nocodazole arrested prometaphase MCF7 cells that expressed GFP-TRIP13 (Fig. 6D, analysis of variance, p > 0.05, n = 35). MAD1 immunoprecipitates also showed comparable MAD2 levels in both uninduced and induced mitotic cell lysates (Fig. 6E). It is likely that a mitotic checkpoint fully activated by nocodazole treatment could override the effects of TRIP13 overexpression (see “Discussion”). There is technical difficulty in evaluating TRIP13 effects on kinetochore intensities of MAD1 and MAD2 in prometaphase cells without drug treatment because the signals naturally vary spanning a wide range, depending on the kinetochore-microtubule binding status at individual kinetochores. Regardless, a conservative conclusion is that TRIP13 AAA-ATPase overexpression has the potential to disrupt the MAD1·C-MAD2 complex.
TRIP13 Is Required for Cell Proliferation
Considering characteristic overexpression of TRIP13 in breast cancer cells (10, 11), we examined whether TRIP13 is required for MCF7 cell growth and proliferation. MCF7 cells treated with TRIP13 siRNA barely showed any cell proliferation in regular cell culture plates over the course of 8 days (Fig. 7A). Similar results were obtained for MCF10A and T47D cells (data not shown). In addition, TRIP13 siRNA-treated MCF7 cells produced only one-third the number of colonies in soft agar plates compared with control cells, suggesting dramatically compromised anchorage-independent cell growth (Fig. 7B). It remains to be seen whether any of the colonies in TRIP13 siRNA-treated wells arose from untransfected MCF7 cells. However, the above results are consistent with the idea that TRIP13 is essential for cell proliferation.
FIGURE 7.
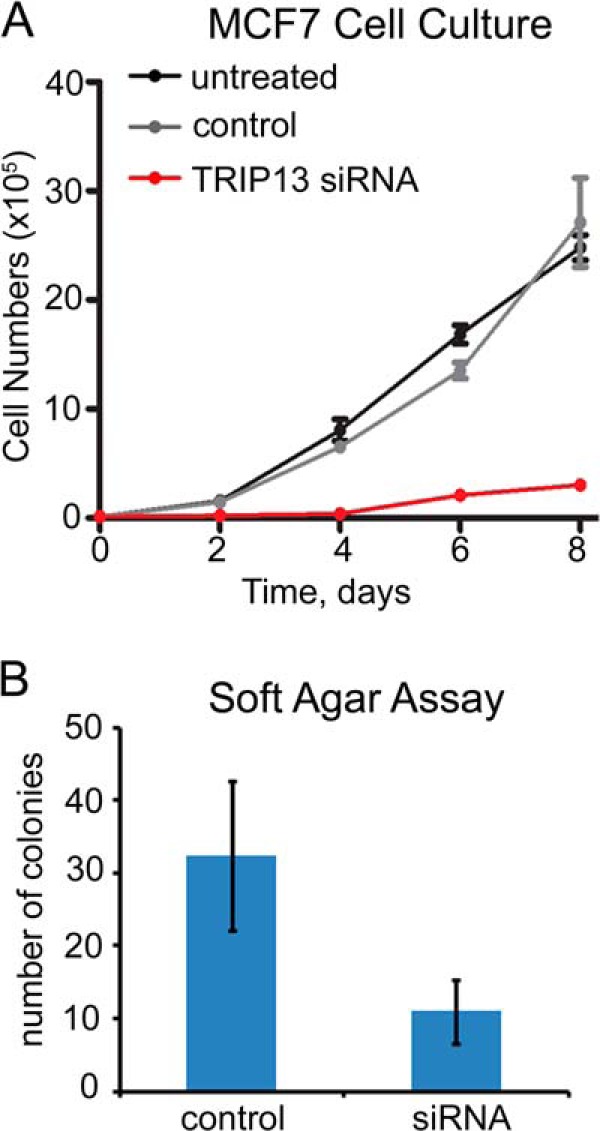
TRIP13 knockdown inhibits MCF7 proliferation. A, MCF7 proliferation assay in regular cell culture. After siRNA knockdown or mock treatment for 48 h, 105 cells were plated in 6-cm plates. Cells were counted in triplicates every 2 days for 8 days. B, comparison of the numbers of colonies in soft agar assays of MCF7 cells after control or TRIP13 siRNA treatment. Error bars, S.E.
DISCUSSION
TRIP13 Is a Novel Mitotic Checkpoint-silencing Protein
Silencing the mitotic checkpoint, just like activating the checkpoint, involves events at two venues: at kinetochores and in the cytoplasm (23, 46). “Wait anaphase” signals (predominantly C-MAD2) need to be terminated at kinetochores that are fully attached with spindle microtubules and bioriented with tension. The termination may happen through dynein-mediated stripping of the MAD1·C-MAD2 catalyst from kinetochores (47). Proteins, such as protein phosphatase 1 and Spindly, may facilitate the stripping by altering protein modification or composition at kinetochores to concurrently weaken the MAD1·C-MAD2 complex associating with its uncharacterized kinetochore receptor and enhance its interaction with the dynein·dynactin complex (48, 49). Although p31comet and TRIP13 are localized at prometaphase kinetochores, it is still unclear how significant a role they play in terminating the “wait anaphase” signal (30, 31, 50) (Fig. 1). Indeed, although we have detected disruption of the MAD1·C-MAD2 complex in interphase cells when TRIP13 was overexpressed, the fully activated checkpoint triggered by nocodazole treatment seemed to be able to override the TRIP13 effects in mitotic cells (Fig. 6). Nevertheless, activation and silencing of the mitotic checkpoint are more and more appreciated as not temporally separated but rather in a constant “tug of war” type of dynamic equilibrium so that cells can respond quickly to internal and external cues to either activate/reactivate or silence the checkpoint (14, 51). In addition, a recent report suggested that interphase MAD1·C-MAD2 complex is also important for preventing premature APC/C activation, probably by maintaining a basal level of C-MAD2 in cells (52).
In the cytoplasm, mitotic checkpoint silencing requires disassembly of the APC/C·MCC supracomplex and the MCC itself. We have shown previously that both the MCC and the MCC·APC/C complexes are stable for at least 24 h if no disassembly factors are present (36). A recently identified APC/C subunit APC15 and ubiquitylation of CDC20 are deemed critical for dissociating MCC from APC/C (43, 53–56). p31comet, on the other hand, was shown to be important for disassembling the MCC (29, 30). It was demonstrated using clever biochemistry methods that ATP hydrolysis at the β-γ high energy phosphodiester bond is essential for the full activity of p31comet, but how the ATP is consumed was not well understood (29, 44). The results presented in this paper provide evidence that TRIP13 AAA-ATPase may coordinate with p31comet to disassemble the MCC with energy input from ATP hydrolysis, hence helping to dismantle the mitotic checkpoint.
TRIP13 ATPase and Energy Requirement for Mitotic Checkpoint Silencing
Many AAA-ATPases form hexameric ring-shaped structures and couple the energy released from ATP hydrolysis with unfolding and folding of proteins or assembly and disassembly of macromolecular complexes (35). How TRIP13 ATPase mechanistically works with p31comet to disassemble the MCC remains to be investigated. In analogy with other AAA-ATPases, it is tempting to hypothesize that TRIP13 uses its own ATPase activity and p31comet as an adapter protein to extract C-MAD2 from various complexes required for an active mitotic checkpoint, such as MCC, MCC·APC/C, or MAD1·MAD2 complexes. All of the complexes only incorporate C-MAD2; therefore, the extraction may be accompanied by a C to O conformational change of MAD2, resulting in disruption of the complexes. If confirmed, the hypothesis explains why the majority of MAD2 in interphase cells is in the O conformation although C-MAD2 is of relatively lower energy and thus at a thermodynamically more stable state (22, 25, 57). Once the energy input converts C-MAD2 to O-MAD2, the latter is kinetically trapped until next mitosis, when a functional MAD1·MAD2 catalyst helps to overcome the activation energy barrier. Interestingly, partial unfolding of MAD2 has been proposed before to explain reversible conversion between its O and C conformers (58, 59). TRIP13 ATPase is a wonderful candidate to facilitate MAD2 conformational change.
There are at least three questions that remain to be answered in light of what is known about AAA-ATPases. First, does TRIP13 function as an oligomer like most other AAA-ATPases? Transfection of cells with both FLAG- and GFP-tagged TRIP13 and subsequent immunoprecipitations showed that only a small fraction of TRIP13 forms oligomers (data not shown). There are precedents that oligomerization of certain AAA-ATPases requires high protein concentrations or is significantly enhanced by co-factors, substrates, ATP, or low salt conditions (35, 60). How TRIP13 becomes activated in cells needs to be carefully studied. Second, in terms of adapter proteins, although in vitro binding with recombinant proteins clearly demonstrated direct interaction between TRIP13 and p31comet, under our experimental conditions, the interaction in cell lysates was only detectable when both proteins were overexpressed (31). However, the interaction had been detected in proteomic scale protein-protein interaction studies using both yeast two-hybrid and mass spectrometry-based strategies in human and mouse cells (32–34). We surmise that the intracellular interaction may also depend upon local concentrations of both proteins. Third, the protein level of TRIP13 seemed constant throughout the cell cycle (data not shown). This raised the question of whether and how its activity is temporally regulated in order for mitotic checkpoint silencing to become dominant at the right time. Several serine/threonine sites in TRIP13 have been found to be phosphorylated during mitosis (61–63). Whether phosphorylation at these sites affects TRIP13-p31comet interaction or its ATPase activity warrants further investigation.
TRIP13 Overexpression in Cancer Cells
Chromosomal instability and aneuploidy are common in solid tumor cells (64). Surprisingly, engineered yeast or mammalian aneuploid cell lines containing extra chromosomes showed proliferative disadvantages (65–67). Therefore, aneuploid cancer cells must have acquired additional changes in the genomes to achieve uncontrolled proliferation while maintaining aneuploidy (68, 69). Genome-wide transcriptional profiling has listed TRIP13 as one of the top ranking overexpressed genes in cancer cells with a CIN phenotype (number 12 in the CIN70 signature) (13). Due to intimate connections between CIN and tumor initiation and progression, it is not surprising that TRIP13 was also found among a 22-gene signature for breast cancers with poor prognosis (10) and among another list of genes that are overexpressed in undifferentiated breast cancers (11).
Taking into consideration the mitotic checkpoint-silencing function of TRIP13 reported here, we propose that TRIP13 overexpression results in untimely mitotic checkpoint silencing, driving CIN and cancer development in mammary epithelial cells or cells of other tissue origins. Another mitotic checkpoint-silencing protein, CUEDC2, was also overexpressed in many tumor samples (70). It is likely that overactive mitotic checkpoint silencing may induce aneuploidy and CIN just as defective mitotic checkpoint activation. We noticed that overexpression of TRIP13 protein was not dramatic in the examined breast cancer cell lines (Fig. 1); however, a partially compromised mitotic checkpoint may be perfect for balancing cell survival and evolution of aneuploid karyotypes to promote tumorigenesis (64). Due to its nature as an enzyme and its requirement for cell proliferation (Fig. 7), TRIP13 is a possible target to develop novel drugs against aneuploid cancers. It will be interesting to examine whether nontumorigenic and tumorigenic mammary epithelial cells respond to TRIP13 inhibition differentially.
Acknowledgments
We sincerely thank Dr. Jerome B. Rattner for the anti-centromere autoimmune serum and Dr. Elena Pugacheva for DNA constructs. We also thank Wenbin Ji for discussion and for providing some mitotic extracts.
This work was supported, in whole or in part, by National Institutes of Health Grants CA169706 and CA06927 (to T. J. Y.) and R01CA169500 (to S. T. L.). This work was also supported by an operating grant from the Canadian Breast Cancer Foundation, Prairies/NWT region (to G. K. C.); the Fox Chase Cancer Center Board of Associates and an appropriation from the Commonwealth of Pennsylvania (to T. J. Y.); and National Science Foundation Grant MCB-1052413 (to S. T. L.).
A. Hershko, personal communication.
- CIN
- chromosomal instability
- AAA-ATPase
- ATPase associated with diverse cellular activities
- APC/C
- anaphase-promoting complex/cyclosome
- C- and O-MAD2
- closed and open MAD2, respectively
- MCC
- mitotic checkpoint complex.
REFERENCES
- 1. Lee J. W., Choi H. S., Gyuris J., Brent R., Moore D. D. (1995) Two classes of proteins dependent on either the presence or absence of thyroid hormone for interaction with the thyroid hormone receptor. Mol. Endocrinol. 9, 243–254 [DOI] [PubMed] [Google Scholar]
- 2. Yasugi T., Vidal M., Sakai H., Howley P. M., Benson J. D. (1997) Two classes of human papillomavirus type 16 E1 mutants suggest pleiotropic conformational constraints affecting E1 multimerization, E2 interaction, and interaction with cellular proteins. J. Virol. 71, 5942–5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. San-Segundo P. A., Roeder G. S. (1999) Pch2 links chromatin silencing to meiotic checkpoint control. Cell 97, 313–324 [DOI] [PubMed] [Google Scholar]
- 4. Li X. C., Li X., Schimenti J. C. (2007) Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet. 3, e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roig I., Dowdle J. A., Toth A., de Rooij D. G., Jasin M., Keeney S. (2010) Mouse TRIP13/PCH2 is required for recombination and normal higher-order chromosome structure during meiosis. PLoS Genet. 6, e1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wojtasz L., Daniel K., Roig I., Bolcun-Filas E., Xu H., Boonsanay V., Eckmann C. R., Cooke H. J., Jasin M., Keeney S., McKay M. J., Toth A. (2009) Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet. 5, e1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bhalla N., Dernburg A. F. (2005) A conserved checkpoint monitors meiotic chromosome synapsis in Caenorhabditis elegans. Science 310, 1683–1686 [DOI] [PubMed] [Google Scholar]
- 8. Joyce E. F., McKim K. S. (2009) Drosophila PCH2 is required for a pachytene checkpoint that monitors double-strand-break-independent events leading to meiotic crossover formation. Genetics 181, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miao C., Tang D., Zhang H., Wang M., Li Y., Tang S., Yu H., Gu M., Cheng Z. (2013) Central region component1, a novel synaptonemal complex component, is essential for meiotic recombination initiation in rice. Plant Cell 25, 2998–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martin K. J., Patrick D. R., Bissell M. J., Fournier M. V. (2008) Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. PLoS One 3, e2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rhodes D. R., Yu J., Shanker K., Deshpande N., Varambally R., Ghosh D., Barrette T., Pandey A., Chinnaiyan A. M. (2004) Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl. Acad. Sci. U.S.A. 101, 9309–9314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carter S. L., Eklund A. C., Kohane I. S., Harris L. N., Szallasi Z. (2006) A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048 [DOI] [PubMed] [Google Scholar]
- 14. Lara-Gonzalez P., Westhorpe F. G., Taylor S. S. (2012) The spindle assembly checkpoint. Curr. Biol. 22, R966–R980 [DOI] [PubMed] [Google Scholar]
- 15. Barford D. (2011) Structural insights into anaphase-promoting complex function and mechanism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 3605–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sudakin V., Chan G. K., Yen T. J. (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154, 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tipton A. R., Tipton M., Yen T., Liu S. T. (2011) Closed MAD2 (C-MAD2) is selectively incorporated into the mitotic checkpoint complex (MCC). Cell Cycle 10, 3740–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang Z., Bharadwaj R., Li B., Yu H. (2001) Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell 1, 227–237 [DOI] [PubMed] [Google Scholar]
- 19. Kulukian A., Han J. S., Cleveland D. W. (2009) Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev. Cell 16, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herzog F., Primorac I., Dube P., Lenart P., Sander B., Mechtler K., Stark H., Peters J. M. (2009) Structure of the anaphase-promoting complex/cyclosome interacting with a mitotic checkpoint complex. Science 323, 1477–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Musacchio A., Salmon E. D. (2007) The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 [DOI] [PubMed] [Google Scholar]
- 22. Luo X., Yu H. (2008) Protein metamorphosis: the two-state behavior of Mad2. Structure 16, 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hardwick K. G., Shah J. V. (2010) Spindle checkpoint silencing: ensuring rapid and concerted anaphase onset. F1000 Biol. Rep. 2, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Foley E. A., Kapoor T. M. (2013) Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 14, 25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luo X., Tang Z., Xia G., Wassmann K., Matsumoto T., Rizo J., Yu H. (2004) The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol. 11, 338–345 [DOI] [PubMed] [Google Scholar]
- 26. Fava L. L., Kaulich M., Nigg E. A., Santamaria A. (2011) Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. EMBO J. 30, 3322–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Habu T., Kim S. H., Weinstein J., Matsumoto T. (2002) Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J. 21, 6419–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang M., Li B., Tomchick D. R., Machius M., Rizo J., Yu H., Luo X. (2007) p31comet blocks Mad2 activation through structural mimicry. Cell 131, 744–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Teichner A., Eytan E., Sitry-Shevah D., Miniowitz-Shemtov S., Dumin E., Gromis J., Hershko A. (2011) p31comet promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc. Natl. Acad. Sci. U.S.A. 108, 3187–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Westhorpe F. G., Tighe A., Lara-Gonzalez P., Taylor S. S. (2011) p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J. Cell Sci. 124, 3905–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tipton A. R., Wang K., Oladimeji P., Sufi S., Gu Z., Liu S. T. (2012) Identification of novel mitosis regulators through data mining with human centromere/kinetochore proteins as group queries. BMC Cell Biol. 13, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stelzl U., Worm U., Lalowski M., Haenig C., Brembeck F. H., Goehler H., Stroedicke M., Zenkner M., Schoenherr A., Koeppen S., Timm J., Mintzlaff S., Abraham C., Bock N., Kietzmann S., Goedde A., Toksöz E., Droege A., Krobitsch S., Korn B., Birchmeier W., Lehrach H., Wanker E. E. (2005) A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968 [DOI] [PubMed] [Google Scholar]
- 33. Rual J. F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., Goldberg D. S., Zhang L. V., Wong S. L., Franklin G., Li S., Albala J. S., Lim J., Fraughton C., Llamosas E., Cevik S., Bex C., Lamesch P., Sikorski R. S., Vandenhaute J., Zoghbi H. Y., Smolyar A., Bosak S., Sequerra R., Doucette-Stamm L., Cusick M. E., Hill D. E., Roth F. P., Vidal M. (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 34. Suzuki H., Fukunishi Y., Kagawa I., Saito R., Oda H., Endo T., Kondo S., Bono H., Okazaki Y., Hayashizaki Y. (2001) Protein-protein interaction panel using mouse full-length cDNAs. Genome Res. 11, 1758–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neuwald A. F., Aravind L., Spouge J. L., Koonin E. V. (1999) AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43 [PubMed] [Google Scholar]
- 36. Tipton A. R., Ji W., Sturt-Gillespie B., Bekier M. E., 2nd, Wang K., Taylor W. R., Liu S. T. (2013) Monopolar spindle 1 (MPS1) kinase promotes production of closed MAD2 (C-MAD2) conformer and assembly of the mitotic checkpoint complex. J. Biol. Chem. 288, 35149–35158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K., Kaufman P. D. (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tipton A. R., Wang K., Link L., Bellizzi J. J., Huang H., Yen T., Liu S. T. (2011) BUBR1 and closed MAD2 (C-MAD2) interact directly to assemble a functional mitotic checkpoint complex. J. Biol. Chem. 286, 21173–21179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jia L., Li B., Warrington R. T., Hao X., Wang S., Yu H. (2011) Defining pathways of spindle checkpoint silencing: functional redundancy between Cdc20 ubiquitination and p31(comet). Mol. Biol. Cell 22, 4227–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Braunstein I., Miniowitz S., Moshe Y., Hershko A. (2007) Inhibitory factors associated with anaphase-promoting complex/cylosome in mitotic checkpoint. Proc. Natl. Acad. Sci. U.S.A. 104, 4870–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rape M., Kirschner M. W. (2004) Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 432, 588–595 [DOI] [PubMed] [Google Scholar]
- 42. Varetti G., Guida C., Santaguida S., Chiroli E., Musacchio A. (2011) Homeostatic control of mitotic arrest. Mol. Cell 44, 710–720 [DOI] [PubMed] [Google Scholar]
- 43. Mansfeld J., Collin P., Collins M. O., Choudhary J. S., Pines J. (2011) APC15 drives the turnover of MCC-CDC20 to make the spindle assembly checkpoint responsive to kinetochore attachment. Nat. Cell Biol. 13, 1234–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miniowitz-Shemtov S., Eytan E., Ganoth D., Sitry-Shevah D., Dumin E., Hershko A. (2012) Role of phosphorylation of Cdc20 in p31(comet)-stimulated disassembly of the mitotic checkpoint complex. Proc. Natl. Acad. Sci. U.S.A. 109, 8056–8060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen R. H., Brady D. M., Smith D., Murray A. W., Hardwick K. G. (1999) The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol. Biol. Cell 10, 2607–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vanoosthuyse V., Hardwick K. G. (2009) Overcoming inhibition in the spindle checkpoint. Genes Dev. 23, 2799–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Howell B. J., McEwen B. F., Canman J. C., Hoffman D. B., Farrar E. M., Rieder C. L., Salmon E. D. (2001) Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J. Cell Biol. 155, 1159–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kops G. J., Shah J. V. (2012) Connecting up and clearing out: how kinetochore attachment silences the spindle assembly checkpoint. Chromosoma 121, 509–525 [DOI] [PubMed] [Google Scholar]
- 49. Maldonado M., Kapoor T. M. (2011) Moving right along: how PP1 helps clear the checkpoint. Dev. Cell 20, 733–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hagan R. S., Manak M. S., Buch H. K., Meier M. G., Meraldi P., Shah J. V., Sorger P. K. (2011) p31(comet) acts to ensure timely spindle checkpoint silencing subsequent to kinetochore attachment. Mol. Biol. Cell 22, 4236–4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jia L., Kim S., Yu H. (2013) Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem. Sci. 38, 302–311 [DOI] [PubMed] [Google Scholar]
- 52. Rodriguez-Bravo V., Maciejowski J., Corona J., Buch H. K., Collin P., Kanemaki M. T., Shah J. V., Jallepalli P. V. (2014) Nuclear pores protect genome integrity by assembling a premitotic and mad1-dependent anaphase inhibitor. Cell 156, 1017–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Foster S. A., Morgan D. O. (2012) The APC/C subunit Mnd2/Apc15 promotes Cdc20 autoubiquitination and spindle assembly checkpoint inactivation. Mol. Cell 47, 921–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Uzunova K., Dye B. T., Schutz H., Ladurner R., Petzold G., Toyoda Y., Jarvis M. A., Brown N. G., Poser I., Novatchkova M., Mechtler K., Hyman A. A., Stark H., Schulman B. A., Peters J. M. (2012) APC15 mediates CDC20 autoubiquitylation by APC/C(MCC) and disassembly of the mitotic checkpoint complex. Nat. Struct. Mol. Biol. 19, 1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stegmeier F., Rape M., Draviam V. M., Nalepa G., Sowa M. E., Ang X. L., McDonald E. R., 3rd, Li M. Z., Hannon G. J., Sorger P. K., Kirschner M. W., Harper J. W., Elledge S. J. (2007) Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature 446, 876–881 [DOI] [PubMed] [Google Scholar]
- 56. Reddy S. K., Rape M., Margansky W. A., Kirschner M. W. (2007) Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature 446, 921–925 [DOI] [PubMed] [Google Scholar]
- 57. Mapelli M., Musacchio A. (2007) MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr. Opin. Struct. Biol. 17, 716–725 [DOI] [PubMed] [Google Scholar]
- 58. Yang M., Li B., Liu C. J., Tomchick D. R., Machius M., Rizo J., Yu H., Luo X. (2008) Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol. 6, e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Skinner J. J., Wood S., Shorter J., Englander S. W., Black B. E. (2008) The Mad2 partial unfolding model: regulating mitosis through Mad2 conformational switching. J. Cell Biol. 183, 761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wendler P., Ciniawsky S., Kock M., Kube S. (2012) Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 1823, 2–14 [DOI] [PubMed] [Google Scholar]
- 61. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 62. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 63. Daub H., Olsen J. V., Bairlein M., Gnad F., Oppermann F. S., Körner R., Greff Z., Kéri G., Stemmann O., Mann M. (2008) Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 31, 438–448 [DOI] [PubMed] [Google Scholar]
- 64. Weaver B. A., Cleveland D. W. (2008) The aneuploidy paradox in cell growth and tumorigenesis. Cancer Cell 14, 431–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thompson S. L., Compton D. A. (2008) Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol. 180, 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Torres E. M., Sokolsky T., Tucker C. M., Chan L. Y., Boselli M., Dunham M. J., Amon A. (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317, 916–924 [DOI] [PubMed] [Google Scholar]
- 67. Williams B. R., Prabhu V. R., Hunter K. E., Glazier C. M., Whittaker C. A., Housman D. E., Amon A. (2008) Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322, 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sheltzer J. M., Blank H. M., Pfau S. J., Tange Y., George B. M., Humpton T. J., Brito I. L., Hiraoka Y., Niwa O., Amon A. (2011) Aneuploidy drives genomic instability in yeast. Science 333, 1026–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Torres E. M., Dephoure N., Panneerselvam A., Tucker C. M., Whittaker C. A., Gygi S. P., Dunham M. J., Amon A. (2010) Identification of aneuploidy-tolerating mutations. Cell 143, 71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gao Y. F., Li T., Chang Y., Wang Y. B., Zhang W. N., Li W. H., He K., Mu R., Zhen C., Man J. H., Pan X., Li T., Chen L., Yu M., Liang B., Chen Y., Xia Q., Zhou T., Gong W. L., Li A. L., Li H. Y., Zhang X. M. (2011) Cdk1-phosphorylated CUEDC2 promotes spindle checkpoint inactivation and chromosomal instability. Nat. Cell Biol. 13, 924–933 [DOI] [PubMed] [Google Scholar]