

Background: Rough-type LPS of C. canimorsus is biologically active, whereas lipid A is not.
Results: Purified C. canimorsus rough-type LPS could be analyzed in intact form by NMR.
Conclusion: Inactive lipid A becomes active in case the core oligosaccharide is attached.
Significance: This study provides evidence that lipid A is not the sole part of the LPS structure responsible for endotoxic activity.
Keywords: Glycolipid Structure, Gram-negative Bacteria, High-performance Liquid Chromatography (HPLC), Lipopolysaccharide (LPS), Nuclear Magnetic Resonance (NMR), Capnocytophaga canimorsus, DHPC Micelles
Abstract
We here describe the NMR analysis of an intact lipopolysaccharide (LPS, endotoxin) in water with 1,2-dihexanoyl-sn-glycero-3-phosphocholine as detergent. When HPLC-purified rough-type LPS of Capnocytophaga canimorsus was prepared, 13C,15N labeling could be avoided. The intact LPS was analyzed by homonuclear (1H) and heteronuclear (1H,13C, and 1H,31P) correlated one- and two-dimensional NMR techniques as well as by mass spectrometry. It consists of a penta-acylated lipid A with an α-linked phosphoethanolamine attached to C-1 of GlcN (I) in the hybrid backbone, lacking the 4′-phosphate. The hydrophilic core oligosaccharide was found to be a complex hexasaccharide with two mannose (Man) and one each of 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo), Gal, GalN, and l-rhamnose residues. Position 4 of Kdo is substituted by phosphoethanolamine, also present in position 6 of the branched ManI residue. This rough-type LPS is exceptional in that all three negative phosphate residues are “masked” by positively charged ethanolamine substituents, leading to an overall zero net charge, which has so far not been observed for any other LPS. In biological assays, the corresponding isolated lipid A was found to be endotoxically almost inactive. By contrast, the intact rough-type LPS described here expressed a 20,000-fold increased endotoxicity, indicating that the core oligosaccharide significantly contributes to the endotoxic potency of the whole rough-type C. canimorsus LPS molecule. Based on these findings, the strict view that lipid A alone represents the toxic center of LPS needs to be reassessed.
Introduction
Capnocytophaga canimorsus is a Gram-negative bacterium of the family Flavobacteriaceae, commonly found in the mouths of dogs and causing rare but serious human infections resulting from bites, scratches, or even licks (1–4). The infection rapidly progresses to life-threatening septicemia (5). Although it is not perfectly clear yet why these bacteria can be so aggressive, a number of virulence factors have been described recently. First, C. canimorsus bacteria have the unusual capacity to deglycosylate human glycoproteins, including IgGs and cell surface glycoproteins from phagocytes (6–8). In addition, in vitro, they resist phagocytosis (2) and killing by complement (9). The lipopolysaccharide (LPS, endotoxin) representing one of the most potent pathogen-associated molecular patterns of Gram-negative bacteria (10) is the key compound to the latter properties (3, 9).
Recently, we analyzed the lipid A structure and function of C. canimorsus and identified it as a penta-acylated lipid A, which lacks the 4′-phosphate and carries a phosphoethanolamine (P-Etn)4 in position C-1 of the lipid A backbone (11). It has long been known but seldom been addressed that, besides phosphates and fatty acids, the core oligosaccharide also contributes to an enhanced endotoxicity of the lipid A molecule by 1–2 orders of magnitude (12, 13). By contrast, in C. canimorsus, the endotoxicity is enhanced by a factor of 20,000 when the core oligosaccharide is attached to lipid A (11). This is most remarkable because this lipid A contains only five fatty acids and only one phosphate residue attached to the lipid A backbone (13, 14). Which functional group in the lipid A and/or in the proximal core oligosaccharide of C. canimorsus might be able to contribute to this unexpected enhancement in endotoxicity?
To answer this question, we first isolated the LPS-derived oligosaccharides by conventional approaches using (i) acid hydrolysis (leading to core oligosaccharide 1; Fig. 1) or (ii) strong hydrazinolysis for deacylation of the LPS/lipid A (leading to core backbone oligosaccharide 2; Fig. 1) and determined the complete structures by ESI-MS and NMR in D2O. By contrast, when rough-type LPS, which still contains the hydrophobic lipid A moiety, was subjected to NMR analysis, only complex organic solvent mixtures, such as chloroform/methanol/water, were able to give well resolved NMR signals (15). However, conformational studies of the LPS molecule expressing relevant biological (i.e. endotoxic active) conformations in an aqueous solvent system cannot be achieved by this approach (16).
FIGURE 1.

LPS and part structures of C. canimorsus. Shown are structures of the core hexasaccharide (1) obtained after acid hydrolysis (AcOH), isolated core backbone octasaccharide (2) obtained after strong hydrazinolysis (N2H4), and the complete rough-type LPS (3).
Although the acyl chains and phosphates have been proven to be extremely important for the LPS/lipid A structure-function relationship (12, 17, 18), the contribution of the core oligosaccharide has not been considered so far. It is generally accepted that the lipid A represents the endotoxic moiety of the whole LPS molecule (14), and other components, like the inner core oligosaccharide, are often considered to be less or even not important. By contrast, in C. canimorsus LPS, we obtained strong evidence that indeed the core oligosaccharide can contribute to its endotoxicity (11). We found that the negatively charged carboxyl group of the Kdo adjacent to the lipid A backbone can replace the function of the 4′-phosphate that is sterically close to it. Therefore, an analytical tool enabling the analysis of the conformation of the core oligosaccharide when attached to the lipid A by NMR in a water mimetic is expected to shed more light onto the LPS structure-function relationship. By this approach, we aimed to explain interactions of intact rough-type LPS with natural pathogen-related receptors (MD-2 and TLR4) under nearly natural conditions, where conformational aspects become important for the understanding of various biological functions.
Here we describe an analytical approach using HPLC purification of C. canimorsus rough-type LPS (3, Fig. 1) followed by NMR analysis in a water mimetic. This way we could show that not only the primary structure but also the conformation of LPS could successfully be analyzed by NMR spectroscopy, avoiding expensive 13C,15N labeling. Our data give new insights into the conformational flexibility of the core oligosaccharide, which can be helpful to explain the difference between the low activity of free lipid A of C. canimorsus on the one hand and the high endotoxic activity of the rough-type LPS on the other (11).
MATERIALS AND METHODS
Bacterial Strains, Growth Conditions, and Extraction of LPS
C. canimorsus 5 (Cc5) (3) and its complement-sensitive Y1C12 mutant (9) were grown on heart infusion agar 5% sheep blood supplemented with gentamicin and, in the case of Y1C12, with erythromycin. The Cc5-based Y1C12 mutant has a transposon insertion within a predicted glycosyltranferase-encoding gene, probably the N-acetylfucosamine transferase WbuB, necessary for the formation of the O-antigen (9). Bacteria were harvested by scraping off colonies from the agar surface of 500 plates, washed, and resuspended in PBS. Selective agents were added at the following concentrations: erythromycin, 10 mg/ml; gentamicin, 20 mg/ml.
Bacterial cells were washed with ethanol, acetone, diethylether (500 ml each) and centrifuged to give 11.2 g (Cc5) and 30.8 g (Y1C12) of air-dried bacteria, respectively. Both LPSs were extracted with phenol/water (19), whereby the crude LPS of WT Cc5 was identified in the water, and that of the mutant was identified in the phenol phase. Extraction by the phenol/chloroform/petroleum ether method (20), RNase/DNase treatment (30 mg; Sigma; 24 h at room temperature), proteinase K digestion (30 mg; 16 h at room temperature), extended dialysis (2 days at 4 °C), and lyophilization revealed 188 mg of Cc5 (1.7%, w/w) and 200 mg of Y1C12 LPS (0.6%, w/w). Compositional analysis of the LPS from the WT strain showed it to be highly contaminated with glucose from amylopectin (21), flavolipin (22), and capnin (23, 24), known to be present in Capnocytophaga spp. and Flavobacteriaceae. In contrast, the LPS from the Y1C12 mutant was devoid of such contaminating material. Because the compositional analysis of the core obtained from the WT strain Cc5 LPS and that of Y1C12 revealed no differences with respect to their sugars and fatty acids, the Y1C12 mutant was considered to be the strain of choice to isolate and analyze the intact rough-type LPS in detail.
Preparation and Purification of 1 after Acid Hydrolysis
LPS isolated from the WT bacteria (25 mg) and Y1C12 mutant (50 mg) was heated in 10 ml of 2% AcOH for 1.5 h at 100 °C. Precipitating lipid A was removed by centrifugation, and the core oligosaccharide in the supernatant was lyophilized and further purified by GPC on a column (2.5 × 120 cm, Toyopearl (HW-40S) Tosoh, Bioscience, Germany), eluted with pyridine/acetic acid/water (8:20:2000, v/v/v, pH ∼4.7) at a flow rate of 0.7 ml/min. Oligosaccharides were monitored by a Knauer differential refractometer. Appropriate fractions (7.5 ml) containing the core sugars (as detected by GLC-MS) were collected and lyophilized. The yield of the GPC-purified 1 was 5.8 mg (25%) for the WT strain Cc5 and 12 mg (24%) for the Y1C12 mutant. Compositional analysis by GLC-MS of both core oligosaccharide fractions resulted in identical sugar residues being present in comparable proportions. As expected, the core oligosaccharide fraction of the mutant did not contain contaminating amylopectin (21) and sugars from the O-chain (9). Thus, the Y1C12 mutant was found to be representative for further structural analysis of the complete rough-type C. canimorsus LPS (3).
Preparation and Purification of 2 after Strong Hydrazinolysis
LPS of the Y1C12 mutant (50 mg) was completely deacylated by strong hydrazinolysis (1.0 ml of N2H4, 100 °C, 16 h). The product was precipitated from cold acetone (10 ml) and dried under a stream of nitrogen. Liberated fatty acids were removed from the sediment by repeated extraction from water/chloroform (3 × 15 ml). The combined water phases were centrifuged (15 min, 7,000 rpm, 4 °C) and lyophilized to give 28.7 mg of crude core backbone oligosaccharide, which was further purified by GPC as described above, whereby a low but acceptable yield (2.2 mg, 4.4% w/w) of 2 was obtained.
Preparation and HPLC Purification of the Intact Rough-type LPS
The rough-type LPS was isolated by chance from an LPS preparation of the Y1C12 mutant (51.3 mg) because mild acetate buffer hydrolysis, which we tried first (0.1 m NaOAc, pH 4.4, containing 1% SDS (25) at 100 °C for 3 h under stirring), did not liberate free lipid A from the core oligosaccharide. Because ESI-MS analysis of this hydrolysate revealed that it still contained a considerable amount of unhydrolyzed and intact rough-type LPS (> 60%), this preparation was considered to be suitable for the purification of compound 3 by preparative HPLC.
To this end, an Abimed-Gilson HPLC system equipped with a semipreparative Kromasil C18 column (5 μm, 100 Å, 10 × 250 mm, MZ-Analysentechnik) was loaded with about 2–5 mg of the crude LPS. Lipid A and LPS fractions were eluted using a gradient of methanol/chloroform/water (57:12:31, v/v/v) containing 10 mm NH4OAc as mobile phase A and chloroform/methanol (70.2:29.8, v/v) with 50 mm NH4OAc as mobile phase B. The initial solvent system consisted of 2% B and was maintained for 20 min, followed by a three-step linear gradient rising from 2 to 17% B (20–50 min), from 17 to 27% B (50–85 min), and from 27 to 100% B (85–165 min). The solvent was held at 100% B for 12 min, and the column was re-equilibrated during 10 min to 2% B and held there for an additional 20 min before the next injection. The flow rate for preparative runs was 2 ml/min (∼80 bar) using a splitter (∼1:35) between the evaporative light-scattering detector (ELSD) and the fraction collector.
The smaller part of the eluate was split by a Sedex model 75C ELSD (S.E.D.E.R.E., Alfortville, France) equipped with a low flow nebulizer. Fractions were collected in 1- and 0.75-min intervals, depending on the peak elution profile. Nitrogen (purity 99.996%) was used as gas to nebulize the post-column flow stream at 3.5 bar into the detector at 50 °C, setting the photomultiplier gain to 9. The detector signal was transferred to the Gilson HPLC Chemstation (Trilution LC, version 2.1) for detection and integration of the ELSD signals.
According to the ELSD detector response, fractions representing the three main peaks in the HPLC chromatogram (Fig. 2) were combined. From one injection (5 mg of hydrolyzed LPS) the following pools were obtained: pool 1 (70–77 min, yield 0.81 mg, 16%), pool 2 (79–90 min, yield 1.45 mg, 30%), and pool 3 (115.5–118 min, yield 0.62 mg, 12%). Besides the LPS of Y1C12, containing a short O-chain oligosaccharide (data not shown), ornithine lipids (30), capnin (24), flavolipin (31), and other lipids were identified in pool 1. The main fraction (pool 2, yield 1.45 mg) (Fig. 2) represented the purified and complete rough-type LPS of C. canimorsus (3). The third fraction, pool 3 (yield 0.62 mg), was free lipid A as determined by ESI-MS ([M] = 1717.308 units). Its structural analysis and exceptional biological properties have been described elsewhere (11).
FIGURE 2.

Reversed phase HPLC profile obtained from the C. canimorsus Y1C12 mutant LPS after mild acetate buffer hydrolysis. Note that only ∼10% of the free lipid A (pool 3) was obtained under the conditions used, whereas the intact rough-type LPS (3, pool 2) predominated (>60%) and could be separated to homogeneity. ELS Detector, evaporative light-scattering detector.
Analytical Procedures
SDS-PAGE
SDS-PAGE of LPS isolated from the WT strain Cc5 and the Y1C12 mutant was run on 18% polyacrylamide gel (26) and silver-stained as described (27).
GLC-MS Analysis
Sugar and fatty acid derivatives were analyzed by GLC-MS on a 5975 inert XL Mass Selective Detector (Agilent Technologies) equipped with a 30-m HP-5MS column (Hewlett-Packard) using a temperature gradient of 150 °C (3 min) → 320 °C at 5 °C/min.
Compositional and Methylation Analyses
Compositional analysis was done by GLC-MS. Glycolipid and oligosaccharide samples (0.1 mg) were subjected to methanolysis (0.5 m HCl/MeOH, 45 min, 85 °C) and per-O-acetylated (15 μl of pyridine, 15 μl of Ac2O, 85 °C, 15 min). The configuration of the aldopyranosides was confirmed by GLC of acetylated (R)-2-octyl glycosides (28), whereby Rha was found to be l-configured and all other sugars d-configurated. Methylation analysis of the isolated core oligosaccharides was carried out after N-acetylation (Ac2O/NaOH) (29), HF treatment (48% aqueous HF, room temperature, 2 h), permethylation, hydrolysis (2 m TFA at 100 °C at 1.5 h and/or 4 m TFA at 100 °C for 6 h), reduction (NaBD4) and per-O-acetylation followed by GLC-MS analysis.
ESI-Mass Spectrometry
ESI-MS analyses of compounds 1, 2, and 3 were performed in the negative ion mode on a high resolution Fourier transform ion cyclotron resonance mass spectrometer (Apex Qe, Bruker Daltonics, Billerica, MA), equipped with a 7-tesla superconducting magnet and an Apollo dual ESI/MALDI ion source. Sample preparation and instrumental settings were recorded as described previously (11).
NMR Spectroscopy
The NMR spectra of oligosaccharides 1 and 2 were recorded at 300 K on a Bruker 600-MHz (AvanceII) NMR spectrometer equipped with a 5-mm quadruple resonance (1H,13C,15N,31P) probe with Z-gradient. Samples were dissolved in 0.75 ml of D2O (99.9% D, Cambridge Isotopes, Andover, MA) and lyophilized twice, and spectra were recorded in 0.50 ml of D2O (99.98% D). Signals were referenced to internal acetone (δH 2.225 ppm, δC 30.89 ppm), and Bruker standard software xwinnmr (version 3.5) was used to acquire and process these data.
NMR spectroscopic measurements of 3 were performed on a Bruker AvanceIII 700-MHz spectrometer equipped with an inverse 5-mm quadruple resonance Z-Grad cryoprobe (or with an inverse 1.7-mm triple resonance Z-Grad microcryoprobe). HPLC-purified compound 3 was dissolved in 25 mm deuterated sodium phosphate buffer (pH 6.8) containing 1.5% (w/v) DHPC-d40 (98% D, Cambridge Isotopes) and transferred to 5-mm Shigemi (1.3 mg in 325 μl) or 1.7-mm NMR microtubes (0.25 mg in 40 μl) (Bruker), depending on the sample material available. Spectra were recorded under conditions described earlier (32) with minor modifications. Elevated temperatures (325 K) for increasing spectral resolution and lower concentration of the detergent were used. 1H,13C signals were referenced via selected internal DHPC-d40 signals (H-2/C-2Gro; δH 5.27 ppm, δC 71.4 ppm) after external calibration with acetone (δH 2.225 ppm, δC 30.89 ppm) recorded under identical conditions. Signals of the H-3Gro in DHPC-d40 have been chosen to reference the 1H,31P HMQC NMR spectra, whereas the one-dimensional 31P NMR spectra were referenced to external aqueous 85% H3PO4 (δP 0.0 ppm). A mixing time of 200 ms was used in ROESY experiments. For the one-dimensional 1H NMR spectra, shown as projections of the F2 axis, the FID signal was multiplied by a Gaussian function (exponential broadening factor −2 Hz, Gaussian broadening factor 0.2 Hz) prior to Fourier transformation. 1H NMR assignments were confirmed by two-dimensional 1H,1H COSY and TOCSY experiments, and 13C NMR assignments were done by two-dimensional 1H,13C HSQC, based on the 1H NMR assignments. Interresidual connectivity and further evidence for 13C assignment were obtained from two-dimensional 1H,13C heteronuclear multiple-bond correlation and 1H,13C HSQC-TOCSY. Connectivity of phosphate groups was assigned by two-dimensional 1H,31P HMQC and 1H,31P HMQC-TOCSY. Bruker software Topspin version 3.1 was used to acquire and process these data.
RESULTS
The Core Oligosaccharide in the Y1C12 Mutant Is Representative of Rough-type C. canimorsus 5 WT LPS
The LPS isolated from the Y1C12 mutant was chosen for the structural analysis of the core oligosaccharide for reasons given below. SDS-PAGE analysis (data not shown) revealed that both WT and Y1C12 mutant contained a significant amount of rough-type LPS. In order to confirm the results obtained by SDS-PAGE, we performed compositional analysis of the core oligosaccharides from both LPS preparations (WT and mutant). They were prepared by acid hydrolysis in 2% AcOH followed by GPC under identical conditions and analyzed separately. Upon GLC-MS analysis, the core oligosaccharide fractions obtained from both strains showed the same retention time upon GPC, and no differences in their composition and proportions of sugars appeared. Both core structures were found to be composed of the same monosaccharides, suggesting that they are identical in both strains due to their genetic relationship. The only difference was the presence of components in the WT Cc5 LPS originating from the O-chain, which eluted prior to the core oligosaccharide (data not shown). As expected, the Y1C12 mutant gave better yield and higher purity for the core oligosaccharide preparation; therefore, the structural analysis described here has been performed with compounds isolated from the mutant, which was regarded to be representative of C. canimorsus LPS.
Isolation and Structural Analysis of the Core Oligosaccharide (1)
To investigate the structure of the core oligosaccharide, LPS of the Y1C12 mutant was hydrolyzed in AcOH, and the core oligosaccharide was purified by GPC. The core oligosaccharide was further analyzed by GLC-MS, ESI-MS, and NMR spectroscopy as described below. Earlier eluting higher molecular fractions contained minor amounts of sugars derived from the O-chain. GLC-MS analysis of the core oligosaccharide (methanolysis, per-O-acetylation) revealed the following monosaccharides: Rha, Gal, GalN, and Man in molar ratios of ∼1:1:1:2. Kdo and P-Etn could not be determined and/or quantified using this methodology.
The charge-deconvoluted ESI-MS spectrum of the core oligosaccharide sample recorded in the negative ion mode showed two major peaks, one having a mass of 1321.384 units, which was assigned to the intact core oligosaccharide composed of 3 hexoses, 1 hexosamine, 1 deoxyhexose, and 1 Kdo with two P-Etn residues (calc: 1321.335 units) being in full agreement with structure 1 (Fig. 1). The second major peak (1158.338 units) was identified to originate from 1 after loss of phosphoethanolamine and water (1-(P-Etn-H2O)) (calc.: 1158.334 units).
To determine the linkages and substitution pattern, 1 was subjected to methylation analysis. Two partially methylated sugar alditol acetates were identified, 2,3,4-Me3-Rha-ol and 3,4,6-Me3-GalNAc-ol, indicating that Rha and GalN represent two terminal sugar residues. Other sugars in the chain were 2,4,6-Me3-Gal-ol (1,3-linked Gal), 2,3,4-Me3-Hex-ol (1,6-linked hexose), and 2,6-Me2-Hex-ol (1,3,4-linked hexose). No sugar fragments of the Kdo were identified because these Kdo derivatives rapidly decompose under the conditions used.
The structure of the core oligosaccharide was further analyzed in detail by homo- and heteronuclear one- and two-dimensional NMR spectroscopy. Complete 1H and 13C signal assignments are given in Table 1. With the exception of β-Gal (δH 4.54 ppm, J1,2 8.1 Hz), all sugars were found to be present in α-linkage because their anomeric protons (H-1) resonate in the range between δH 5.0 and 5.26 ppm. The linkages between all sugars in 1 could be completely determined by ROESY and HMBC experiments (Table 2). The connectivity between H-1 of α-GalN (δH 5.21 ppm) and H-6a/H-6b of ManII (at δH 3.65/4.16 ppm) in the ROESY spectrum was indicative of the α-(1→6) linkage of the terminal GalN in agreement with the data of the methylation analysis. The anomeric proton of the second terminal Rha sugar residue (H-1, δH 5.01 ppm) showed NOE cross correlation to H-3 of Gal (δH 3.73 ppm). The 3-substituted Gal and the 6-substituted ManII were found both to be linked to the same sugar residue, namely ManI, thus representing a branching point in the core oligosaccharide. This was deduced from H-1 of β-Gal (δH 4.54 ppm) showing a strong and significant NOE to H-4 of ManI (δH 4.25 ppm) for one branch and from H-1 of ManII (δH 5.26 ppm) showing an intense NOE to H-3 of ManI (δH 4.17 ppm) representing the second branch. Taken together, the first mannose (ManI) is disubstituted: in position 3 with α-d-GalN-(1→6)-α-d-ManII-(1→ and in position 4 with α-l-Rha-(1→3)-β-d-Gal-(1→ (Fig. 1). The last anomeric NOE signal of H-1 (ManI, δH 5.05 ppm) showed a connectivity to H-5 (δH 4.24 ppm) of Kdo, which represents the “reducing end” of 1 with Kdo being exclusively in the α-anomeric form. All assignments of the NOE signals were further corroborated by data obtained from the HMBC spectrum (Table 2). The positions of the two P-Etn residues in 1 were identified from the chemical shifts of the carbon resonances in the HSQC spectrum (Table 1) in which the signal of Kdo H-4 was shifted to lower field due to substitution with P-Etn (δH 4.50 ppm/δC 72.2 ppm). The same observation was made for ManI H-6 (δH 4.22, 4.31 ppm/δC 64.9 ppm), which also is P-Etn-substituted. In addition, these signals showed cross-peaks in the 1H,31P HMQC and 1H,31P HMQC-TOCSY (Table 3) spectra, further corroborating this interpretation.
TABLE 1.
1H and 13C NMR data of the isolated core oligosaccharide (1), core backbone oligosaccharide (2), and rough-type LPS (3) from the C. canimorsus Y1C12 mutant
Compounds 1 and 2 were measured in D2O (300 K), whereas 3 was recorded in 25 mm Na-P buffer, pH 6.8, containing 1.5% DHPC-d40 at 325 K. For further details, see “Materials and Methods.” n.d., not determined.
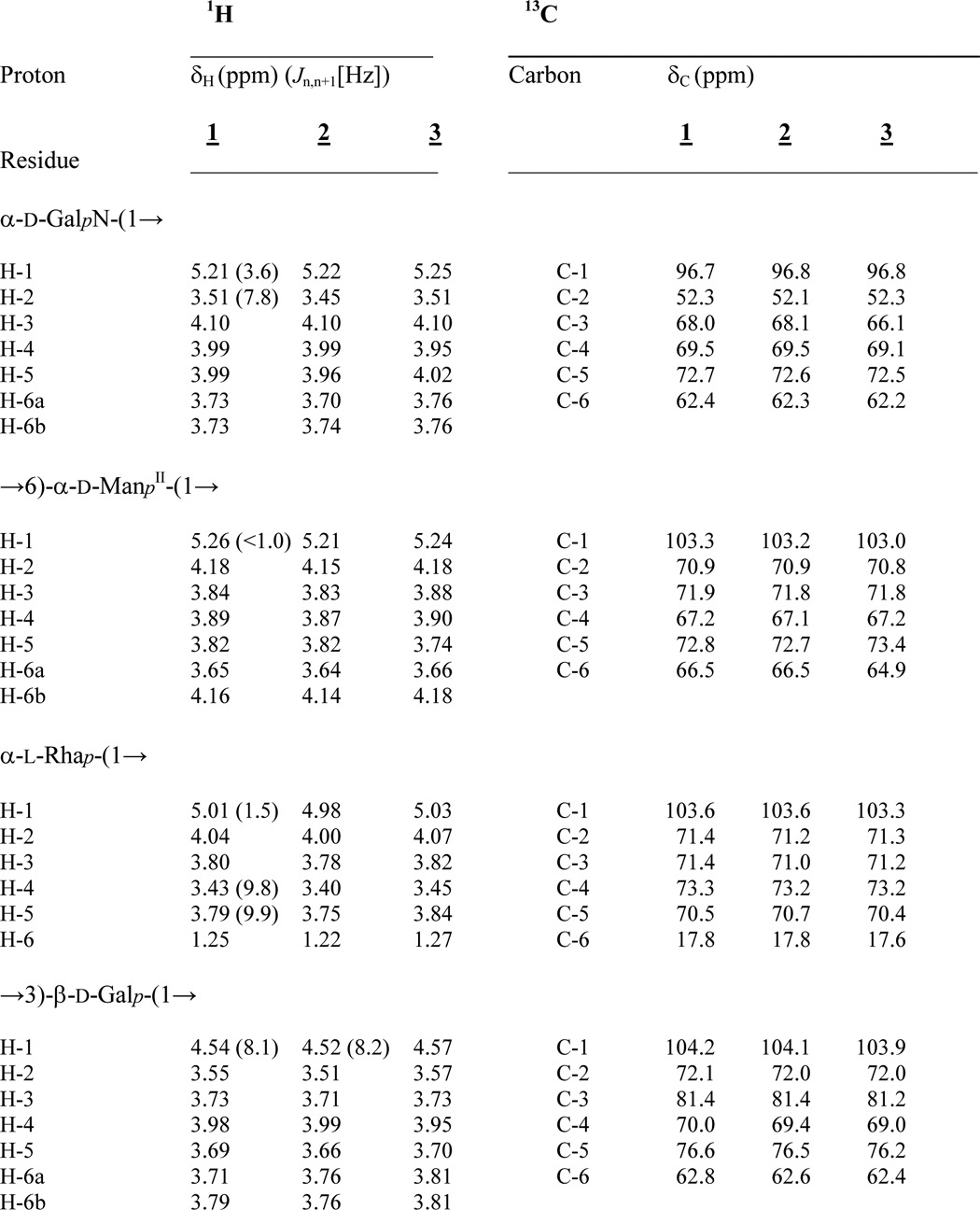
& 3Jl,p (6.8 Hz).
* Non-resolved multiplet.
** Lacking due to assignment by HSQC.
TABLE 2.
NOE and HMBC connectivities for compounds 1, 2, and 3
NOEs were classified into strong (s), medium (m), weak (w), and very weak (vw) after calibration to intraresidual NOEs (not listed in this table, except H-3 for Kdo (§). n.d., not detectable.

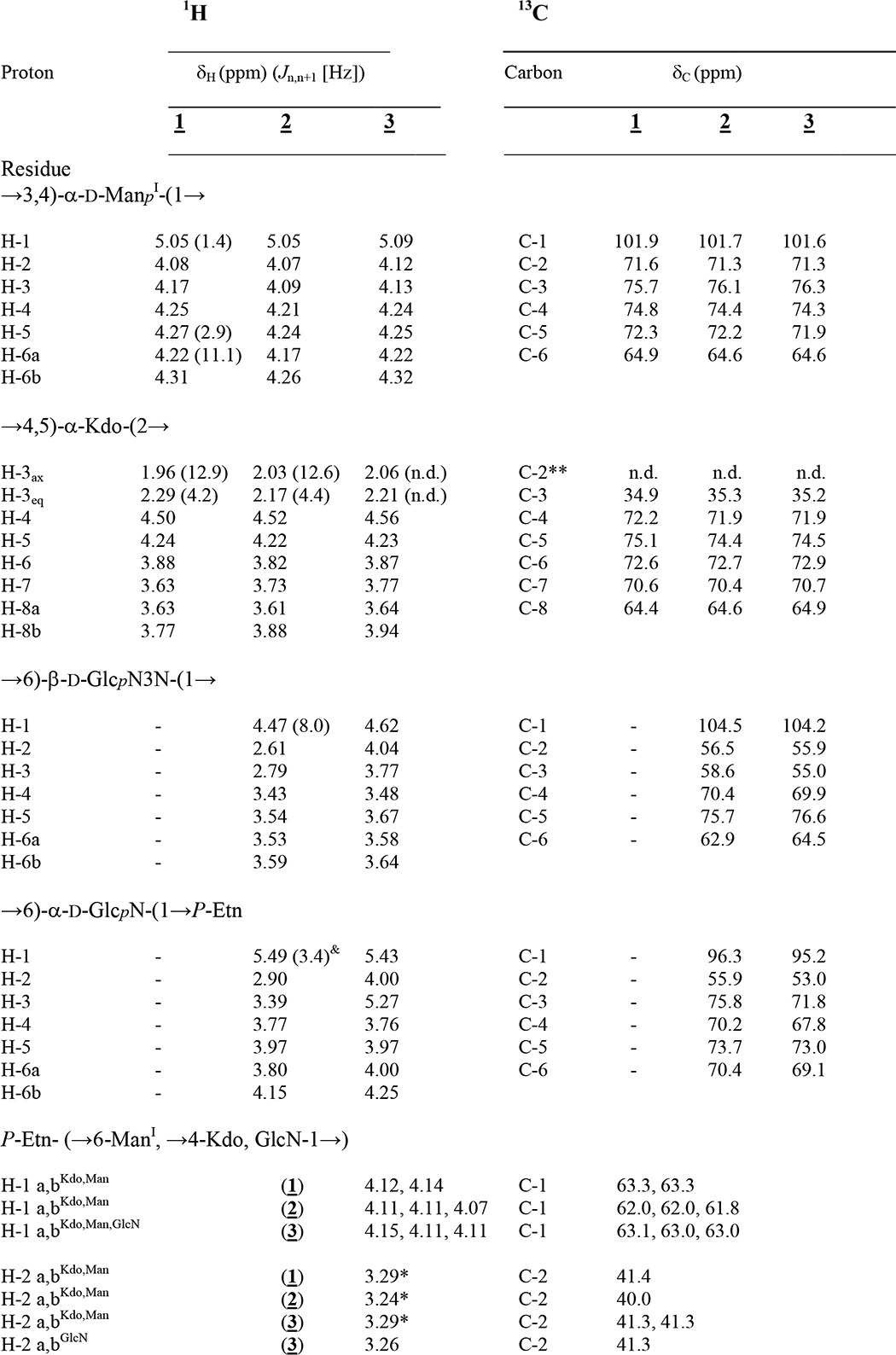
TABLE 3.
31P NMR (283.7 MHz) data of compounds 1, 2, and 3
31P signal resonances were assigned according to 1H,31P HMQC and 1H,31P HMQC-TOCSY spectroscopy.
Isolation and Structural Analysis of the Core Backbone Oligosaccharide (2)
Strong hydrazinolysis was used to isolate the core oligosaccharide with the lipid A backbone disaccharide attached. The oligosaccharide obtained from the completely deacylated LPS (50 mg) was purified by GPC. It resulted in 2 (Fig. 1) in low but acceptable yield. By using negative ion mode ESI-MS, an octasaccharide was identified consisting of Hex3-(HexN)2-HexN3N-dHex-Kdo (found 1721.537 u; calc. for C56H106O47N7P3 1721.517 units) corresponding to structure 2. The HSQC data of 2 are given in Table 1. Again, with the exception of β-Gal (δH 4.52 ppm, J1,2 8.2 Hz, δC 104.1 ppm) and GlcN3N (δH 4.47 ppm, J1,2 8.0 Hz, δC 104.5 ppm), all sugars were found to be α-linked. ROESY and HMBC experiments showed identical linkages as identified in the NMR spectra of the hexasaccharide 1 described above. The structural difference between 1 and 2 is the presence of the lipid A backbone [β-d-GlcpN3N-(1′→6)-α-d-GlcpN-1-P-Etn] to which the core oligosaccharide (1) is attached (at position 6′ of GlcpN3N).
The positions of the three P-Etn-residues were identified from the chemical shifts of the carbon resonances in the HSQC spectrum as well as by 1H,31P HMQC and 1H,31P HMQC-TOCSY spectra (Tables 1 and 3). The third phosphate was found attached to the lipid A hybrid backbone as P-Etn residue at GlcN C-1 (H-1/C-1 δH 5.49 ppm/δC 96.3 ppm). The typical dd signal of H-1 at GlcN (I) showed two coupling constants, one (3J1,2 3.4 Hz) indicating α-configuration and the second (3J1,P 6.8 Hz) characteristic of the α-anomeric phosphate substitution in the lipid A backbone (33). Furthermore, a weak NOE signal could be identified between GlcN H-1 (δH 5.49 ppm) and H-1a,b, the methylene protons of H-1a,b-P-Etn (δH 4.15, 4.11 ppm) (Table 1), indicating that P-Etn is α-glycosidically bound to C-1 of GlcN of the lipid A hybrid backbone (11).
Isolation and Structural Analysis of the Intact Rough-type LPS
The intact rough-type LPS (3) (Fig. 1) was obtained from the LPS preparation of the Y1C12 mutant by preparative HPLC (Fig. 2). As mentioned above, this rough-type LPS represents a significant molecular species present in the native WT LPS. The HPLC analysis of the mild acetate buffer-treated LPS resulted in three major fractions (pools 1, 2, and 3) (Fig. 2). Pool 2 was found to contain the intact rough-type LPS (3) and was further analyzed by MS and NMR, as described here in more detail. The other pools were identified as crude and contaminated S-form LPS (pool 1) and lipid A (pool 3), respectively, the analysis and structure of which is described elsewhere (11).
The ESI-MS spectrum of 3 is shown in Fig. 3. The LPS is composed of a hexasaccharide (1) (Fig. 1) with a penta-acylated lipid A attached, this way representing a rough-type LPS. The molecular mass identified (found: 2976.680 units; Fig. 3) was in full agreement with the structure 3, being composed of Hex3-(HexN)-dHex-Kdo with two P-Etn residues in the core and the complete penta-acylated lipid A with one P-Etn at the C-1 of GlcN in the hybrid lipid A backbone (11) (calc. for C136H256O56N7P3 [M] = 2976.661 units and [M + Na] = 2998.639 units). The ESI-MS of 3 also showed further heterogeneity in the acylation of the lipid A (i.e. the chain length of the fatty acids differing by one or two methylene group(s)) (-CH2-, Δm/z = 14 units), which was also previously found in the free lipid A (11).
FIGURE 3.

Charge-deconvoluted mass spectrum of 3 obtained by ESI Fourier transform ion cyclotron resonance MS in the negative ion mode. The monoisotopic mass found (2976.680 Da) is in excellent agreement with structure 3 (Fig. 1). The inlet shows an enlargement of the isotopic peaks obtained from the most abundant peak. The calculated ion intensity profile (red) is nearly identical to the measured one (blue), indicating the heterogeneity in the hybrid lipid A backbone (GlcN3N-GlcN) versus E. coli-type backbone (GlcN-GlcN) to be minor (∼1–2%). Mass values in the inset represent the calculated masses for 3.
The HPLC purification of C. canimorsus rough-type LPS (3) revealed a degree of purity sufficient for structural and conformational studies using high-field NMR spectroscopy without 13C,15N-labeling. To this end, homonuclear two-dimensional techniques (COSY, TOCSY, and ROESY) and heteronuclear two-dimensional spectra 1H,13C HSQC, 1H,13C HMBC, 1H,31P HMQC, and 1H,31P HMQC-TOCSY of compound 3 in small DHPC-d40 micelles could be obtained with rather good resolution of all signals (Fig. 4). Fig. 5 shows the most complex but well resolved part of the 1H,13C HSQC spectrum of 3 in more detail. Because water was used as solvent, sugar resonances and assignments of the glycolipid can be compared directly with those of the deacylated and purified oligosaccharides 1 and 2 representing partial structures of this LPS (Table 1). The terminal (non-reducing) part of both oligosaccharides (1 and 2) had almost identical chemical shifts and coupling constants when compared with those of the native rough-type LPS (3). Notably, 1H,13C signals from atoms of the hybrid lipid A backbone (GlcpN3N-GlcpN disaccharide) embedded in the micellar membrane (Table 1) as well as the signals of the fatty acids (Table 4) were well resolved in the HSQC spectrum of 3 (Figs. 4–6). However, as can be seen in Fig. 5, the ring protons/carbons of the lipid A backbone appear significantly reduced (∼50% of intensity) as compared with those obtained from the core oligosaccharide due to an impaired flexibility and resultant line broadening in the NMR spectrum. Despite the fact that the peak resolution of signals obtained from compound 3 were slightly reduced, some diagnostic 3J-coupling constants in the flexible core oligosaccharide could nevertheless be identified (Fig. 5 and Table 1). For the complete assignment of the HSQC spectrum of 3, data obtained from oligosaccharides 1 and 2 were extremely helpful. Especially the assignment of positions of the charged, zwitterionic P-Etn residues could be determined by changes in the chemical shift values and by the NOE connectivities (Tables 1 and 2). Of note is the fact that all negatively charged phosphate residues were found to be “neutralized” by positively charged ethanolamine groups (P-O-CH2-CH2-NH3+). This fact holds true not only for the three P-Etn residues but also for the carboxyl group of Kdo, which is “neutralized” by the positively charged GalpNH3+ residue in the core oligosaccharide when considering the complete rough-type LPS structure of the Y1C12 mutant.
FIGURE 4.

HSQC-contour plot of the complete spectrum (1H 6.0–0.5 ppm, 13C 120–5 ppm) in which all three distinct regions of 3 can be clearly distinguished. Ring protons/carbon signals and those of fatty acids are enlarged in Fig. 5 and 6, to demonstrate good signal resolution. Signals from the DHPC-d40 (98% D) detergent are shown in orange. The 1H NMR spectrum is shown along the F2 axis at the top.
FIGURE 5.

Section of the 1H,13C-HSQC NMR (700 MHz) spectrum showing ring proton/carbon signals of 3. The spectrum was recorded at 325 K in D2O containing 1.5% DHPC-d40. Selected 1H,13C ring signals and their assignments are indicated. 1H,13C signals from the DHPC-d40 detergent (98% D) are shown in orange. The one-dimensional 1H spectrum is shown as a projection of the F2 axis.
TABLE 4.
1H and 13C NMR data of the fatty acids in 3
Data were taken from HSQC spectra. Signals of C-1 carbons are not indicated.
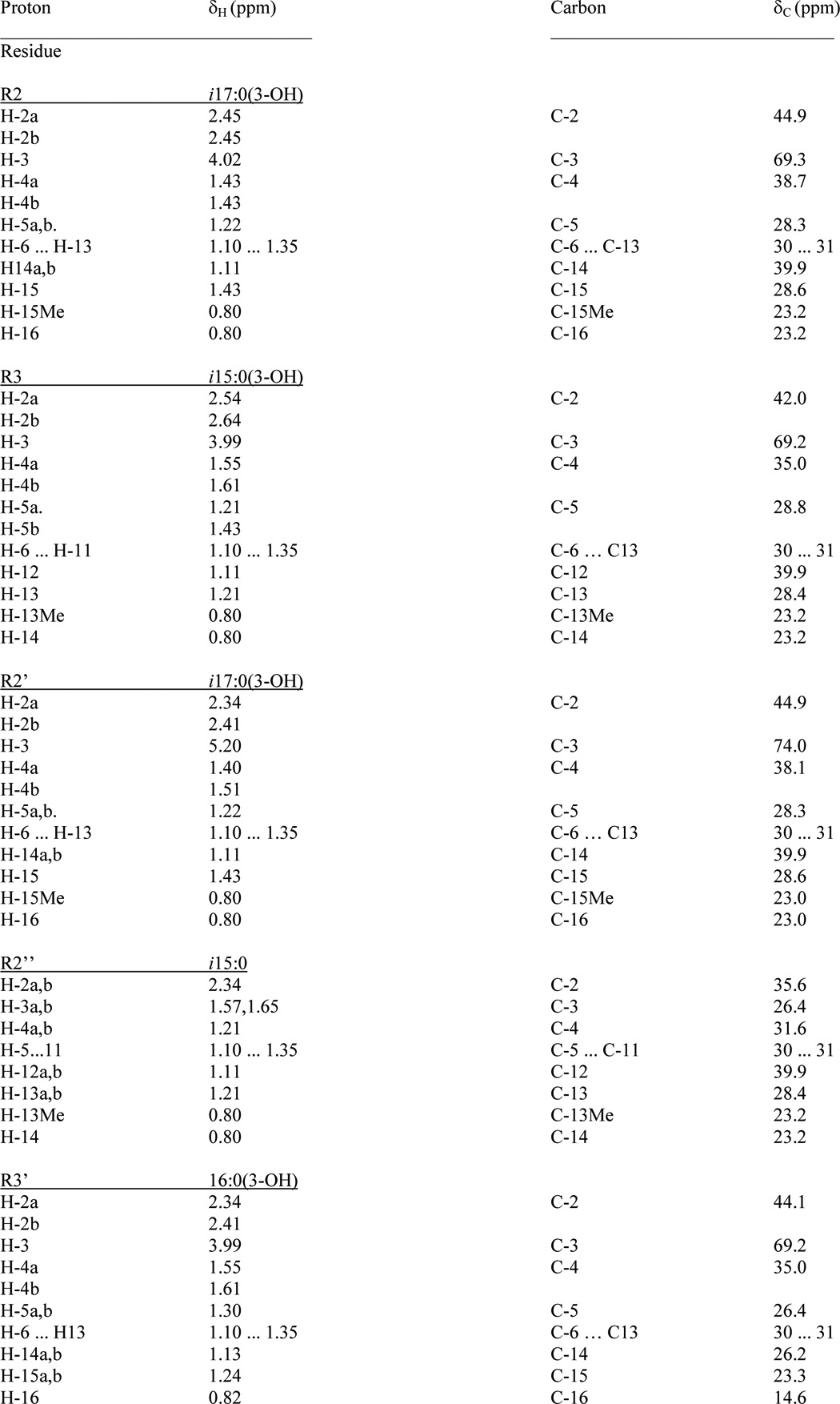
FIGURE 6.

HSQC contour plot showing the region with fatty acid signals H-2/C-2 to H-ω/C-ω as well as H-3ax,eq/C-3 Kdo and H-6/C-6 of l-Rha sugars. Signals assigned to one acyloxyacyl residue (R2″-R2′), (3R)-3-(15-methyl-hexadecanoyloxy)-13-methyl-tetradecanoic acid, attached to position 2′ of GlcpN3N (II) as well as those originating from the (R)-3-hydroxy-hexadecanoic acid (R3′) are indicated. Signals from the DHPC-d40 detergent are shown in orange.
Concerning substitution of the phosphate residues, all three were present in the form of P-Etn attached to the core backbone oligosaccharide. The resonances of the H-6a,b/C-6 signals of ManI could be clearly distinguished from the unsubstituted H-6a,b/C-6 signals of Man attached to Kdo (34). ManI signals at δH 4.32 and 4.22 ppm/C-6: δC 64.6 ppm indicated the 6-position to be substituted by P-Etn. The assignment of the P-Etn residue resulted from a weak NOE signal of α-ManI (H-1 δH 5.09 ppm) to -CH2-NH3+ of P-Etn (H-2a,b δH 3.29 ppm) (Table 3).
The second phosphate in 3 was found attached to C-4 of Kdo (H-4/C-4 δH 4.56 ppm/δC 71.9 ppm). Its position was deduced from a weak NOE signal identified between Kdo H-4 and H-1a,b (δH ∼4.11 ppm) as well as H-2a,b (δH ∼3.29 ppm), both representing methylene protons of the Etn residue attached to the 4-P of Kdo.
The third phosphate was found to be linked to C-1 of the lipid A backbone GlcN (H-1/C-1 δH 5.43 ppm/δC 95.2 ppm). Its position was deduced from a weak NOE signal identified between GlcN H-1 (δH 5.43 ppm) and the methylene protons H-1a,b (δH ∼4.15 ppm) of 1-P-Etn (Table 1), indicating that P-Etn was α-glycosidically bound to C-1 of GlcN of the lipid A hybrid backbone (11).
Two-dimensional 1H,31P NMR spectra (HMQC and HMQC-TOCSY) were used to analyze all phosphate positions in more detail. Despite the disadvantage that the 31P signals of the phosphate buffer (25 mm) and DHPC-d40 detergent (3.3 mm) were more intense, compared with phosphate signals of 3 (1.3 mm), the HMQC-TOCSY spectra allowed us to assign all signals from the individual sugar residues unequivocally (Table 1).
The ROESY spectrum of 3 revealed correlations of the anomeric protons between all sugars by which substitution and sequence of the complete core oligosaccharide could be determined (Fig. 7). Comparison with those ROESY signals of the oligosaccharides 1 and 2, lacking the fatty acids of the lipid A, revealed identical but also different conformations (Table 2). Of utmost interest was the structural and conformational analysis of compound 3. It deserves special mention that some selected ROESY signals identified there were lacking in 1 and 2 and vice versa.
FIGURE 7.

NOE connectivities detected in 3 by which the linkages of sugars and P-Etn residues were assigned. Some of the anomeric protons (e.g. H-1 of l-Rha and ManII) show NOE cross-peaks (Table 2) not only to the substituting but also to the neighboring sugar protons to which they are bound, indicating a rather twisted conformation of the branched oligosaccharide. The fatty acid residues attached to the lipid A backbone (R3′, R2″-R2′, R3, and R2) are abbreviated for clarity.
Of special note is one specific conformation expressed by the core oligosaccharide in 3, which was only observed in this compound. Here weak but detectable NOE signals between the axial proton of Kdo (H-3ax) above and those two axial protons in ManI (H-3 and H-5) below the pyranose ring indicate a significant rotational freedom around the C1-ManI-O-C5-Kdo bond in the core oligosaccharide (Fig. 8). This is unexpected because the glycolipid part of the rough-type LPS is embedded into the micellar membrane of DHPC-d40, resulting in a steric hindrance of the free rotation around this bond between ManI and Kdo.
FIGURE 8.

Special conformation adopted by the intact core when bound to the lipid A. Shown is part of the ROESY spectrum of 3 with NOE signals showing interresidual connectivities between H-3ax of Kdo and H-3/H-5 of ManI. This indicates a free rotation in the core oligosaccharide of about 180° (gray arrow) around the ManI-C1-O-C5-Kdo bond axis as shown in the bottom part together with the membrane matrix of the DHPC micelles. Indicated are the two disaccharides attached to ManI (i.e. α-l-Rhap-(1→3)-β-d-Galp-(1→ (OR1) and α-d-GalNp-(1→6)-α-d-ManpII-(1→4) (OR2)).
DISCUSSION
The structural analysis of a complex glycolipid by NMR in water described here represents the first example of a natural abundant highly purified intact rough-type LPS embedded in small micelles in a water mimetic. Such NMR spectra could so far only be obtained with expensive 13C,15N-labeled rough-type LPS (32). The high purity of the LPS preparation permits the use of NMR spectroscopy for a thorough and complete analysis of not only the primary but also the secondary structure (i.e. the conformation of natural abundant LPS molecules when present in water). It is well documented that the conformations of LPS are critical for the interaction with its biological partners, initial binding proteins (LBP (36) and CD14 (37)) and also with the final target receptors (MD-2/TLR4 (17)). Using the approach described here, LPS conformational analysis now becomes accessible for its investigation in water, which cannot be analyzed by x-ray crystallography due to the extreme high flexibility of the sugars of the core oligosaccharides (17). Another advantage of this approach is the fact that, after extensive and non-destructive NMR analysis, the LPS preparation under investigation remains available for further biological studies.
In order to demonstrate the feasibility and power of this NMR method, we have chosen a rough-type LPS of C. canimorsus, the lipid A structure and biological (endotoxic) activities of which have recently been described (11). In this previous study, only the lipid A and not the structures of the core oligosaccharide (1) and the complete rough-type LPS (3) were analyzed. In the present study, we aim to close this gap. We could confirm the impact of our method in comparing it with the classical way of determining the core oligosaccharide by cleaving the LPS molecule in two parts and separating the resulting core oligosaccharide from the water-insoluble free lipid A. After mild acid treatment, a hexasaccharide (1) could be isolated in pure form. In a second approach, strong hydrazinolysis to deacylate the LPS revealed an octasaccharide (2), representing the carbohydrate part of the rough-type LPS (core oligosaccharide with a hybrid lipid A backbone (GlcpN3N-GlcpN) attached).
We also detected trace amounts (1–2%) of the E. coli-type backbone (GlcpN-GlcpN). This lipid A backbone type has also been found in the LPS of Flavobacterium meningosepticum, representing another species of Flavobacteriaceae to which C. canimorsus belongs. However, in F. menigosepticum, the E. coli-type backbone dominated, and the proportion of both lipid A backbones was inverted (38), thus being different from the lipid A backbone identified here.
Both oligosaccharides 1 and 2 have been extensively analyzed by NMR spectroscopy. Because NMR spectra for 3 were also recorded in water, a direct comparison of the NMR resonances from all 1H,13C and 31P signals (and even, in some cases, coupling constants of 1H signals (3Jn,n + 1)) could be made. In addition, the comparability of NMR data facilitates further LPS structural analysis considerably, because many NMR data of various linear and branched polysaccharides are referenced and available via internet databases and utilities (e.g. CASPER) (39).
Phosphorylation of Kdo at position C-4 appears not to be unique for C. canimorsus, where it is present in its P-Etn form. It is also found in core oligosaccharides from other Gram-negative bacteria, such as Haemophilus influenzae I-69 (40) and in non-typeable H. influenzae, where this position of Kdo carries a pyrophosphoethanolamine residue (41). Additionally, such phosphorylation has been identified in the inner core oligosaccharide of Bordetella pertussis (42) and Vibrio cholerae (43). The 4-P at KdoI can, by means of charge, replace the carboxyl group of the second Kdo (KdoII), usually present in the LPS of Enterobacteriaceae. This second negative charge represents an important and conserved structural feature in nearly all Gram-negative bacteria. However, in the case of C. canimorsus LPS, the 4-P does not contribute to the second negative charge because of its positively charged Etn substituent.
We expected that the zwitterionic residues of P-Etn at C-4 of Kdo may influence the conformation of the oligosaccharide considerably, both in the free oligosaccharide forms (1 and 2) and in complete rough-type LPS (3). Diagnostic NOEs were found to be indicative of different conformers, which are influenced by zwitterionic interactions, because they also influence the freedom in adopting various conformations of this branched core hexasaccharide. Such specific NOEs are shown in Figs. 7 and 8, and corresponding NMR data are given in Table 2. Surprisingly, we found a high flexibility for the core oligosaccharide even when attached to the lipid A anchor, which is expected to refit the conformational freedom of the inner core oligosaccharide when embedded via the lipid A anchor into the bacterial outer membrane. The spectrum and interpretation of the ROESY signals given in Fig. 8 show one extremely distorted conformer. It is most likely that this conformational freedom of the core oligosaccharide is also realized in case where the LPS molecule is located in the outer leaflet of the outer bacterial membrane.
In agreement with the x-ray crystallographic data, this extreme kind of conformational freedom of the core oligosaccharide in the native form is advantageous for a specific interaction of charged and polar groups of the LPS with those in the corresponding endotoxin target proteins, such as MD-2, TLR4, CD-14, and LBP (17). But most interestingly, this special role of the core oligosaccharide for endotoxin/receptor interaction cannot be observed by x-ray crystallographic studies. NMR conformational analysis, as used here, might be considered to represent a complementary method to investigate carbohydrate conformations and dynamics, which has not been possible to describe in detail so far in these molecules (17, 44, 45).
Recently, we published the structure-function analysis of lipid A isolated from the LPS of C. canimorsus 5 and found that this lipid A is penta-acylated and lacks the 4′-phosphate in the hybrid lipid A backbone (11). Because the 4′-phosphate-deprived lipid A is known to reduce the endotoxic activity significantly (12), we here try to describe a model that can explain why the C. canimorsus LPS nevertheless is able to induce an overwhelming sepsis. The impact for endotoxicity of this negatively charged 4′-phosphate came from data obtained by x-ray crystallography on the TLR4·MD-2·LPS complex (17) as well as from functional studies (46) and genome-wide profiling (47). This ionic interaction is important because it is critical for the ligand affinity of lipid A, enabling formation of a hexameric (TLR4·MD-2·LPS)2 complex, which is necessary for TLR4-mediated signaling. Therefore, we raised the question of whether the negatively charged carboxylate group in position C-1 of the Kdo might be able to substitute the lacking 4′-phosphate in the lipid A backbone. This assumption is based on the complex model of Park et al. (17) as well as our own modeling calculations (11).
We concluded that the negatively charged carboxyl group at C-1 of the Kdo can adopt the function of the negative charge of the 4′-P attached to the non-reducing part of the lipid A backbone. Based on our model, the lacking 4′-P and, hence, its negative charge can be structurally and functionally replaced by the carboxy group of Kdo, this way resuming the electrostatic binding to the positively charged amino acids identified for binding to TLR4 (Arg-264 and Lys-362) and MD-2 (Ser-118 and Lys-58) (Protein Data Bank code 3FXI) (17). The importance of the charged groups for LPS·TLR4·MD-2 has also been identified by Meng et al. (46) for the binding of lipid IVA to MD-2 and TLR4. Molecular modeling revealed that this negative charge might indeed be an essential structural feature for this kind of binding (core oligosaccharide to MD-2 (11)). Notably, in human MD-2, three positively charged amino acids (Lys-122, Lys-125, and Lys-58) have been identified by functional studies to be important for TLR4-independent LPS recognition by MD-2. Of these, Lys-58 is in spatial proximity to the 4′-P of the lipid A backbone. We have previously shown by molecular modeling that C. canimorsus lipid A is probably binding to human MD-2 in a very similar way as E. coli hexa-acylated lipid A (11). This further supports the idea that the negatively charged carboxylate group of the proximate Kdo may indeed compensate for the lack of the 4′-P in the lipid A backbone of C. canimorsus (48). The outcome of this ionic compensation might explain the strongly enhanced endotoxic activity, when comparing C. canimorsus free lipid A with rough-type LPS, differing by a factor of 20,000 (11). The fact that the negative charge of a carboxymethyl group in lipid A indeed can mimic that of a phosphate residue has been shown for different synthetic lipid A structures (49), further supporting our interpretation.
In this context, another important structural feature of the rough-type LPS of C. canimorsus deserves special mention. The innate immune defense takes advantage of antimicrobial peptides (AMPs), which represent one of the first lines of defense against pathogenic bacteria. AMPs are present in phagocytic cells on body surfaces, skin, and mucosa (50). In most cases, AMPs represent positively charged peptides and act as neutralizing agents able to kill bacterial pathogens by insertion into the membrane and disruption (51). The main targets of AMPs in Gram-negative bacteria, therefore, are negatively charged polar lipids on the surface of the bacterial outer membrane, especially LPS (52). However, due to the zero net charge of the LPS/lipid A structure described here, C. canimorsus is able to avoid this kind of ionogenic attack by AMPs. Therefore, this structural element provides an advantage against immunological defense based on positively charged antimicrobial peptides secreted by the host, especially in the dog's mouth, which represents the natural habitat of C. canimorsus bacteria.
Acknowledgments
We are thankful to Y. A. Knirel, O. Bystrova, and E. Zdorovenko (N. D. Zelinsky Institute, Moscow, Russia) for assistance at the beginning of the project; E. Vinogradov (National Research Council, Ottawa, Canada) for help with interpretation of NMR spectra; and S. Meier (Department of Chemistry, Technical University of Denmark, Lyngby, Denmark) for critically reading the manuscript. The skillful assistance of B. Kunz with ESI-MS analysis is also gratefully acknowledged.
This work was supported in part by the Cluster of Excellence “Inflammation at Interfaces” (EXC306).
- P-Etn
- phosphoethanolamine
- DHPC
- 1,2-dihexanoyl-sn-glycero-3-phosphocholine
- DHPC-d40
- perdeutero-DHPC
- ELSD
- evaporative light-scattering detector
- Gal
- galactose
- GalN
- 2-amino-2-deoxy-d-galactose
- GLC
- gas-liquid chromatography
- GlcN
- 2-amino-2-deoxy-d-glucose
- GlcN3N
- 2,3-diamino-2,3-dideoxy-d-glucose
- GPC
- gel permeation chromatography
- Gro
- glycerol
- HMBC
- heteronuclear multiple-bond correlation
- HMQC
- heteronuclear multiple quantum coherence
- HSQC
- heteronuclear single quantum coherence
- Kdo
- 3-deoxy-d-manno-oct-2-ulosonic acid
- Man
- mannose
- MD-2
- myeloid differentiation factor 2
- P
- phosphate
- Rha
- rhamnose
- ROESY
- rotating-frame nuclear Overhauser effect spectroscopy
- TLR2 and TLR4
- Toll-like receptor 2 and 4, respectively
- TOCSY
- total correlated spectroscopy
- Cc5
- C. canimorsus 5
- ESI
- electrospray ionization
- AMP
- antimicrobial peptide.
REFERENCES
- 1. Oehler R. L., Velez A. P., Mizrachi M., Lamarche J., Gompf S. (2009) Bite-related and septic syndromes caused by cats and dogs. Lancet Infect. Dis. 9, 439–447 [DOI] [PubMed] [Google Scholar]
- 2. Janda J. M., Graves M. H., Lindquist D., Probert W. S. (2006) Diagnosing Capnocytophaga canimorsus infections. Emerg. Infect. Dis. 12, 340–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shin H., Mally M., Kuhn M., Paroz C., Cornelis G. R. (2007) Escape from immune surveillance by Capnocytophaga canimorsus. J. Infect. Dis. 195, 375–386 [DOI] [PubMed] [Google Scholar]
- 4. Mally M., Paroz C., Shin H., Meyer S., Soussoula L. V., Schmiediger U., Saillen-Paroz C., Cornelis G. R. (2009) Prevalence of Capnocytophaga canimorsus in dogs and occurrence of potential virulence factors. Microbes Infect. 11, 509–514 [DOI] [PubMed] [Google Scholar]
- 5. Pers C., Gahrn-Hansen B., Frederiksen W. (1996) Capnocytophaga canimorsus septicemia in Denmark, 1982–1995: review of 39 cases. Clin. Infect. Dis. 23, 71–75 [DOI] [PubMed] [Google Scholar]
- 6. Mally M., Shin H., Paroz C., Landmann R., Cornelis G. R. (2008) Capnocytophaga canimorsus: a human pathogen feeding at the surface of epithelial cells and phagocytes. PLoS Pathog. 4, e1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Manfredi P., Renzi F., Mally M., Sauteur L., Schmaler M., Moes S., Jenö P., Cornelis G. R. (2011) The genome and surface proteome of Capnocytophaga canimorsus reveal a key role of glycan foraging systems in host glycoproteins deglycosylation. Mol. Microbiol. 81, 1050–1060 [DOI] [PubMed] [Google Scholar]
- 8. Renzi F., Manfredi P., Mally M., Moes S., Jenö P., Cornelis G. R. (2011) The N-glycan glycoprotein deglycosylation complex (Gpd) from Capnocytophaga canimorsus deglycosylates human IgG. PLoS Pathog. 7, e1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shin H., Mally M., Meyer S., Fiechter C., Paroz C., Zaehringer U., Cornelis G. R. (2009) Resistance of Capnocytophaga canimorsus to killing by human complement and polymorphonuclear leukocytes. Infect. Immun. 77, 2262–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alexander C., Rietschel E. T. (2001) Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 7, 167–202 [PubMed] [Google Scholar]
- 11. Ittig S., Lindner B., Stenta M., Manfredi P., Zdorovenko E., Knirel Y. A., dal Peraro M., Cornelis G. R., Zähringer U. (2012) The lipopolysaccharide from Capnocytophaga canimorsus reveals an unexpected role of the core-oligosaccharide in MD-2 binding. PLoS Pathog. 8, e1002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Loppnow H., Brade H., Dürrbaum I., Dinarello C. A., Kusumoto S., Rietschel E. T., Flad H.-D. (1989) IL-1 induction-capacity of defined lipopolysaccharide partial structures. J. Immunol. 142, 3229–3238 [PubMed] [Google Scholar]
- 13. Rietschel E. T., Kirikae T., Schade F. U., Mamat U., Schmidt G., Loppnow H., Ulmer A. J., Zähringer U., Seydel U., Di Padova F. (1994) Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 8, 217–225 [DOI] [PubMed] [Google Scholar]
- 14. Zähringer U., Lindner B., Rietschel E. T. (1999) Chemical structure of lipid A: recent advances in structural analysis of biologically active molecules. Brade H., Opal S., Vogel S., Morrison D. C. (eds) Endotoxin in Health and Disease, pp. 93–114, Marcel Dekker, New York [Google Scholar]
- 15. Raetz C. R. H., Garrett T. A., Reynolds C. M., Shaw W. A., Moore J. D., Smith D. C., Jr., Ribeiro A. A., Murphy R. C., Ulevitch R. J., Fearns C., Reichart D., Glass C. K., Benner C., Subramaniam S., Harkewicz R., Bowers-Gentry R. C., Buczynski M. W., Cooper J. A., Deems R. A., Dennis E. A. (2006) Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J. Lipid Res. 47, 1097–1111 [DOI] [PubMed] [Google Scholar]
- 16. Richter W., Vogel V., Howe J., Steiniger F., Brauser A., Koch M. H. J., Roessle M., Gutsmann T., Garidel P., Mäntele W., Brandenburg K. (2011) Morphology, size distribution, and aggregate structure of lipopolysaccharide and lipid A dispersions from enterobacterial origin. Innate Immun. 17, 427–438 [DOI] [PubMed] [Google Scholar]
- 17. Park B. S., Song D. H., Kim H. M., Choi B.-S., Lee H., Lee J.-O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 [DOI] [PubMed] [Google Scholar]
- 18. DeMarco M. L., Woods R. J. (2011) From agonist to antagonist: structure and dynamics of innate immune glycoprotein MD-2 upon recognition of variably acylated bacterial endotoxins. Mol. Immunol. 49, 124–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Westphal O., Jann K. (1965) Bacterial lipopolysaccharides. Extraction with phenol-water and further applications of the procedure. Methods Carbohydr. Chem. 5, 83–91 [Google Scholar]
- 20. Galanos C., Lüderitz O., Westphal O. (1969) A new method for the extraction of R lipopolysaccharides. Eur. J. Biochem. 9, 245–249 [DOI] [PubMed] [Google Scholar]
- 21. Vinogradov E., Perry M. B., Conlan J. W. (2002) Structural analysis of Francisella tularensis lipopolysaccharide. Eur. J. Biochem. 269, 6112–6118 [DOI] [PubMed] [Google Scholar]
- 22. Kawai Y., Yano I., Kaneda K. (1988) Various kinds of lipoamino acids including a novel serine-containing lipid in an opportunistic pathogen Flavobacterium: their structure and biological activities on erythrocytes. Eur. J. Biochem. 171, 73–80 [DOI] [PubMed] [Google Scholar]
- 23. Godchaux W., 3rd, Leadbetter E. R. (1980) Capnocytophaga spp. contain sulfonolipids that are novel in procaryotes. J. Bacteriol. 144, 592–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Godchaux W., 3rd, Leadbetter E. R. (1984) Sulfonolipids of gliding bacteria. Structure of the N-acylaminosulfonates. J. Biol. Chem. 259, 2982–2990 [PubMed] [Google Scholar]
- 25. Caroff M., Tacken A., Szabó L. (1988) Detergent-accelerated hydrolysis of bacterial endotoxins and determination of the anomeric configuration of the glycosyl phosphate present in the “isolated lipid A” fragment of the Bordetella pertussis endotoxin. Carbohydr. Res. 175, 273–282 [DOI] [PubMed] [Google Scholar]
- 26. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 27. Tsai C. M., Frasch C. E. (1982) A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem. 119, 115–119 [DOI] [PubMed] [Google Scholar]
- 28. Leontein K., Lindberg B., Lönngren J. (1978) Assignment of absolute configuration of sugars by g.l.c. of their acetylated glycosides formed from chiral alcohols. Carbohydr. Res. 62, 359–362 [Google Scholar]
- 29. Hase S., Rietschel E. T. (1976) Isolation and analysis of the lipid A backbone. Lipid A structure of lipopolysaccharides from various bacterial groups. Eur. J. Biochem. 63, 101–107 [DOI] [PubMed] [Google Scholar]
- 30. Pitta T. P., Leadbetter E. R., Godchaux W., 3rd (1989) Increase of ornithine amino lipid content in a sulfonolipid-deficient mutant of Cytophaga johnsonae. J. Bacteriol. 171, 952–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gomi K., Kawasaki K., Kawai Y., Shiozaki M., Nishijima M. (2002) Toll-like receptor 4-MD-2 complex mediates the signal transduction induced by flavolipin, an amino acid-containing lipid unique to Flavobacterium meningosepticum. J. Immunol. 168, 2939–2943 [DOI] [PubMed] [Google Scholar]
- 32. Wang W., Sass H.-J., Zähringer U., Grzesiek S. (2008) Structure and dynamics of 13C,15N-labeled lipopolysaccharide in a membrane mimetic. Angew. Chem. Int. Ed. Engl. 47, 9870–9874 [DOI] [PubMed] [Google Scholar]
- 33. Zähringer U., Sinnwell V., Peter-Katalinic J., Rietschel E. T., Galanos C. (1993) Isolation and characterization of the tetrasaccharide (bis)phosphate from the glycosyl backbone of Salmonella minnesota and Escherichia coli Re-mutant lipopolysaccharides. Tetrahedron 49, 4193–4200 [Google Scholar]
- 34. Moll H., Knirel Y. A., Helbig J. H., Zähringer U. (1997) Identification of an α-d-Manp-(1→8)-Kdo disaccharide in the inner core region and the structure of the complete core region of the Legionella pneumophila serogroup 1 lipopolysaccharide. Carbohydr. Res. 304, 91–95 [DOI] [PubMed] [Google Scholar]
- 35. Kondakova A., Lindner B. (2005) Structural characterization of complex bacterial glycolipids by Fourier transform mass spectrometry. Eur. J. Mass Spectrom. 11, 535–546 [DOI] [PubMed] [Google Scholar]
- 36. Kohara J., Tsuneyoshi N., Gauchat J.-F., Kimoto M., Fukudome K. (2006) Preparation and characterization of truncated human lipopolysaccharide-binding protein in Escherichia coli. Protein Expr. Purif. 49, 276–283 [DOI] [PubMed] [Google Scholar]
- 37. Kim J.-I., Lee C. J., Jin M. S., Lee C.-H., Paik S.-G., Lee H., Lee J.-O. (2005) Crystal structure of CD14 and its implications for lipopolysaccharide signaling. J. Biol. Chem. 280, 11347–11351 [DOI] [PubMed] [Google Scholar]
- 38. Kato H., Haishima Y., Iida T., Tanaka A., Tanamoto K. (1998) Chemical structure of lipid A isolated from Flavobacterium meningosepticum lipopolysaccharide. J. Bacteriol. 180, 3891–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jansson P. E., Kenne L., Widmalm G. (1989) Computer-assisted structural analysis of polysaccharides with an extended version of casper using 1H- and 13C-n.m.r. data. Carbohydr. Res. 188, 169–191 [DOI] [PubMed] [Google Scholar]
- 40. Helander I. M., Lindner B., Brade H., Altmann K., Lindberg A. A., Rietschel E. T., Zähringer U. (1988) Chemical structure of the lipopolysaccharide of Haemophilus influenzae strain I-69 Rd−/b+: Description of a novel deep-rough chemotype. Eur. J. Biochem. 177, 483–492 [DOI] [PubMed] [Google Scholar]
- 41. Schweda E. K. H., Twelkmeyer B., Li J. (2008) Profiling structural elements of short-chain lipopolysaccharide of non-typeable Haemophilus influenzae. Innate Immun. 14, 199–211 [DOI] [PubMed] [Google Scholar]
- 42. Lebbar S., Caroff M., Szabó L., Mérienne C., Szilógyi L. (1994) Structure of a hexasaccharide proximal to the hydrophobic region of lipopolysaccharides present in Bordetella pertussis endotoxin preparations. Carbohydr. Res. 259, 257–275 [DOI] [PubMed] [Google Scholar]
- 43. Bock K., Vinogradov E. V., Holst O., Brade H. (1994) Isolation and structural analysis of oligosaccharide phosphates containing the complete carbohydrate chain of the lipopolysaccharide from Vibrio cholerae strain H11 (non-O1). Eur. J. Biochem. 225, 1029–1039 [DOI] [PubMed] [Google Scholar]
- 44. Ohto U., Fukase K., Miyake K., Satow Y. (2007) Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 316, 1632–1634 [DOI] [PubMed] [Google Scholar]
- 45. Kim H. M., Park B. S., Kim J.-I., Kim S. E., Lee J., Oh S. C., Enkhbayar P., Matsushima N., Lee H., Yoo O. J., Lee J.-O. (2007) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130, 906–917 [DOI] [PubMed] [Google Scholar]
- 46. Meng J., Lien E., Golenbock D. T. (2010) MD-2-mediated ionic interactions between lipid A and TLR4 are essential for receptor activation. J. Biol. Chem. 285, 8695–8702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meng J., Gong M., Björkbacka H., Golenbock D. T. (2011) Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J. Immunol. 187, 3683–3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vasl J., Oblak A., Gioannini T. L., Weiss J. P., Jerala R. (2009) Novel roles of lysines 122, 125, and 58 in functional differences between human and murine MD-2. J. Immunol. 183, 5138–5145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu W. C., Oikawa M., Fukase K., Suda T., Kusumoto S. (1999) A divergent synthesis of lipid A and its chemically stable unnatural analogues. Bull. Chem. Soc. Jpn. 72, 1377–1385 [Google Scholar]
- 50. Gruenheid S., Le Moual H. (2012) Resistance to antimicrobial peptides in Gram-negative bacteria. FEMS Microbiol. Lett. 330, 81–89 [DOI] [PubMed] [Google Scholar]
- 51. Hancock R. E. W., Sahl H.-G. (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 [DOI] [PubMed] [Google Scholar]
- 52. Chamorro C., Boerman M. A., Arnusch C. J., Breukink E., Pieters R. J. (2012) Enhancing membrane disruption by targeting and multicalent presentation of antimicrobial peptides. Biochim. Biophys. Acta 1818, 2171–2174 [DOI] [PubMed] [Google Scholar]