

Background: UPR-associated death signals are activated during plasma cell differentiation.
Results: Bcl-xL sequesters Bim during plasma cell differentiation, preventing apoptosis.
Conclusion: Bcl-xL dependence during plasma cell differentiation protects the cell from the UPR activation necessary for ER remodeling.
Significance: Bcl-2 family dynamics are crucial for both the development of plasma cells and the treatment of diseases of abnormal plasma cells.
Keywords: Apoptosis, Bcl-2 Proteins, Differentiation, ER Stress, Lymphocyte, UPR, Plasma Cell
Abstract
Although it is known that the unfolded protein response (UPR) plays a significant role in the process of plasma cell differentiation, the contribution of the individual sensors of the UPR to this process remains unclear. In this study we examine the death signals and compensatory survival signals activated during B cell activation and the first stages of plasma cell differentiation. During in vitro differentiation of both primary murine B cells and the Bcl1 cell line, we demonstrate that in addition to activation of the physiological UPR, changes in the expression of several Bcl-2 proteins occur, which are consistent with a lowering of the apoptotic threshold of the cell. Specifically, we observed decreased expression of Bcl-2 and Mcl-1 and increased expression of the proapoptotic protein Bim. However, these changes were countered by Bcl-xL induction, which is necessary to protect differentiating cells both from ER stress-induced death by tunicamycin and from the death signals inherent in differentiation. Consistent with differentiating cells becoming dependent on Bcl-xL for survival, the addition of ABT-737 resulted in apoptosis in differentiating cells through the inhibition of sequestration of Bim. Confirming this result, differentiation in the context of RNAi-mediated Bcl-xL knockdown also induced apoptosis. This cell death is C/EBP homologous protein (CHOP)-dependent, connecting these events to the UPR. Thus plasma cell differentiation proceeds through a Bcl-xL-dependent intermediate.
Introduction
Plasma cell differentiation involves striking changes in the morphology and physiology of the cell. To become a professional antibody-secreting cell, the B cell must increase the size and capacity of the endoplasmic reticulum (ER)2 and secretory apparatus (1). To accomplish this end, the cell activates a set of signaling pathways known as the unfolded protein response (UPR) (2). During periods of ER stress when the protein load in the ER outweighs its folding capacity, three sensors of ER stress, activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and PKR-like ER kinase (PERK), are activated (3). These sensors reside across the ER membrane with their luminal tails bound to the ER chaperone Bip (GRP78) keeping them in an inactive state. When unfolded protein builds in the ER, Bip is titrated off the luminal tails of these signaling molecules. ATF6 translocates to the Golgi where it is cleaved to yield p50ATF6, a transcription factor that up-regulates ER quality control and capacity (4–8). IRE1 oligomerizes and activates its ribonuclease, which splices x-box-binding protein 1 (XBP1) mRNA (9–13). This spliced XBP1 mRNA yields a highly active transcription factor that up-regulates chaperones and expands ER function and capacity (14, 15). PERK oligomerizes and phosphorylates eukaryotic translation initiation factor 2 α (eIF2α) (16). Phosphorylated eIF2α inhibits global protein translation while favoring translation of activating transcription factor 4 (ATF4) mRNA (17). ATF4 also increases ER capacity as well as strongly inducing C/EBP homologous protein (CHOP) (17–19). Activation of these three signaling molecules results in the up-regulation of ER protein folding capacity, quality control, and ER-associated degradation while delaying further translation of mRNA. If ER stress is not resolved through the UPR, CHOP induces apoptosis by inhibition of Bcl-2 and induction of Bim (20, 21).
The expansion of the secretory capacity necessary for a B cell to become a professional antibody-secreting cell makes use of the UPR machinery already present in all cells. It must however overcome the effects of PERK activation including translation inhibition and apoptosis. ATF6 and IRE1 are activated and XBP1 is spliced during plasma cell differentiation, and they are necessary to form plasma cells in vivo as animals deficient in any of these genes lack plasma cells (2, 22–24). Conversely, perk−/− animals have plasma cells, and PERK is not significantly activated during plasma cell differentiation (25, 26). Although it is true that ATF4, p50ATF6, and XBP1s all bind to the promoter of CHOP, only ATF4 has been shown to be necessary for CHOP activation (18). Therefore it stands to reason that inactivation of PERK would inhibit ATF4 and CHOP, relieving the cell of translation inhibition and the proapoptotic effects of CHOP. However, it has also been shown that some activation of CHOP is necessary for production of a maximally efficient plasma cell as Chop−/− animals have plasma cells that secrete immunoglobulin at a lower rate than wild type animals (27). Therefore the proapoptotic aspects of CHOP signaling must be overcome for optimal plasma cell differentiation.
During all stages of B cell development, there are prosurvival and proapoptotic proteins responsible for determining which B cells are propagated and which are deleted from the repertoire. The Bcl-2 family of proteins responsible for regulating the intrinsic pathway of apoptosis plays a major role in this survival signaling (28–30). This is true in mature B cells and in plasma cells, but less is known about this pathway during the transition between these cell types. The multidomain antiapoptotic proteins Bcl-2, Mcl-1, and Bcl-xL are important in B cell survival. They function by binding and sequestering proapoptotic BH3-only proteins, such as Bim (31, 32). This prevents Bim from activating Bax and Bak, which when active induce mitochondrial outer membrane permeabilization, resulting in cytochrome c release that leads to downstream caspase activation and apoptosis. This function of antiapoptotic proteins can now be targeted by a class of drugs called BH3 mimetics. These are small molecules that bind to and antagonize the antiapoptotic proteins. ABT-737, a mimetic of the BH3-only protein Bad, binds to Bcl-xL, Bcl-2, and Bcl-w, displacing Bim and leading to apoptosis in cells that are dependent on one of these proteins for survival (33). ABT-737 does not bind Mcl-1 and therefore will not cause apoptosis in a cell that is dependent on Mcl-1 for survival. In murine immunization models, it has been shown that germinal center B cells and existing plasma cells are insensitive to ABT-737 (34). Accordingly, it has also been shown that these cell types are dependent on Mcl-1 for survival (30, 35). These studies did show that there was a deficit in newly formed plasma cells in the presence of ABT-737; however, the molecular basis for this deficit was not fully defined. In this study we define the molecular basis of differential Bcl-2 family dependence during plasma cell differentiation.
EXPERIMENTAL PROCEDURES
Cell Culture
The Bcl1 cell line, clone CW13.20.3B3, was acquired from ATCC (CRL-1699). Primary murine B cells were prepared from splenocytes isolated from C57BL/6 spleens and depleted of non-B cells and activated B cells by magnetic bead column separation (Miltenyi B cell isolation kit 130-090-862, LS columns). All cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 2 μm l-glutamine, 100 IU penicillin/streptomycin, 10 mm HEPES buffer, 1 mm sodium pyruvate, nonessential amino acids, and 50 μm 2-mercaptoethanol.
In Vitro Differentiation
Bcl1 cells were cultured at a concentration of 0.3 × 106 cells/ml in complete growth medium supplemented with 10 ng/ml IL-5 (R&D Systems) and 10 μg/ml lipopolysaccharide (Sigma L-4391) for up to 96 h. UPR was activated with 0.5 μg/ml tunicamycin (Sigma T7765) followed by replacement with complete growth medium. Primary C57BL/6 B cells were cultured at a concentration of 1 × 106 cells/ml in complete growth medium supplemented with 20 ng/ml IL-4 (PeproTech) and 20 μg/ml lipopolysaccharide (Sigma L-6216) for up to 96 h. ABT-737 and ABT-199 were a generous gift of Abbvie (North Chicago, IL).
Lentiviral Knockdown
Virus was prepared in 293T cells using the MISSION shRNA TORC1 system (Sigma SHCLNG-NM_004083) or with pLKO.1 vector control. Bcl1 cells were then infected and selected with puromycin. Transductants were kept in selection during passage and verified by Western blot and quantitative RT-PCR. Experiments were carried out in the absence of puromycin.
Flow Cytometry
Cells were collected at the various time points and treatments. 0.25–0.5 million cells were washed with PBS and resuspended in 100 μl of FACS buffer (1% BSA in PBS containing 0.01% sodium azide) and the appropriate amount of antibody for 30 min at 4 °C. Cells were then washed in FACS buffer and resuspended in 0.5 ml of FACS buffer with 5 μl of 7-aminoactinomycin D and incubated at room temperature for 5 min. Samples were then assayed on a BD FACSCanto II.
CD19-V450 (560375), CD138-PE (553714), and CD44-FITC (553133) antibodies and 7-aminoactinomycin D were purchased from BD Biosciences. IgM-FITC (69819) was purchased from Santa Cruz Biotechnology. Apoptosis was assayed with annexin V-FITC (BioVision 1001-1000) and propidium iodide (2 μg/ml Sigma) staining and measured with a BD FACSCanto II as described previously (36).
Immunoblotting and Immunoprecipitation
Western blotting was performed on lysates collected at the various time points and treatments as described previously (36). Samples were run on 4–15% Bio-Rad TGX mini gels. Gels were transferred to nitrocellulose membrane and blocked as described previously (36).
ATF4 rabbit polyclonal antibody was purchased from Abcam. Bcl-2 hamster (3F11) and Syrian and Armenian hamster-HRP mouse antibodies were purchased from BD Biosciences. Bcl-2 rabbit (50E3), Bip rabbit (C50B12), CHOP rabbit (D46F1), phospho-eIF2α (Ser-51) rabbit (D9G8), phospho-eIF2α (Ser-51) rabbit (119A11), eIF2α mouse (L57A5), GRP94 rabbit polyclonal antibodies and p58IPK rabbit (C56E7) and Mcl-1 rabbit (D35A5) antibodies were purchased from Cell Signaling Technology. Rabbit IgG-HRP donkey and mouse-IgG-HRP sheep antibodies were purchased from GE Healthcare. Bim rabbit polyclonal antibody was purchased from Millipore. β-Actin mouse (AC-15) and β-actin rabbit polyclonal antibodies were purchased from Sigma Aldrich. Bcl-xL mouse (2A1) and Bcl-xL rabbit polyclonal antibodies (13.6) have been previously described (37).
Co-immunoprecipitation was performed with the ImmunoCruz Optima C kit (Santa Cruz Biotechnology) as described previously (36). Lysates were prepared in 2% CHAPS buffer. 100 μg of protein was used for each antibody for each sample. Samples were precleared with protein G and preclearing matrix for 2 h. Immunoprecipitation matrices were prepared using 3 μg of the appropriate antibody. Precleared lysates were rocked in immunoprecipitation matrices overnight. Samples were eluted in 2:1 radioimmunoprecipitation assay buffer: 6× loading dye under nonreducing conditions after a 5-min incubation at 95 °C.
Mcl-1 mouse (B6) antibody was purchased from Santa Cruz Biotechnology. Bcl-2 mouse (Bcl/10C4) antibody was purchased from Novus Biologicals. Bcl-xL mouse (7B2.5) was previously described (37).
Gene Expression
cDNA was prepared from RNA harvested at specified time points using the ABI high capacity cDNA kit (Applied Biosystems). Real-time PCR was performed using TaqMan gene expression master mix (ABI 4368814) with an ABI 9600 Fast thermocycler as described previously (38). The following probes were used: Mcl-1 (mcl1) Mm00725832_s1, Bcl-2 (bcl2) Mm00477631_m1, Bcl-xL (bcl2l1) Mm00437783_m1, Bim (bcl2l11) Mm00437796_m1, Blimp-1 (prdm1) Mm004761289_m1, Bcl-6 (bcl6) Mm00477633_m1, and GAPDH 4352932-0912031 were purchased from Applied Biosystems.
ELISA
ELISA was performed according to protocol using the mouse IgM ELISA kit (Bethyl Laboratories). Supernatants were diluted 1:4 and loaded at 25 μl/well and then calibrated to live cell number counted using trypan blue exclusion. Lysates were loaded at 2.5 μg of total protein/well. 3,3′, 5,5;-Tetramethylbenzidine (TMB) substrate (Invitrogen) and color change were used to measure concentration. Color change was measured as A450–A550. IgM concentrations were reported as IgM per cells in culture or IgM per cells contributing to the lysate loaded and were calculated against the four parameter logistic curve fit of dilutions of standard mouse serum with known IgM concentration.
XBP1 Splicing Assay
cDNA was prepared from RNA collected from samples at specified time points with the various treatments. The region of the splice site was amplified using primers flanking the splice site. Products were amplified using the DreamTaq kit (Fermentas) and a custom PCR routine: 94 °C/4 min, 40 cycles of 94 °C/30 s, 65 °C/30 s, 72 °C/30 s, and then 72 °C/10 min. PCR products were run on 3% high resolution agarose gels (Invitrogen). 10-cm gels were run at 50 V for 6 h. Primers were: forward, 5′-GAACACGCTTGGGAATGGACAC-3′, and reverse, 5′-AGAAAGGGAGGCTGGTAAGGAAC-3′.
RESULTS
IL-5 and LPS Treatment Induces Differentiation in Bcl1 Cells
The murine B cell leukemia cell line Bcl1 can be stimulated to differentiate to an antibody-secreting cell using lipopolysaccharide (LPS) and cytokines (39, 40). We tested combinations of previously described differentiation stimuli including IL-2, IL-5, and LPS and found that the combination of IL-5 and LPS was as efficient a differentiation stimulus as the combination of all three (not shown). Consistent with published data (40), this cell line has high basal expression of the IL-5 receptor (not shown). Differentiating Bcl1 cells display phenotypic changes associated with the early stages of plasma cell differentiation including up-regulation of CD138, CD19, and CD44 along with down-regulation of surface IgM (Fig. 1A). To examine the effects of UPR activation on plasma cell differentiation, we induced ER stress with an inhibitor of N-linked glycosylation, tunicamycin. A 5-h pulse of tunicamycin activated the UPR as demonstrated by increased expression of CHOP and XBP1 (not shown). These cells rapidly underwent apoptosis unless LPS was included in the differentiation stimulus following the tunicamycin pulse (Fig. 1B, and not shown). Although IL-5 was not sufficient on its own to protect cells, only those cells that received both LPS and IL-5 produced and secreted antibody at a rate similar or greater than cells that did not receive tunicamycin pretreatment (Fig. 1, C and D, and not shown). These data demonstrate that Bcl1 cells are sensitive to ER stress-induced cell death yet are protected by LPS signaling during differentiation.
FIGURE 1.

Bcl1 cells differentiate with IL-5 and LPS treatment. A, Bcl1 cells were treated with IL-5 and LPS for 72 h prior to collection and staining. B cell and plasma cell markers were measured by flow cytometry. B–D, Bcl1 cells were untreated (NT) or pretreated with tunicamycin (Tm) for 5 h prior to treatment with IL-5 and/or LPS for 96 h. B, apoptosis was measured by annexin V and propidium iodide staining. C, IgM was measured by ELISA of the supernatant of the various treatment groups and is reported as a function of the live cell number. D, IgM was measured by ELISA of the lysates of the various treatment groups and is reported as a function of the live cell number contributing to the lysate. Data are represented as the mean ± S.E. of three independent experiments (*, p < 0.05, **, p < 0.01).
Differentiating Bcl1 Cells Activate CHOP Independent of PERK
Because plasma cell differentiation induces parts of the unfolded protein response, we chose to further examine the activation of this pathway during ER stress and differentiation. All cells treated with the combination of LPS and IL-5 displayed a large increase in the ER chaperones GRP94 and GRP78 (Bip) regardless of tunicamycin treatment (Fig. 2A). Consistent with this observation, activation of IRE1, as measured by XBP1 splicing, was highest in cells treated with both LPS and IL-5 throughout the time course (Fig. 2B). Together these data suggest that differentiation of Bcl1 cells results in the activation of a physiologic UPR and that tunicamycin does not influence this response. In cells pulsed with tunicamycin for 5 h, we observed phosphorylation of eIF2α at higher levels than untreated cells at 24- and 48-h time points by Western blot (Fig. 2A). Phosphorylated eIF2α was not observed when cells were differentiated in the absence of tunicamycin, a finding consistent with PERK activation not being part of plasma cell differentiation. Protein levels of ATF4 and CHOP, both downstream effectors of the PERK arm of the UPR, were also increased by tunicamycin addition. Cells that received tunicamycin without differentiation stimulus do not show as robust activation of this arm downstream of eIF2α phosphorylation, but it is important to note that the viability of these cells was very low at these time points (Fig. 1B). Surprisingly, all cells treated with LPS and IL-5 in the absence of tunicamycin also demonstrated robust and prolonged expression of both ATF4 and CHOP in the absence of eIF2α phosphorylation. Moreover, cells receiving both IL-5 and LPS also showed induction of the negative regulator of PERK, p58ipk (Fig. 2A). These data demonstrate that during differentiation, Bcl1 cells activate a robust UPR including downstream elements of XBP1 activation and the ATF4/CHOP arm of the UPR, without measurable activation of PERK.
FIGURE 2.

Differentiating Bcl1 cells activate the unfolded protein response and up-regulate Bcl-xL. Bcl1 cells were pretreated with tunicamycin (Tm) for 5 h prior to treatment with IL-5, LPS, or a combination of both for 96 h. A, protein levels of UPR-related proteins were determined by Western blot. B, XBP1 splicing was determined by amplification of the spliced region and running the PCR products on agarose gels. U and S designate unspliced and spliced PCR products, respectively. C, Bcl1 cells were treated IL-5 and LPS for 96 h. Quantitative RT-PCR was performed on RNA collected at 24-h periods. Cycle numbers were normalized to GAPDH and presented as relative quantitation (RQ) as a function of untreated samples. Data are represented as the mean ± S.E. of three independent experiments. D, Bcl1 cells were pretreated with tunicamycin for 5 h prior to treatment with IL-5, LPS, or a combination of both for 96 h. Protein levels of Bcl-2 family proteins were determined by Western blot.
Bcl-xL Is Induced during Cytokine- and LPS-driven Differentiation
Having observed CHOP activation in differentiating Bcl1 cells and given that CHOP induction can lead to apoptosis, we investigated the survival signaling inherent to the differentiation stimuli. During a 4-day time course of IL-5- and LPS-driven differentiation in Bcl1 cells, Bcl-xL mRNA increased more than 10-fold, whereas mRNA for both Bcl-2 and Mcl-1 decreased (Fig. 2C). At the protein level, Mcl-1 decreased dramatically when cells were treated with both IL-5 and LPS, whereas Bcl-xL reciprocally increased (Fig. 2D). Bcl-2 protein levels did not change significantly. Although IL-5 and LPS treatment alone did not induce Bim, cells that were pulsed with tunicamycin prior to IL-5 and LPS treatment displayed large increases in Bim protein by 72 h. Interestingly, this was not sufficient to cause significant apoptosis in cells also treated with LPS and IL-5, suggesting that induction of Bcl-xL was sufficient to protect these cells.
Bcl-xL Sequestration of Bim Protects Differentiating Bcl-1 Cells
Because the expression patterns of the Bcl-2 family of proteins are consistent with B cells traversing a Bcl-xL-dependent state during plasma cell differentiation, we determined the effects of the Bcl-xL inhibitor ABT-737 on Bcl1 differentiation. ABT-737 binds to the BH3 binding pocket of Bcl-2 and Bcl-xL but not Mcl-1, blocking the interaction with proapoptotic factors such as Bim. Bcl1 cells were pulsed with tunicamycin for 5 h and then treated with IL-5, LPS, or a combination of both in the presence or absence of 600 nm ABT-737. At 24 h after treatment, LPS was able to protect cells from tunicamycin-induced apoptosis (Fig. 3A). This LPS-mediated protection was completely abrogated in the presence of ABT-737, demonstrating that the protective factor was Bcl-xL or Bcl-2. We then examined the survival signaling during differentiation in the absence of tunicamycin. Bcl1 cells were given IL-5 and LPS over a 96-h time course. Cells were treated with varying doses of ABT-737 only during the last 24 h prior to collection, and apoptosis was determined. ABT-737 sensitivity increased as the cells differentiated through the 72- and 96-h time points, suggesting that the cells become more Bcl-2- or Bcl-xL-dependent as they differentiate (Fig. 3B).
FIGURE 3.

Bcl-xL mediates protection from ER stress- and differentiation-induced apoptosis in Bcl1 cells. A, Bcl1 cells were pretreated with tunicamycin (Tm) for 5 h prior to treatment with IL-5, LPS, or a combination of both for 24 h with or without 600 nm ABT-737. Apoptosis was measured using annexin V and propidium iodide staining. B, Bcl1 cells were treated with IL-5 and LPS for 96 h. Samples were treated with varying doses of ABT-737 for the final 24 h prior to collection, and apoptosis was measured by annexin V and propidium iodide staining. C, Bcl-1 cells were treated with IL-5 and LPS with or without 600 nm ABT-737 for 72 h. Immunoprecipitation (IP) was performed on lysates prepared from individual treatments. IB, immunoblot. Data are presented as the mean ± S.E. of three independent experiments (*, p < 0.05, **, p < 0.01, ***, p < 0.001).
To interrogate the mechanism behind this protection, co-immunoprecipitation was performed on cells treated with IL-5 and LPS for 72 h in the presence or absence of ABT-737. We immunoprecipitated the antiapoptotic proteins Bcl-xL, Mcl-1, and Bcl-2 and blotted for Bim to determine the Bim binding pattern (Fig. 3C). In untreated Bcl1 cells, Bim was primarily bound to Mcl-1 and Bcl-2. However, in cells treated with IL-5 and LPS, Bim was almost completely bound to Bcl-xL. Interestingly, this was not due to changes in Bcl-2 expression, which are unchanged during differentiation (Fig. 2D). Association of Bim with Bcl-xL suggests that these cells are Bcl-xL-dependent, a finding that is consistent with the change in ABT-737 sensitivity during differentiation. To further investigate Bcl-xL dependence during B cell differentiation, we determined the effects of ABT-737 on Bim binding to Bcl-2 proteins during differentiation. As seen in Fig. 3C, the addition of ABT-737 in undifferentiated cells resulted in a shift of Bim from being bound by both Bcl-2 and Mcl-1 to exclusive binding to Mcl-1. This explains why these cells are not sensitive to ABT-737 (Fig. 3A). In contrast when cells are differentiated, Bim is now exclusively bound to Bcl-xL, and the addition of ABT-737 results in release of Bim. However, in these cells, there is no Mcl-1 available to sequester Bim, allowing for induction of apoptosis.
Bcl-xL Induction in Differentiating Primary Murine B Cells
To validate these findings in a non-transformed cell, we performed in vitro differentiation of primary murine B cells. Of the various treatments examined for differentiation, IL-4 and LPS in combination yielded the most robust response as shown by increased Blimp-1 and Bcl-xL expression (not shown). Purified primary resting B cells were cultured with IL-4 and LPS for up to 96 h to induce differentiation of antibody-secreting cells. We first examined the mRNA levels of the Bcl-2 family proteins. As seen with Bcl1 cells, mRNA for both Mcl-1 and Bcl-2 decreased and Bcl-xL mRNA increased with treatment (Fig. 4A). Increased Blimp-1 expression and decreased Bcl-6 expression were also observed (Fig. 4B). Western blotting demonstrated a loss in Bcl-2 protein accompanied by a reciprocal increase in Bcl-xL protein (Fig. 4C). Mcl-1 protein expression did not decrease along with the loss of mRNA or as it had in the Bcl1 cell line. Also somewhat unlike the Bcl1 cell line, differentiation stimulus alone was sufficient to induce Bim in primary cells. ATF4 and CHOP protein expression was observed by Western blot after 72 and 96 h of differentiation, respectively, indicating UPR activation (Fig. 4D). Intracellular light chain expression also increased throughout the treatment course, indicating an increase in immunoglobulin production (Fig. 4D). We performed a co-immunoprecipitation to examine Bim binding in these differentiating primary cells (Fig. 4E). In primary murine B cells, at time of harvest, Bim was bound to Bcl-2. After 48 h of treatment with IL-4 and LPS, Bim was bound to Bcl-2 and to a lesser extent Mcl-1. After 72 h of treatment, Bim was found almost exclusively on Bcl-xL. Importantly, when ABT-737 was added during the last 24 h of treatment, Bim displaced from Bcl-2 by ABT-737 was able to bind Mcl-1 at 48 h. However, at 72 h, Bim displaced from Bcl-xL by ABT-737 did not in great measure bind to Mcl-1.
FIGURE 4.

Bcl-xL is induced during plasma cell differentiation in primary B cells. A and B, primary murine B cells were treated with IL-4 and LPS for 96 h. Quantitative RT-PCR was performed on RNA prepared from samples collected at 24-h intervals. Cycle numbers were normalized to GAPDH and presented as relative quantitation (RQ) as a function of untreated time 0 samples. Data are represented as the mean ± S.E. of three independent experiments. C and D, primary murine B cells were treated with IL-4 and LPS for 4 days. 600 nm ABT-737 was added to cultures 24 h prior to collection. Bcl-2 family members were assayed by Western blot at the indicated times. E, primary murine B cells were treated with IL-4 and LPS for 72 h. ABT-737 was added to the cultures in the final 24 h of treatment. Co-immunoprecipitation (IP) was used to assess Bim binding at 0-, 48-, and 72-h time points. IB, immunoblot.
We then examined the ABT-737 sensitivity of these differentiating primary B cells. Primary B cells were treated with IL-4 and LPS for a period of 72 h. ABT-737 was added to the cells during the last 24 h of differentiation. ABT-737 treatment caused a loss of large, differentiating, CD138+ cells and a gain in small B cells in the live cell subset (Fig. 5, A and B). Furthermore, the viability of total CD138+ cells was significantly decreased, whereas the change in viability of CD138− and total cells did not meet significance (Fig. 5, C–E). This indicates an increase in ABT-737 sensitivity during plasma cell differentiation.
FIGURE 5.

ABT-737 sensitizes primary B cells to differentiation-induced apoptosis. Primary murine B cells were treated with IL-4 and LPS for 96 h. ABT-737 was added during the final 24 h prior to collection. A and B, live cells determined by negative 7-aminoactinomycin D (7AAD) staining were gated into small B cells or large differentiating cells (representative experiment in A) and are quantified in B. C and D, total cells were gated into CD138-positive and CD138-negative fractions (C is representative of gating), and 72-h cell death was measured as the proportion of 7-aminoactinomycin D-positive (7AAD-pos.) cells (representative experiment in D) and is quantified in E. Data in B and E are presented as the mean ± S.E. of six independent experiments. (*, p < 0.05, **, p < 0.01). n.s., not significant.
ABT-737-induced Apoptosis Is CHOP-dependent
To investigate the link between UPR activation and Bcl-xL dependence during differentiation, we silenced ATF4 and CHOP in Bcl1 cells. Sets of five short hairpin lentiviral constructs were stably infected and tested for both ATF4 and CHOP. The two constructs that displayed the best knockdown of ATF4 or CHOP following treatment with IL-5 and LPS were used for further experiments (not shown). The cells were treated with IL-5 and LPS for 72 h. ABT-737 was added to the cells 24 h prior to collection. Knockdown of ATF4 was able to block a significant, although modest amount of ABT-737-induced apoptosis in differentiating cells (Fig. 6A), whereas knockdown of CHOP reduced apoptosis in ABT-737-treated differentiating cells from 50% in pLKO.1 vector control cells to 30% in shCHOP cells (Fig. 6B). This protection from ABT-737-induced apoptosis corresponded to the level of knockdown of ATF4 and CHOP as measured by Western blot (Fig. 6, C and D). This indicates that activation of ATF4/CHOP is responsible for cell death by Bcl-xL/Bcl-2 inhibition during LPS- and cytokine-driven differentiation.
FIGURE 6.

Knockdown of CHOP abrogates differentiation induced ABT-737 sensitivity. Bcl1 cells infected with shATF4, shCHOP, or pLKO1 vector control lentivirus were treated with IL-5 and LPS for 72 h. 600 nm ABT-737 was added 24 h prior to collection. A–D, apoptosis was measured at 72 h. Western blots were prepared at 72 h (representative experiments in C and D). Data are presented as four independent experiments. (*, p < 0.05, **, p < 0.01). E and F, Bcl1 cells stably infected with shBcl-xL or shGFP lentivirus were untreated or treated with IL-5 and LPS for 72 h. ABT-737 (600 nm) or ABT-199 (600 nm) was added 24 h prior to collection. E, Western blots were prepared at 72- and 96-h time points to verify knockdown. F, apoptosis was measured at 72 h. Data in F are presented as the mean ± S.E. of three independent experiments. (*, p < 0.05, **, p < 0.01).
Bcl-xL Silencing Sensitizes Cells to Differentiation-induced Apoptosis
To verify that the protective factor induced by differentiation was Bcl-xL, we used shRNA to knock down Bcl-xL expression in Bcl1 cells. We tested five shRNA hairpins for Bcl-xL knockdown during differentiation against a hairpin targeting GFP as a control and chose the two with the best knockdown for further experiments (Fig. 6E and not shown). Differentiation with IL-5 and LPS treatment alone induced apoptosis in Bcl1 cells expressing shRNA to Bcl-xL but not in control cells expressing shRNA to GFP (Fig. 6F). In shBcl-xL cells but not shGFP cells, the addition of ABT-199, which binds to and inhibits only Bcl-2 (41), increased apoptosis to levels comparable with cells treated with ABT-737 during differentiation regardless of hairpin. This suggests that although differentiation favors Bim sequestration by Bcl-xL, in conditions where Bcl-xL expression is abrogated, Bim can bind to available Bcl-2.
DISCUSSION
Although it is established that the UPR is integral to the process of plasma cell differentiation, the precise role and activation of components of this pathway have not been completely elucidated. It stands to reason that activation of the downstream effectors of the ATF6 and IRE1/XBP1 arms of the UPR is beneficial to the process of becoming a professional protein-secreting cell such as a plasma cell. However, activation of the PERK arm includes aspects of the UPR that are in direct contrast to the needs of the plasma cell, namely translation inhibition and apoptosis. Consistent with this model, PERK is neither necessary nor is it activated during plasma cell differentiation despite having an activation step nearly identical to IRE1 (25, 26). However, it has been shown that one of the main downstream effectors of PERK, CHOP, is necessary for a maximally efficient plasma cell (27). Activation of CHOP has also been shown to induce Bim and inhibit Bcl-2, leading to apoptosis (20, 21). In this study we have verified that the UPR of the differentiating Bcl1 cell includes activation of ATF4 and CHOP but lacks phosphorylation of eIF2α (Fig. 2A). Examining the effects of PERK activation on this differentiation, we demonstrated that Bcl1 cells were very sensitive to tunicamycin-induced apoptosis, yet were protected when they were subsequently given LPS as a differentiation stimulus (Fig. 3A). This led us to hypothesize that there was survival signaling inherent in the differentiation contributing to this protection. We examined the Bcl-2 family of antiapoptotic proteins and showed that Bcl-xL was induced by differentiation both in the Bcl1 cell line and in primary C57BL/6 B cells (Figs. 2, B and D, and 4, A and C). Additionally, in primary B cells, we saw the decrease of Bcl-2 and increase of Bim levels along with CHOP activation (Fig. 4D). These data suggest that the differentiating B cells become dependent on Bcl-xL, and consistent with this possibility, differentiating cells are more sensitive to ABT-737. Although LPS treatment provided immediate protection from ER stress-induced apoptosis (Fig. 3A), only after 72 h of stimulation was there a shift to Bcl-xL dependence (Fig. 3B), suggesting that differentiation is necessary for this shift. This is consistent with previous studies demonstrating that newly formed plasma cells are lost in animals treated with ABT-737, whereas no loss occurs in mature B cells in the germinal centers and spleen or in long lived bone marrow plasma cells (30, 34, 35). This would suggest that Bcl-xL dependence only occurs at an intermediate stage of plasma cell differentiation. The sensitive population of differentiating primary B cells were CD138+, CD19High, indicative of an early plasma blast phenotype (Fig. 5 and data not shown). The molecular basis for the change in ABT-737 sensitivity was due to a change in sequestration of Bim by Bcl-2 and Mcl-1 to Bcl-xL during this stage of differentiation.
From these data we propose a model in which differentiation induces a loss of Mcl-1 and Bcl-2 and induction of Bim associated with UPR activation. Although we have not directly demonstrated the mechanism of differentiation-induced apoptosis, we have shown that it is at least partially dependent on ATF4 and CHOP, which are part of the UPR. However, differentiation also induces Bcl-xL, which is sufficient to sequester Bim, preventing apoptosis (Fig. 7). Presumably, this stage is short lived as it has been shown that long lived plasma cells are reliant on enforced expression of Mcl-1 along with survival signals provided by their stromal niche (35). Although some of this change in dependence is certainly due to changes in expression, there is evidence that expression alone does not determine Bim binding (38). This may explain why in normal B cell differentiation, Bim released from Bcl-xL is unable to bind to Mcl-1, yet prior to this point in differentiation, Bim released from Bcl-2 can bind to Mcl-1. It may also explain why we see changes in the expression of antiapoptotic Bcl-2 proteins throughout lymphocyte development and differentiation, although these proteins all have the same basic function in regulating apoptosis.
FIGURE 7.
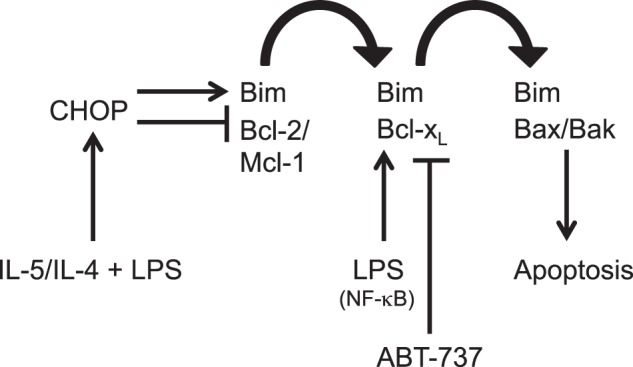
Schematic of proposed Bcl-2 family BH3 protein interactions in differentiation. LPS- and cytokine-driven differentiation induces Bim while decreasing expression of Mcl-1 and Bcl-2. Bcl-xL is induced and binds Bim that is displaced. ABT-737 blocks Bcl-xL, releasing Bim, which is then free to activate Bax and Bak, resulting in the induction of apoptosis.
Acknowledgments
We thank Ned Waller and Kasia Darlak for assistance with mouse spleens and the Boise laboratory for critical feedback on the data.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA127910 and R01 CA129968 as well as funding from the T. J. Martell Foundation (to L. H. B.).
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- CHOP
- C/EBP homologous protein
- C/EBP
- CCAAT-enhancer-binding protein
- PERK
- PKR-like ER kinase.
REFERENCES
- 1. Gass J. N., Gunn K. E., Sriburi R., Brewer J. W. (2004) Stressed-out B cells? Plasma-cell differentiation and the unfolded protein response. Trends Immunol. 25, 17–24 [DOI] [PubMed] [Google Scholar]
- 2. Gass J. N., Gifford N. M., Brewer J. W. (2002) Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J. Biol. Chem. 277, 49047–49054 [DOI] [PubMed] [Google Scholar]
- 3. Schröder M., Kaufman R. J. (2005) ER stress and the unfolded protein response. Mutat. Res. 569, 29–63 [DOI] [PubMed] [Google Scholar]
- 4. Yoshida H., Haze K., Yanagi H., Yura T., Mori K. (1998) Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins: involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749 [DOI] [PubMed] [Google Scholar]
- 5. Haze K., Yoshida H., Yanagi H., Yura T., Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shen J., Chen X., Hendershot L., Prywes R. (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 3, 99–111 [DOI] [PubMed] [Google Scholar]
- 7. Kokame K., Kato H., Miyata T. (2001) Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 276, 9199–9205 [DOI] [PubMed] [Google Scholar]
- 8. Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., Mori K. (2001) Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6α and 6β that activates the mammalian unfolded protein response. Mol. Cell. Biol. 21, 1239–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shamu C. E., Walter P. (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15, 3028–3039 [PMC free article] [PubMed] [Google Scholar]
- 10. Bertolotti A., Zhang Y., Hendershot L. M., Harding H. P., Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 [DOI] [PubMed] [Google Scholar]
- 11. Liu C. Y., Xu Z., Kaufman R. J. (2003) Structure and intermolecular interactions of the luminal dimerization domain of human IRE1α. J. Biol. Chem. 278, 17680–17687 [DOI] [PubMed] [Google Scholar]
- 12. Sidrauski C., Walter P. (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039 [DOI] [PubMed] [Google Scholar]
- 13. Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 [DOI] [PubMed] [Google Scholar]
- 14. Clauss I. M., Chu M., Zhao J. L., Glimcher L. H. (1996) The basic domain/leucine zipper protein hXBP-1 preferentially binds to and transactivates CRE-like sequences containing an ACGT core. Nucleic Acids Res. 24, 1855–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee A. H., Iwakoshi N. N., Glimcher L. H. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harding H. P., Zhang Y., Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 [DOI] [PubMed] [Google Scholar]
- 17. Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 [DOI] [PubMed] [Google Scholar]
- 18. Ma Y., Brewer J. W., Diehl J. A., Hendershot L. M. (2002) Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318, 1351–1365 [DOI] [PubMed] [Google Scholar]
- 19. Jiang H. Y., Wek S. A., McGrath B. C., Lu D., Hai T., Harding H. P., Wang X., Ron D., Cavener D. R., Wek R. C. (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 24, 1365–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCullough K. D., Martindale J. L., Klotz L. O., Aw T. Y., Holbrook N. J. (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Puthalakath H., O'Reilly L. A., Gunn P., Lee L., Kelly P. N., Huntington N. D., Hughes P. D., Michalak E. M., McKimm-Breschkin J., Motoyama N., Gotoh T., Akira S., Bouillet P., Strasser A. (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 [DOI] [PubMed] [Google Scholar]
- 22. Reimold A. M., Iwakoshi N. N., Manis J., Vallabhajosyula P., Szomolanyi-Tsuda E., Gravallese E. M., Friend D., Grusby M. J., Alt F., Glimcher L. H. (2001) Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300–307 [DOI] [PubMed] [Google Scholar]
- 23. Gunn K. E., Gifford N. M., Mori K., Brewer J. W. (2004) A role for the unfolded protein response in optimizing antibody secretion. Mol. Immunol. 41, 919–927 [DOI] [PubMed] [Google Scholar]
- 24. Lee K., Tirasophon W., Shen X., Michalak M., Prywes R., Okada T., Yoshida H., Mori K., Kaufman R. J. (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gass J. N., Jiang H. Y., Wek R. C., Brewer J. W. (2008) The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol. Immunol. 45, 1035–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma Y., Shimizu Y., Mann M. J., Jin Y., Hendershot L. M. (2010) Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones 15, 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Masciarelli S., Fra A. M., Pengo N., Bertolotti M., Cenci S., Fagioli C., Ron D., Hendershot L. M., Sitia R. (2010) CHOP-independent apoptosis and pathway-selective induction of the UPR in developing plasma cells. Mol. Immunol. 47, 1356–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Strasser A., Bouillet P. (2003) The control of apoptosis in lymphocyte selection. Immunol. Rev. 193, 82–92 [DOI] [PubMed] [Google Scholar]
- 29. Veis D. J., Sorenson C. M., Shutter J. R., Korsmeyer S. J. (1993) Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75, 229–240 [DOI] [PubMed] [Google Scholar]
- 30. Vikstrom I., Carotta S., Lüthje K., Peperzak V., Jost P. J., Glaser S., Busslinger M., Bouillet P., Strasser A., Nutt S. L., Tarlinton D. M. (2010) Mcl-1 is essential for germinal center formation and B cell memory. Science 330, 1095–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Danial N. N., Korsmeyer S. J. (2004) Cell death: critical control points. Cell 116, 205–219 [DOI] [PubMed] [Google Scholar]
- 32. Cory S., Adams J. M. (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–656 [DOI] [PubMed] [Google Scholar]
- 33. Oltersdorf T., Elmore S. W., Shoemaker A. R., Armstrong R. C., Augeri D. J., Belli B. A., Bruncko M., Deckwerth T. L., Dinges J., Hajduk P. J., Joseph M. K., Kitada S., Korsmeyer S. J., Kunzer A. R., Letai A., Li C., Mitten M. J., Nettesheim D. G., Ng S., Nimmer P. M., O'Connor J. M., Oleksijew A., Petros A. M., Reed J. C., Shen W., Tahir S. K., Thompson C. B., Tomaselli K. J., Wang B., Wendt M. D., Zhang H., Fesik S. W., Rosenberg S. H. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 [DOI] [PubMed] [Google Scholar]
- 34. Carrington E. M., Vikstrom I. B., Light A., Sutherland R. M., Londrigan S. L., Mason K. D., Huang D. C., Lew A. M., Tarlinton D. M. (2010) BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc. Natl. Acad. Sci. U.S.A. 107, 10967–10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peperzak V., Vikström I., Walker J., Glaser S. P., LePage M., Coquery C. M., Erickson L. D., Fairfax K., Mackay F., Strasser A., Nutt S. L., Tarlinton D. M. (2013) Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 14, 290–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morales A. A., Gutman D., Lee K. P., Boise L. H. (2008) BH3-only proteins Noxa, Bmf, and Bim are necessary for arsenic trioxide-induced cell death in myeloma. Blood 111, 5152–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boise L. H., Minn A. J., Noel P. J., June C. H., Accavitti M. A., Lindsten T., Thompson C. B. (1995) CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xL. Immunity 3, 87–98 [DOI] [PubMed] [Google Scholar]
- 38. Morales A. A., Kurtoglu M., Matulis S. M., Liu J., Siefker D., Gutman D. M., Kaufman J. L., Lee K. P., Lonial S., Boise L. H. (2011) Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood 118, 1329–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blackman M. A., Tigges M. A., Minie M. E., Koshland M. E. (1986) A model system for peptide hormone action in differentiation: interleukin 2 induces a B lymphoma to transcribe the J chain gene. Cell 47, 609–617 [DOI] [PubMed] [Google Scholar]
- 40. Mita S., Harada N., Naomi S., Hitoshi Y., Sakamoto K., Akagi M., Tominaga A., Takatsu K. (1988) Receptors for T cell-replacing factor/interleukin 5: specificity, quantitation, and its implication. J. Exp. Med. 168, 863–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Souers A. J., Leverson J. D., Boghaert E. R., Ackler S. L., Catron N. D., Chen J., Dayton B. D., Ding H., Enschede S. H., Fairbrother W. J., Huang D. C., Hymowitz S. G., Jin S., Khaw S. L., Kovar P. J., Lam L. T., Lee J., Maecker H. L., Marsh K. C., Mason K. D., Mitten M. J., Nimmer P. M., Oleksijew A., Park C. H., Park C. M., Phillips D. C., Roberts A. W., Sampath D., Seymour J. F., Smith M. L., Sullivan G. M., Tahir S. K., Tse C., Wendt M. D., Xiao Y., Xue J. C., Zhang H., Humerickhouse R. A., Rosenberg S. H., Elmore S. W. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 [DOI] [PubMed] [Google Scholar]