

Background: Bitopic ligands bind concomitantly to orthosteric and allosteric receptor sites.
Results: Residues affecting binding and biased signaling of the selective agonists TBPB and 77-LH-28-1 were identified at the M1 muscarinic receptor.
Conclusion: Novel bitopic ligand binding poses and mechanisms of receptor activation were identified.
Significance: Understanding the basis of bitopic ligand mechanisms can enable the design of selective ligands.
Keywords: Allosteric Regulation, Drug Discovery, Mutagenesis, Pharmacology, Receptor Structure-Function, Bitopic Ligand, Muscarinic Acetylcholine Receptors
Abstract
TBPB and 77-LH-28-1 are selective agonists of the M1 muscarinic acetylcholine receptor (mAChR) that may gain their selectivity through a bitopic mechanism, interacting concomitantly with the orthosteric site and part of an allosteric site. The current study combined site-directed mutagenesis, analytical pharmacology,and molecular modeling to gain further insights into the structural basis underlying binding and signaling by these agonists. Mutations within the orthosteric binding site caused similar reductions in affinity and signaling efficacy for both selective and prototypical orthosteric ligands. In contrast, the mutation of residues within transmembrane helix (TM) 2 and the second extracellular loop (ECL2) discriminated between the different classes of ligand. In particular, ECL2 appears to be involved in the selective binding of bitopic ligands and in coordinating biased agonism between intracellular calcium mobilization and ERK1/2 phosphorylation. Molecular modeling of the interaction between TBPB and the M1 mAChR revealed a binding pose predicted to extend from the orthosteric site up toward a putative allosteric site bordered by TM2, TM3, and TM7, thus consistent with a bitopic mode of binding. Overall, these findings provide valuable structural and mechanistic insights into bitopic ligand actions and receptor activation and support a role for ECL2 in dictating the active states that can be adopted by a G protein-coupled receptor. This may enable greater selective ligand design and development for mAChRs and facilitate improved identification of bitopic ligands.
Introduction
The M1 muscarinic acetylcholine receptor (mAChR)4 is a prototypical member of the family of rhodopsin-like G protein-coupled receptors and a target for the treatment of disorders of cognition and memory. Targeting the M1 mAChR has proved promising; the M1/M4 mAChR-preferring agonist, xanomeline, improved cognitive behaviors in both Alzheimer and schizophrenic patients (1, 2). However, like all mAChR-based therapeutics, xanomeline targets the ACh (orthosteric) binding site, which is highly conserved across the mAChR family (3). As such, the effectiveness of xanomeline is limited by inadequate subtype selectivity and adverse effects via peripheral M2 and M3 mAChRs (1, 2).
Targeting topographically distinct allosteric binding sites on the receptor can afford better subtype-selective ligands. Molecules that interact with allosteric sites have the potential to modulate the binding and function of co-bound orthosteric ligands as well as activating receptor signaling in their own right (4). Accordingly, the recent surge in the number of highly M1 mAChR-selective ligands has often been ascribed to their engagement with allosteric binding sites (5–9), but such a mechanism remains ambiguous for many of these ligands. For example, although the M1 mAChR-selective agonist N-desmethylclozapine was originally suggested to be allosteric, definitive evidence for an allosteric interaction with the receptor is lacking (10–12). Moreover, novel bitopic ligands have recently been described, which concomitantly associate with both the orthosteric binding site and an allosteric binding site (13–19); it is possible that previous studies of selective agonists have classified such bitopic ligands as purely allosteric.
Indeed, we recently proposed that the M1 mAChR-selective agonists TBPB (1-(1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one), AC-42 (4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl] piperidine), and 77-LH-28-1 (1-[3-(4-butyl-1-piperidinyl)propyl]-3,4-dihydro-2(1H)-quinolinone), initially classified as allosteric agonists, are likely bitopic agonists (15, 19). Consistent with a bitopic mechanism of action (13, 14, 19), reverse engineering of TBPB by removal of an allosteric pharmacophore resulted in the generation of an agonist fragment, VCP794 (1-(1-cyclohexylpiperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one), that lost agonist selectivity for the M1 mAChR (19). Moreover, compared with the agonism of TBPB, VCP794 displays a different bias, one toward the activation of receptor-mediated phosphorylation of extracellular signal-regulated kinases 1 and 2 (pERK1/2) over intracellular calcium ([Ca2+]i) mobilization (19). These findings highlight a potential for developing or manipulating bitopic agonists to selectively target receptor subtypes and/or signaling pathways.
In the current study, we sought to investigate how such bitopic ligands potentially engage with the receptor. Prior mutational studies have provided some insight into bitopic mechanisms of action and the engagement of mAChRs by the structural congeners AC-42 and 77-LH-28-1 (10, 15, 20–22). A more recent mutagenesis investigation focuses on the pharmacology of TBPB and AC-42 (23). However, this latter study does not differentiate among the effects of mutations on the affinity, efficacy, and signaling bias of the selective agonists. Therefore, to investigate our hypothesis that TBPB and 77-LH-28-1 share similar bitopic modes of interaction at the M1 mAChR, we examined the effects of mutations of residues implicated in the orthosteric binding site or neighboring putative allosteric binding sites in the M1 mAChR (5, 15, 22, 24–28) on agonist affinity, efficacy, and signaling bias. By comparing the mutational effects on the pharmacology of TBPB and 77-LH-28-1 with orthosteric ligands, as well as fragment derivatives of TBPB (19), we have detailed their bitopic mechanism of interaction at the M1 mAChR. Furthermore, we have identified both common and divergent activation mechanisms exploited by bitopic ligands from orthosteric ligands and provided the first quantification of mutational effects on biased agonism at this receptor subtype. These findings warrant further consideration for the development and study of subtype selective agonists of the mAChRs.
EXPERIMENTAL PROCEDURES
Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were from Invitrogen and JRH Biosciences (Lenexa, KS), respectively. Hygromycin was purchased from Roche Applied Science. The AlphaScreenTM SureFireTM pERK1/2 reagents were kindly donated by Drs. Michael Crouch and Ron Osmond (TGR Biosciences, South Australia, Australia). The AlphaScreen streptavidin donor beads and anti-IgG (protein A) acceptor beads used for pERK1/2 detection, 384-well ProxiPlates, and [3H]N-quinuclidinyl benzilate ([3H]QNB; specific activity, 50.5 Ci/mmol) were purchased from PerkinElmer Life Sciences. Fluo-4-AM was purchased from Invitrogen. 77-LH-28-1, TBPB, and its fragment derivatives, VCP794 and VCP813, were synthesized in-house as described previously (8, 19). All other chemicals were from Sigma or as otherwise stated below.
cDNA Constructs and Generation of Stable Cell Lines
cDNA in pcDNA3.1+ encoding the wild-type human M1 mAChR was obtained from Missouri University of Science and Technology. Oligonucleotides used for mutagenesis were purchased from GeneWorks (Hindmarsh, South Australia, Australia). An N-terminal c-Myc epitope tag (EQKLISEEDL) was introduced to the sequence of the M1 mAChR, along with flanking AttB sites, by overlap extension polymerase chain reaction using the following primers: 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCCACCATGGAGCAGAAGCTTATCAGCGAGGAGGACCTGaacacttcagccccacctgctgtc-3′ (N-terminal forward primer) and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTCAGCATTGGCGGGAGGGAGTGCG-3′ (C-terminal reverse primer). The construct produced was cloned into the pDONR201TM plasmid using the BP clonase enzyme mix according to manufacturer's instructions (Invitrogen). The desired mutations were then introduced into the construct using the QuikChangeTM site-directed mutagenesis kit (Agilent, Melbourne, Australia). All M1 mAChR receptor constructs in pDONR201TM were subsequently transferred into the pEF5/frt/V5/dest vector using the LR clonase enzyme mix (Invitrogen). Receptor constructs in pEF5/frt/V5/dest were used to isogenically transfect Flp-In CHO cells (Invitrogen) as described previously (21). Cells were selected using 600 μg/ml hygromycin to generate cell lines stably expressing each mutant receptor construct. Cells were maintained and cultured in high glucose DMEM supplemented with 10% FBS, 16 mm HEPES, and 600 μg/ml hygromycin at 37 °C under a humidified atmosphere containing 5% CO2.
Membrane Preparations
Membranes of cells expressing each M1 mAChR variant were prepared as described previously (19, 29). Briefly, following culture, cells were harvested with PBS (136.89 mm NaCl, 2.68 mm KCl, 3.21 mm Na2HPO4, and 1.47 mm KH2PO4) supplemented with 2 mm EDTA and resuspended in homogenization buffer containing 20 mm HEPES and 10 mm EDTA, pH 7.4. Cells were then homogenized using a Polytron homogenizer (PT1200 CL, Kinematica, Basel, Switzerland) on ice. Membranes were then separated via centrifugation and the final pellet resuspended in 5 ml of buffer (20 mm HEPES and 0.1 mm EDTA, pH 7.4).
Flow Cytometric Detection of Cell Surface Receptor Expression
Cells were seeded in a 24-well plate at a density of 150,000 cells/well and cultured overnight. The following day, the medium was aspirated, and cells were harvested with PBS supplemented with 2 mm EDTA. Cell pellets were resuspended in assay buffer (PBS, 0.1% BSA, and 2 mm EDTA) and transferred to a 96-well v-bottomed plate. Cells were then centrifuged at 250 × g at 4 °C for 3 min and resuspended in 100 μl of blocking buffer (PBS, 5% BSA, and 2 mm EDTA). After incubation on ice for 30 min, cells were incubated with mouse monoclonal 9E10 antibody (prepared in-house) targeted to the c-Myc epitope tag and diluted in assay buffer at 2 μg/ml for 60 min on ice. Cells were subsequently washed twice with assay buffer and incubated with a secondary goat anti-mouse IgG antibody conjugated to Alexa Fluor 647 (2 μg/ml, Molecular Probes, Invitrogen) for 30 min on ice. A single wash followed, and cells were resuspended in assay buffer containing 0.05 μg/ml propidium iodide with the fluorescence signal quantified using a FACSCantoII flow cytometer (BD Biosciences).
Saturation Radioligand Binding
Saturation binding studies were performed using [3H]QNB to estimate the affinity (equilibrium dissociation constant (KD)) of the radioligand and receptor expression levels. Membrane preparations were diluted to 3–20 μg/tube (except for membranes expressing c-Myc M1 D1053.32N mAChR, which were diluted to 50 μg/tube) using binding buffer and incubated with 0.01–10 nm [3H]QNB in a final assay volume of 1 ml/tube for 3 h at 37 °C in a shaking water bath. Following incubation, the assay was terminated by rapid filtration onto GF/B grade filter paper (Whatman) using a Brandel harvester followed by three washes with ice-cold NaCl (0.9%). Filters were dried under a heat lamp and prepared for liquid scintillation counting by transfer to scintillation vials and the addition of 4 ml of Ultima GoldTM scintillation mixture to each vial. Radioactivity was measured on a Tri-Carb 2900TR liquid scintillation analyzer (PerkinElmer Life Sciences) after incubation for at least 30 min.
Inhibition Radioligand Binding
Equilibrium radioligand binding assays were performed to estimate the binding affinity of non-radiolabeled compounds at the M1 mAChR. The final assay volume was either 1 or 2 ml depending on the concentrations of [3H]QNB used. Membrane preparations were diluted to 3–44 μg/tube in binding buffer with 100 μm Gpp(NH)p. Membrane preparations of the M1 mAChRs were co-incubated with an approximate KD concentration of [3H]QNB and varying concentrations (10 pm-10 mm) of the following ligands: atropine, ACh, xanomeline, 77-LH-28-1, TBPB, VCP794, and VCP813. The assay was terminated and measurements conducted as described above.
Intracellular Calcium ([Ca2+]i) Mobilization
Cells were cultured overnight on transparent 96-well cell culture plates at 3 × 104 cells/well and assayed as described previously (19). Briefly, cells were washed once using HEPES-buffered saline solution (HBS; 150 mm NaCl, 2.2 mm CaCl2, 2.6 mm KCl, 1.18 mm MgCl2, 10 mm HEPES, and 10 mm d-glucose) containing 0.5% BSA and 4 mm probenecid at pH 7.4 and then treated with (1 μm) Fluo-4-AM (in HBS/BSA/probenecid) for 60 min at 37 °C in 5% CO2 in the dark. Cells were then further washed twice and placed in HBS/BSA/probenecid solution for ligand addition and assay in a FlexStation 3 plate reader (Molecular Devices, Sunnyvale, CA). For receptor alkylation experiments, following incubation with Fluo-4-AM, cells were treated with varying concentrations of phenoxybenzamine for 30 min at 37 °C in the dark. Cells were then washed (three times) with assay buffer prior to stimulation with agonists. For all experiments, peak changes in fluorescence signal were normalized to the cellular response to 100 μm ATP, which was used as an internal positive control.
Measurement of pERK1/2
Cells were seeded on transparent 96-well cell culture plates at 3 × 104 cells/well. After 4 h, cells were washed once with PBS and then incubated in serum-free DMEM (supplemented with 16 mm HEPES) overnight. Receptor-mediated pERK1/2 was determined using the AlphaScreenTM ERK1/2 SureFireTM kit as described previously (19). For all experiments, 10% FBS was used as an internal positive control to stimulate pERK1/2, for which the maximal responses were used for normalization of data, and vehicle was used as a negative control. For the receptor alkylation experiments, cells were treated with varying concentrations (1, 3, and 10 μm) of phenoxybenzamine for 15–60 min at 37 °C in 5% CO2. Cells were then washed three times with PBS and again incubated in serum-free DMEM for 4 h prior to assay.
Computational Methods for the Model of the Ligand-Receptor Complex
For residues located in transmembrane domains, Ballesteros and Weinstein numbers are provided (in superscript) to indicate the relative position of residues within the transmembrane domains (30). To distinguish residues within the three extracellular loops, residue numbers are followed by an E number (in superscript), where the number denotes the number of the extracellular loop (e.g. Tyr-179E2). To generate a homology model of the M1 mAChR, the sequence of the human M1 mAChR was first retrieved from the Swiss-Prot database. ClustalX software (31) was used to align the sequence with the crystal structure of the M3 mAChR (Protein Data Bank ID: 4DAJ) (32). The model of the receptor was built using the Modeler v9.12 suite of programs (33), which yielded 15 candidate models. The conserved disulfide bond between Cys-983.25 at the beginning of TM3 and Cys-178E2 present in the template structure was also built and maintained as a constraint for geometric optimization. The best structure was selected from these candidates according to the Modeler DOPE (discrete optimized protein energy) assessment score and visual inspection. The resulting receptor structure was optimized using the force field described by Duan et al. (34) and the general AMBER force field (GAFF), and HF/6–31G*-derived RESP atomic charges were used for the ligands (35).
The docking of TBPB was performed with the MOE (molecular operating environment) software package (Chemical Computing Group, Inc.). TBPB was docked into the receptor model with its protonated nitrogen, proximal to the benzimidazolone moiety, and conserved in the structure of VCP794, interacting with Asp-1053.32, a residue conserved in all aminergic receptors and known to be important for ligand interaction (36, 37). The best docking solution was selected according to the scoring function of MOE and the results of the experimental data.
The complex obtained was subjected to refinement using MOE in a 200-ps molecular dynamics simulation (MMF94x force field, 300 K, Born solvation, 2-fs time step). Further optimization of the complex was performed by molecular dynamics simulation over 200 ps in which the ligand, the binding site (10 Å around the ligands), and the ECL2 were kept flexible while protein backbone atoms were kept fixed (MMFF94 force field, Born solvation, 300 K, 2-fs time step). The final complex was subsequently energy-minimized by applying gradient minimization until the root mean square gradient was lower than 0.001 kcal/mol Å and embedded in a pre-equilibrated lipid bilayer (∼280 molecules of 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) and ∼215,000 water molecules and counterions). Simulations were carried out using the NAMD2.9 (38) package with the TIP3 water model and the CHARMM27 all-hydrogen force field (39) for protein and lipids and using the particle mesh Ewald (PME) method (40) to evaluate electrostatic interactions. After an equilibration period of 1 ns (where we applied a positional restraint of 10 kcal mol−1 Å−2 to the Cα of the receptor structure), the molecular dynamics simulations were performed over 20 ns using a 1-fs integration time step, constant pressure, and constant temperature of 310 K.
Data Analysis
All data were analyzed using GraphPad Prism 6.02 (GraphPad Software, San Diego, CA). Data sets from [3H]QNB binding assays and agonist concentration-response curves were analyzed as described previously (19) to derive estimates of the radioligand dissociation constant (KD), maximal density of binding sites (Bmax), inhibitor dissociation constants (KI), agonist potencies (EC50), and maximal agonist responses (Emax).
To compare agonist profiles between the wild-type M1 mAChR and mutant receptors, agonist concentration-response curves were also fitted to the operational model of agonism below (41),
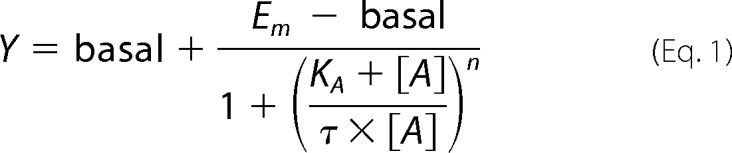 |
where Em is the maximal response of the system; KA denotes the functional equilibrium dissociation constant of the agonist (A); τ is an index of coupling efficiency (efficacy) of the agonist and is defined as RT/KE (where RT is the total concentration of receptors and KE is the concentration of agonist-receptor complex that yields half the Em); and n is the slope of the transducer function that links occupancy to response. For agonists that did not produce the maximal system response, the KA value was estimated directly from the functional data via Equation 1. In cases where agonists exhibited high efficacy that yielded the maximal responses, KA values were constrained to the respective KI values determined from radioligand binding studies (see “Results”) in order for the nonlinear regression algorithm to converge. The exception to this path was the determination of KA values for 77-LH-28-1, TBPB, VCP794, or xanomeline at the wild-type M1 mAChR through receptor occlusion by phenoxybenzamine alkylation, which resulted in significant reductions in the maximal responses. Given the proportional relationship of RT to measured τ, KA is invariant with receptor depletion. Hence, unique estimates of KA could be obtained by direct operational model fitting of the family of concentration-response curves for each agonist (42).
Accounting for different magnitudes of expression between receptor variants, the corrected τ values (τc) were calculated to facilitate more accurate efficacy comparisons. Receptor expression values normalized to wild-type expression determined from flow cytometric data (see below) were utilized to calculate τc as follows (equation 2).
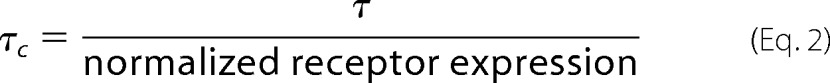 |
To quantify biased agonism, agonist concentration-response curves for [Ca2+]i mobilization or pERK1/2 were refitted using the following reparameterization of the operational model as described by Kenakin et al. (43) (Equation 3),
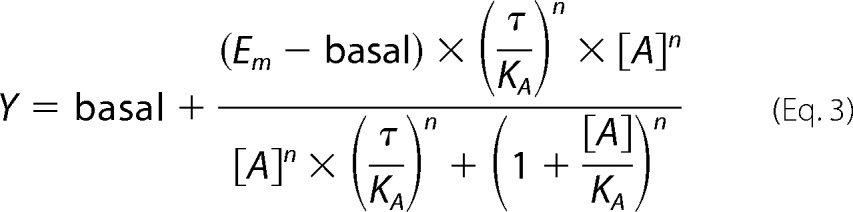 |
where the “transduction coefficient,” τ/KA, is estimated as a single fitted value.
Bias values were calculated as described previously (19, 20); the transduction coefficients of each test agonist were first normalized to that determined for the reference agonist, ACh.
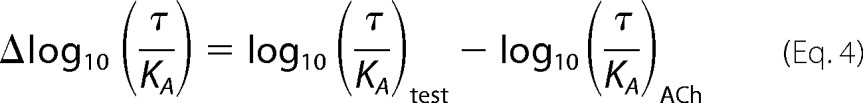 |
Normalized transduction coefficients (Δlog10 (τ/KA)) were then used to calculate the bias factor for each agonist between pERK1/2 and [Ca2+]i mobilization,
 |
where
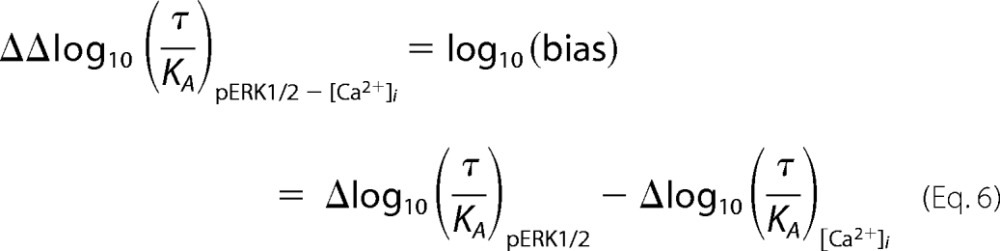 |
With the determination of composite parameters, the associated propagation of error in Equations 4–6 was accounted through application of the following equation,
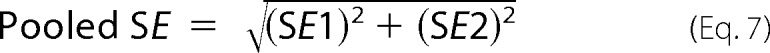 |
Flow cytometric data sets of cell samples were analyzed using FlowJo 10.0.6 (Tree Star, Inc., Ashland, OR) to select for live cells followed by the determination of mean Alexa Fluor 647 fluorescence. Specific Alexa Fluor 647 labeling was quantified by subtracting the mean fluorescence values of cell controls where 9E10 antibody was absent. These values were then normalized to those from cells without the epitope-tagged receptor and the c-Myc-tagged wild-type M1 mAChR.
Potency (EC50), affinity (KD, KI, and KA), and efficacy (τ) parameters were estimated as logarithms (44). Statistical analyses of data were performed, as appropriate, using Student's t test or one-way ANOVA with Dunnett's post-hoc tests, and significance was taken as p < 0.05. To minimize the impact of propagation of error from multiple steps of data manipulation in bias value calculations, for each agonist except ACh the normalized transduction coefficients (Δlog10 (τ/KA)) for pERK1/2 and [Ca2+]i mobilization (Equation 4) were used for statistical comparison to determine agonist bias.
RESULTS
Selection of Amino Acid Mutations and Generation of Cell Lines Stably Expressing the M1 mAChR
We selected a range of amino acid residues of the M1 mAChR involved (or predicted to be) in the binding and function of orthosteric and allosteric ligands (depicted in the snake plot in Fig. 1). We generated alanine substitutions of Tyr-1063.33 and Tyr-3816.51 within the orthosteric binding site. This was based on the prior use of these mutations to pharmacologically differentiate between orthosteric and novel, putatively allosteric, ligands (24, 26). Asp-1053.32 plays key roles in the binding of the amine head group of canonical orthosteric ligands, as well as in receptor activation (25, 26, 45). Because the D1053.32A mutation markedly reduces orthosteric ligand binding affinity and efficacy, we generated glutamate and asparagine mutations to facilitate better probes for ligand interactions with this residue. In addition, Leu-1023.29 and Trp-1013.28 were selected based on their role in stabilizing the binding of orthosteric ligands (26). Moreover, we investigated W1013.28A and F772.56I because of their unique discriminatory effects on the function of TBPB and 77-LH-28-1 relative to other prototypical ligands (15, 22, 23).
FIGURE 1.

Structures of orthosteric and bitopic agonists and fragment derivatives of TBPB and a snake diagram of the M1 mAChR highlighting residues mutated in the study.
The extracellular domains of the mAChRs interact with a variety of allosteric ligands. As such, we generated a range of alanine mutations of residues within these domains. We generated the Y822.61A, Y852.64A, W4007.35A, and E4017.36A mutations to investigate the roles of transmembrane helical domains 2 and 7 (TM2 and TM7), which have been implicated in the binding and function of bitopic ligands, such as 77-LH-28-1, and allosteric modulators (5, 15, 22, 28, 46). Additionally, given their proximity to the orthosteric binding site, we also investigated the role of the second extracellular loop (ECL2) residues, Tyr-179E2, Ile-180E2, Gln-181E2, Phe-182E2, and Leu-183E2. This region is involved with the stabilization of orthosteric ligand binding and receptor activation (47, 48) as well as the interaction of allosteric and bitopic ligands (14, 46). The nonconserved residues of ECL1 (His-90E1) and ECL3 (Lys-392E3 and Asp-393E3) were also included based on the previous suggestion of involvement with allosteric agonist binding (27).
Effects of Mutations on Expression of the M1 mAChR
Saturation binding using the radiolabeled orthosteric antagonist [3H]QNB revealed reductions in receptor expression for all of the mutations investigated, although the levels of L183E2A and K392E3A M1 mAChRs did not significantly differ from the wild-type receptor (p > 0.05, one-way ANOVA and Dunnett's post-hoc test; Tables 1 and 2). An alternative quantification of cell surface receptor expression, flow cytometry of antibody binding to the c-Myc epitope, was performed to detect immunolabeled cell surface-expressed receptors on live cells. Although a comparison of the normalized estimates found significantly greater expression levels for the F772.56I, D1053.32N, and Y3816.51A mutants from flow cytometry compared with radioligand binding (Student's t test, p < 0.05), the overall data from flow cytometric analysis agreed well with saturation binding data.
TABLE 1.
Level of expression of M1 mAChR mutant constructs to wild-type M1 mAChR determined from radioligand binding and flow cytometric analyses
Values represent the percentage of expression relative to wild-type M1 mAChR and are the mean ± S.E. of 3–4 separate experiments.
| M1 mAChR | Receptor expression |
|
|---|---|---|
| [3H]QNB binding | Flow cytometry | |
| F772.56I | 42.3 ± 3.38a,b | 53.7 ± 2.35b,c |
| Y822.61A | 51.6 ± 11.3a | 35.3 ± 1.49c |
| Y852.64A | 26.3 ± 5.42a | 25.6 ± 0.83c |
| H90E1A | 16.6 ± 1.93a | 16.4 ± 0.87c |
| W1013.28A | 18.7 ± 3.73a | 14.2 ± 0.51c |
| L1023.29A | 31.3 ± 8.24a | 16.4 ± 1.01c |
| D1053.32E | 46.7 ± 5.53a | 43.9 ± 2.36c |
| D1053.32N | 26.2 ± 2.54a,b | 38.2 ± 0.99b,c |
| Y1063.33A | 24.5 ± 1.39a | 29.1 ± 1.45c |
| Y179E2A | 32.3 ± 5.30a | 35.6 ± 0.69c |
| I180E2A | 33.8 ± 6.51a | 40.1 ± 1.84c |
| Q181E2A | 33.3 ± 6.34a | 31.7 ± 0.62c |
| F182E2A | 41.8 ± 10.2a | 32.2 ± 1.04c |
| L183E2A | 65.3 ± 11.9 | 46.6 ± 3.70c |
| Y3816.51A | 9.9 ± 2.56a,b | 18.0 ± 0.93b,c |
| K392E3A | 68.8 ± 24.0 | 44.2 ± 1.18c |
| D393E3A | 48.1 ± 8.28a | 35.0 ± 1.22c |
| W4007.35A | 28.6 ± 6.71a | 18.9 ± 1.58c |
| E4017.36A | 27.5 ± 2.92a | 27.3 ± 1.17c |
a Values are significantly different compared with wild-type M1 mAChR; p < 0.05, one-way ANOVA with Dunnett's post-hoc test.
b Normalized estimates from different methods are significantly different; p < 0.05, unpaired Student's t test.
c Raw values are different compared with wild-type M1 mAChR control for each separate experiment; p < 0.05, one-way ANOVA with Dunnett's post-hoc test.
TABLE 2.
Radioligand binding parameter estimates for ligands at M1 mAChR variants
Values represent the mean ± S.E. from 3–4 separate experiments conducted in duplicate. Mutations are listed in order of sequence from the N terminus. ND, not determined.
| M1 mAChR | [3H]QNB | Atropine | ACh | TBPB | VCP794 | 77-LH-28-1 | Xanomeline | VCP813 | |
|---|---|---|---|---|---|---|---|---|---|
| pKDa | Bmaxb | pKIc | |||||||
| WT | 10.76 ± 0.07 | 1.96 ± 0.17 | 8.86 ± 0.04 | 4.66 ± 0.17 | 7.06 ± 0.12 | 6.29 ± 0.03 | 6.26 ± 0.07 | 6.79 ± 0.09 | 4.92 ± 0.08 |
| F772.56I | 10.83 ± 0.14 | 0.83 ± 0.07 | 8.76 ± 0.04 | 4.48 ± 0.09 | 6.89 ± 0.13 | 5.68 ± 0.09d | 5.99 ± 0.03d | 6.83 ± 0.10 | 4.94 ± 0.11 |
| Y822.61A | 10.01 ± 0.17d | 1.01 ± 0.22 | 8.19 ± 0.07d | 3.54 ± 0.03d | 5.97 ± 0.03d | 4.83 ± 0.06d | 5.79 ± 0.09d | 6.18 ± 0.07d | 4.26 ± 0.04d |
| Y852.64A | 10.49 ± 0.12 | 0.51 ± 0.11 | 8.47 ± 0.04d | 3.54 ± 0.003d | 6.29 ± 0.05d | 4.91 ± 0.010d | 5.63 ± 0.02d | 6.33 ± 0.04d | 4.48 ± 0.007d |
| H90E1A | 10.76 ± 0.10 | 0.33 ± 0.04 | 8.79 ± 0.04 | 4.17 ± 0.02d | 7.08 ± 0.03 | 5.96 ± 0.03d | 6.26 ± 0.03 | 6.66 ± 0.01 | 4.84 ± 0.02 |
| W1013.28A | 9.50 ± 0.09d | 0.37 ± 0.07 | 8.11 ± 0.04d | 3.42 ± 0.05d | 8.57 ± 0.04d | 7.99 ± 0.03d | 8.00 ± 0.06d | 5.45 ± 0.13d | 4.39 ± 0.10d |
| L1023.29A | 9.37 ± 0.01d | 0.61 ± 0.16 | 7.61 ± 0.06d | 2.89 ± 0.02d | 6.78 ± 0.02d | 5.36 ± 0.002d | 5.71 ± 0.001d | 5.21 ± 0.13d | 4.46 ± 0.03d |
| D1053.32E | 10.43 ± 0.04 | 0.91 ± 0.11 | 8.91 ± 0.03 | 3.36 ± 0.08d | 7.55 ± 0.02d | 6.09 ± 0.03d | 6.26 ± 0.07 | 6.29 ± 0.08d | 5.05 ± 0.05 |
| D1053.32N | 8.19 ± 0.13d | 0.51 ± 0.05 | ND | ND | ND | ND | ND | ND | ND |
| Y1063.33A | 9.22 ± 0.15d | 0.48 ± 0.03 | 7.42 ± 0.01d | 2.96 ± 0.17d | 6.40 ± 0.01d | 5.65 ± 0.07d | 5.66 ± 0.14d | ND | 5.09 ± 0.17 |
| Y179E2A | 10.39 ± 0.15 | 0.63 ± 0.10 | 8.35 ± 0.009d | 3.99 ± 0.06d | 7.97 ± 0.10d | 6.37 ± 0.02 | 6.59 ± 0.03d | 6.33 ± 0.04d | 4.60 ± 0.04 |
| I180E2A | 10.57 ± 0.14 | 0.66 ± 0.13 | 8.65 ± 0.07d | 3.80 ± 0.11d | 7.58 ± 0.01d | 6.93 ± 0.08d | 6.97 ± 0.04d | 5.85 ± 0.02d | 4.91 ± 0.02 |
| Q181E2A | 10.64 ± 0.17 | 0.65 ± 0.12 | 8.67 ± 0.03 | 4.57 ± 0.09 | 7.30 ± 0.03d | 6.24 ± 0.04 | 6.43 ± 0.03 | 6.60 ± 0.02d | 4.76 ± 0.02 |
| F182E2A | 10.62 ± 0.10 | 0.82 ± 0.20 | 8.36 ± 0.02d | 4.13 ± 0.11d | 7.18 ± 0.01 | 6.35 ± 0.010 | 6.26 ± 0.03 | 6.39 ± 0.005d | 4.47 ± 0.005d |
| L183E2A | 10.20 ± 0.02d | 1.28 ± 0.23 | 8.29 ± 0.02d | 4.19 ± 0.13d | 7.33 ± 0.01d | 6.48 ± 0.04d | 6.47 ± 0.06 | 6.64 ± 0.03d | 4.66 ± 0.03 |
| Y3816.51A | 10.44 ± 0.03 | 0.19 ± 0.05 | 6.75 ± 0.04d | 3.02 ± 0.05d | 8.36 ± 0.02d | 6.75 ± 0.04d | 6.94 ± 0.03d | 7.01 ± 0.02d | 5.22 ± 0.02 |
| K392E3A | 10.61 ± 0.30 | 1.35 ± 0.47 | 8.70 ± 0.05 | 4.21 ± 0.07d | 6.78 ± 0.04d | 5.84 ± 0.03d | 5.98 ± 0.06d | 6.71 ± 0.07 | 4.72 ± 0.01 |
| D393E3A | 10.66 ± 0.15 | 0.94 ± 0.16 | 8.75 ± 0.06 | 4.42 ± 0.07 | 6.82 ± 0.04d | 5.92 ± 0.03d | 6.12 ± 0.01 | 6.75 ± 0.07 | 4.81 ± 0.03 |
| W4007.35A | 10.49 ± 0.18 | 0.56 ± 0.13 | 8.53 ± 0.04d | 3.41 ± 0.02d | 6.19 ± 0.02d | 5.15 ± 0.02d | 5.30 ± 0.04d | 6.88 ± 0.06 | 4.60 ± 0.01 |
| E4017.36A | 10.56 ± 0.29 | 0.54 ± 0.06 | 8.66 ± 0.05d | 4.10 ± 0.09d | 6.79 ± 0.03d | 5.88 ± 0.02d | 6.16 ± 0.02 | 6.67 ± 0.08 | 4.98 ± 0.22 |
a Negative logarithm of the radioligand equilibrium dissociation constant.
b Maximum density of binding sites determined as pmol/mg of protein.
c Negative logarithm of the ligand equilibrium dissociation constant.
d Significantly different compared with wild-type receptor value; p < 0.05, one-way ANOVA with Dunnett's post-hoc test.
Effects of Mutations on Binding Affinity of Ligands to the M1 mAChR
Equilibrium binding studies were performed to examine and compare the effects of mutations on the affinity of orthosteric ([3H]QNB, ACh, atropine, and xanomeline) and bitopic ligands (77-LH-28-1 and TBPB). Additionally, the agonist and antagonist fragment derivatives of TBPB, VCP794 and VCP813, respectively, were also investigated (19). The concentration-dependent displacement of [3H]QNB from the M1 mAChRs by each ligand (Fig. 2) was indistinguishable from a competitive interaction with the receptor, suggesting either orthosteric antagonism or allosteric modulation characterized by very high negative cooperativity. Irrespectively, the data were analyzed by fitting them to one-site inhibition mass action curves to determine estimates of equilibrium binding dissociation constants (Table 2), because highly negative allosteric modulation is indistinguishable from simple competition at equilibrium.
FIGURE 2.

Differential effects of selected mutations on the binding of orthosteric and bitopic ligands to the M1 mAChR. Shown are data for the concentration-dependent inhibition of [3H]QNB binding by atropine (A), ACh (B), xanomeline (C), TBPB (D), 77-LH-28-1 (E), VCP794 (F), and VCP813 (G) at the wild-type, F772.56I, W1013.28A, Y179A, I180A, and Y3816.51A variants of the M1 mAChR. Data represent the mean ± S.E. of at least three experiments performed in duplicate.
As expected, the orthosteric binding site mutations (W1013.28A,L1023.29A, D1053.32E, Y1063.33A, and Y3816.51A) resulted in overall reductions in the affinities of the selected orthosteric ligands (Fig. 3). However, there were some exceptions. In contrast to the orthosteric agonists (ACh and xanomeline), the antagonists [3H]QNB and atropine were both unaffected by the D1053.32E mutation (Table 2 and Fig. 3, A and C). Additionally, for the Y3816.51A mutation, [3H]QNB affinity was unaffected, whereas xanomeline affinity was modestly enhanced. For the D1053.32N mutant, a marked reduction (over 400-fold) in the binding affinity of [3H]QNB was observed (Table 2 and Fig. 3A). Together with the lack of detectable agonist responses in subsequent functional assays (data not shown) and findings from previous studies of mAChRs (10, 26, 45), this mutation was not investigated further.
FIGURE 3.

Bitopic ligands display different binding profiles compared with orthosteric ligands. Bars represent the change in equilibrium binding affinity (pKD or pKI), compared with the wild-type M1 mAChR, of orthosteric ligands [3H]QNB, atropine, ACh, and xanomeline (A–D, respectively), bitopic ligands TBPB and 77-LH-28-1 (E and F, respectively), and fragment derivatives of TBPB, VCP794, and VCP813 (G and H, respectively). In cases where an estimate could not be determined for a ligand at a mutation, the x axis label is shown faded. *, significant change in affinity relative to wild-type receptor, p < 0.05, one-way ANOVA with Dunnett's post-test.
Consistent with previous reports (15, 22, 23), improved binding of TBPB and 77-LH-28-1 was detected at the W1013.28A receptor. This was also observed for the agonist fragment derivative of TBPB (VCP794), and all three ligands exhibited improved binding at the Y3816.51A receptor (Fig. 2, D–F). Interestingly, the effect of D1053.32E was different among TBPB, VCP794, and 77-LH-28-1, improving, reducing, or not altering the affinities of the ligands, respectively. The other mutations within the orthosteric binding site resulted in reductions in ligand affinities (Table 2). In the case of the antagonist fragment VCP813, its binding was largely unchanged at most mutants and only modestly reduced at the W1013.28A and L1023.29A mutants.
An examination of the other mutated residues within the heptahelical bundle outside of the orthosteric site found similar reductions in binding affinity of all agonist ligands at the Y822.61A, Y852.64A, W4007.35A, and E4017.36A mutations (Table 2 and Fig. 3). Exceptions were found with 77-LH-28-1 and xanomeline, both of which maintained affinity for the E4017.36A mutant, whereas xanomeline was also unaffected by the W4007.35A mutation. Intriguingly, although 77-LH-28-1 exhibited a small but significant reduction in affinity at the F772.56I mutation, consistent with previous findings (15), it also exhibited reduced affinity (∼3-fold) at the Y822.61A receptor (Fig. 3D), in contrast to an earlier report (22). Furthermore, unlike the structurally similar TBPB, VCP794 displayed reduced affinity for the F772.56I mutant (Fig. 3F), whereas the affinities of the remainder of the tested ligands were unaltered.
Alanine substitutions of nonconserved residues in ECL1 and ECL2 (His-90E1, Lys-392E3, and Asp-393E3) had little effect on the binding affinities of most agonists tested (Table 2 and Fig. 3). However, mutation of conserved residues in ECL2 revealed surprisingly divergent behaviors of orthosteric and bitopic agonists. Although the affinities of ACh (Fig. 3E) and xanomeline (Fig. 3G) were significantly decreased at these mutations, bitopic ligands exhibited either equivalent or improved binding affinities (Fig. 3, B and D). However, these effects were not observed at the Q181E2A mutant, which largely had no effect on the binding of each of the ligands tested except for subtle improvement or reduction in the estimated affinities of TBPB and xanomeline, respectively. Surprisingly, VCP813 maintained its binding affinity at the mutations of the extracellular loop, with only a small reduction found at the F182E2A receptor.
Signaling of Agonists at Mutant M1 mAChRs
To examine and compare the effect of mutations on the agonist profiles of ACh, xanomeline, 77-LH-28-1, TBPB, VCP794, and VCP813, concentration-response curves were constructed for M1 mAChR-mediated [Ca2+]i mobilization and ERK1/2 phosphorylation. VCP813 did not exhibit any agonist activity at the wild-type receptor, consistent with previous identification that it is an antagonist (19), nor at any other M1 mAChR mutant (data not shown) and was not studied further.
An examination of the agonist responses in [Ca2+]i mobilization experiments at M1 mAChR mutants revealed striking effects at the F772.56I, Y822.61A, Y852.64A, W1013.28A, I180E2A, and L183E2A mutants (Fig. 4). These mutations had differential effects on the orthosteric relative to the bitopic agonists tested. For the F772.56I, Y822.61A, and Y852.64A mutants within TM2 (Fig. 4, B–D), the agonism of the ligands TBPB, 77-LH-28-1, and VCP794 was reduced compared with the orthosteric agonists ACh and xanomeline. This was most apparent with the Y822.61A and Y852.64A mutants. In contrast, the W1013.28A, I180E2A, and L183E2A mutants resulted in enhancements in bitopic agonist potencies relative to the orthosteric agonists (Fig. 4, E–G). pERK1/2 experiments revealed similar marked changes in agonist potencies at mutants relative to wild-type M1 mAChR to those observed in [Ca2+]i mobilization (Fig. 5). Intriguingly, although agonist potencies for pERK1/2 were lower overall compared with the respective [Ca2+]i mobilization experiments, more marked losses in the agonist responses of TBPB and VCP794 were found at the Y179E2A and F182E2A mutants, respectively (Fig. 5, F and H), for agonist stimulation of pERK1/2.
FIGURE 4.

Agonist stimulation of mutant M1 mAChR-mediated [Ca2+]i mobilization. Shown are data for the wild-type (A), F772.56I (B), Y822.61A (C), Y852.64A (D), W1013.28A (E), I180A (F), and L183A (G) mutants. Data represent the mean ± S.E. of at least three experiments performed in duplicate. The error bars not shown lie within the dimensions of the symbol.
FIGURE 5.

Agonist stimulation of mutant M1 mAChR-mediated pERK1/2. Shown are data for the wild-type (A), F772.56I (B), Y822.61A (C), Y852.64A (D), W1013.28A (E), Y179A (F), I180A (G), F182A (H), and L183A (I) mutants. Data represent the mean ± S.E. of at least three experiments performed in duplicate. The error bars not shown lie within the dimensions of the symbol.
There was little difference in the relative rank order of potencies at the wild-type M1 mAChR between the two cellular responses (Table 3). To more quantitatively assess the agonist profiles, data sets were fitted to the operational model of agonism to derive estimates of the functional affinity (KA) underlying each signaling state, as well as operational measures of efficacy (τ). Because all of the agonists yielded the maximal system response at the wild-type M1 mAChR, the functional affinity estimates in this instance were determined via operational model fitting to curves established in the absence and presence of irreversible occlusion of the receptors by incubation with phenoxybenzamine (Table 4).
TABLE 3.
Potencies of agonists at Flp-In CHO cells expressing the wild-type M1 mAChR
Values are mean ± S.E. of 3–6 separate experiments.
| Agonist | pEC50a |
|
|---|---|---|
| [Ca2+]i mobilization | pERK1/2 | |
| Ach | 8.91 ± 0.07 | 7.76 ± 0.06 |
| TBPB | 8.14 ± 0.07 | 7.22 ± 0.08b |
| VCP794 | 7.90 ± 0.09 | 7.50 ± 0.14 |
| 77-LH-28-1 | 8.28 ± 0.03 | 7.71 ± 0.05 |
| Xanomeline | 8.33 ± 0.04 | 7.55 ± 0.07 |
a Negative logarithm of the concentration of agonist that elicits 50% of its maximal response.
b Maximal response of the agonist was lower compared with Ach.
TABLE 4.
Affinity estimates of agonists for the wild-type M1 mAChR and mutant variants determined from signaling assays
Values are the mean ± S.E. from 3–5 separate experiments. Mutations are listed in order of sequence from the N terminus. NR, no response or insufficient response detected for determination of affinity estimate. ND, not determined due to maximal response not different from ACh.
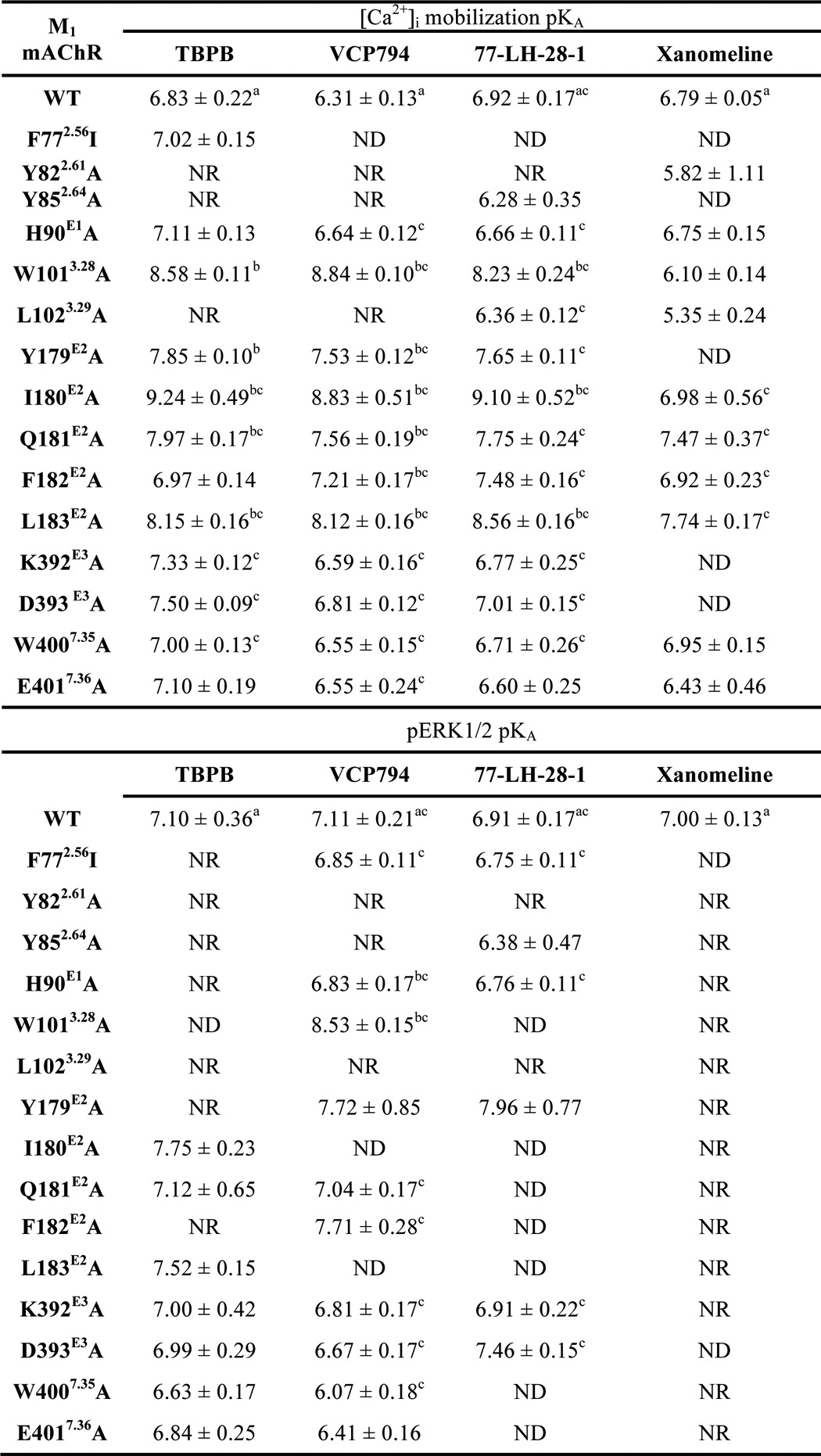
a Data were determined from irreversible occlusion of receptors by phenoxybenzamine.
b Significantly different (p < 0.05) from wild-type receptor by one-way ANOVA analysis with Dunnett's post-hoc test.
c Significantly different (p < 0.05) from value determined through radioligand binding experiments by one-way ANOVA analysis with Dunnett's post-hoc test.
An important observation resulting from this analysis was that, in general, the estimated agonist functional affinities (Table 4) were substantially greater than the respective affinity estimates determined from radioligand binding experiments (Table 2). This was particularly so with VCP794 and 77-LH-28-1, which exhibited an average 10-fold greater functional affinity compared with radioligand binding affinity estimates. These discrepancies between functional and binding affinity estimates may be due to overall differences in the assay conditions between the binding and signaling experiments, or may be indicative of agonist engagement of alternate receptor conformations driven by transducer-specific affinities that are operative at the level of the intact cell but not captured in the biochemical radioligand binding experiments.
If the differences in affinity estimates were predominantly due to differences in assay conditions, then one could expect changes in absolute affinity values but not in the overall rank orders, such that a significant correlation would exist between binding and functional affinity values. Thus, the functional and binding affinity estimates (for those mutants where these values could be derived) were subjected to a correlation analysis. In all cases, the analyses of functional affinity estimates from [Ca2+]i mobilization (Fig. 6) and pERK1/2 studies (Fig. 7) revealed a significant correlation with the affinity values from the radioligand binding studies (p < 0.05), suggesting that the major source of discrepancy is likely due to the assay conditions (e.g. ionic strength, temperature, and time). However, there were some exceptions, derived from the comparison of [Ca2+]i mobilization and radioligand binding data. Specifically, we identified outliers in each data set, as defined by their location outside of the boundaries of the 95% confidence bands for the line of best fit. Exclusion of these outliers resulted in substantially improved R2 values of 0.78, 0.88, 0.75, and 0.86 for TBPB, VCP794, 77-LH-28-1, and xanomeline, respectively. Interestingly, of the outliers, two mutations, I180E2A and L183E2A, were common across all four agonists, suggesting that in these two instances the agonist functional affinity estimates may reflect the stabilization of transducer-specific receptor conformations that were not detected in the binding assays. These changes in agonist functional affinity estimates at mutations are further illustrated in Figs. 8 and 9. Despite fewer determined functional affinity estimates from pERK1/2 experiments, the comparison with estimates from [Ca2+]i mobilization experiments revealed divergent affinity effects at the mutations of ECL2, including the I180E2A and L183E2A mutants. At these mutations, substantial and significant improvements to the functional affinities of TBPB, VCP794, and 77-LH-28-1 were observed in [Ca2+]i mobilization experiments (Fig. 8) but not pERK1/2 experiments (Fig. 9).
FIGURE 6.

Correlation analysis of functional affinity estimates from [Ca2+]i mobilization experiments with affinity estimates from radioligand binding experiments for TBPB (A), VCP794 (B), 77-LH-281 (C), and xanomeline (D). Linear regression curves depicted are the line of best fit (solid line) and the 95% confidence bands (dotted line curve). For reference, the WT receptor value is shown as an open symbol, in addition to the line of unity with radioligand binding affinity estimates (dashed line). Outliers, located outside the 95% confidence bands, are labeled with their respective mutations.
FIGURE 7.
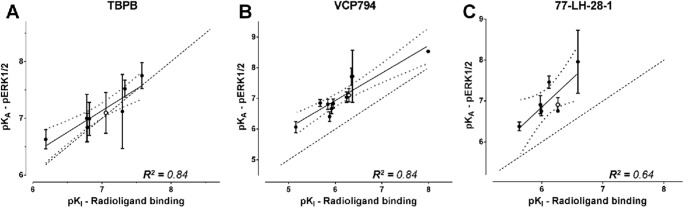
Correlation analysis of functional affinity estimates from pERK1/2 experiments with affinity estimates from radioligand binding experiments for A) TBPB, B) VCP794 and C) 77-LH-281. Linear regression curves depicted are the line of best fit (solid line) and the 95% confidence bands (dotted line curve). For reference, the WT receptor value is shown as an open symbol, in addition to the line of unity with radioligand binding affinity estimates (dashed line).
FIGURE 8.

Effects of mutations on agonist affinities estimated from [Ca2+]i mobilization experiments. Bars represent the change in functional affinity estimates (pKA) relative to the wild-type M1 mAChR for TBPB (A), 77-LH-28-1 (B), VCP794 (C), and xanomeline (D). In cases where an estimate could not be determined for an agonist at a mutation, the x axis label is shown faded. Estimates for ACh and the D105E and Y381A mutations were not determined and as a consequence are not included. *, significant change in affinity relative to wild-type receptor, p < 0.05, one-way ANOVA with Dunnett's post-test.
FIGURE 9.
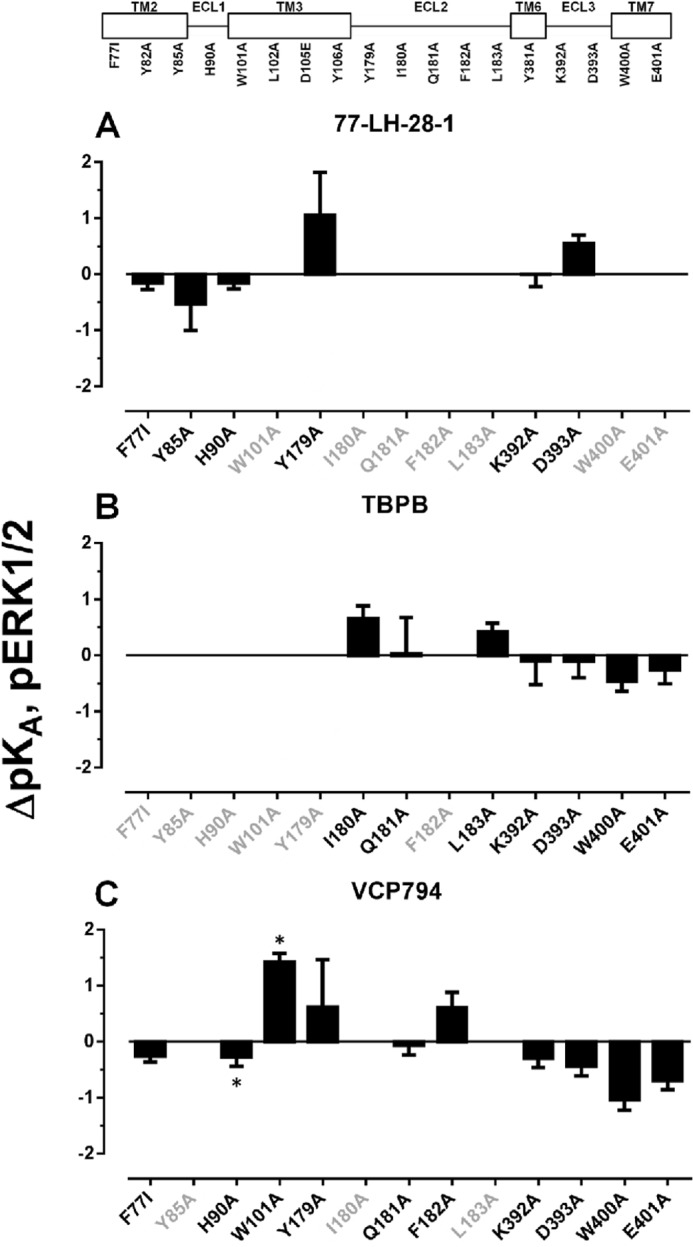
Effects of mutations on agonist affinities estimated from pERK1/2 experiments. Bars represent the change in functional affinity estimates (pKA), compared with the wild-type M1 mAChR, of agonists 77-LH-28-1 (A), TBPB (B), and VCP794 (C). In cases where an estimate could not be determined for an agonist at a mutation, the x axis label is shown faded. Estimates for ACh and mutations Y82A, L102A, D105E, and Y381A were not determined and as a consequence are not included. *, significant change in affinity relative to wild-type receptor, p < 0.05, one-way ANOVA with Dunnett's post-test.
Along with the estimation of functional affinity (KA), application of the operational model of agonism also yielded estimates of efficacy (τ). Given that the measured efficacy of agonist responses is affected by differences in receptor expression (41), the τ values estimated from operational model fitting of agonist functional data at each mutant were corrected for receptor expression (from flow cytometric analysis), yielding corrected efficacy estimates (τc). Due to cases where the agonist functional affinity could not be determined (i.e. full or insufficient agonist responses), efficacy estimates were derived by constraining the functional affinity value (KA) to the affinity value estimated from radioligand binding studies (KI). However, as described above, the KA of an agonist may be divergent from its KI estimate. Consequently, this constraint to agonist KI values yielded estimates of τ that are reflective of changes in both efficacy and functional affinity (KA), as was the case with ACh. However, although there is an intrinsic level of error in this approach, the positive correlations between the mutational effects on functional and binding affinity estimates suggest that the estimates of efficacy for these cases will be indicative of the general direction of change relative to the wild-type receptor.
On analysis of agonist efficacies (Table 5 and Figs. 10 and 11), reductions in signaling efficacy were detected for agonists at most of the mutations within and neighboring the orthosteric site (W1013.28A, L1023.29A, D1053.32E, Y1063.33A, and Y3816.51A). As described previously, W1013.28A was a notable exception to this group. Analyses determined a reduction of ACh signaling efficacy by about 3-fold (Figs. 10A and 11A) but not of the other agonists tested. In contrast, significant reductions in efficacy and loss of detectable agonist responses were found with all agonists at the Y3816.51A mutant, despite significantly enhanced binding affinity exhibited by TBPB, VCP794, 77-LH-28-1, and xanomeline. However, agonist functional affinity estimates could not be determined for the Y3816.51A mutant due to a lack of reliable determination of the maximal agonist response.
TABLE 5.
Relative efficacies of agonists for the wild-type M1 mAChR and mutant variants determined from signaling assays
Values are the mean ± S.E. from 3–4 separate experiments. Mutations are listed in order of sequence from the N terminus. ND, not determined.
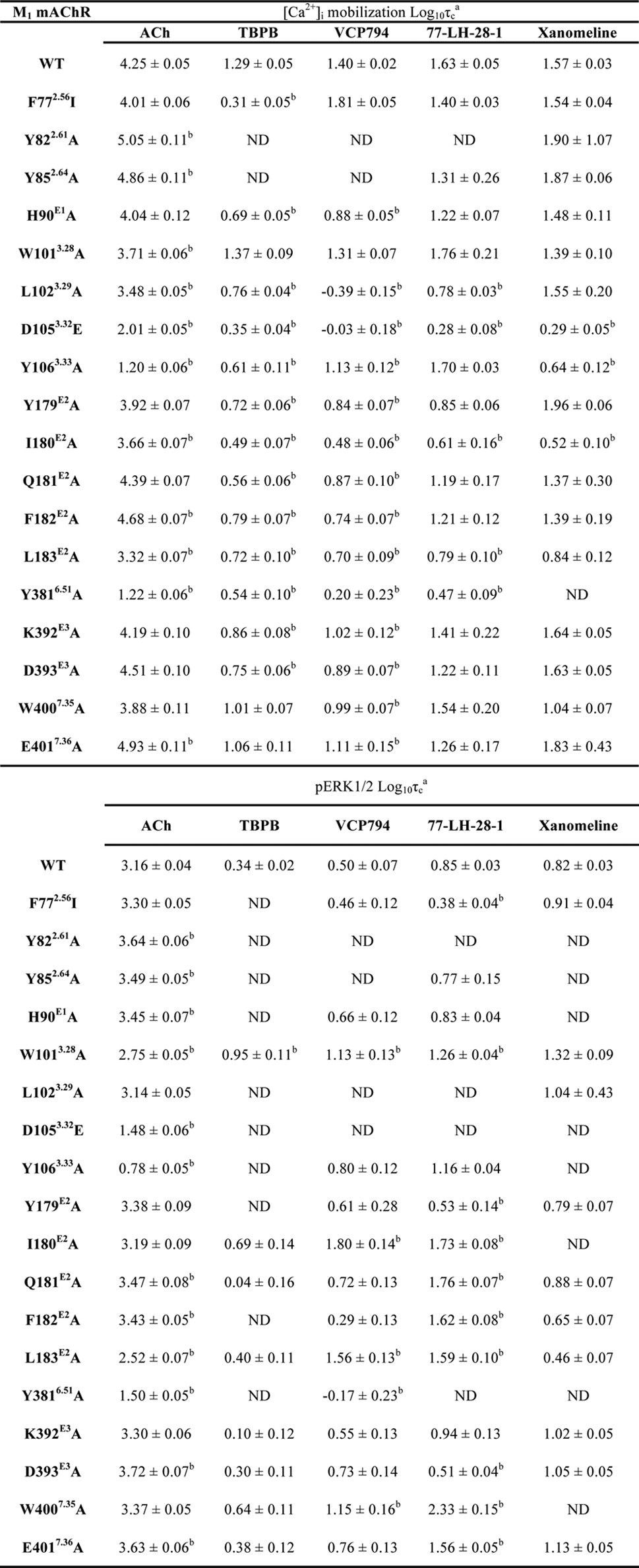
a Relative efficacy parameter, τ, was determined via nonlinear regression of the data to an operational model of agonism and corrected for receptor expression levels, relative to reference wild-type M1 mAChR control cells, to yield a corrected τc parameter.
b Significantly different compared with wild-type receptor, p < 0.05, one-way ANOVA with Dunnett's post-hoc test.
FIGURE 10.

Effect of mutations on agonist signaling efficacies for M1 mAChR-mediated [Ca2+]i mobilization. Bars represent the change in logτc (efficacy estimates corrected for receptor expression level) compared with the wild-type M1 mAChR for ACh (A), TBPB (B), 77-LH-28-1 (C), VCP794 (D), and xanomeline (E). Values were derived from an operational model of agonism (see “Experimental Procedures”). In cases where an estimate could not be determined for an agonist at a mutation, the x axis label is shown faded. *, significant change in efficacy relative to wild-type receptor, p < 0.05, one-way ANOVA with Dunnett's post-test.
FIGURE 11.

Effect of mutations on agonist signaling efficacies for M1 mAChR-mediated pERK1/2. Bars represent the change in logτc (efficacy estimates corrected for receptor expression level) compared with the wild-type M1 mAChR for ACh (A), TBPB (B), 77-LH-28-1 (C), VCP794 (D), and xanomeline (E). Values were derived from an operational model of agonism (see “Experimental Procedures”). In cases where an estimate could not be determined for an agonist at a mutation, the x axis label is shown faded. *, significant change in efficacy relative to wild-type receptor, p < 0.05, one-way ANOVA with Dunnett's post-test.
Differential effects on agonist efficacies were observed with mutations of TM2. Although the F772.56I mutant significantly reduced the efficacy of TBPB (Figs. 10B and 11B), the efficacy of VCP794 was unchanged (Figs. 10D and 11D). The difference between TBPB and VCP794 is interesting given the similarities in chemical structure. In comparison, the efficacy of 77-LH-28-1-activated pERK1/2 was reduced at the F772.56I mutant (Fig. 10C), consistent with previous reports (15, 22, 23). However, its efficacy in [Ca2+]i mobilization experiments was unchanged (Fig. 11C). In the case of the Y822.61A and Y852.64A mutants, the loss of bitopic agonist responses likely emanates from marked reductions in efficacy; the reductions in the bitopic ligand binding affinities and receptor expression are insufficient to account for these observations. Indeed, this is highlighted by the different profile of 77-LH-28-1, which had unchanged efficacy at the Y852.64A mutant (Table 5).
Although mutations in ECL2 enhanced the binding and/or functional affinity of the bitopic agonists, a general reduction in agonist efficacies was observed at these mutants. This indicates that the greater potencies of bitopic agonists at these mutants, relative to ACh, are the result of greater affinity for the receptor. However, at the Y179E2A, Q181E2A, and F182E2A mutants, and the mutations of ECL1 and ECL3, the efficacies of ACh, xanomeline, and 77-LH-28-1 were enhanced overall or unchanged, in contrast to TBPB and VCP794.
Identification of Receptor Domains Involved with Agonist Bias
Given the diverse changes in agonist functional affinities and/or efficacies at the receptor observed for both [Ca2+]i mobilization and pERK1/2, these could affect the agonist bias profile for the two signaling pathways. We thus utilized the operational model (43), which accounts for changes in both agonist parameters via derivation of composite τ/KA values, to analyze agonist bias profiles at each mutation relative to the preferences of ACh (Table 6). At the wild-type M1 mAChR, VCP794, 77-LH-28-1, and xanomeline were identified as biased agonists for pERK1/2. The profiles of bias for each agonist relative to ACh across the investigated mutations are presented in Fig. 12; values greater than 1 indicate -fold bias toward pERK1/2, and values less than 1 indicate the inverse of the -fold bias toward [Ca2+]i mobilization. Unfortunately, the bias factors of agonists at several mutations could not be determined due to a loss of detectable agonist pERK1/2 responses and thus are not shown.
TABLE 6.
Bias factors of agonists for [Ca2+]i mobilization and pERK1/2
Data were analyzed using an operational model of agonism (see “Experimental Procedures”). Agonist transduction coefficients (logτ/KA) were each normalized to the values determined for ACh for each signaling pathway (pERK1/2 and [Ca2+]i mobilization). To avoid the propagation of error from multiple data manipulation steps, the normalized transduction coefficients for the two pathways were compared statistically to determine signaling bias compared with ACh (see “Experimental Procedures”). Values are expressed as mean ± S.E. of 3–6 independent experiments conducted in duplicate. ND, not determined.
| Mutation | ACh | TBPB | VCP794 | 77-LH-28-1 | Xanomeline |
|---|---|---|---|---|---|
| log bias pERK1/2 − [Ca2+]i mobilization | |||||
| WT | 0.00 ± 0.08 | 0.02 ± 0.08 | 0.47 ± 0.08a | 0.42 ± 0.08a | 0.43 ± 0.08a |
| F772.56I | 0.00 ± 0.05 | ND | 0.57 ± 0.09a | 0.49 ± 0.10a | 0.14 ± 0.05 |
| Y822.61A | 0.00 ± 0.11 | ND | ND | ND | 0.75 ± 0.12a |
| Y852.64A | 0.00 ± 0.08 | ND | ND | 0.78 ± 0.15a | 0.46 ± 0.10a |
| H90E1A | 0.00 ± 0.05 | ND | 0.50 ± 0.20 | 0.27 ± 0.12 | 0.05 ± 0.07 |
| W1013.28A | 0.00 ± 0.06 | 0.20 ± 0.06 | 0.33 ± 0.07a | 0.30 ± 0.06a | ND |
| Y1063.33A | 0.00 ± 0.05 | ND | 0.40 ± 0.06a | 0.38 ± 0.03a | ND |
| Y179E2A | 0.00 ± 0.08 | ND | −0.02 ± 0.10 | 0.13 ± 0.15 | −0.72 ± 0.08a |
| I180E2A | 0.00 ± 0.07 | −0.66 ± 0.35 | −0.05 ± 0.36 | −0.45 ± 0.26 | ND |
| Q181E2A | 0.00 ± 0.10 | −0.42 ± 0.82 | 0.28 ± 0.19 | 0.18 ± 0.13 | −0.61 ± 0.11a |
| F182E2A | 0.00 ± 0.07 | ND | 0.88 ± 0.43a | ND | −0.01 ± 0.22 |
| L183E2A | 0.00 ± 0.06 | −0.17 ± 0.16 | 0.19 ± 0.10 | −0.21 ± 0.08 | −0.50 ± 0.15a |
| Y3816.51A | 0.00 ± 0.31 | ND | 0.62 ± 0.49 | ND | ND |
| K392E3A | 0.00 ± 0.06 | −0.08 ± 0.45 | 0.71 ± 0.14a | 0.10 ± 0.07 | 0.27 ± 0.07a |
| D393E3A | 0.00 ± 0.06 | −0.02 ± 0.31 | 0.62 ± 0.13a | 0.67 ± 0.16a | 0.38 ± 0.06a |
| W4007.35A | 0.00 ± 0.06 | 0.00 ± 0.27 | 0.37 ± 0.17 | 0.06 ± 0.09 | 0.17 ± 0.19 |
| E4017.36A | 0.00 ± 0.09 | 0.47 ± 0.32 | 0.95 ± 0.19a | 1.45 ± 0.11a | 0.77 ± 0.11a |
a Normalized agonist transduction coefficients for pERK1/2 and [Ca2+]i mobilization were significantly different; p < 0.05, Student's t test.
FIGURE 12.

Effect of mutations on agonist bias of M1 mAChR-mediated pERK1/2 and [Ca2+[rsqb]i mobilization. Radial plot of agonist bias factors (the ratio of the transduction coefficient for pERK1/2 to the coefficient for [Ca2+]i mobilization, each normalized to the values determined for ACh) derived from an operational model of agonism (see “Experimental Procedures”) are plotted for each M1 mAChR variant. Values greater than 1 denote bias toward pERK1/2, and values less than 1 denote bias toward [Ca2+]i mobilization relative to ACh signaling. Dashed lines indicate mutations for which an estimate could not be determined.
In the case of the first cluster of mutants encompassing TM2, ECL1, and TM3 mutations, bias factors could not be determined in a number of instances because of an overall loss in signaling efficacy due to the mutation. For those instances where bias factors were determined, however, it was noted that the bias toward pERK1/2 over [Ca2+]i mobilization was largely maintained, if not slightly exaggerated, with the only notable exception being the loss of xanomeline bias at the F772.56I and H90A mutations. For the second cluster of mutants, namely those in ECL2, a clear pattern emerged, namely that the bias moved away from pERK1/2 and toward [Ca2+]i mobilization. This was particularly significant for xanomeline at the Y179E2A, Q181E2A, and L183E2A mutations but was also evident for the other agonists. The only exception to this was the effect of F182E2A on VCP794.
Furthermore, in the case of TBPB, it was unable to stimulate pERK1/2 at Y179E2A and F182E2A mutations. Given that radioligand binding studies demonstrate that TBPB exhibits enhanced or unchanged affinity at these mutations, respectively, the loss in pERK1/2 activity likely reflects marked reductions in pERK1/2 efficacy and/or functional affinity compared with [Ca2+]i mobilization and a substantial change in bias toward this latter response.
The final cluster of mutants encompassed TM6, ECL3, and TM7, where it was noted that the effect of most mutations was to reduce the pERK1/2 bias for one or more agonists. The major exceptions to this were D393A, which had no effect on the bias for any agonist, and E4017.36A, which strikingly increased the bias toward pERK1/2 for all agonists.
Modeling of TBPB and VCP794 Binding to the M1 mAChR
The recent solution of crystal structures of the M2 and M3 mAChRs (32, 49, 50) provides insights into the structure of mAChRs. A homology model of the M1 mAChR was generated based on the M3 mAChR, due to the shared sequence homology and functional coupling profiles of the two subtypes. Molecular dynamics simulations of TBPB and VCP794 binding to the solvated M1 mAChR predict distinct interactions with the receptor. Final poses after 20 and 19 ns of simulation, respectively, are depicted in Fig. 13. From these in silico predictions, both ligands share similar poses within a solvated pocket of the receptor. Although there are differences in size and structure between TBPB and VCP794, the residues engaged by these ligands within the binding pocket are largely similar. This pocket is largely enclosed by hydrophobic and aromatic residues that were observed to surround QNB, iperoxo, and tiotropium in the M2 and M3 mAChRs, respectively (32, 49, 50). However, the common benzimidazolone portion of both ligands is positioned between TM2, TM3, and TM7, with the agonist molecules extending from this moiety down into the heptahelical bundle (Fig. 13, A and B). This yields a larger pocket that is also bordered by Ser-782.57, Tyr-822.61, Tyr-852.64, Trp-1013.28, Leu-1023.29, Tyr-179, and Ile-180.
FIGURE 13.

Molecular modeling suggests novel binding poses of TBPB and VCP794 at the M1 mAChR. Shown are side views of the M1 mAChR following molecular dynamics simulation with TBPB and VCP794 after 20 and 19 ns, respectively (A and B). Ribbon representations of views of TBPB (A, light green) and VCP794 (B, dark green) from the TM4 and TM5 face (helices and parts of ECL2 were removed for clarity). Residues that exhibited different positions compared with equivalent residues at the M2 (magenta) and M3 (blue) mAChR crystal structures (Haga et al. (49) and Kruse et al. (32)) are shown in C, with comparative superimposition of the TBPB-bound M1 mAChR. The inner surface of the M1 mAChR, from the TM6 and TM7 faces (helices removed for clarity) following simulation with TBPB is shown in D. The poses of TBPB, VCP794, QNB, and tiotropium (white) (Haga et al. (49) and Kruse et al. (32)) are shown for comparison. Residues mutated in this study are colored based on location: TM2 (yellow), TM3 (red), and ECL2 (cyan).
These predictions appear consistent with the affinity reductions of the bitopic ligands observed at most of the mutants of these residues. For instance, the rotamerization of Trp-1013.28 away from the orthosteric pocket, compared with the M2 and M3 mAChR crystal structures (32, 49), serves to accommodate the binding of TBPB (Fig. 13C) and VCP794 and is concordant with the effects of alanine substitution of this residue. However, although a gauche+ conformation of Trp-1013.28 has been suggested as permitting 77-LH-28-1 binding to the M1 mAChR (10, 15, 22), our simulations did not predict such a pronounced rotamerization of the residue. In agreement with the enhanced bitopic ligand binding affinities at the Y3816.51A mutant, the final ligand poses reveal only Tyr-3816.51 positioned away from the orthosteric pocket (Fig. 13C), compared with the resolved structures of the M2 and M3 mAChRs (32, 49, 50). This movement of Tyr-3816.51 away from the binding pocket may be a consequence of steric interactions between the residue and bitopic ligands. Indeed, this residue is further away from the pocket when TBPB is present, compared with VCP794, which also correlates with their relative changes in affinity at the Y3816.51A mutation.
Although both TBPB and VCP794 occupy the orthosteric binding site, this engagement is different than QNB and tiotropium bound to the M2 and M3 mAChRs, as the bitopic ligands reside some distance away from TM5 (Fig. 13D). In particular, residues surrounding the benzimidazolone portion of TBPB and VCP794, such as Tyr-179E2 and Ile-180E2, are oriented to enclose the space that this moiety occupies. In addition to extending into the orthosteric site, TBPB and VCP794 appear to extend into an extracellular region between TM2, TM3, and TM7. Although this region plays an indirect role in the stabilization of the orthosteric binding site (51), it is also directly involved with the binding of prototypical allosteric modulators such as gallamine (28). Moreover, for both ligands, a prominent rotameric movement is observed of Tyr-4047.39 away from the center of the heptahelical bundle toward TM6. Interestingly, this movement may also assist in the movement of Tyr-3816.51 to accommodate TBPB binding, with weak hydrogen bonding between Tyr-4047.39 and Tyr-3816.51 observed with simulation.
DISCUSSION
Bitopic ligands provide an interesting approach to selectively targeting receptor subtypes and biasing intracellular signaling. Through mutagenesis of the orthosteric binding domain and neighboring allosteric domain(s) of the M1 mAChR, we provide further evidence for the bitopic engagement of the M1 mAChR by TBPB and 77-LH-28-1. Furthermore, we have identified residues of the M1 mAChR that are involved with bitopic ligand signaling responses into cells.
Various investigations of selective ligands at the M1 mAChR have utilized sensitivity to the Y3816.51A mutation as evidence of engagement, or lack thereof, with the orthosteric binding domain (6, 7, 11, 52). However, our findings support bitopic ligand engagement with the orthosteric site for such ligands. The differential effects of the W1013.28A and Y3816.51A mutants also highlight the different occupation of the orthosteric site by bitopic ligands compared with purely orthosteric ligands such as ACh. Furthermore, this illustrates the limitation of relying on a single mutation to discriminate between purely orthosteric ligands, purely allosteric ligands, and bitopic ligands in receptors where these sites are in relatively close apposition, such as the mAChRs.
Apart from mutations within the orthosteric binding site, the largest reductions in binding affinity of the bitopic ligands were found at the Y822.61A, Y852.64A, and W4007.35A mutants. Furthermore, the marked reductions in bitopic agonist efficacies at mutants of TM2 indicate that this region is necessary for receptor activation by these ligands. This is consistent with previous findings at the M1 and M2 mAChRs and the reported mode of binding of 77-LH-28-1 (10, 15, 22), suggesting a different role for TM2 in bitopic ligand efficacy compared with orthosteric ligands (51). Indeed, previous structure-activity and truncation studies of TBPB (19, 53) have demonstrated that modification or removal of the benzimidazolone moiety of TBPB impairs the agonist activity of the molecule. This appears consistent with the predicted engagement of the moiety of TM2. Moreover, the structurally similar dihydroquinolone of 77-LH-28-1 is predicted to engage TM2 within both the M1 and M2 mAChRs (10, 15, 22). This suggests that the common features of these moieties could be exploited to develop ligands that engage with this domain of the receptor.
We have also identified novel roles for ECL2 in bitopic ligand binding and function at the M1 mAChR. In contrast to the orthosteric ligands, the greater binding affinities of bitopic ligands exhibited at mutations of ECL2 suggest that these residues impede the binding of the bitopic ligands, probably to the orthosteric site. These enhanced affinities are also similarly observed in functional studies. Moreover, we identified the I180E2A and L183E2A mutants as eliciting even larger improvements of bitopic agonist functional affinity in [Ca2+]i mobilization studies compared with binding studies. This indicates that these agonists could stabilize high affinity conformations of the M1 mAChR for this functional pathway, suggesting a role for these residues (and ECL2) in functional signaling of the receptor.
Indeed, regarding M1 mAChR signaling, ECL2 appears to play a role in signaling bias, evident in the mutational changes in the bias profiles of the bitopic ligands (Fig. 12). This finding is consistent with prior studies of ECL2 as a possible constraint for the active conformations naturally adopted by the M2 mAChR (10, 20), and suggest that the region may play a general role as a molecular “microswitch” in more than one receptor. Furthermore, it appears that the enhanced functional affinities of agonists at mutations of ECL2, relative to radioligand binding estimates, bears considerable weight in signaling bias; compared with the wild-type M1 mAChR, agonists were biased away from pERK1/2 at the I180E2A and L183E2A mutants, despite agonist efficacies for [Ca2+]i mobilization being reduced. Consequently, this highlights the importance of accounting for functional affinities that diverge from affinity estimates from binding assays (54).
Although mutations altering the signaling bias of agonists have been reported for the M2 mAChR, glucagon-like peptide type 1 receptor and dopamine D2L receptor (10, 20, 55–57), ours is the first study to quantitatively characterize this phenomenon at the M1 mAChR using an operational approach. Furthermore, it is interesting to note that the blunting of agonist signaling to pERK1/2 observed at the Y177E2A M2 mAChR mutant (20) bears a similarity to our examination of the equivalent Y179E2A mutant at the M1 mAChR. Given that ECL2 of mAChRs is involved with the binding of allosteric ligands (21, 50, 58–60), along with its potential involvement in biased signaling, further scrutiny into the bias profiles of mAChR allosteric ligands may be warranted.
From the crystal structures of the inactive state M2 and M3 mAChRs and the active state M2 mAChR, an aromatic cage enclosing the bound orthosteric antagonist or agonist is evident (32, 49, 50). However, this by no means indicates that the boundary of the orthosteric site is rigid and stable. In addition to previous investigation with 77-LH-28-1 (15), our predicted poses of TBPB and VCP794 at the M1 mAChR suggest that the flexibility of amino acid residues in the aromatic cage can accommodate bitopic ligands. Furthermore, previous reports have indicated that residues of the orthosteric site aromatic cage are required for receptor activation (23, 24, 61). Hence, the lower efficacies of bitopic agonists may be due to the disruption of the aromatic cage as they extend up toward an allosteric site and, consequently, the disruption of key activation networks.
Surprisingly, our molecular dynamics simulations of TBPB predicted that the ortho-tolyl moiety, shared with its antagonist derivative VCP813, was engaged with the orthosteric binding domain of the receptor. This is interesting given that VCP813 interacts allosterically and the ortho-tolyl moiety conveys agonist selectivity for the M1 mAChR (19). Also, the modest reductions in VCP813 affinity observed at several mutations do not support direct interactions with the bitopic space occupied by TBPB and VCP794. This suggests that the antagonist may interact solely with a different, yet undefined, allosteric binding site. Although VCP813 is derived from TBPB, truncation of a ligand does not guarantee a fragment derivative that will bind in the same domain of the target protein (62). However, such a purely allosteric interaction by VCP813 may also represent a secondary mode of binding for TBPB. This mode would support the noncompetitive and purely allosteric retardation of [3H]N-methylscopolamine dissociation from the M1 mAChR by TBPB (19, 23).
In conclusion, this study has characterized the bitopic mechanism of action of TBPB and 77-LH-28-1 at the M1 mAChR. We propose that these ligands share a similar binding domain, which includes both the orthosteric binding site and a putative allosteric region, to engage and activate the M1 mAChR. Furthermore, we have identified regions of the M1 mAChR that have distinct roles and differential effects in the affinity and efficacy of bitopic ligands compared with orthosteric ligands. This knowledge can be exploited potentially for the development of bitopic and/or allosteric ligands that exhibit greater selectivity for the M1 mAChR and/or select signaling pathways.
Acknowledgments
We are grateful for the assistance of Dr. Nathan E. Hall for help with the selection of some of the mutations for this study and to Drs. Michael Crouch and Ron Osmond (TGR Biosciences) for the generous supply of the SureFire ERK1/2 kit. The computational studies were supported by Resource Allocation Scheme Grant VR0024 from the Victorian Life Sciences Computation Initiative (VLSCI) on its Peak Computing Facility at the University of Melbourne.
This work was supported by Program Grant APP1055134 from the National Health and Medical Research Council (NHMRC) of Australia and Discovery Grant DP110100687 from the Australian Research Council.
- mAChR
- M1 muscarinic acetylcholine receptor
- ACh
- acetylcholine
- QNB
- N-quinuclidinyl benzilate
- Gpp(NH)p
- guanosine 5′-(β,γ-imido)triphosphate
- TM
- transmembrane
- ANOVA
- analysis of variance.
REFERENCES
- 1. Bodick N. C., Offen W. W., Levey A. I., Cutler N. R., Gauthier S. G., Satlin A., Shannon H. E., Tollefson G. D., Rasmussen K., Bymaster F. P., Hurley D. J., Potter W. Z., Paul S. M. (1997) Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch. Neurol. 54, 465–473 [DOI] [PubMed] [Google Scholar]
- 2. Shekhar A., Potter W. Z., Lightfoot J., Lienemann J., Dubé S., Mallinckrodt C., Bymaster F. P., McKinzie D. L., Felder C. C. (2008) Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry 165, 1033–1039 [DOI] [PubMed] [Google Scholar]
- 3. Langmead C. J., Watson J., Reavill C. (2008) Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 117, 232–243 [DOI] [PubMed] [Google Scholar]
- 4. Keov P., Sexton P. M., Christopoulos A. (2011) Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 60, 24–35 [DOI] [PubMed] [Google Scholar]
- 5. Ma L., Seager M. A., Seager M., Wittmann M., Jacobson M., Bickel D., Burno M., Jones K., Graufelds V. K., Xu G., Pearson M., McCampbell A., Gaspar R., Shughrue P., Danziger A., Regan C., Flick R., Pascarella D., Garson S., Doran S., Kreatsoulas C., Veng L., Lindsley C. W., Shipe W., Kuduk S., Sur C., Kinney G., Seabrook G. R., Ray W. J. (2009) Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 15950–15955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marlo J. E., Niswender C. M., Days E. L., Bridges T. M., Xiang Y., Rodriguez A. L., Shirey J. K., Brady A. E., Nalywajko T., Luo Q., Austin C. A., Williams M. B., Kim K., Williams R., Orton D., Brown H. A., Lindsley C. W., Weaver C. D., Conn P. J. (2009) Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of Activity. Mol. Pharmacol. 75, 577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones C. K., Brady A. E., Davis A. A., Xiang Z., Bubser M., Tantawy M. N., Kane A. S., Bridges T. M., Kennedy J. P., Bradley S. R., Peterson T. E., Ansari M. S., Baldwin R. M., Kessler R. M., Deutch A. Y., Lah J. J., Levey A. I., Lindsley C. W., Conn P. J. (2008) Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J. Neurosci. 28, 10422–10433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Langmead C. J., Austin N. E., Branch C. L., Brown J. T., Buchanan K. A., Davies C. H., Forbes I. T., Fry V. A., Hagan J. J., Herdon H. J., Jones G. A., Jeggo R., Kew J. N., Mazzali A., Melarange R., Patel N., Pardoe J., Randall A. D., Roberts C., Roopun A., Starr K. R., Teriakidis A., Wood M. D., Whittington M., Wu Z., Watson J. (2008) Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist 77-LH-28-1. Br. J. Pharmacol. 154, 1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Budzik B., Garzya V., Shi D., Walker G., Woolley-Roberts M., Pardoe J., Lucas A., Tehan B., Rivero R. A., Langmead C. J., Watson J., Wu Z., Forbes I. T., Jin J. (2010) Novel N-substituted benzimidazolones as potent, selective, CNS-penetrant, and orally active M1 mAChR agonists. ACS Med. Chem. Lett. 1, 244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gregory K. J., Hall N. E., Tobin A. B., Sexton P. M., Christopoulos A. (2010) Identification of orthosteric and allosteric site Mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J. Biol. Chem. 285, 7459–7474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sur C., Mallorga P. J., Wittmann M., Jacobson M. A., Pascarella D., Williams J. B., Brandish P. E., Pettibone D. J., Scolnick E. M., Conn P. J. (2003) N-Desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-d-aspartate receptor activity. Proc. Natl. Acad. Sci. U.S.A. 100, 13674–13679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spalding T. A., Ma J. N., Ott T. R., Friberg M., Bajpai A., Bradley S. R., Davis R. E., Brann M. R., Burstein E. S. (2006) Structural requirements of transmembrane domain 3 for activation by the M1 muscarinic receptor agonists AC-42, AC-260584, clozapine, and N-desmethylclozapine: evidence for three distinct modes of receptor activation. Mol. Pharmacol. 70, 1974–1983 [DOI] [PubMed] [Google Scholar]
- 13. Antony J., Kellershohn K., Mohr-Andrä M., Kebig A., Prilla S., Muth M., Heller E., Disingrini T., Dallanoce C., Bertoni S., Schrobang J., Tränkle C., Kostenis E., Christopoulos A., Höltje H. D., Barocelli E., De Amici M., Holzgrabe U., Mohr K. (2009) Dualsteric GPCR targeting: a novel route to binding and signaling pathway selectivity. FASEB J. 23, 442–450 [DOI] [PubMed] [Google Scholar]
- 14. Valant C., Gregory K. J., Hall N. E., Scammells P. J., Lew M. J., Sexton P. M., Christopoulos A. (2008) A novel mechanism of G protein-coupled receptor functional selectivity. J. Biol. Chem. 283, 29312–29321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Avlani V. A., Langmead C. J., Guida E., Wood M. D., Tehan B. G., Herdon H. J., Watson J. M., Sexton P. M. (2010) Orthosteric and allosteric modes of interaction of novel selective agonists of the M1 muscarinic acetylcholine receptor. Mol. Pharmacol. 78, 94–104 [DOI] [PubMed] [Google Scholar]
- 16. Narlawar R., Lane J. R., Doddareddy M., Lin J., Brussee J., Ijzerman A. P. (2010) Hybrid ortho/allosteric ligands for the adenosine A1 receptor. J. Med. Chem. 53, 3028–3037 [DOI] [PubMed] [Google Scholar]
- 17. Disingrini T., Muth M., Dallanoce C., Barocelli E., Bertoni S., Kellershohn K., Mohr K., De Amici M., Holzgrabe U. (2006) Design, synthesis, and action of oxotremorine-related hybrid-type allosteric modulators of muscarinic acetylcholine receptors. J. Med. Chem. 49, 366–372 [DOI] [PubMed] [Google Scholar]
- 18. Steinfeld T., Mammen M., Smith J. A., Wilson R. D., Jasper J. R. (2007) A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol. Pharmacol. 72, 291–302 [DOI] [PubMed] [Google Scholar]
- 19. Keov P., Valant C., Devine S. M., Lane J. R., Scammells P. J., Sexton P. M., Christopoulos A. (2013) Reverse engineering of the selective agonist TBPB unveils both orthosteric and allosteric modes of action at the M1 muscarinic acetylcholine receptor. Mol. Pharmacol. 84, 425–437 [DOI] [PubMed] [Google Scholar]
- 20. Gregory K. J., Sexton P. M., Tobin A. B., Christopoulos A. (2012) Stimulus bias provides evidence for conformational constraints in the structure of a G protein-coupled receptor. J. Biol. Chem. 287, 37066–37077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. May L. T., Avlani V. A., Langmead C. J., Herdon H. J., Wood M. D., Sexton P. M., Christopoulos A. (2007) Structure-function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol. Pharmacol. 72, 463–476 [DOI] [PubMed] [Google Scholar]
- 22. Lebon G., Langmead C. J., Tehan B. G., Hulme E. C. (2009) Mutagenic mapping suggests a novel binding mode for selective agonists of M1 muscarinic acetylcholine receptors. Mol. Pharmacol. 75, 331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacobson M. A., Kreatsoulas C., Pascarella D. M., O'Brien J. A., Sur C. (2010) The M1 muscarinic receptor allosteric agonists AC-42 and 1-[1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one bind to a unique site distinct from the acetylcholine orthosteric site. Mol. Pharmacol. 78, 648–657 [DOI] [PubMed] [Google Scholar]
- 24. Ward S. D., Curtis C. A., Hulme E. C. (1999) Alanine-scanning mutagenesis of transmembrane domain 6 of the M1 muscarinic acetylcholine receptor suggests That Tyr381 plays key roles in receptor function. Mol. Pharmacol. 56, 1031–1041 [DOI] [PubMed] [Google Scholar]
- 25. Spalding T. A., Birdsall N. J., Curtis C. A., Hulme E. C. (1994) Acetylcholine mustard labels the binding site aspartate in muscarinic acetylcholine receptors. J. Biol. Chem. 269, 4092–4097 [PubMed] [Google Scholar]
- 26. Lu Z. L., Hulme E. C. (1999) The functional topography of transmembrane domain 3 of the M1 muscarinic acetylcholine receptor, revealed by scanning mutagenesis. J. Biol. Chem. 274, 7309–7315 [DOI] [PubMed] [Google Scholar]
- 27. Lebois E. P., Bridges T. M., Lewis L. M., Dawson E. S., Kane A. S., Xiang Z., Jadhav S. B., Yin H., Kennedy J. P., Meiler J., Niswender C. M., Jones C. K., Conn P. J., Weaver C. D., Lindsley C. W. (2010) Discovery and characterization of novel subtype-selective allosteric agonists for the investigation of M1 receptor function in the central nervous system. ACS Chem. Neurosci. 1, 104–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsui H., Lazareno S., Birdsall N. J. (1995) Probing of the location of the allosteric site on m1 muscarinic receptors by site-directed mutagenesis. Mol. Pharmacol. 47, 88–98 [PubMed] [Google Scholar]
- 29. Nawaratne V., Leach K., Suratman N., Loiacono R. E., Felder C. C., Armbruster B. N., Roth B. L., Sexton P. M., Christopoulos A. (2008) New insights into the function of M4 muscarinic acetylcholine receptors gained using a novel allosteric modulator and a DREADD (designer receptor exclusively activated by a designer drug). Mol. Pharmacol. 74, 1119–1131 [DOI] [PubMed] [Google Scholar]
- 30. Ballesteros J. A., Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors in Methods in Neurosciences (Sealfon S. C., ed), Vol 25, pp. 366–428, Academic Press [Google Scholar]
- 31. Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kruse A. C., Hu J., Pan A. C., Arlow D. H., Rosenbaum D. M., Rosemond E., Green H. F., Liu T., Chae P. S., Dror R. O., Shaw D. E., Weis W. I., Wess J., Kobilka B. K. (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 34. Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., Caldwell J., Wang J., Kollman P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 [DOI] [PubMed] [Google Scholar]
- 35. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 36. Mansour A., Meng F., Meador-Woodruff J. H., Taylor L. P., Civelli O., Akil H. (1992) Site-directed mutagenesis of the human dopamine D2 receptor. Eur. J. Pharmacol. 227, 205–214 [DOI] [PubMed] [Google Scholar]
- 37. Wang C. D., Gallaher T. K., Shih J. C. (1993) Site-directed mutagenesis of the serotonin 5-hydroxytrypamine2 receptor: identification of amino acids necessary for ligand binding and receptor activation. Mol. Pharmacol. 43, 931–940 [PubMed] [Google Scholar]
- 38. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 40. Patra M., Karttunen M., Hyvönen M. T., Falck E., Lindqvist P., Vattulainen I. (2003) Molecular dynamics simulations of lipid bilayers: major artifacts due to truncating electrostatic interactions. Biophys. J. 84, 3636–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Black J. W., Leff P. (1983) Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 220, 141–162 [DOI] [PubMed] [Google Scholar]
- 42. Black J. W., Leff P., Shankley N. P., Wood J. (1985) An operational model of pharmacological agonism: the effect of E/[A] curve shape on agonist dissociation constant estimation. Br. J. Pharmacol. 84, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kenakin T., Watson C., Muniz-Medina V., Christopoulos A., Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Christopoulos A. (1998) Assessing the distribution of parameters in models of ligand-receptor interaction: to log or not to log. Trends Pharmacol. Sci. 19, 351–357 [DOI] [PubMed] [Google Scholar]
- 45. Leach K., Davey A. E., Felder C. C., Sexton P. M., Christopoulos A. (2011) The role of transmembrane domain 3 in the actions of orthosteric, allosteric, and atypical agonists of the M4 muscarinic Acetylcholine receptor. Mol. Pharmacol. 79, 855–865 [DOI] [PubMed] [Google Scholar]
- 46. Voigtländer U., Jöhren K., Mohr M., Raasch A., Tränkle C., Buller S., Ellis J., Höltje H. D., Mohr K. (2003) Allosteric site on muscarinic acetylcholine receptors: identification of two amino acids in the muscarinic M2 receptor that account entirely for the M2/M5 subtype selectivities of some structurally diverse allosteric ligands in N-methylscopolamine-occupie. Mol. Pharmacol. 64, 21–31 [DOI] [PubMed] [Google Scholar]
- 47. Goodwin J. A., Hulme E. C., Langmead C. J., Tehan B. G. (2007) Roof and floor of the muscarinic binding pocket: variations in the binding modes of orthosteric ligands. Mol. Pharmacol. 72, 1484–1496 [DOI] [PubMed] [Google Scholar]
- 48. Scarselli M., Li B., Kim S. K., Wess J. (2007) Multiple residues in the second extracellular loop are critical for M3 muscarinic acetylcholine receptor activation. J. Biol. Chem. 282, 7385–7396 [DOI] [PubMed] [Google Scholar]
- 49. Haga K., Kruse A. C., Asada H., Yurugi-Kobayashi T., Shiroishi M., Zhang C., Weis W. I., Okada T., Kobilka B. K., Haga T., Kobayashi T. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482, 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kruse A. C., Ring A. M., Manglik A., Hu J., Hu K., Eitel K., Hübner H., Pardon E., Valant C., Sexton P. M., Christopoulos A., Felder C. C., Gmeiner P., Steyaert J., Weis W. I., Garcia K. C., Wess J., Kobilka B. K. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bee M. S., Hulme E. C. (2007) Functional analysis of transmembrane domain 2 of the M1 muscarinic acetylcholine receptor. J. Biol. Chem. 282, 32471–32479 [DOI] [PubMed] [Google Scholar]
- 52. Nathan P. J., Watson J., Lund J., Davies C. H., Peters G., Dodds C. M., Swirski B., Lawrence P., Bentley G. D., O'Neill B. V., Robertson J., Watson S., Jones G. A., Maruff P., Croft R. J., Laruelle M., Bullmore E. T. (2013) The potent M1 receptor allosteric agonist GSK1034702 improves episodic memory in humans in the nicotine abstinence model of cognitive dysfunction. Int. J. Neuropsychopharmacol. 16, 721–731 [DOI] [PubMed] [Google Scholar]
- 53. Bridges T. M., Brady A. E., Kennedy J. P., Daniels R. N., Miller N. R., Kim K., Breininger M. L., Gentry P. R., Brogan J. T., Jones C. K., Conn P. J., Lindsley C. W. (2008) Synthesis and SAR of analogues of the M1 allosteric agonist TBPB. Part I: exploration of alternative benzyl and privileged structure moieties. Bioorg. Med. Chem. Lett. 18, 5439–5442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kenakin T., Christopoulos A. (2013) Signalling bias in new drug discovery: detection, quantification, and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216 [DOI] [PubMed] [Google Scholar]
- 55. Koole C., Wootten D., Simms J., Savage E. E., Miller L. J., Christopoulos A., Sexton P. M. (2012) Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) differentially regulates orthosteric but not allosteric agonist binding and function. J. Biol. Chem. 287, 3659–3673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koole C., Wootten D., Simms J., Miller L. J., Christopoulos A., Sexton P. M. (2012) Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) has a critical role in GLP-1 peptide binding and receptor activation. J. Biol. Chem. 287, 3642–3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tschammer N., Bollinger S., Kenakin T., Gmeiner P. (2011) Histidine 6.55 is a major determinant of ligand-biased signaling in dopamine D2L receptor. Mol. Pharmacol. 79, 575–585 [DOI] [PubMed] [Google Scholar]
- 58. Leppik R. A., Miller R. C., Eck M., Paquet J. L. (1994) Role of acidic amino acids in the allosteric modulation by gallamine of antagonist binding at the m2 muscarinic acetylcholine receptor. Mol. Pharmacol. 45, 983–990 [PubMed] [Google Scholar]
- 59. Krejcí A., Tucek S. (2001) Changes of cooperativity between N-methylscopolamine and allosteric modulators alcuronium and gallamine induced by mutations of external loops of muscarinic M3 receptors. Mol. Pharmacol. 60, 761–767 [PubMed] [Google Scholar]
- 60. Avlani V. A., Gregory K. J., Morton C. J., Parker M. W., Sexton P. M., Christopoulos A. (2007) Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J. Biol. Chem. 282, 25677–25686 [DOI] [PubMed] [Google Scholar]
- 61. Trumpp-Kallmeyer S., Hoflack J., Bruinvels A., Hibert M. (1992) Modeling of G-protein-coupled receptors: application to dopamine, adrenaline, serotonin, acetylcholine, and mammalian opsin receptors. J. Med. Chem. 35, 3448–3462 [DOI] [PubMed] [Google Scholar]
- 62. Babaoglu K., Shoichet B. K. (2006) Deconstructing fragment-based inhibitor discovery. Nat. Chem. Biol. 2, 720–723 [DOI] [PubMed] [Google Scholar]