

Background: Deficiency or nonsense mutation of CRBN causes memory deficits.
Results: Truncated CRBN has insufficient affinity for AMPKα and cannot modulate the AMPK-mTOR pathway.
Conclusion: CRBN modulates protein synthesis through the AMPK-mTOR pathway, and may be critical for certain forms of memory encoding.
Significance: Our findings suggest the first plausible mechanism for the phenotype resulting from the CRBN mutation.
Keywords: AMP-activated Kinase (AMPK), Gene Knockout, Mammalian Target of Rapamycin (mTOR), Protein Synthesis, Signal Transduction, Cereblon, Mental Deficit, Nonsense Mutation
Abstract
Initially identified as a protein implicated in human mental deficit, cereblon (CRBN) was recently recognized as a negative regulator of adenosine monophosphate-activated protein kinase (AMPK) in vivo and in vitro. Here, we present results showing that CRBN can effectively regulate new protein synthesis through the mammalian target of rapamycin (mTOR) signaling pathway, a downstream target of AMPK. Whereas deficiency of Crbn repressed protein translation via activation of the AMPK-mTOR cascade in Crbn-knock-out mice, ectopic expression of the wild-type CRBN increased protein synthesis by inhibiting endogenous AMPK. Unlike the wild-type CRBN, a mutant CRBN found in human patients, which lacks the last 24 amino acids, failed to rescue mTOR-dependent repression of protein synthesis in Crbn-deficient mouse fibroblasts. These results provide the first evidence that Crbn can activate the protein synthesis machinery through the mTOR signaling pathway by inhibiting AMPK. In light of the fact that protein synthesis regulated by mTOR is essential for various forms of synaptic plasticity that underlie the cognitive functions of the brain, the results of this study suggest a plausible mechanism for CRBN involvement in higher brain function in humans, and they may help explain how a specific mutation in CRBN can affect the cognitive ability of patients.
Introduction
Cereblon (CRBN),3 a gene on human chromosome 3p26.2, was initially reported as a candidate gene for a mild form of autosomal recessive non-syndromic mental retardation (ARNSMR) (1). Subsequently, the CRBN protein has been characterized in several different cellular contexts. CRBN interacts with the cytoplasmic region of large-conductance calcium-activated potassium (BKCa) channels to regulate surface expression of the channel protein (2). In addition, CRBN is the primary target of thalidomide-induced teratogenicity, and is thought to function as a substrate receptor of an E3 ubiquitin ligase complex (3). A recent study showed that CRBN interacts with the α subunit of adenosine monophosphate-activated protein kinase (AMPK) and inhibits the activation of AMPK in vitro as well as in vivo (4, 5).
AMPK, a master sensor of cellular energy balance, increases ATP-producing catabolic pathways and inhibits ATP-consuming anabolic pathways. AMPK, a serine/threonine protein kinase, is a heterotrimer consisting of a catalytic α subunit and two regulatory subunits, β and γ. AMPK activity can be modulated by phosphorylation on a threonine residue (Thr-172) by upstream kinases such as liver kinase B1 (LKB1). AMPK activation inhibits energy-consuming anabolic processes such as protein translation (6–10) and accomplishes these effects largely through inhibition of the mammalian target of rapamycin (mTOR) signaling (11).
The conserved serine-threonine protein kinase mTOR regulates cell growth, proliferation, and synaptic plasticity by controlling protein synthesis. Activation of mTOR acts on one of the primary triggers for the initiation of cap-dependent translation through the phosphorylation and activation of S6 kinase (S6K1), and through the phosphorylation and inactivation of a repressor of mRNA translation, eukaryotic initiation factor 4E-binding protein (4E-BP1) (12–15). Two biochemically distinct mTOR complexes, mTORC1 and mTORC2, are found in mammalian cells, and the activity of mTORC1 is regulated by AMPK. AMPK can suppress the activity of mTORC1 by directly phosphorylating at least two regulator proteins, tuberous sclerosis 2 (TSC2) and raptor.
Despite the significance of CBRN in brain function, suggested by clinical and experimental evidence (1, 16), the molecular etiology of the cognitive phenotypes resulting from CRBN mutation has not been elucidated. In this study, we investigated the functional roles of CRBN as an upstream regulator of the mTOR signaling pathway. Our results show that CRBN can up-regulate cap-dependent translation by inhibiting AMPK. Unlike the wild-type (WT) CRBN, a mutant CRBN lacking the C-terminal 24 amino acids (R419X) was unable to regulate the mTOR pathway, due to its inability to suppress AMPK activity. Because new protein synthesis is essential for different forms of synaptic plasticity in the brain (15, 17–21), defects in CRBN-dependent regulation of mTOR signaling may represent the molecular mechanism underlying learning and memory defects associated with the CRBN mutation.
EXPERIMENTAL PROCEDURES
Experimental Animals
Male mice were used in this study. Animals were maintained under specific pathogen-free conditions. All experiments were approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee.
Antibodies
The following antibodies were used in this study: monoclonal anti-AMPK α (Invitrogen), rabbit polyclonal anti-phospho-AMPK α (Cell Signaling), rabbit polyclonal anti-AMPK β (Cell Signaling), rabbit polyclonal anti-AMPK γ1 (C terminus) (Epitomics), rabbit monoclonal anti-raptor (Cell Signaling), rabbit polyclonal anti-phospho-raptor (Ser-792) (Cell Signaling), rabbit polyclonal anti-mTOR (Cell Signaling), rabbit polyclonal anti-phospho-mTOR (Cell Signaling), rabbit polyclonal anti-S6K (Cell Signaling), mouse monoclonal anti-phospho-S6K (Cell Signaling), mouse monoclonal anti-S6 (Cell Signaling), rabbit polyclonal anti-phospho-S6 (Cell Signaling), rabbit polyclonal anti-4EBP1 (Cell Signaling), rabbit polyclonal anti-phospho-4EBP1 (Cell Signaling), mouse monoclonal anti-HA (Cell Signaling), mouse monoclonal anti-BKCa (BD Transduction LaboratoriesTM), and rabbit polyclonal anti-GAPDH (Abfrontier, Seoul, Korea). Rabbit polyclonal anti-CRBN antibody was described previously (4).
Plasmid Construction and Transfection
Plasmids encoding the HA-tagged human CRBN (HA-CRBN) and mouse Crbn (HA-CRBN) were described previously (4). HA-CRBN R419X (human) and HA-Crbn R422X (mouse) were constructed as described in the previous report (22). Cells were transfected using LipofectamineTM LTX (Invitrogen), and then cells were seeded 24 h before lysate preparation. A small amount of a plasmid expressing EGFP was co-transfected to validate equivalent expression of exogenous proteins in cells.
RT-PCR Experiments
Total RNA was isolated from brain tissues of the indicated mice using the TRIzol reagent (Invitrogen). The sequences of the primers used in the PCR experiments were described previously (5).
Cell Culture
SH-SY5Y cells and mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco's modified Eagle's medium (DMEM, GIBCO) with 10% (v/v) fetal bovine serum (FBS, Hyclone). Crbn +/+, Crbn +/−, and Crbn −/− MEFs were isolated from E14.5 embryos born to heterozygous intercrosses and assayed at passages 3–6, as previously described (23).
Tissue Lysate Preparation
Hippocampal tissues were obtained from 9-week-old male mice. Hippocampal tissues were homogenized in ice-chilled buffer (20 mm Tris-HCl, pH 7.4, 0.32 m sucrose, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, 10 μg/ml aprotinin, 15 μg/ml leupeptin, 50 mm NaF, and 1 mm sodium orthovanadate), as previously described (24).
Co-immunoprecipitation
Cells were solubilized in lysis buffer (RIPA buffer: 20 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 1% Nonidet P-40, 1% sodium deoxycholate, 2 mm Na3VO4, 100 mm NaF, 1 mm PMSF, protease inhibitor mixture). The supernatant was incubated with various primary antibodies, e.g. anti-AMPK α or anti-HA antibodies, overnight at 4 °C. Antibody-protein complexes were precipitated with equilibrated protein G beads (Amersham Biosciences) at 4 °C for 3 h, followed by incubation with lysis buffer at 37 °C for 15 min.
Analysis of Protein Synthesis
Analysis of protein synthesis was examined as previously described (25). Briefly, cells were labeled with [35S]methionine (10 mCi/ml) for 30 min in methionine-free minimal essential medium. After being washed with PBS, cell extracts were prepared by lysing the cells with Nonidet P-40 lysis buffer (2% Nonidet P-40, 80 mm NaCl, 100 mm Tris-HCl, 0.1% SDS).
Translation Assay
Translation was measured by luciferase reporter activity using the pRMF reporter, kindly provided to us by Dr. Sung Key Jang (Pohang University of Science and Technology, Korea). Equal amounts of extract were used to assay cap-dependent translation of Renilla luciferase (R-Luc) or IRES-dependent translation of firefly luciferase (F-Luc), using a dual-luciferase reporter assay system. Cap-dependent translation was calculated by normalizing the R-Luc activity to the F-Luc activity, as described previously (26, 27).
Statistical Analysis
All displayed values represent means ± S.E. Significant differences between groups were determined using two-tailed unpaired Student's t-tests, and multiple comparisons were performed using one-way ANOVA or two-way repeated-measures ANOVA. Differences with p < 0.05 were considered statistically significant, and are indicated in the figure legends.
RESULTS
Crbn Deficiency Reduces the Activity of mTOR in the Brain
The importance of neuronal protein synthesis in memory formation has been well established in numerous experimental systems (17, 18, 28–30). De novo protein synthesis underlying long-term synaptic plasticity is primarily regulated by the mTOR signaling pathway (15, 17–21). Active mTOR phosphorylates and activates the downstream effector S6K1, which then phosphorylates its downstream target, ribosomal protein S6; by contrast, mTOR phosphorylation of 4EBP1 results in inhibition of that protein (12–15). Phosphorylation of these two translational regulators by mTOR increases the overall translation capacity of the cell (15, 18, 31).
Because CRBN negatively regulates AMPK (4, 5) and AMPK activation can suppress the activity of mTOR (6–10), we wondered whether deficiency of Crbn would affect mTOR signaling in the mouse brain. In a recent report, we described the generation of Crbn-knock-out (Crbn-KO) mice, in which the Crbn gene is deleted throughout the body (5). To validate the deficiency of Crbn in the brain, we measured levels of the Crbn mRNA by reverse transcription-polymerase chain reaction (RT-PCR) using total RNA extracted from the brains of WT (Crbn +/+), heterozygote KO (Crbn +/−), and homozygote KO (Crbn −/−) mice (Fig. 1A). Deficiency of Crbn protein in the brains of Crbn-KO mice was also confirmed by Western blot analysis (Fig. 1B). CRBN-specific polyclonal antibody detected a protein band with the expected molecular mass (53 kDa) in the brains of WT mice, whereas no immunoreactivity was detected in brain lysates from Crbn homozygous KO (Fig. 1, B and C). Expression of Crbn was reduced by 44% in the brains of heterozygous KO mice.
FIGURE 1.
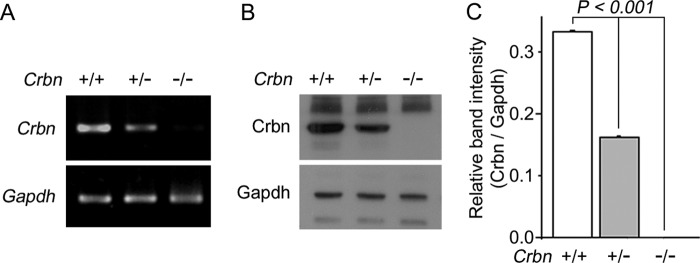
Confirmation of Crbn deficiency in the brain of Crbn-KO mice. A, Crbn mRNA levels, as determined by RT-PCR analysis, from brain tissues of the indicated mice. Gapdh was used as an internal control. A reduced level of Crbn transcription is evident in the Crbn +/− mice (n = 4 per group). B, endogenous levels of Crbn protein, as determined by Western blotting of the brain lysates of the indicated mice. Gapdh was used as the loading control (n = 4 per group). C, relative band intensities, as determined by densitometric analysis, of the blot shown in B. Results were obtained from four independent experiments. Error bars represent S.E.
We then measured the phosphorylation level of AMPK in the hippocampi of WT and KO mice. As expected, the levels of AMPK α subunit phosphorylated at Thr-172 (P-AMPKα) in the hippocampi of Crbn +/− and Crbn −/− mice were significantly increased relative to the level in Crbn +/+ mice (Fig. 2, A and B). Next, we investigated whether AMPK activation induced by deletion of Crbn can affect mTOR signaling. To this end, we monitored the amount of phosphorylated raptor, mTOR, S6K, S6, and 4EBP1. Higher levels of P-AMPK were accompanied with higher levels of P-raptor but with lower levels of P-mTOR, P-S6K, P-S6, and P-4EBP1 in Crbn +/− and Crbn −/− hippocampi, respectively (Fig. 2, A and C–G). Similar results were also obtained in primary cultures of mouse embryonic fibroblasts (MEFs) (Fig. 3). These findings imply that AMPK activation by Crbn deficiency can reduce cellular translation by inhibiting endogenous mTOR signaling.
FIGURE 2.

Suppression of mTOR signaling pathway in the brain of Crbn-KO mice. A, Western blot analyses of endogenous AMPK α, P-AMPK α, raptor, P-raptor, mTOR, P-mTOR, S6K, P-S6K, S6, P-S6, 4EBP1, and P-4EBP1 in hippocampus tissue lysates. Gapdh was used to confirm equal protein loading. The results shown are representative of four independent experiments. Asterisks denote nonspecific bands. B–F, relative band intensities as determined by densitometric analysis of the blot shown in A. Error bars represent the S.E. (n = 4). G, schematic diagram of the AMPK-mTOR signaling pathway.
FIGURE 3.

Suppression of mTOR signaling pathway in Crbn-deficient mouse embryonic fibroblasts. A, Western blot analysis of AMPK α, P-AMPK α, raptor, P-raptor, mTOR, P-mTOR, S6K, P-S6K, S6, P-S6, 4EBP1, and P-4EBP1 proteins levels in Crbn +/+, Crbn +/−, and Crbn −/− primary MEFs. Gapdh was used as the loading control. The results shown are representative of four independent experiments. Asterisks denote nonspecific bands. B–G, relative band intensities, as determined by densitometric analysis of the blot shown in A. Error bars represent the S.E.
Crbn Deficiency Negatively Regulates Both Protein Synthesis and Cap-dependent Translation
Because Crbn deficiency significantly inhibited mTOR signaling, we next investigated whether Crbn deletion would influence new protein synthesis. Not surprisingly, overall protein synthesis was significantly reduced in Crbn +/− and Crbn −/− MEFs relative to the level in Crbn +/+ MEFs (Fig. 4, A and B). mTORC1 regulates cap-dependent translation through phosphorylation of 4EBP1, which releases 4EBP1 from eIF-4E and promotes translation initiation (32), so we further examined the effects of Crbn deficiency on cap-dependent translation using a relative luciferase assay (26, 27). As shown in Fig. 4C, cap-dependent translation was significantly suppressed in Crbn +/− and Crbn −/− MEFs. These results indicate that Crbn deficiency can inhibit not only the activation of mTOR but also cap-dependent translation, a downstream process regulated by the AMPK-mTOR signaling cascade.
FIGURE 4.
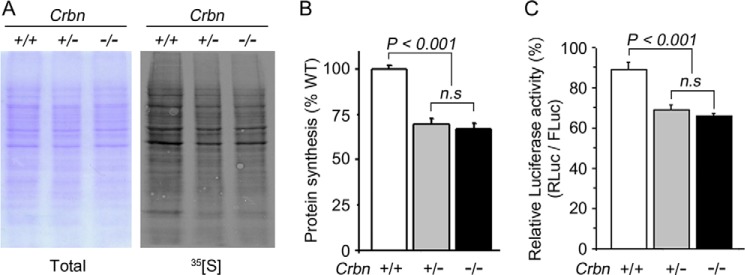
Repression of total protein synthesis and cap-dependent translation in Crbn KO mice. A, new protein synthesis as determined by autoradiography (right panel). A Coomassie Blue stain of the same gel was used to confirm equal loading of total proteins in each lane (left panel). The results shown are representative of four independent experiments. B, differences in protein synthesis, as determined by densitometric analysis of the blot shown in A. Error bars represent the S.E. (n = 4). C, Cap-dependent translation, as measured by dual-luciferase assay using the pRMF reporter. Cap-dependent translation of Renilla luciferase (R-Luc) was normalized against IRES-dependent translation of firefly luciferase (F-Luc). The results shown were obtained from four independent experiments. Error bars represent the S.E. (n = 4).
Exogenous Expression of WT CRBN, but Not the R419X Mutant, Down-regulates AMPK-mTOR Signaling Pathway
Because the mTOR signaling pathway was suppressed by Crbn deficiency and Crbn deficiency resulted in the constitutive activation of AMPK, we wondered whether ectopic expression of CRBN would affect the signal pathway in the opposite manner. Moreover, we also wondered how the human mutation linked to mild mental deficit influences AMPK-mTOR signaling. In ARNSMR patients, the C-terminal 24 amino acids are missing from the full-length protein of 442 amino acids, because of a nonsense mutation in CRBN (R419X) (1). CRBN is highly conserved among higher mammals, with an overall amino acid sequence identity of 95% between human and mouse. In the C-terminal region, which is absent in patients because of a nonsense mutation, 23 out of the 24 amino acid residues are identical between human CRBN and mouse Crbn; the sole non-identical residue is a conservative substitution (Glu to Asp).
To explore the effects of ectopic expression, we transiently transfected WT or CRBN R419X into SH-SY5Y human neuroblastoma cells (Fig. 5A). Western blot analyses revealed that intensity of the P-AMPKα band was significantly reduced upon ectopic expression of WT CRBN, as we previously reported (4). However, the level of P-AMPK α did not change relative to that in mock-transfected cells upon ectopic expression of the R419X mutant (Fig. 5B). In WT CRBN-expressing cells, the decrease in P-AMPKα was accompanied by lower levels of P-raptor, but higher levels of P-mTOR, P-S6K, P-S6, and P-4EBP1. However, expression of the R419X mutant did not significantly alter the phosphorylation level of these proteins relative to the level in mock-transfected cells (Fig. 5, C–G).
FIGURE 5.

Effects of exogenous WT CRBN or the R419X mutation on the AMPK-mTOR signal pathways. A, Western blot analysis of SH-SY5Y cells transiently transfected with HA-CRBN, HA-R419X, or HA empty vector. Cell lysates were immunoblotted with anti-AMPK α, anti-P-AMP α, anti-raptor, anti-P-raptor, anti-mTOR, anti-P-mTOR, anti-S6K, anti-P-S6K, anti-S6, anti-P-S6, anti-4EBP1, anti-P-4EBP1, or anti-HA antibodies. Anti-GAPDH was used to verify equal protein loading. The results shown are representative of four independent experiments. Asterisks denote nonspecific bands. B–F, relative band intensities, as determined by densitometric analysis of the blot shown in A. Error bars represent the S.E. (n = 4).
Next, we examined the effects of WT Crbn and R422X (a mouse mutant corresponding to human CRBN R419X) on the mTOR signaling pathway in WT MEFs and AMPK double-knock-out (DKO) MEFs, which lack the α1 and α2 subunits of AMPK. Consistent with a previous report (33), the levels of P-S6K in mock-transfected AMPK DKO MEFs were suppressed upon nutrient deprivation, although the effect was less than that that seen in mock-transfected WT MEFs (Fig. 6C, compare WT and AMPK DKO under nutrient plus versus nutrient minus conditions, respectively (open bars)). As we previously reported (4), the ectopic expression of WT Crbn in WT MEFs reduced the level of P-AMPK α and increased the level of P-S6K in a nutrient-independent manner; however, there was no significant difference in the levels of P-AMPK and P-S6K upon expression of the R422X mutant compared with the levels in mock-transfected WT MEFs (Fig. 6A). Notably, the expression of WT Crbn or the R422X mutant had no significant effect on the levels of P-S6K in AMPK DKO MEFs relative to those in mock-transfected AMPK DKO MEFs, either in the presence or absence of nutrients (Fig. 6, B and C). These results indicate that Crbn does not affect mTOR signaling in the absence of functional AMPK.
FIGURE 6.
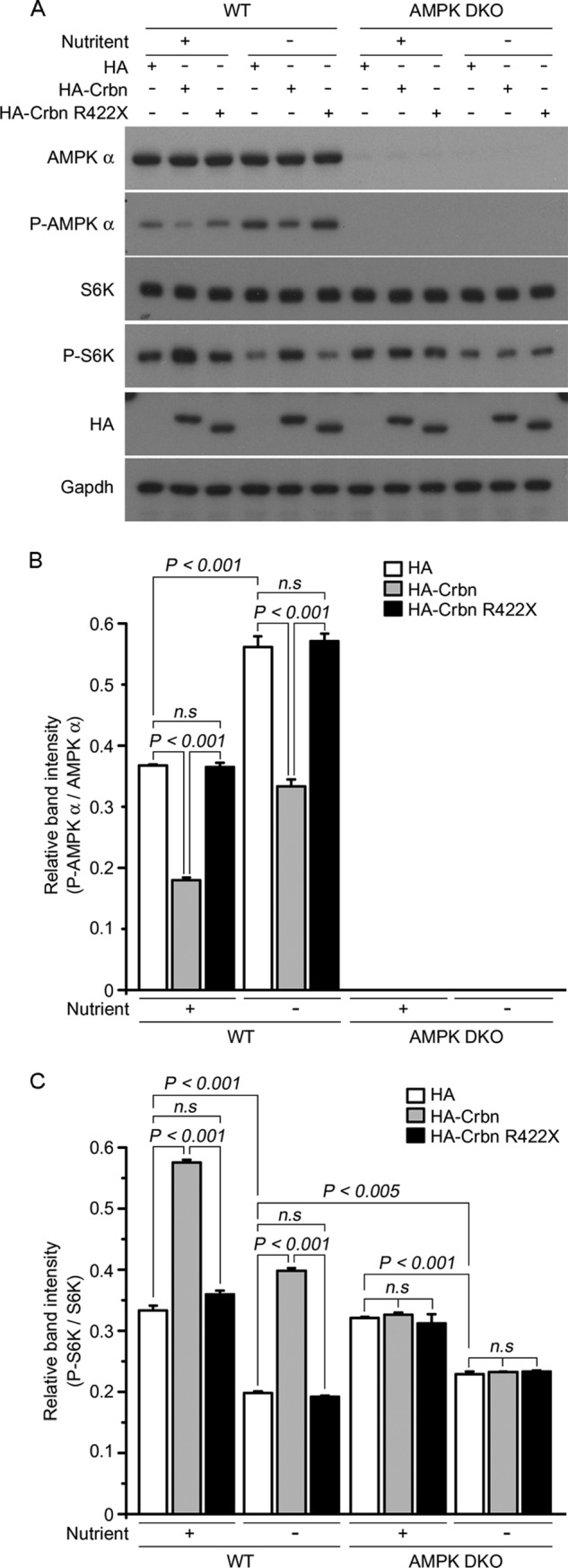
Crbn-dependent regulation of the mTOR signaling pathway is absent in AMPK-deficient MEFs. A, WT and AMPK DKO MEFs were deprived of glucose for 1 h and then re-stimulated for 10 min. Cell lysates were prepared and immunoblotted with anti-AMPK α, anti-P-AMPK α, anti-S6K, anti-P-S6K, or anti-HA antibodies. GAPDH was used as the loading control. The results are representative of four independent experiments. B and C, relative intensities (as determined by densitometric analysis) of the bands on the blot shown in A. Error bars represent the S.E.
CRBN negatively regulates AMPK activation by interacting with the α subunit, which reduces the affinity of the γ subunit for the AMPK complex (4). Therefore, we asked whether CRBN R419X can interact with the AMPK α subunit, and, if so, whether expression of the mutant CRBN can influence the formation of the heterotrimeric complex of AMPK subunits (α, β, and γ). We tested the effects of CRBN R419X expression on the AMPK complex by immunoprecipitating the endogenous AMPK complex from SH-SY5Y cells (Fig. 7A). Although both exogenous WT and CRBN R419X were detected in the AMPK complex, CRBN R419X appeared to interact with the complex with much lower affinity than WT CRBN (Fig. 7D). The intensity of the γ-subunit band in the immunoprecipitate was significantly reduced by exogenous CRBN WT, as previously reported (4). However, no such decrease in the γ-subunit band was observed upon exogenous expression of CRBN R419X (Fig. 7C). In both cases, the intensity of the β-subunit band did not change significantly (Fig. 7B). These observations strongly suggest that CRBN R419X cannot regulate AMPK-mTOR signaling due to its insufficient affinity for the α subunit of AMPK and inability to displace the γ subunit from the AMPK complex.
FIGURE 7.

The α subunit of AMPK has much lower affinity for CRBN R419X than for CRBN WT. A, Western blot analysis of SH-SY5Y cells transfected with empty HA, HA-CRBN, or HA-CRBN R419X. Proteins were immunoprecipitated with anti-AMPK α and probed with anti-AMPK α, anti-AMPK β, anti-AMPK γ1, and anti-HA antibodies. LC indicates the IgG light chain. B–D, relative band intensities, as determined by densitometric analysis of the blot shown in A. The results shown were obtained from four independent experiments. Error bars represent the S.E.
Expression of Crbn WT, but Not Crbn R422X, Rescues the Translational De-repression Induced by Crbn Deficiency
To further validate the functional role of Crbn in translational regulation via AMPK-mTOR signaling, we attempted to rescue the phenotype of the Crbn deficiency by exogenously expressing either Crbn WT or Crbn R422X (Fig. 8A). Constitutive activation of AMPK in Crbn −/− MEF cells was effectively suppressed by exogenous expression of WT Crbn (Fig. 8B). The expression of Crbn WT was also accompanied by higher levels of P-S6, as determined by Western-blot analysis (Fig. 8C), and higher levels of cap-dependent translation, as determined by the relative luciferase assay (Fig. 8D). The exogenous expression of R422X Crbn, however, did not suppress AMPK phosphorylation (Fig. 8B). Accordingly, S6 phosphorylation and translational de-repression were not observed upon expression of the mutant protein. These results further demonstrate that constitutive activation of AMPK is a direct and reversible cellular response induced solely by the loss of Crbn, and that the lack of the endogenous Crbn gene can be rescued by exogenous expression of Crbn WT, but not by Crbn truncated as a result of a nonsense mutation.
FIGURE 8.

Expression of Crbn WT, but not Crbn R422X, restored translational repression induced by the AMPK-mTOR pathway in Crbn-deficient cells. A, Western blotting analysis of AMPK α, P-AMPK α, S6, P-S6 protein levels, and exogenously expressed HA-Crbn and HA-Crbn R422X in Crbn-deficient primary MEFs. Gapdh was used to confirm equal protein loading. The results shown are representative of four independent experiments. B–C, relative band intensities as determined by densitometric analysis of the blot shown in A. Error bars represent the S.E. D, Cap-dependent translation, as measured by dual-luciferase reporter assay. HA, HA-Crbn, or HA- Crbn R422X was transiently co-transfected along with the pRMF vector into Crbn-deficient primary MEFs. The results shown were obtained from four independent experiments. Error bars represent the S.E. (n = 4).
DISCUSSION
It is widely accepted that memory formation requires not only mRNA transcription but also production of new proteins (17, 18, 29, 30). As the central regulator of translational initiation, the mTOR cascade is required for synaptic plasticity and memory processes that are dependent on the protein synthesis machinery (15, 17–21). The activity of mTOR, in turn, can be modulated by several upstream kinases, including AMPK. As the cellular energy sensor and a negative regulator of anabolic processes, activated AMPK phosphorylates mTORC1 and suppresses the synthesis of new cellular proteins (34, 35).
Here we show, for the first time, that the expression level of CRBN, a negative regulator of AMPK, can effectively modulate the mTOR pathway and cellular protein synthesis. We observed that deficiency of endogenous Crbn resulted in constitutive activation of AMPK, thereby suppressing overall protein synthesis (controlled by the mTOR pathway) in the mouse hippocampus (Figs. 2 and 4). Accordingly, ectopic expression of CRBN WT suppressed AMPK activity and activated the mTOR pathway in human neuroblastoma (Fig. 5). Moreover, the AMPK-dependent suppression of protein translation in Crbn −/− MEF cells was rescued by exogenous expression of Crbn WT, resulting in inhibition of endogenous AMPK activity (Fig. 8). These findings not only strengthen the idea that CRBN is an endogenous negative regulator of AMPK (4, 5), but also provide a testable hypothesis regarding the mechanism by which the nonsense mutation in CRBN causes mental deficit in humans (Fig. 9).
FIGURE 9.
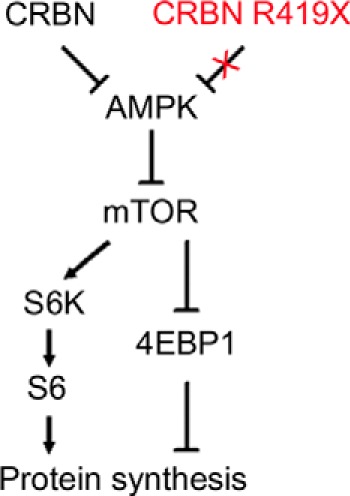
Logical relationships mediated by the AMPK-mTOR cascade between either CRBN or CRBN R419X and the protein synthesis machinery.
Since its initial identification as a candidate protein involved in human mental deficit (1), the significance of CRBN in brain function was further demonstrated using a mouse model in which forebrain-specific deletion of Crbn resulted in significant learning and memory defects (16). Furthermore, in whole-body Crbn-deficient mice, we also observed severe deficits in behaviors involving hippocampal function, but no noticeable abnormalities in motor function.4 Despite its potential involvement in higher brain function, however, the cellular roles of CRBN in the central nervous system are still controversial, and the functional consequences of the C-terminal deletion found in mutant CRBN have never been characterized.
We therefore examined the functional effects of the mutant CRBN, CRBN R419X, on the mTOR-dependent modulation of protein synthesis, and attempted to obtain experimental evidence for the cellular mechanism underlying the phenotypes of this mutant. Unlike CRBN WT, the R419X mutant failed to inhibit endogenous AMPK, because it could not release sufficiently the γ subunit from the AMPK complex (Figs. 5, 6, and 7). It is noteworthy that the truncated CRBN could still interact with AMPK α, albeit with much lower affinity; consequently, the γ subunit was retained in the AMPK complex (Fig. 7, A and D). Furthermore, Crbn R422X was unable to rescue suppression of mTOR-dependent translation by AMPK in Crbn −/− MEF cells (Fig. 8). CRBN R419X was completely ineffective as a regulator of the AMPK-mTOR cascade, despite its appreciable expression level (Fig. 5). Notably, in this regard, the expression level of HA-tagged CRBN R419X was comparable to that of the WT protein (Fig. 5A, lowest panel), strongly suggesting that the abnormalities observed in affected individuals may not be a consequence of CRBN insufficiency, per se, but may rather be the result of the loss of functional activity of the missing C terminus. Therefore, misregulation of the AMPK-mTOR pathway and improper translation of new proteins may be involved in the cellular mechanism underlying the mental defects observed in patients with the CRBN mutation. Our findings are also supported by a previous report showing that activation of AMPK by hippocampal injections of AICAR, a well-known activator of AMPK, reduced memory encoding by reducing the phosphorylation of mTOR cascade components (36).
Although we focused here on the functional roles of CRBN in the AMPK-mTOR pathway, other binding partners of CRBN have been identified. One CRBN-binding protein that has drawn attention is an ion channel known as the large-conductance calcium-activated potassium (BKCa) channel (2), which is widely expressed in central neurons where it modulates their excitability through both pre- and postsynaptic mechanisms (37). By interacting with the C-terminal cytosolic domain, CRBN regulates the assembly and the surface expression of the BKCa channel. Therefore, using co-immunoprecipitation analysis, we examined the binding of WT and mutant CRBN to the channel in COS-7 cells. However, we did not observe any appreciable difference between the affinities of WT and mutant CRBN for the BKCa channel (Fig. 10). However, this result does not entirely rule out the possibility that the BKCa channel is involved in the roles played by CRBN in brain function, because it remains to be seen whether mutant CRBN acts similarly to CRBN WT with respect to regulation of the BKCa channel in vivo.
FIGURE 10.
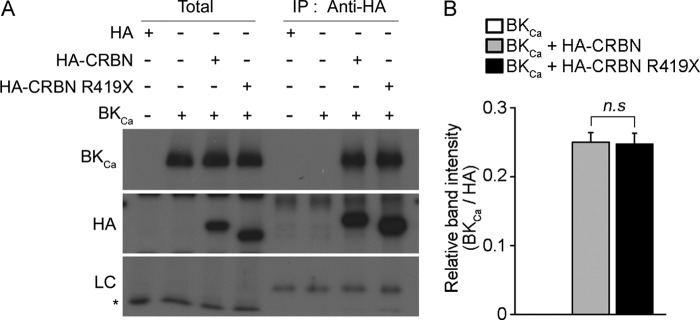
Interaction of the BKCa channel with WT and a truncated CRBN. A, Western blot analysis of COS-7 cells transiently co-transfected with HA, HA-CRBN, or HA-CRBN R419X along with the α subunit of the BKCa channel (BKCa). Cells were harvested after 24 h, and CRBN was immunoprecipitated using an anti-HA antibody. Western blots of the immunoprecipitates were probed with either anti-BKCa channel or anti-HA antibodies. The plus and minus symbols indicate the presence or absence of the indicated genes in transfection samples. The results shown are representative of four independent experiments. Asterisks denote nonspecific bands. B, relative band intensities, as determined by densitometric analysis of the blot shown in A. Error bars represent the S.E. (n = 4).
Although our results strongly suggest that CRBN is of functional importance as an endogenous regulator of mTOR pathway in the brain, several questions remain to be answered. First, we need to elucidate, at the molecular level, why the R419X mutant has much lower binding affinity for the AMPK α subunit. We previously reported that CRBN interacts with the AMPKα via its N-terminal Lon domain (4), located at the other end of the protein. One possibility, of course, is that the loss of the C-terminal 24 amino acids induces some structural changes in the protein, lowering the affinity for the AMPK α subunit. We expect that comparative biochemical and structural studies of the mutant and WT CRBN proteins will provide a straightforward answer to this question. Second, to what extent are cellular proteins affected by CRBN-dependent translational regulation? It will be of great interest to determine whether CRBN regulates overall protein synthesis via the AMPK-mTOR pathway by adjusting its activity to cellular energy status, or instead targets a specific set of proteins. Because CRBN is a relatively newly discovered gene, its expression has not been extensively investigated at either the transcriptional or translational level. Thus, it will be critical to understand the expressional regulation of CRBN in a cellular context. Most importantly, the physiological function of truncated mutant CRBN needs to be elucidated in vivo. Although we demonstrated that the exogenous expression of Crbn R422X could not reverse the suppression of the mTOR cascade in a completely Crbn-null background, this result should be confirmed in vivo by introducing the mutant gene into a Crbn-deficient mouse.
Nonetheless, this study provides the first in vivo evidence that Crbn can regulate the protein synthesis machinery through the AMPK-mTOR pathway, and that the proper expression of functional Crbn may be critical for the encoding of learning and memory in mice. This study also proposes a testable working hypothesis regarding the mechanism by which CRBN is involved in higher brain functions in humans, as well as how a specific mutation in CRBN can affect the cognitive ability of patients.
Acknowledgment
AMPK DKO MEFs were kindly provided by Dr. Benoit Viollet (INSERM, France).
This work was supported by grants for the Korea Healthcare Technology Research and Development Project (HI13C1412), Ministry for Health and Welfare, the National Leading Research Laboratories (2011-0028665), and the Science Research Center of Excellence Program (2007-0056157) of Ministry of Science, ICT & Future Planning/National Research Foundation of Korea (to C. S. P.).
K. M. Lee and C. S. Park, unpublished data.
- CRBN
- Cereblon
- AMPK
- AMP-activated kinase
- mTOR
- mammalian target of rapamycin.
REFERENCES
- 1. Higgins J. J., Pucilowska J., Lombardi R. Q., Rooney J. P. (2004) A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 63, 1927–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jo S., Lee K. H., Song S., Jung Y. K., Park C. S. (2005) Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J. Neurochem. 94, 1212–1224 [DOI] [PubMed] [Google Scholar]
- 3. Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H. (2010) Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 [DOI] [PubMed] [Google Scholar]
- 4. Lee K. M., Jo S., Kim H., Lee J., Park C. S. (2011) Functional modulation of AMP-activated protein kinase by cereblon. Biochim. Biophys. Acta 1813, 448–455 [DOI] [PubMed] [Google Scholar]
- 5. Lee K. M., Yang S. J., Kim Y. D., Choi Y. D., Nam J. H., Choi C. S., Choi H. S., Park C. S. (2013) Disruption of the cereblon gene enhances hepatic AMPK activity and prevents high-fat diet-induced obesity and insulin resistance in mice. Diabetes 62, 1855–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alimova I. N., Liu B., Fan Z., Edgerton S. M., Dillon T., Lind S. E., Thor A. D. (2009) Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 8, 909–915 [DOI] [PubMed] [Google Scholar]
- 7. Dowling R. J., Zakikhani M., Fantus I. G., Pollak M., Sonenberg N. (2007) Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 67, 10804–10812 [DOI] [PubMed] [Google Scholar]
- 8. Inoki K., Zhu T., Guan K. L. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 9. Rocha G. Z., Dias M. M., Ropelle E. R., Osório-Costa F., Rossato F. A., Vercesi A. E., Saad M. J., Carvalheira J. B. (2011) Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin. Cancer Res. 17, 3993–4005 [DOI] [PubMed] [Google Scholar]
- 10. Zakikhani M., Dowling R. J., Sonenberg N., Pollak M. N. (2008) The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev. Res. 1, 369–375 [DOI] [PubMed] [Google Scholar]
- 11. Hardie D. G. (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Chem. Biol. 8, 774–785 [DOI] [PubMed] [Google Scholar]
- 12. Brunn G. J., Hudson C. C., Sekulic A., Williams J. M., Hosoi H., Houghton P. J., Lawrence J. C., Jr., Abraham R. T. (1997) Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 277, 99–101 [DOI] [PubMed] [Google Scholar]
- 13. Burnett P. E., Barrow R. K., Cohen N. A., Snyder S. H., Sabatini D. M. (1998) RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U.S.A. 95, 1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gingras A. C., Raught B., Sonenberg N. (2001) Regulation of translation initiation by FRAP/mTOR. Genes Dev. 15, 807–826 [DOI] [PubMed] [Google Scholar]
- 15. Tsokas P., Ma T., Iyengar R., Landau E. M., Blitzer R. D. (2007) Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J. Neurosci. 27, 5885–5894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rajadhyaksha A. M., Ra S., Kishinevsky S., Lee A. S., Romanienko P., DuBoff M., Yang C., Zupan B., Byrne M., Daruwalla Z. R., Mark W., Kosofsky B. E., Toth M., Higgins J. J. (2012) Behavioral characterization of cereblon forebrain-specific conditional null mice: a model for human non-syndromic intellectual disability. Behav. Brain Res. 226, 428–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stanton P. K., Sarvey J. M. (1984) Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J. Neurosci. 4, 3080–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang S. J., Reis G., Kang H., Gingras A. C., Sonenberg N., Schuman E. M. (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 99, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Costa-Mattioli M., Sossin W. S., Klann E., Sonenberg N. (2009) Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richter J. D., Klann E. (2009) Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 23, 1–11 [DOI] [PubMed] [Google Scholar]
- 21. Ehninger D., Han S., Shilyansky C., Zhou Y., Li W., Kwiatkowski D. J., Ramesh V., Silva A. J. (2008) Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 14, 843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu G., Jiang X., Jaffrey S. R. (2013) A mental retardation-linked nonsense mutation in cereblon is rescued by proteasome inhibition. J. Biol. Chem. 288, 29573–29585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu J. (2005) Preparation, culture, and immortalization of mouse embryonic fibroblasts. Current Protocols in Molecular Biology (Ausubel F. M., ed) Chapter 28, Unit 28 21, Wiley, New York: [DOI] [PubMed] [Google Scholar]
- 24. Alonso M., Vianna M. R., Depino A. M., Mello e Souza T., Pereira P., Szapiro G., Viola H., Pitossi F., Izquierdo I., Medina J. H. (2002) BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus 12, 551–560 [DOI] [PubMed] [Google Scholar]
- 25. Wu S., Hu Y., Wang J. L., Chatterjee M., Shi Y., Kaufman R. J. (2002) Ultraviolet light inhibits translation through activation of the unfolded protein response kinase PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 277, 18077–18083 [DOI] [PubMed] [Google Scholar]
- 26. Chappell S. A., LeQuesne J. P., Paulin F. E., deSchoolmeester M. L., Stoneley M., Soutar R. L., Ralston S. H., Helfrich M. H., Willis A. E. (2000) A mutation in the c-myc-IRES leads to enhanced internal ribosome entry in multiple myeloma: a novel mechanism of oncogene de-regulation. Oncogene 19, 4437–4440 [DOI] [PubMed] [Google Scholar]
- 27. Ling J., Morley S. J., Traugh J. A. (2005) Inhibition of cap-dependent translation via phosphorylation of eIF4G by protein kinase Pak2. EMBO J. 24, 4094–4105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neves G., Cooke S. F., Bliss T. V. (2008) Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75 [DOI] [PubMed] [Google Scholar]
- 29. Casadio A., Martin K. C., Giustetto M., Zhu H., Chen M., Bartsch D., Bailey C. H., Kandel E. R. (1999) A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell 99, 221–237 [DOI] [PubMed] [Google Scholar]
- 30. Kelleher R. J., 3rd, Govindarajan A., Jung H. Y., Kang H., Tonegawa S. (2004) Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116, 467–479 [DOI] [PubMed] [Google Scholar]
- 31. Hay N., Sonenberg N. (2004) Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 [DOI] [PubMed] [Google Scholar]
- 32. Ma X. M., Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 33. Efeyan A., Zoncu R., Chang S., Gumper I., Snitkin H., Wolfson R. L., Kirak O., Sabatini D. D., Sabatini D. M. (2013) Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493, 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hall M. N. (2008) mTOR-what does it do? Transplant. Proc. 40, S5–S8 [DOI] [PubMed] [Google Scholar]
- 35. Sarbassov D. D., Ali S. M., Sabatini D. M. (2005) Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 17, 596–603 [DOI] [PubMed] [Google Scholar]
- 36. Dash P. K., Orsi S. A., Moore A. N. (2006) Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-Mammalian target of rapamycin pathway. J. Neurosci. 26, 8048–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grimes W. N., Li W., Chávez A. E., Diamond J. S. (2009) BK channels modulate pre- and postsynaptic signaling at reciprocal synapses in retina. Nat. Neurosci. 12, 585–592 [DOI] [PMC free article] [PubMed] [Google Scholar]