

Background: Repression of cyclin D1 is required for terminal cell cycle arrest associated with myogenesis.
Results: pRb and MyoD function together to block induction of Fra-1 and in turn cyclin D1.
Conclusion: MyoD and pRb cooperate to effect transcriptional repression during myogenesis.
Significance: The findings reveal a link between MyoD, pRb, Fra-1, and cyclin D1, the latter three of which are often deregulated in cancer.
Keywords: Cell Differentiation, Gene Regulation, Myogenesis, Transcription Enhancer, Transcription Factor, Fra-1, MyoD, pRb, Terminal Cell Cycle Arrest
Abstract
The acquisition of skeletal muscle-specific function and terminal cell cycle arrest represent two important features of the myogenic differentiation program. These cellular processes are distinct and can be separated genetically. The lineage-specific transcription factor MyoD and the retinoblastoma protein pRb participate in both of these cellular events. Whether and how MyoD and pRb work together to effect terminal cell cycle arrest is uncertain. To address this question, we focused on cyclin D1, whose stable repression is required for terminal cell cycle arrest and execution of myogenesis. MyoD and pRb are both required for the repression of cyclin D1; their actions, however, were found not to be direct. Rather, they operate to regulate the immediate early gene Fra-1, a critical player in mitogen-dependent induction of cyclin D1. Two conserved MyoD-binding sites were identified in an intronic enhancer of Fra-1 and shown to be required for the stable repression of Fra-1 and, in turn, cyclin D1. Localization of MyoD alone to the intronic enhancer of Fra-1 in the absence of pRb was not sufficient to elicit a block to Fra-1 induction; pRb was also recruited to the intronic enhancer in a MyoD-dependent manner. These observations suggest that MyoD and pRb work together cooperatively at the level of the intronic enhancer of Fra-1 during terminal cell cycle arrest. This work reveals a previously unappreciated link between a lineage-specific transcription factor, a tumor suppressor, and a proto-oncogene in the control of an important facet of myogenic differentiation.
Introduction
Myogenic differentiation involves the execution of various processes including the acquisition of skeletal muscle-specific function and terminal cell cycle arrest. The principal effectors of these processes are the myogenic basic helix-loop-helix transcription factors: MyoD (MyoD1), Myf5, myogenin (Myog), and MRF4 (Myf6), that affect transcription following binding to specific DNA sequences (CANNTG) termed an E box. Indeed, ectopic expression of a single myogenic transcription factor, MyoD, is sufficient to initiate the myogenic program (1, 2).
When wild-type myoblasts are grown under low mitogen conditions that induce myogenesis, they withdraw from the cell cycle and begin to differentiate. If, following completion of the program, the differentiated myoblasts are restimulated in the presence of high mitogen containing serum, they fail to re-enter the cell cycle—this being a functional definition of terminal cell cycle arrest. How terminal cell arrest is established and maintained is poorly understood.
The retinoblastoma protein, pRb (Rb1), like MyoD, has been shown to play a significant role in several aspects of myogenesis, and this is supported by both in vivo and in vitro data. Skeletal muscle deficient for Rb shows reduced fiber density, a lack of expression of late markers of differentiation (e.g. muscle creatine kinase, MCK (Ckmm)), and ectopic proliferation (3–6). The cell culture correlates of these observations are manifest in Rb-deficient myoblasts by the inability of MyoD to induce MCK expression and the failure to undergo terminal cell cycle arrest (7, 8). The role of pRb in mediating these two processes, however, is distinct, and the same is true of MyoD.
At the level of pRb, its abilities to stimulate skeletal muscle differentiation and bring about terminal cell cycle arrest are genetically separable, as revealed by in vivo and in vitro analyses (6, 9, 10), and both of these processes do not require the well characterized E2F-dependent G1 phase cell cycle control function of pRb (9, 11–14). The role of pRb in MyoD-mediated stimulation of MCK expression has been traced to the transactivation function of MEF2C (6, 15), a member of the MEF2 family of transcription factors that cooperates with MyoD to effect muscle-specific gene activation. With respect to MyoD, its requirements for effecting differentiation and terminal cell cycle arrest are different (16, 17). Notwithstanding these insights, whether MyoD and pRb actually collaborate to effect terminal cell cycle arrest and, if so, the nature of this collaboration are not known.
To gain a greater appreciation for the myogenic program, several groups have defined the changes in gene expression during differentiation on a global scale (18–23). A number of salient observations from these studies are pertinent to the work presented here. In the canonical view of MyoD function, it is portrayed as a transcriptional activator; however, a role for MyoD in directly repressing transcription during myogenesis was revealed (18, 22, 23). In terms of differentiation-associated terminal cell cycle arrest, one study noted that surprisingly few cell cycle regulatory genes were affected by MyoD, leading the authors to propose that MyoD-mediated control of the cell cycle may be achieved indirectly through the regulation of other transcription factors (22). A role for MyoD in inhibiting transcription in a cycling, undifferentiated state has been described (24), but this mechanism is unlikely to operate during myogenic differentiation and terminal cell cycle arrest. Despite these efforts, how MyoD blocks transcription and the relevance of this function to myogenesis remain to be determined.
Although several mechanisms have been proposed for withdrawal from the cell cycle during myogenesis (25–30), a significant event that takes place during terminal cell cycle arrest is the stable repression of cyclin D1 (Ccnd1). The importance of the block to cyclin D1 induction upon restimulation of differentiated myoblasts is indicated by the observation that ectopic expression of cyclin D1 can drive differentiated myoblasts into the S phase by effecting the phosphorylation and inactivation of pRb (31). Further, cyclin D1 has been shown to inhibit the transcriptional activity of MyoD, myogenin, and MEF2C, thereby hindering their ability to execute the myogenic differentiation program (32–35). These observations suggest that the repressed state of cyclin D1 is essential to the maintenance of terminal cell cycle arrest and the faithful execution of the myogenic differentiation program. However, how the repressed state of cyclin D1 is achieved is uncertain.
Here we have explored the role of MyoD and pRb in terminal cell cycle arrest during the myogenic program of differentiation. We report that MyoD and pRb work together to effect terminal cell cycle arrest. Their cooperative actions inhibit the induction of cyclin D1 during mitogenic restimulation of differentiated myoblasts; however, the actions of MyoD and pRb are not direct. Rather, we find that MyoD and pRb directly block the induction of Fra-1 (Fosl1), an immediate early gene product and an upstream participant effecting the expression of cyclin D1. We identified conserved sequences in the Fra-1 gene that are responsible for the recruitment of MyoD, an event that leads to the localization of pRb to the Fra-1 gene; both MyoD and pRb are required for the inhibition of Fra-1 and cyclin D1 induction in differentiated myoblasts. Collectively, these observations reveal a previously unappreciated role for MyoD and pRb in mediating a block to transcription during a salient facet of myogenesis: terminal cell cycle arrest.
EXPERIMENTAL PROCEDURES
Cell Culture
Immortalized 3T3 derivatives of Rb+/+ and Rb−/− mouse embryo fibroblasts have been described (6, 10, 36) and were cultured in DMEM supplemented with 10% FBS. The C2C12 murine myoblast line (37) was cultured in DMEM supplemented with 20% FBS. Rb+/+ and Rb−/− myoblasts (3T3 derivatives infected with retrovirus encoding MyoD) and C2C12 cells were differentiated in DMEM containing 2% horse serum for 6 days with a change in media every 2 days. The cells were restimulated with the addition of DMEM containing 20% FBS. C2C12 cells were rendered quiescent by culturing in 0.1% FBS for 3 days.
Plasmids, Infections, and Antibodies
Retroviral vectors encoding MyoD (a gift from Andrew Lassar) (8) and p16 (38) were used to perform infections as described (10, 39). Murine Fra-1 (a gift from Tim Rothstein) was subcloned into pShuttle-CMV (a gift from Bert Vogelstein), and recombinant adenovirus was prepared as described (40). Ad-GFP was a gift from Kornelia Polyak. Infections were performed at an multiplicity of infection of 100. Antibody against cyclin D1 (Ab-3) was from Lab Vision (Thermo Scientific); antibodies against MyoD (M318, sc-760 and C-20, sc-304), Fra-1 (N-17, sc-183), Fra-2 (Q-20, sc-604), c-Fos (4, sc-52), and c-Jun (N, sc-45) were from Santa Cruz Biotechnology; antibody against pRb (G3-245) was from BD Pharmingen (BD Biosciences); antibody against p16 was a gift from James Decaprio; and antibodies against β-actin (catalog number A5441) and tubulin (catalog number T5168) were from Sigma.
Fra-1 Promoter/Enhancer Luciferase Reporter
A genomic fragment of the murine Fra-1 (Fosl1) gene spanning 5′-upstream sequences, exon 1, intron 1, and part of exon 2 (a gift from Erwin Wagner) (41) was used to create a Fra-1 promoter/enhancer reporter. Sequences running from −930 to +2491 (41) relative to the transcriptional start site were subcloned such that sequences in exon 2 were in frame with luciferase in the pGL-3 Basic vector (Promega). Mutant Fra-1 promoter/enhancer reporters were generated by site-directed mutagenesis (Stratagene) and confirmed by sequencing. The following mutations were introduced: E box 1, CAGGTG > TCAGGC; E box 2, CACGTG > TCAGGC; and E box 3, CACCTG > TCAGGC. To generate stable lines of each promoter/enhancer reporter construct, the Rb+/+ and Rb−/− 3T3 fibroblasts were co-transfected with Fra-1 promoter/enhancer reporter and pBabe-puro plasmid (5:1 ratio) using Superfect transfection reagent (Qiagen). Cells were selected under 3 μg/ml puromycin (Calbiochem) to generate pooled populations of Fra-1 promoter/enhancer reporter lines.
Transcriptional Activation Assays
Stable lines harboring an integrated Fra-1 promoter/enhancer reporter and its mutant derivatives were infected either with retrovirus encoding MyoD or “empty” vector. Forty-eight hours later, the cells were induced to differentiate by culturing in medium containing 2% horse serum for 3 days. The cells were restimulated with media containing 20% FBS, normalized for protein content, and luciferase activity was measured as described (42). Statistical analyses were performed using Student's t test.
Chromatin Immunoprecipitation
ChIP was carried out essentially as described (43). The cells were cross-linked with 1% formaldehyde solution at 37 °C for 10 min followed by two quick washes with cold PBS on ice. The cells were scraped into cold PBS and either stored at −80 °C or used immediately for ChIP. Briefly, the cells were lysed with ChIP lysis buffer (50 mm Tris-HCl, pH 8.0, 1% SDS, 5 mm EDTA, plus protease inhibitors; Roche) on ice for 10–20 min and sonicated five times at 12% amplitude for 15 s (Fisher Scientific model 500 Sonic Dismembrator). The majority of DNA fragments were below 500 bp. Samples were subjected to centrifugation at 16,000 × g for 20 min, and 100 μl of supernatant was mixed with 900 μl of ChIP dilution buffer (20 mm Tris-HCl, pH 8.1, 1% Triton X-100, 2 mm EDTA, 150 mm NaCl, plus protease inhibitors). Lysates were incubated overnight at 4 °C with specific antibodies to MyoD (M318, sc-760; Santa Cruz Biotechnology), pRb (clone G3-245; BD Biosciences), RNA polymerase II (C-21, sc-900; Santa Cruz Biotechnology), or appropriate mouse and rabbit IgG as controls. Immune complexes were incubated with protein A or protein G-Sepharose beads (Santa Cruz Biotechnology) for 1 h and then washed with ChIP wash buffers TSE I (20 mm Tris-HCl, pH 8.1, 0.1% SDS, 1% Triton X-100, 2 mm EDTA, 150 mm NaCl), TSE II (20 mm Tris-HCl, pH 8.1, 0.1% SDS, 1% Triton X-100, 2 mm EDTA, 500 mm NaCl), and buffer III (10 mm Tris-HCl, pH 8.1, 0.25 m LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mm EDTA), followed by two washes with TE (pH 8.1) for 10 min each at 4 °C. Samples were eluted from the beads with 100 μl of 1% SDS, 0.1 m NaHCO3 for 20 min and incubated overnight at 65 °C to reverse cross-linking. DNA was purified using Qiagen PCR purification kit and eluted in 100 μl of elution buffer (Qiagen). PCR was carried out with specific primers using Taq polymerase kit (Qiagen). Primer sequences are as follows: intron 1 of Fra-1 (Fra-1 int 1) +, 5′-AGGTTGCAGGTGTCCATTTCCTGT-3′; intron 1 of Fra-1 (Fra-1 int 1) −, 5′-TGCTTAGGCAAGTCTGGACAGCTA-3′; control 3 kb downstream (Fra-1 3 kb) +, 5′-GCACTGAATGCACAAGGTGCTCAT-3′; control 3 kb downstream (Fra-1 3 kb) −, 5′-ACGGTGGCTCACAACCACCTATAA-3′; transcriptional start site in Fra-1 (Fra-1 TSS) +, 5′-TGTGTTGGGAACCTTGGCTAGTCT-3′; transcriptional start site in Fra-1 (Fra-1 TSS) −, 5′-AAGTTCTTGGGCTGAACCACTTGC-3′; MCK enhancer (MCK) +, 5′-GTCTAGGCTGCCCATGTAAGG-3′ (44); and MCK enhancer (MCK) −, 5′-CAGGCCCAGGAAGGATACAG-3′ (44).
RESULTS
Specific Block to Fra-1 Induction during Terminal Cell Cycle Arrest
Given that members of the AP-1 family of transcription factors are implicated in the mitogenic induction of cyclin D1 (45), we hypothesized that a block to one or more AP-1 members might be responsible for the corresponding inhibition of cyclin D1 induction. C2C12 myoblasts were cultured under conditions known to induce a reversible quiescent state or an irreversible differentiated state and then restimulated by the addition of 20% fetal bovine serum. Next, the induction of the protein products encoded by several immediate early genes (c-Fos, c-Jun, Fra-1, Fra-2, JunB, and JunD) and cyclin D1 was monitored. The expression of all of these proteins was induced when quiescent myoblasts were restimulated (Fig. 1; data not shown). A different scenario emerged when differentiated (as opposed to quiescent) C2C12 cells were restimulated. Here, many of the proteins encoded by immediate early genes were induced (Fig. 1; data not shown; see also Fig. 2), consistent with previous findings (46). This occurred with the striking exception of Fra-1, whose expression was not induced specifically upon restimulation of differentiated myoblasts (Fig. 1). In a similar fashion, cyclin D1 was not induced in differentiated myoblasts upon restimulation; cyclin D1 was induced upon restimulation of quiescent myoblasts (Fig. 1). These observations suggest that Fra-1 is unique among immediate early gene products in its inability to be induced upon serum stimulation of differentiated myoblasts, an event that is coincident with a similar lack of induction of cyclin D1.
FIGURE 1.
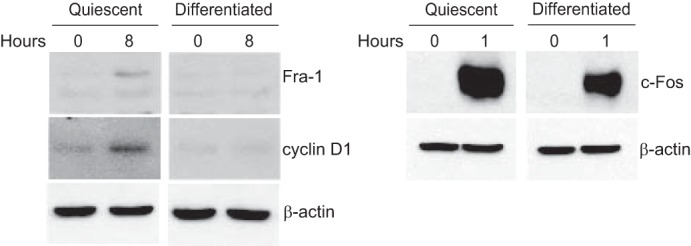
Fra-1 and cyclin D1 are induced upon restimulation of quiescent but not differentiated myoblasts. C2C12 cells were rendered quiescent by culturing in 0.1% FBS for 3 days or differentiated by culturing in 2% horse serum for 6 days. Subsequently, the cells were restimulated with 20% FBS for 8 h (left panel) or 1 h (right panel). The lysates were prepared and subjected to immunoblotting with antibodies to the indicated proteins. β-Actin immunoblots were used to assess protein loading. The results are representative of at least three independent experiments.
FIGURE 2.

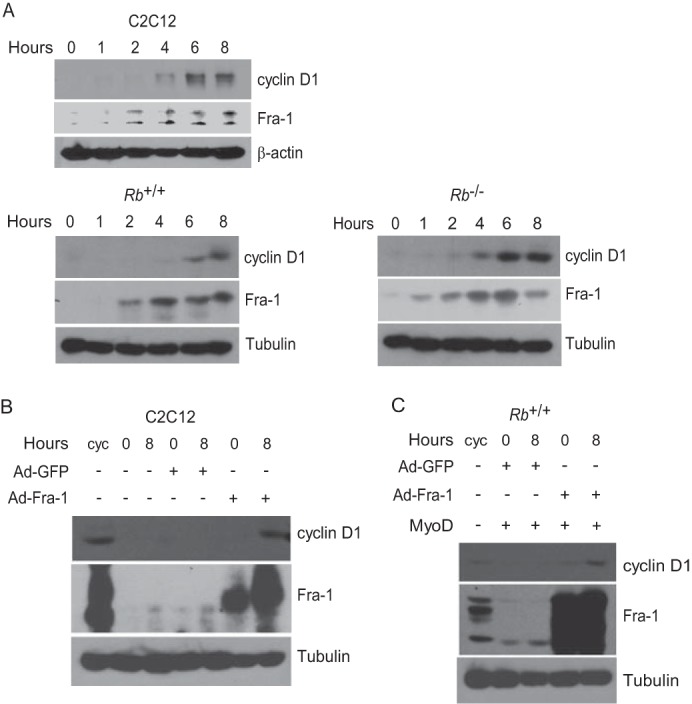
Regulation of Fra-1 and cyclin D1 by MyoD and pRb. A, block to Fra-1 and cyclin D1 induction is MyoD- and pRb-dependent. Rb+/+ and Rb−/− fibroblasts (indicated) were infected with virus directing the expression of MyoD or empty vector and cultured under conditions to induce differentiation (2% horse serum) for 3 days (0-h time point). Subsequently, cells were restimulated for 8 h with 20% FBS. Lysates were prepared and subjected to immunoblotting with antibodies to the indicated proteins. Tubulin immunoblots were used to assess protein loading. The results are representative of at least three independent experiments. B, as in A, except c-Fos was analyzed at 1 h after restimulation. C, p16 induces a G1 arrest independent of down-regulation of Fra-1. Rb+/+ fibroblasts were infected with MyoD- or p16-encoding virus or empty vector and cultured under conditions to induce myogenic differentiation for 3 days (0-h time point). Subsequently, cells were restimulated for 8 h with 20% FBS. Expression of the indicated proteins was analyzed by immunoblotting. Lysates were prepared and subjected to immunoblotting with antibodies to the indicated proteins. Tubulin immunoblots were used to assess protein loading. The results are representative of at least three independent experiments.
MyoD and pRb Cooperate to Effect Terminal Cell Cycle Arrest
Next, we determined whether both MyoD and pRb might participate in the block to induction of Fra-1 and cyclin D1. Rb+/+ and Rb−/− fibroblasts were infected with a retrovirus encoding MyoD or empty vector. Following infection, cells were cultured under conditions known to induce myogenic differentiation. Subsequently, the expression of Fra-1 and cyclin D1 was monitored upon restimulation of the cells. In Rb-deficient myoblasts Fra-1 was induced (Fig. 2A), and correspondingly, cyclin D1 expression also increased, consistent with previous observations (7). Similar observations were made in restimulated Rb-positive cells infected with empty vector (Fig. 2A). By contrast, in the presence of both MyoD and pRb, Fra-1 and cyclin D1 were not induced (Fig. 2A). When assayed at later times in Rb+/+ myoblasts, induction of Fra-1 and cyclin D1 was still not detected, thus ruling out the possibility that their expression was delayed (data not shown). The effect of MyoD and pRb on Fra-1 induction was specific, because proteins encoded by other immediate early genes were induced regardless of the presence of MyoD or pRb (Fig. 2, A and B). A consistent decline in c-Fos was noted in the presence of MyoD, but independent of pRb, in agreement with previous findings (26). Further, ectopic expression of the cdk4 and cdk6 inhibitor p16 (Cdkn2a) did not prevent the induction of either Fra-1 or cyclin D1 (Fig. 2C), reinforcing the notion that a G1 arrest mediated by pRb-E2F complexes is distinct from the irreversible arrest brought about by pRb during myogenic differentiation. Together, these observations suggest that pRb and MyoD establish and perhaps maintain terminal cell cycle arrest, in part, by preventing the induction of Fra-1 and cyclin D1.
Fra-1 Acts Upstream of Cyclin D1
The results presented above, together with the observation that AP-1 family members participate in the serum induction of cyclin D1, suggest that the block to Fra-1 induction is causally related to the corresponding block to cyclin D1 induction. Consistent with this possibility and in keeping with previous observations (47, 48), expression of Fra-1 preceded that of cyclin D1 following restimulation of quiescent Rb+/+ and Rb−/− fibroblasts, as well as C2C12 cells (Fig. 3A). That Fra-1 and cyclin D1 were induced earlier in Rb−/− compared with Rb+/+ fibroblasts is consistent with the observation that the former traverse G1 faster (49).
FIGURE 3.
Fra-1 participates in the induction of cyclin D1. A, kinetics of Fra-1 and cyclin D1 induction. C2C12 cells (upper panel) were rendered quiescent by culturing in 0.1% FBS for 3 days and then restimulated by the addition of 20% FBS. Additionally, Rb+/+ (lower left panel) and Rb−/− (lower right panel) fibroblasts were cultured under differentiation conditions for 3 days (which in the absence of MyoD renders these cells quiescent) and then restimulated by the addition of 20% FBS. At the indicated times, lysates were prepared and subjected to immunoblotting with antibodies to the indicated proteins. The results are representative of at least three independent experiments. B, Fra-1 operates upstream of cyclin D1 to induce its expression. C2C12 cells were cultured under conditions to induce their differentiation and then infected with adenovirus directing the expression of Fra-1 or GFP (0-h time point). Subsequently, cells were restimulated for 8 h with 20% FBS. A cycling (cyc) population of C2C12 cells was included for comparison. Expression of the indicated proteins at the indicated times was analyzed by immunoblotting. Tubulin immunoblots were used to assess protein loading. The results are representative of at least three independent experiments. C, as in B except Rb+/+ myoblasts were used. The results are representative of at least three independent experiments.
To determine whether Fra-1 operates upstream of and in series with cyclin D1, we asked whether ectopic expression of Fra-1 in differentiated myoblasts can induce the expression of cyclin D1 upon serum restimulation. To this end, recombinant adenoviruses were used to ectopically express Fra-1 or GFP as a control in differentiated C2C12 cells. Upon serum restimulation, cyclin D1 was induced in cells expressing Fra-1, but not GFP (Fig. 3B), suggesting that the block to Fra-1 induction is responsible for the lack of cyclin D1 induction. Similar observations were made with wild-type Rb myoblasts (Fig. 3C). As anticipated, cyclin D1 was not induced under differentiation conditions but required serum restimulation. This follows from the fact that Fra-1 on its own cannot mediate transcriptional induction, because it lacks a transactivation domain and must heterodimerize with mitogen-induced Jun family members to affect transcription (50–52). Together with the findings noted above, these observations suggest that the cyclin D1 gene is not rendered refractory to induction following myogenic differentiation. Rather, the actions of MyoD and pRb in effecting the block to cyclin D1 induction are indirect and mediated at the level of the Fra-1 gene.
MyoD Binds to Sites Located in Intron 1 of Fra-1
To determine how MyoD and pRb impose a block to Fra-1 induction, we considered a region previously identified as an intronic enhancer in the Fra-1 gene responsible for its basal and serum-inducible expression. Specifically, three closely spaced AP-1 (and AP-1 like) sites located in the first intron of the Fra-1 gene have been shown to play a significant role in the induction of the Fra-1 during mitogen-induced cell cycle re-entry from a quiescent state (52, 53). We searched for other possible conserved regulatory elements within the first 2000 bp of the first intron in the Fra-1 gene comparing murine and human sequences using Bayes block aligner (54) (murine and human Fra-1 intron 1 are 2105 and 3234 bps in length, respectively). This analysis revealed that the largest region displaying the highest degree of conservation contained the 5′-most AP-1 element (Fig. 4A); the other two AP-1 elements were located downstream in the adjacent conserved region. Of these two conserved regions identified in intron 1 of Fra-1, in the larger, 5′-most one, we noted two potential MyoD-binding sites (E boxes, CANNTG) juxtaposed to the intronic enhancer of Fra-1 (Fig. 4A), and the sequence of each of these two E box elements is identical among murine, rat, human, and chimp species.
FIGURE 4.

MyoD is localized to E boxes located in the intronic enhancer of Fra-1 during differentiation. A, first 2000 bp of murine and human intron 1 of Fra-1 were analyzed with Bayes block aligner (top). Probability of conservation: 0.8–0.999 (red), 0.6–0.799 (blue), 0.4–0.599 (yellow), 0.2–0.399 (green), and 0–0.199 (black). The largest region showing the highest degree of conservation is shown at the bottom, and E boxes and AP-1 sites are underlined (coordinates are defined by setting the first nucleotide of intron 1 to 1). B, MyoD localization to intronic E boxes of Fra-1 during differentiation. ChIP analysis of cycling and differentiated C2C12 cells was performed with antibody to MyoD (M) or IgG as control. For the precipitated DNA, sequences spanning the E boxes in intron 1 of Fra-1 (Fra-1 int 1), ∼3 kb downstream of the Fra-1 intronic E boxes (Fra-1 3 kb), and the MCK enhancer (MCK) were analyzed by PCR. One-tenth of the lysates used for ChIP were also subjected to PCR (input, In). The results are representative of at least three independent experiments. C, as in B, except Rb-positive fibroblasts were infected with MyoD-encoding virus (left panel) or empty vector (right panel). The results are representative of at least three independent experiments. D, as in C, except Rb-deficient fibroblasts were employed. The results are representative of at least three independent experiments.
MyoD-Binding Sites in Fra-1 Are Required to Block Its Induction
To evaluate the possible relevance of the conserved intronic E boxes, we asked whether MyoD localized to sequences in the first intron of the Fra-1 gene using ChIP assays. C2C12 cells were differentiated, and soluble chromatin was prepared from formaldehyde-treated cells. Antibody specific for MyoD was then used to precipitate genomic DNA fragments bound to MyoD. This DNA was analyzed by PCR using a primer pair spanning the two E boxes identified in intron 1 of the Fra-1 gene. A primer pair located 3 kb downstream of the E boxes was used as a negative control, and a primer pair to sequences in the muscle creatine kinase (MCK) enhancer, a known target of MyoD, was used as a positive control. These analyses revealed that MyoD was specifically bound to sequences in the first intron of the Fra-1 gene in differentiated, but not cycling, myoblasts (Fig. 4B). Similar observations were made with Rb-positive and -negative myoblasts (Fig. 4, C and D). Results were confirmed with two different antibodies to MyoD, and specificity was assessed with a lack of a signal in identically treated Rb+/+ and Rb−/− fibroblasts (i.e. in the absence of MyoD) (Fig. 4, C and D; data not shown). Consistent with the presence of two conserved E boxes in intron 1 of the Fra-1 gene, these results suggest that MyoD occupies these sites during differentiation and does so independent of Rb status.
To assess the functional significance of binding of MyoD to the conserved E box elements in the first intron of Fra-1, we generated a Fra-1 promoter/enhancer reporter. Sequences from a murine genomic fragment extending from upstream of the transcriptional start sites through exon 1, intron 1, and part of exon 2 were cloned upstream of luciferase (Fig. 5A). In addition to this wild-type promoter/enhancer reporter, three mutants were generated in which sequences of the two conserved E boxes and one nonconserved E box were altered in such a way that they would be predicted to disrupt MyoD binding. The wild-type promoter/enhancer reporter was stably integrated into Rb+/+ and Rb−/− fibroblasts and the mutant promoter/enhancer reporters into Rb+/+ fibroblasts. Subsequently, cells were infected with a retrovirus encoding MyoD or empty vector, differentiated, and then restimulated. As anticipated, under these conditions, the wild-type promoter/enhancer reporter was not induced following restimulation in an pRb- and MyoD-dependent manner (Fig. 5, B and C). By contrast, mutation of either or both of the conserved E boxes resulted in induction of the promoter/enhancer reporter regardless of whether MyoD was present (Fig. 5, D–F); the promoter/enhancer reporter bearing a mutation in the nonconserved E box behaved like the wild-type reporter (Fig. 5G). Together with our observation suggesting that MyoD can be localized to sequences in intron 1 of the Fra-1 gene (Fig. 4), these findings provide functional evidence suggesting that MyoD binding to the conserved intronic E-boxes participates in imposing a block to Fra-1 induction following restimulation of differentiated myoblasts. Further, MyoD localization to intron 1 of Fra-1 alone is not sufficient to inhibit its induction; pRb must also be present, consistent with the results presented in Fig. 2A.
FIGURE 5.
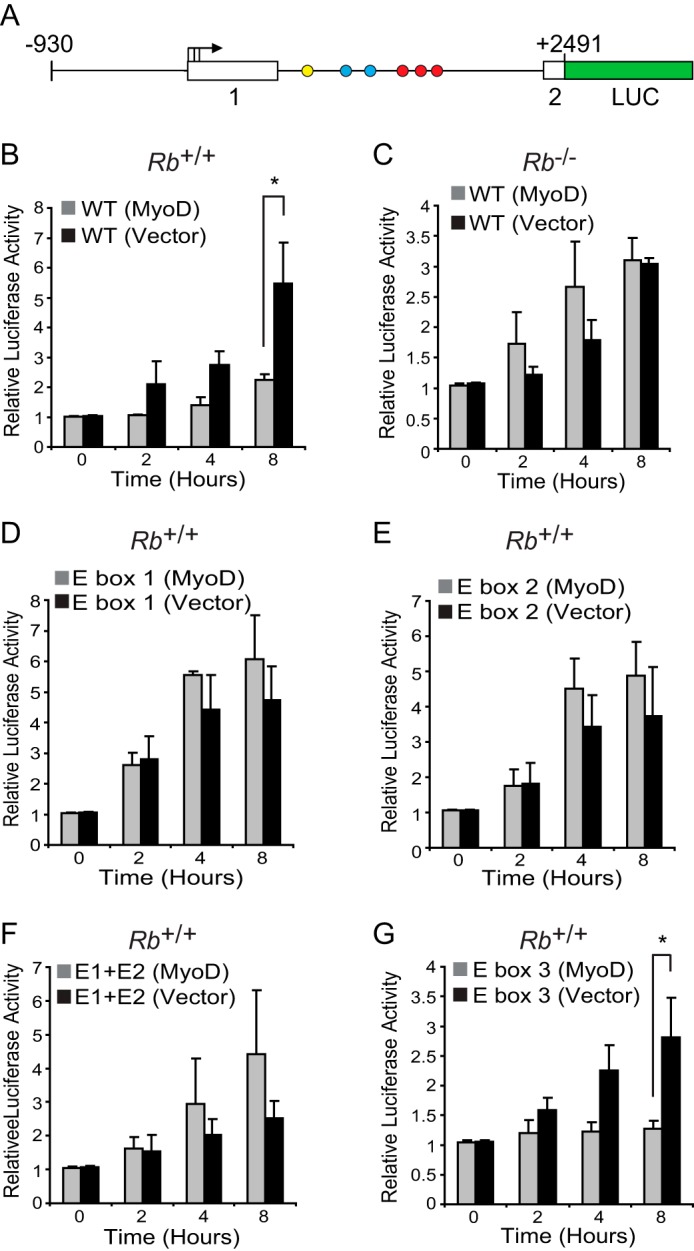
Block to Fra-1 induction by MyoD is mediated through conserved E boxes located in the intronic enhancer of Fra-1. A, schematic diagram of Fra-1 promoter reporter with intronic E boxes (blue circles), AP-1 elements (red circles), and a nonconserved E box (yellow circle) indicated. B and C, block to Fra-1 promoter induction is pRb-dependent. Rb+/+ (B) and Rb−/− (C) fibroblasts with a stably integrated wild-type Fra-1 promoter reporter were infected with MyoD-encoding virus or empty vector and cultured under conditions to induce differentiation. Subsequently, cells were restimulated, and promoter activity was normalized for protein content determined at the times indicated. Two independent sets of promoter reporter lines were made, and each yielded similar results. The results are means ± S.D. of three independent experiments, each with triplicate determinations. *, p = 0.007. D–G, block to Fra-1 promoter induction is dependent on conserved E boxes located in intron 1. The results shown are as in B, except E box mutants of the Fra-1 promoter reporter were analyzed in Rb+/+ myoblasts. Mutants included were the individually mutated conserved E boxes, E box 1 (D), E box 2 (E), the double mutant, E box 1 and E box 2 (E1 + E2) (F), and a nonconserved E box, E box 3 (G). Two independent sets of promoter reporter lines were made, and each yielded similar results. The results are means ± S.D. of three independent experiments, each with triplicate determinations. *, p = 0.032.
pRb Is Recruited to Intron 1 of Fra-1 in a MyoD-dependent Manner
The results presented above suggest that inhibition of Fra-1 induction following restimulation of differentiated myoblasts involves the actions of MyoD at the intronic enhancer. Further, pRb, in addition to MyoD, is required to impose the block to Fra-1 expression, leaving open the question as to what the role of pRb is in this process. Given the well documented role of pRb in repressing transcription (55) and the reported interaction between MyoD and pRb (25), we entertained the possibility that pRb is also recruited to the intronic enhancer of Fra-1. By ChIP analysis, we found that pRb was localized to the MyoD-binding sites within the intronic enhancer of Fra-1 in differentiated, but not cycling, C2C12 cells (Fig. 6A), suggesting that MyoD and pRb act together at the intronic enhancer of Fra-1.
FIGURE 6.
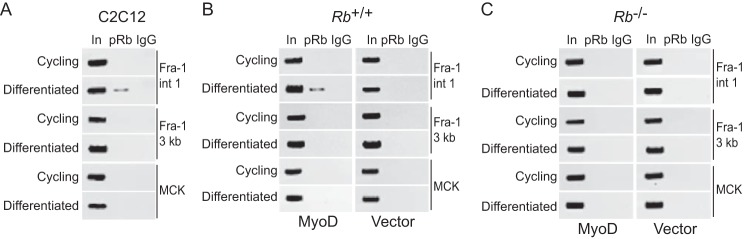
pRb recruitment to the intronic enhancer of Fra-1 is MyoD-dependent. A, pRb is recruited to intron 1 of Fra-1 during differentiation. ChIP analysis of cycling and differentiated C2C12 cells was performed with antibody to pRb or IgG as control. Sequences spanning the E boxes in intron 1 of Fra-1 (Fra-1 int 1), ∼3 kb downstream of the Fra-1 intronic E boxes (Fra-1 3 kb), and the MCK enhancer (MCK) were analyzed by PCR. One-tenth of the lysates used for ChIP were also subjected to PCR (input, In). The results are representative of at least three independent experiments. B, pRb recruitment to intron 1 of Fra-1 is MyoD-dependent. The results shown are as in A, except Rb-positive fibroblasts were infected with MyoD-encoding virus (left panel) or empty vector (right panel). The results are representative of at least three independent experiments. C, as in B, except Rb-deficient fibroblasts were employed. The results are representative of at least three independent experiments.
Next, we asked whether recruitment of pRb to the Fra-1 gene was dependent of MyoD. Rb+/+ fibroblasts were transduced with a MyoD-encoding retrovirus or empty vector. We found that under differentiation conditions, pRb was localized to the intronic enhancer of Fra-1, but only in the presence of MyoD (Fig. 6B). Localization of pRb was not observed in cycling cells either in the presence or absence of MyoD (Fig. 6B). The specificity of the ChIP for pRb was confirmed by performing similar experiments in Rb−/− myoblasts and fibroblasts (Fig. 6C). These observations are consistent with a model in which MyoD recruits pRb to the intronic enhancer of Fra-1, an event dependent on the conserved MyoD-binding sites located in Fra-1.
Recruitment of RNA Polymerase II to Fra-1 Is Inhibited during Differentiation
We also explored the functional consequences of recruitment of MyoD and pRb to the intronic enhancer of Fra-1 during differentiation. Various models have been proposed to explain how enhancers operate (56), and in each of them there exists an interaction of the enhancer with the promoter allowing RNA polymerase II (Pol II)2 loaded at the enhancer to be transferred to the promoter (43, 56). Accordingly, we predicted that recruitment of MyoD and pRb to Fra-1 during differentiation might prevent the localization of Pol II to the intronic enhancer of Fra-1. C2C12 myoblasts were cultured under conditions to effect a reversible quiescent state or an irreversible differentiated state and then restimulated (Fig. 1). We found that in restimulated quiescent myoblasts (i.e. conditions in which Fra-1 is induced), Pol II was localized to the intronic enhancer of Fra-1, as well as the transcriptional start site (Fig. 7). By contrast, in differentiated myoblasts that were restimulated (i.e. conditions where Fra-1 is not induced), we found that Pol II failed to localize to the intronic enhancer and promoter of Fra-1 (Fig. 7). As a control, the MCK enhancer was analyzed. As anticipated, Pol II failed to localize to the MCK enhancer in quiescent and restimulated myoblasts. By contrast, differentiated myoblasts displayed localization of Pol II to the MCK enhancer. That localization of Pol II to the MCK enhancer was not observed when differentiated myoblasts were restimulated was expected, because this scenario has been shown to result in the suppression of MCK mRNA levels (57). These observations suggest that the intronic enhancer of Fra-1 is rendered inactive with respect to Pol II recruitment upon differentiation of myoblasts.
FIGURE 7.
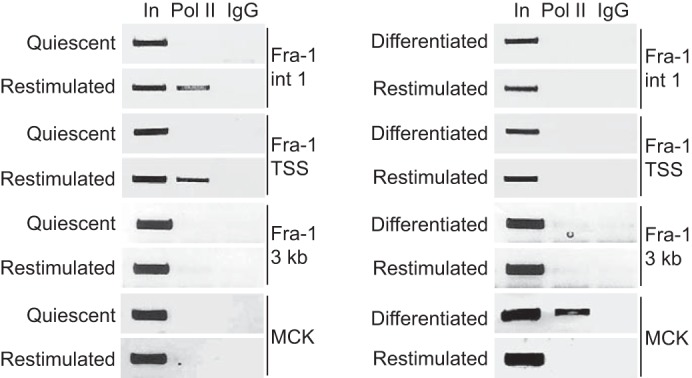
Block to RNA polymerase II recruitment to Fra-1 during differentiation. C2C12 cells were rendered quiescent (left panel) by culturing in 0.1% FBS for 3 days or differentiated (right panel) by culturing in 2% horse serum for 6 days. Subsequently, the cells were restimulated for 8 h in the presence of 20% FBS. ChIP analysis was performed with antibody to Pol II or IgG as control. Sequences spanning the E boxes in intron 1 of Fra-1 (Fra-1 int1), the transcriptional start site of Fra-1 (Fra-1 TSS), ∼3 kb downstream of the Fra-1 intronic E boxes (Fra-1 3 kb), and the MCK enhancer (MCK) were analyzed by PCR. One-tenth of the lysates used for ChIP were also subjected to PCR (input, In). The results are representative of at least two independent experiments.
DISCUSSION
Although it is accepted that MyoD and pRb have essential roles in establishing and perhaps maintaining terminal cell cycle arrest during myogenesis, whether and how they cooperate and the specific events they govern during this process have been unclear. We have provided evidence that MyoD and pRb cooperate to block the induction of cyclin D1, an inhibitor of both MyoD and pRb, during restimulation of differentiated myoblasts. However, the means by which MyoD and pRb impose a block to cyclin D1 induction is indirect. Our findings suggest that together MyoD and pRb render the Fra-1 gene, an important upstream regulator of cyclin D1 expression, refractory to induction. These findings establish a previously unappreciated functional link between a lineage-specific transcription factor, a tumor suppressor, and a proto-oncogene. Further, they provide a mechanism by which MyoD can inhibit transcription and suggest that this function of MyoD contributes to an important component of the myogenic differentiation program.
Previous studies established that MyoD is recruited in a temporal fashion to a large cohort of genes during myogenesis, allowing for the orchestration of the differentiation program (18, 22). Several genes affected directly by MyoD are activated; however, a subset of these genes was repressed, and our findings bear on how MyoD might effect transcriptional repression and the biological significance of this phenomenon. The cis-regulatory elements of many, but not all genes activated by MyoD, contain two E boxes, and this allows for cooperative binding of MyoD (58). The same is likely occurring at the intronic E boxes of Fra-1, because the two conserved E boxes are required for silencing of this gene (Fig. 5). These observations suggest that recruitment of MyoD to intron 1 of Fra-1 is the initiating event affecting gene expression. At a global level, the effect of MyoD on the Fra-1 gene occurs early during differentiation; this follows from the observation that Fra-1 is among the genes most rapidly repressed during myogenic differentiation (19, 20). Thus, the timing of recruitment of MyoD to the Fra-1 gene appears to effect two outcomes. First, the early localization of MyoD at the Fra-1 gene brings about the repression of Fra-1 during the initial phase of the myogenic differentiation program, thereby decreasing the expression of cyclin D1, which in turn allows the appropriate functioning of MyoD. In this regard, it is noteworthy that the kinetics of repression of cyclin D1 is delayed compared with Fra-1 (19), consistent with our data suggesting that loss of Fra-1 expression is causally related to the effect on cyclin D1 expression. Second, recruitment of MyoD effects the stable repression of the Fra-1 gene, rendering it, and consequently cyclin D1, immune to growth factor stimulation. Again, by preventing the induction of cyclin D1, this allows MyoD and other myogenic factors to maintain the mature differentiated characteristics of myotubes, because perturbing the function of myogenic transcription factors is thought to allow dedifferentiation and re-entry into the cell cycle (59).
Although it is generally agreed that pRb is essential for the establishment of an irreversible cell cycle arrest during myogenesis, attempts to determine whether pRb is required for maintenance of terminal cell cycle arrest have led to contradictory results. Specifically, inactivation of a conditional Rb allele in terminally differentiated myoblasts followed by serum restimulation failed to induce entry into the S phase (60, 61). Others, using a different strategy, RNAi, to deplete pRb in terminally differentiated myoblasts arrived at the opposite conclusion (62). We believe that our findings bear on this issue. The kinetics of down-regulation of Fra-1 and cyclin D1 during myogenesis (19, 20) together with our data suggest that the cooperative actions of MyoD and pRb at Fra-1 is an early event during the myogenic differentiation program. This suggests that the epigenetic regulation effected by MyoD and pRb at Fra-1 is stable and participates in the maintenance of the stable repression of Fra-1 and, in turn, cyclin D1, although it cannot be ruled out that other events independent of MyoD and pRb are involved (see below). Consistent with these possibilities, cyclin D1 is not induced upon restimulation of differentiated myoblasts in which Rb has been inactivated (61).
The ability of MyoD to induce the cdk inhibitor p21 is often regarded as the mechanism underlying irreversible cell cycle arrest of differentiated myoblasts (27, 63, 64). However, p21 is induced by MyoD in Rb-deficient myoblasts but fails to bring about terminal cell cycle arrest in this setting (8), and ectopic expression of p21 in Rb−/− myoblasts fails to inhibit DNA synthesis (8). Further, p21, like p16, would be predicted to effect a late G1 arrest during restimulation of differentiated myoblasts. However, previous studies have suggested that restimulated differentiated myoblasts are blocked in mid G1 (46), and our findings indicate that an important component of the immediate early response is inhibited, suggesting the block is in early G1. Based on these observations, we suggest a role for p21 induction in effecting the accumulation of unphosphorylated active pRb, thereby facilitating its ability to participate in the block to Fra-1 expression.
How MyoD is recruited to the intronic E boxes of Fra-1 as a function of differentiation is not known, but this also remains to be determined for muscle-specific genes containing two E boxes. The localization of MyoD to intron 1 of Fra-1 allows for the recruitment of pRb (Fig. 6). The interaction between MyoD and pRb has been reported (25) and reproduced by others (65), but nonetheless remains controversial (29, 65). This has largely been due to the inability to place the interaction between pRb and MyoD into a physiological setting of one or more of the events that occur during myogenesis. For example, although pRb is clearly required for the ability of MyoD to induce MCK expression, the contribution of pRb in this setting is indirect (6, 15), consistent with our finding that pRb is not recruited to this gene (Fig. 6). Thus, there does not appear to be a motivation or need to invoke an interaction between pRb and MyoD in effecting myogenic differentiation independent of their role in terminal cell cycle arrest. Our findings suggest that the interaction between pRb and MyoD does occur, but in a gene-specific manner to effect terminal cell cycle arrest. How this promoter/enhancer selectively occurs remains to be determined. An attractive hypothesis might be that MyoD-pRb complexes are stabilized at intron 1 of Fra-1 by a factor already present at the Fra-1 gene, similar to what has been shown for the homeodomain protein Pbx (44). Related to this possibility, it is noteworthy that the intronic enhancer in Fra-1 is embedded within a CpG island, suggesting a possible role for the context of the sequence in which the MyoD-binding sites in Fra-1 are located. Regardless of the details, our findings suggest a physiological context in which the cooperative actions of MyoD and pRb during myogenesis congregate at intron 1 of Fra-1.
Previous studies, albeit conflicting, have suggested that histone modifications at the cyclin D1 promoter render it refractory to restimulation. In one study, it was suggested that methylation of lysine 9 in histone H3 (H3K9me3) was responsible for the repression of cyclin D1 (66). By contrast, another group failed to detect H3K9me3 at the cyclin D1 promoter in differentiated myoblasts and suggested that the disparate results may be due to the use of improved reagents (62). Rather, they found that cyclin D1 was marked by H3K27me3 in differentiated myoblasts and that this occurred even in the absence of pRb (62), suggesting an event independent of the cooperative actions of MyoD and pRb described here. It is noteworthy, however, that a promoter marked by H3K27me3 is not necessarily inactive if it is bivalent, i.e. marked by both H3K4me3 and H3K27me3 (67, 68). Along these lines, in embryonic fibroblasts, similar to one of the experimental systems employed here, cyclin D1 displays bivalent histone marks (68). This suggests that although cyclin D1 harbors the repressive H3K27me3 mark, it may not be sufficient to silence cyclin D1. Further, our finding that ectopic expression of Fra-1 in differentiated myoblasts is capable of inducing cyclin D1 to a level seen in cycling myoblasts (Fig. 3), suggests that cyclin D1 is not rendered refractory to induction during the state of differentiation. Here, it is noteworthy that C2C12 cell ChIP-seq data at the ENCODE Project at UCSC reveals localization of Fra-1 to the cyclin D1 promoter, suggesting a direct effect of Fra-1. Further studies will be required to determine how MyoD and pRb affect the activity of the intronic enhancer of Fra-1 to, for example, influence the recruitment of RNA polymerase II and whether and how the bivalent state of cyclin D1 is resolved during differentiation. Notwithstanding, we cannot rule out the possibility that methylation events at cyclin D1 might facilitate its silencing during the maintenance of a terminally differentiated state in an pRb-independent manner.
Our findings suggest that pRb and MyoD, by imposing a block in early G1, prevent S phase re-entry of differentiated myoblasts. This is distinct from fibroblasts exiting quiescence where pRb acts in late G1 at a time that temporally and functionally coincides with the restriction point (49). However, in both settings, cyclin D appears to be the target: in fibroblasts, p16, a cyclin D-cdk4/6 inhibitor, effects an pRb-dependent late G1 block, and restimulation of differentiated myoblasts does not progress past early G1 because cyclin D1 is not synthesized. Why restimulated differentiated myoblasts choose to effect their arrest in G1 prior to the synthesis of cyclin D1 is not clear but can perhaps be rationalized. Cyclin D1 can inhibit several myogenic transcription factors including MyoD (see Introduction); thus, blocking the synthesis of cyclin D1 is necessary for the establishment and maintenance of the differentiated phenotype. Hence, it appears that the timely coordination of terminal cell cycle arrest with differentiation is not just coincidence but rather a necessary component of the myogenic differentiation program. Further, Fra-1 is a proto-oncogene (52), and it is likely that cyclin D1 does not represent its sole effector of transformation and that other targets of Fra-1 are deleterious to myogenesis. In this context, it is noteworthy that ectopic expression c-Jun, the product of another immediate early gene and heterodimeric partner of Fra-1, inhibits myogenesis (69). Regardless, the choice to target Fra-1, as opposed to cyclin D1, may reflect Fra-1 as being a nodal point whose expression must be blocked for the appropriate execution of differentiation.
Acknowledgments
We thank Andrew Lassar, Tim Rothstein, Bert Vogelstein, Kornelia Polyak, James Decaprio, and Erwin Wagner for providing reagents and Myles Brown for advice with chromatin immunoprecipitation. We thank members of the Ewen lab for advice and comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 CA065842 (to M. E. E.). This work was also supported by Department of Defense Breast Cancer Research Program Fellowship DAMD17-01-1-0224 (to H. N. R.).
- Pol II
- RNA polymerase II.
REFERENCES
- 1. Davis R. L., Weintraub H., Lassar A. B. (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000 [DOI] [PubMed] [Google Scholar]
- 2. Weintraub H., Davis R., Tapscott S., Thayer M., Krause M., Benezra R., Blackwell T. K., Turner D., Rupp R., Hollenberg S. (1991) The myoD gene family: nodal point during specification of the muscle cell lineage. Science 251, 761–766 [DOI] [PubMed] [Google Scholar]
- 3. Zacksenhaus E., Jiang Z., Chung D., Marth J. D., Phillips R. A., Gallie B. L. (1996) pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 10, 3051–3064 [DOI] [PubMed] [Google Scholar]
- 4. Wu L., de Bruin A., Saavedra H. I., Starovic M., Trimboli A., Yang Y., Opavska J., Wilson P., Thompson J. C., Ostrowski M. C., Rosol T. J., Woollett L. A., Weinstein M., Cross J. C., Robinson M. L., Leone G. (2003) Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 421, 942–947 [DOI] [PubMed] [Google Scholar]
- 5. de Bruin A., Wu L., Saavedra H. I., Wilson P., Yang W., Rosol T. J., Weinstein M., Rosinson M. L., Leone G. (2003) Rb function in extraembryonic lineages suppresses apoptosis in CNS of Rb-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 100, 6546–6551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takahashi C., Bronson R. T., Socolovsky M., Contreras B., Lee K. Y., Jacks T., Noda M., Kucherlapati R., Ewen M. E. (2003) Rb and N-ras function together to control differentiation in the mouse. Mol. Cell Biol. 23, 5256–5268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schneider J. W., Gu W., Zhu L., Mahdavi V., Nadal-Ginard B. (1994) Reversal of terminal differentiation mediated by p107 in Rb−/− muscle cells. Science 264, 1467–1471 [DOI] [PubMed] [Google Scholar]
- 8. Novitch B. G., Mulligan G. J., Jacks T., Lassar A. B. (1996) Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J. Cell Biol. 135, 441–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen T.-T., Wang J. Y. (2000) Establishment of irreversible growth arrest in myogenic differentiation requires the RB LXCXE-binding function. Mol. Cell Biol. 20, 5571–5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takahashi C., Contreras B., Bronson R. T., Loda M., Ewen M. E. (2004) Genetic interaction between Rb and K-ras in the control of differentiation and tumor suppression. Mol. Cell Biol. 24, 10406–10415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sellers W. R., Novitch B. G., Miyake S., Heith A., Otterson G. A., Kaye F. J., Lassar A. B., Kaelin W. G., Jr. (1998) Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor growth. Genes Dev. 12, 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsai K. Y., Hu Y., Macleod K. F., Crowley D., Yamasaki L., Jacks T. (1998) Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell 2, 293–304 [DOI] [PubMed] [Google Scholar]
- 13. Ziebold U., Reza T., Caron A., Lees J. A. (2001) E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 15, 386–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saavedra H. I., Wu L., de Bruin A., Timmers C., Rosol T. J., Weinstein M., Robinson M. L., Leone G. (2002) Specificity of E2F1, E2F2 and E2F3 in mediating phenotypes induced by loss of Rb. Cell Growth Differ. 13, 215–225 [PubMed] [Google Scholar]
- 15. Novitch B. G., Spicer D. B., Kim P. S., Cheung W. L., Lassar A. B. (1999) pRb is required for MEF2-dependent gene expression as well as cell-cycle arrest during skeletal muscle differentiation. Curr. Biol. 9, 449–459 [DOI] [PubMed] [Google Scholar]
- 16. Puri P. L., Avantaggiati M. L., Balsano C., Sang N., Graessmann A., Giordano A., Levrero M. (1997) p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J. 16, 369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de la Serna I. L., Roy K., Carlson K. A., Imbalzano A. N. (2001) MyoD can induce cell cycle arrest but not muscle differentiation in the presence of dominant negative SWI/SNF chromatin remodeling enzymes. J. Biol. Chem. 276, 41486–41491 [DOI] [PubMed] [Google Scholar]
- 18. Bergstrom D. A., Penn B. H., Strand A., Perry R. L., Rudnicki M. A., Tapscott S. J. (2002) Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol. Cell 9, 587–600 [DOI] [PubMed] [Google Scholar]
- 19. Tomczak K. K., Marinescu V. D., Ramoni M. F., Sanoudou D., Montanaro F., Han M., Kunkel L. M., Kohane I. S., Beggs A. H. (2004) Expression profiling and identification of novel genes involved in myogenic differentiation. FASEB J. 18, 403–405 [DOI] [PubMed] [Google Scholar]
- 20. Moran J. L., Li Y., Hill A. A., Mounts W. M., Miller C. P. (2002) Gene expression changes during mouse skeletal myoblast differentiation revealed by transcriptional profiling. Physiol. Genomics 10, 103–111 [DOI] [PubMed] [Google Scholar]
- 21. Shen X., Collier J. M., Hlaing M., Zhang L., Delshad E. H., Bristow J., Bernstein H. S. (2003) Genome-wide examination of myoblast cell cycle withdrawal during differentiation. Dev. Dyn. 226, 128–138 [DOI] [PubMed] [Google Scholar]
- 22. Blais A., Tsikitis M., Acosta-Alvear D., Sharan R., Kluger Y., Dynlacht B. D. (2005) An initial blueprint for myogenic differentiation. Genes Dev. 19, 553–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cao Y., Kumar R. M., Penn B. H., Berkes C. A., Kooperberg C., Boyer L. A., Young R. A., Tapscott S. J. (2006) Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J. 25, 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mal A., Harter M. L. (2003) MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 1735–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gu W., Schneider J. W., Condorelli G., Kaushal S., Mahdavi V., Nadal-Ginard B. (1993) Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell 72, 309–324 [DOI] [PubMed] [Google Scholar]
- 26. Trouche D., Grigoriev M., Lenormand J.-L., Robin P., Leibovitch S. A., Sassone-Corsi P., Harel-Bellan A. (1993) Repression of c-fos promoter by MyoD on muscle cell differentiation. Nature 363, 79–82 [DOI] [PubMed] [Google Scholar]
- 27. Halevy O., Novitch B. G., Spicer D. B., Skapek S. X., Rhee J., Hannon G. J., Beach D., Lassar A. B. (1995) Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 267, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 28. Franklin D. S., Xiong Y. (1996) Induction of p18INK4c and its predominant association with CDK4 and CDK6 during myogenic differentiation. Mol. Biol. Cell 7, 1587–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J. M., Zhao X., Wei Q., Paterson B. M. (1999) Direct inhibition of G1 cdk kinase activity by MyoD promotes myoblast cell cycle withdrawal and terminal differentiation. EMBO J. 18, 6983–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cenciarelli C., De Santa F., Puri P. L., Mattei E., Ricci L., Bucci F., Felsani A., Caruso M. (1999) Critical role played by cyclin D3 in the MyoD-mediated arrest of cell cycle during myoblast differentiation. Mol. Cell Biol. 19, 5203–5217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Latella L., Sacco A., Pajalunga D., Tiainen M., Macera D., D'Angelo M., Felici A., Sacchi A., Crescenzi M. (2001) Reconstitution of cyclin D1-associated kinase activity drives terminally differentiated cells into the cell cycle. Mol. Cell Biol. 21, 5631–5643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Skapek S. X., Rhee J., Spicer D. B., Lassar A. B. (1995) Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science 267, 1022–1024 [DOI] [PubMed] [Google Scholar]
- 33. Skapek S. X., Rhee J., Kim P. S., Novitch B. G., Lassar A. B. (1996) Cyclin-mediated inhibition of muscle gene expression via a mechanism that is independent of pRB hyperphosphorylation. Mol. Cell Biol. 16, 7043–7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rao S. S., Chu C., Kohtz D. S. (1994) Ectopic expression of cyclin D1 prevents activation of gene transcription by myogenic basic helix-loop-helix regulators. Mol. Cell Biol. 14, 5259–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lazaro J. B., Bailey P. J., Lassar A. B. (2002) Cyclin D-cdk4 activity modulates the subnuclear localization and interaction of MEF2 with SRC-family coactivators during skeletal muscle differentiation. Genes Dev. 16, 1792–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peeper D. S., Upton T. M., Ladha M. H., Neuman E., Zalvide J., Bernards R., DeCaprio J. A., Ewen M. E. (1997) Ras signalling linked to the cell-cycle machinery by the retinoblastoma protein. Nature 386, 177–181 [DOI] [PubMed] [Google Scholar]
- 37. Blau H. M., Pavlath G. K., Hardeman E. C., Chiu C. P., Silberstein L., Webster S. G., Miller S. C., Webster C. (1985) Plasticity of the differentiated state. Science 230, 758–766 [DOI] [PubMed] [Google Scholar]
- 38. Shapiro G. I., Park J. E., Edwards C. D., Mao L., Merlo A., Sidransky D., Ewen M. E., Rollins B. J. (1995) Multiple mechanisms of p16INK4A inactivation in non-small cell lung cancer cell lines. Cancer Res. 55, 6200–6209 [PubMed] [Google Scholar]
- 39. Lee K. Y., Ladha M. H., McMahon C., Ewen M. E. (1999) The retinoblastoma protein is linked to the activation of Ras. Mol. Cell Biol. 19, 7724–7732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He T.-C., Zhou S., da Costa L. T., Yu J., Kinzler K. W., Vogelstein B. (1998) A simplified system for generation recombinant adenovirus. Proc. Natl. Acad. Sci. U.S.A. 95, 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schreiber M., Poirier C., Franchi A., Kurzbauer R., Guenet J.-L., Carle G. F., Wagner E. F. (1997) Structure and chromosomal assigment of the mouse fra-1 gene, and its exclusion as a candidate gene for oc (osteosclerosis). Oncogene 15, 1171–1178 [DOI] [PubMed] [Google Scholar]
- 42. Lamb J., Ladha M. H., McMahon C., Sutherland R. L., Ewen M. E. (2000) Regulation of the functional interaction between cyclin D1 and the estrogen receptor. Mol. Cell Biol. 20, 8667–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Q., Carroll J. S., Brown M. (2005) Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 19, 631–642 [DOI] [PubMed] [Google Scholar]
- 44. Berkes C. A., Bergstrom D. A., Penn B. H., Seaver K. J., Knoepfler P. S., Tapscott S. J. (2004) Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol. Cell 14, 465–477 [DOI] [PubMed] [Google Scholar]
- 45. Brown J. R., Nigh E., Lee R. J., Ye H., Thompson M. A., Saudou F., Pestell R. G., Greenberg M. E. (1998) Fos family members induce cell cycle entry by activating cyclin D1. Mol. Cell Biol. 18, 5609–5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tiainen M., Pajalunga D., Ferrantelli F., Soddu S., Salvatori G., Sacchi A., Crescenzi M. (1996) Terminally differentiated skeletal myotubes are not confined to G0 but can enter G1 upon growth factor stimulation. Cell Growth Differ. 7, 1039–1050 [PubMed] [Google Scholar]
- 47. Murphy L. O., MacKeigan J. P., Blenis J. (2004) A network of immediate early gene products propogates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell Biol. 24, 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burch P. M., Yuan Z., Loonen A., Heintz N. H. (2004) An extracellular signal-regulated kinase 1- and 2-dependent program of chromatin trafficking of c-Fos and Fra-1 is required for cyclin D1 expression during cell cycle reentry. Mol. Cell Biol. 24, 4696–4709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Herrera R. E., Sah V. P., Williams B. O., Mäkelä T. P., Weinberg R. A., Jacks T. (1996) Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol. Cell Biol. 16, 2402–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cohen D. R., Ferreira P. C., Gentz R., Franza B. R., Jr., Curran T. (1989) The product of the fos-related gene, fra-1, binds cooperatively to the AP-1 site with Jun: transcription factor AP-1 is comprised of multiple protein complexes. Genes Dev. 3, 173–184 [DOI] [PubMed] [Google Scholar]
- 51. Wisdon R., Verma I. M. (1993) Transformation by Fos proteins requires a C-terminal transactivation domain. Mol. Cell Biol. 13, 7429–7438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bergers G., Graninger P., Braselmann S., Wrighton C., Busslinger M. (1995) Transcriptional activation of the fra-1 gene by AP-1 is mediated by regulatory sequences in the first intron. Mol. Cell Biol. 15, 3748–3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Casalino L., De Cesare D., Verde P. (2003) Accumulation of Fra-1 in ras-transformed cells depends on both transcriptional autoregulation and MEK-dependent postranslational stabilization. Mol. Cell Biol. 23, 4401–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhu J., Liu J. S., Lawrence C. E. (1998) Bayesian adaptive sequence alignment algorithms. Bioinformatics 14, 25–39 [DOI] [PubMed] [Google Scholar]
- 55. Frolov M. V., Dyson N. J. (2004) Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 117, 2173–2181 [DOI] [PubMed] [Google Scholar]
- 56. Bulger M., Groudine M. (2011) Functional and mechanistic diversity of distal transcription enhancers. Cell 144, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spizz G., Roman D., Strauss A., Olson E. N. (1986) Serum and fibroblast growth factor inhibit myogenic differentiation through a mechanism dependent on protein synthesis and independent of cell proliferation. J. Biol. Chem. 261, 9483–9488 [PubMed] [Google Scholar]
- 58. Weintraub H., Davis R., Lockshon D., Lassar A. (1990) MyoD binds cooperatively to two sites in a target enhancer sequence: occupancy of two sites is required for activation. Proc. Natl. Acad. Sci. U.S.A. 87, 5623–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Odelberg S. J., Kollhoff A., Keating M. T. (2000) Dedifferentiation of mammalian myotubes induced by msx1. Cell 103, 1099–1109 [DOI] [PubMed] [Google Scholar]
- 60. Huh M. S., Parker M. H., Scimè A., Parks R., Rudnicki M. A. (2004) Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J. Cell Biol. 166, 865–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Camarda G., Siepi F., Pajalunga D., Bernardini C., Rossi R., Montecucco A., Meccia E., Crescenzi M. (2004) A pRb-independent mechanism preserves the postmitotic state in terminally differentiated skeletal muscle cells. J. Cell Biol. 167, 417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Blais A., van Oevelen C. J., Margueron R., Acosta-Alvear D., Dynlacht B. D. (2007) Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J. Cell Biol. 179, 1399–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guo K., Wang J., Andrés V., Smith R. C., Walsh K. (1995) MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myoctye terminal differentiation. Mol. Cell Biol. 15, 3823–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang P., Wong C., Liu D., Finegold M., Harper J. W., Elledge S. J. (1999) p21CIP1 and p57KIP2 control muscle differentiation at the myogenin step. Genes Dev. 13, 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lassar A. B., Skapek S. X., Novitch B. (1994) Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr. Opin. Cell Biol. 6, 788–794 [DOI] [PubMed] [Google Scholar]
- 66. Ait-Si-Ali S., Guasconi V., Fritsch L., Yahi H., Sekhri R., Naguibneva I., Robin P., Cabon F., Polesskaya A., Harel-Bellan A. (2004) A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not cycling cells. EMBO J. 23, 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bernstein B. E., Mikkelsen T. S., Xie X., Kamal M., Huebert D. J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Jaenisch R., Wagschal A., Feil R., Schreiber S. L., Lander E. S. (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 [DOI] [PubMed] [Google Scholar]
- 68. Mikkelsen T. S., Ku M., Jaffe D. B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T. K., Koche R. P., Lee W., Mendenhall E., O'Donovan A., Presser A., Russ C., Xie X., Meissner A., Wernig M., Jaenisch R., Nusbaum C., Lander E. S., Bernstein B. E. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bengal E., Ransone L., Scharfmann R., Dwarki V. J., Tapscott S. J., Weintraub H., Verma I. M. (1992) Functional antagonism between c-Jun and MyoD proteins: a direct physical association. Cell 68, 507–519 [DOI] [PubMed] [Google Scholar]