

Background: HLA-DM-mediated peptide exchange is a key factor in epitope selection, but how HLA-DM selects peptides for editing is not known.
Results: Peptide complexes sensitive to HLA-DM editing exhibited conformational alterations.
Conclusion: HLA-DM efficiently identifies unstable complexes by sensing MHCII-peptide conformations.
Significance: These data emphasize HLA-DM as a conformational editor and provide novel mechanistic insight into its function.
Keywords: Antigen Presentation, Conformational Change, Enzyme Kinetics, Enzyme Mechanism, Major Histocompatibility Complex (MHC), Protein-Protein Interaction, DM Susceptibility
Abstract
HLA-DM mediates the exchange of peptides loaded onto MHCII molecules during antigen presentation by a mechanism that remains unclear and controversial. Here, we investigated the sequence and structural determinants of HLA-DM interaction. Peptides interacting nonoptimally in the P1 pocket exhibited low MHCII binding affinity and kinetic instability and were highly susceptible to HLA-DM-mediated peptide exchange. These changes were accompanied by conformational alterations detected by surface plasmon resonance, SDS resistance assay, antibody binding assay, gel filtration, dynamic light scattering, small angle x-ray scattering, and NMR spectroscopy. Surprisingly, all of those changes could be reversed by substitution of the P9 pocket anchor residue. Moreover, MHCII mutations outside the P1 pocket and the HLA-DM interaction site increased HLA-DM susceptibility. These results indicate that a dynamic MHCII conformational determinant rather than P1 pocket occupancy is the key factor determining susceptibility to HLA-DM-mediated peptide exchange and provide a molecular mechanism for HLA-DM to efficiently target unstable MHCII-peptide complexes for editing and exchange those for more stable ones.
Introduction
Antigen presentation to CD4+ T cells by MHCII molecules is a key step in control of adaptive immune responses. Proteolytic cleavage of exogenous and endogenous proteins results in peptides that are loaded onto MHCII molecules for presentation at the cell surface (1–3). Nascent MHCII are chaperoned to specialized endosomal compartments by the class II associated invariant chain, which is processed to leave a nested set of peptides, known as CLIP,2 remaining bound in the MHCII-peptide binding groove. The exchange of CLIP for antigenic peptides is catalyzed by HLA-DM (DM), a nonclassical MHC II molecule (4–6). The differential effect of DM on different peptides has been shown to play a key role in antigen presentation (7–9) and epitope selection (10–15), but the determinants of DM-mediated peptide exchange remain elusive and controversial (16–26).
Previous studies proposed that DM targets the conserved hydrogen bonds between MHCII and peptide in the vicinity of the P1 pocket (17, 18), although others found that these interactions are dispensable for DM action (20, 21, 23, 27). Peptide side chain pocket interactions also have been implicated in DM susceptibility, particularly emphasizing the importance of pocket 1 near the N terminus of the bound peptide (22, 28). In contrast, two studies of MHCII and peptide variants suggested that interactions along the entire peptide binding groove contribute to DM susceptibility (21, 29). Two recent crystal structures of DM complexes have again focused attention on the MHCII pocket 1 region. DM bound to DR1 carrying a covalently bound, N-terminally truncated peptide shows extensive rearrangement of the MHCII pocket 1 (24). DM bound to DO, a natural inhibitor that acts as a DM substrate mimic, shows similar rearrangements in the same region (30). Both structures were interpreted as representing a transient intermediate in the DM-MHCII-peptide exchange reaction, which would resolve upon peptide binding and displacement of DM. In the DM-DR1 structure report, a model was presented in which peptides that lack an optimum pocket 1 residue would not be able to displace DM from the DM-DR complex, tying DM susceptibility to the nature of the residue occupying pocket 1 (24).
Despite extensive efforts and progress, the determinants of DM susceptibility are still unclear, and the role of the peptide pocket 1 residue in DM-mediated peptide exchange has not been systematically addressed. In this study, we directly evaluated the role of the side chain occupying the pocket 1 in determining susceptibility to DM-mediated peptide exchange. We characterized the determinants of DM susceptibility using DR1 and sets of peptides derived from the immunodominant alloantigen HLA-A2(104–117) (31–34). We weakened the MHCII-peptide interaction by substitution of the key pocket 1 residue and then attempted to reconstitute tight binding and DM resistance by systematic substitution at other positions. Intriguingly, we found that substitution with an optimal residue (leucine) at position 9 can counteract the low binding affinity, low kinetic stability, and high DM susceptibility of peptides with nonoptimal alanine, threonine, or valine residues at position 1. Moreover, the DM binding activity, SDS sensitivity, recognition by a conformation-specific antibody, hydrodynamic properties, and radius of gyration of DR1 complexes carrying peptides with nonoptimal alanine at pocket 1 could be altered by change of the pocket 9 residue. NMR spectroscopy indicated that the pocket 9 substitution decreased conformational variability in the pocket 1 region. Similarly, MHCII substitutions away from pocket 1 and the DM interaction site altered DM binding and peptide exchange activity. Taken together, these results suggest that multiple substitutions along the entire peptide binding groove can contribute to DM susceptibility and that a dynamic MHCII-peptide conformation determined by interactions throughout the peptide-binding site is the important determinant of DM susceptibility.
EXPERIMENTAL PROCEDURES
Peptide Synthesis and Labeling
MHCI protein HLA-A2(104–117)-derived peptide GSDWRFLRGYHQYA and HA(306–318)-derived peptide PKYVKQNTLKLAT and analogs were synthesized (21st Century Biochemicals, Marlboro, MA) with biotin linked to the peptide N terminus via a tetrapolyethylene oxide linker for IC50 and dissociation kinetics assays and with acetylated N termini for SPR and crystallization studies. For competition binding studies, the N-terminally acetylated HA analog (Ac-PRFVKQNTLRLAT) and CLIP analog (Ac-VSKMRMATPLLMQ) were labeled with Alexa488 tetrafluorophenyl ester (Invitrogen) through the primary amine of K5 (HA) and K3 (CLIP).
Protein Expression and Purification
Soluble extracellular domains of recombinant HLA-DR1 (DRA*0101/DRB1*010101) and HLA-DM were expressed in Drosophila S2 cells and purified by immunoaffinity chromatography followed by Superdex200 (GE Healthcare) size exclusion chromatography as described (4, 35). For NMR analysis, single subunits were recombinantly expressed in Escherichia coli as inclusion bodies, folded in vitro, and purified as described previously (36, 37). For reduction of signal complexity for subsequent NMR analysis, only the α-subunit was labeled with 15N. Empty 15Nα-labeled DR1 molecules were loaded a posteriori with a 20-fold molar peptide excess for 48 h at 37 °C in the presence of 2 mm of the loading enhancer Ac-FR-NH2. For mutagenesis studies, wild-type and mutant DR1 subunits were expressed in E. coli and folded and purified as described above.
Fluorescence Polarization Assay
Fluorescence polarization assay was used to measure the IC50 value of each peptide, using Alexa488-HA(306–318) as probe peptide as described previously (13, 38).
Peptide Dissociation Assay and DM Susceptibility Calculation
Peptide dissociation kinetics was measured by europium time-resolved fluorescence as described previously (13). DM susceptibility, the specific rate enhancement defined as the slope of the off-rate versus DM concentration curve, was calculated as (koff, DM − koff, in)/[DM], where koff, in is the intrinsic off-rate; koff, DM is the DM-mediated off-rate; and [DM] is concentration of DM.
Peptide Exchange Assay
DR1 was loaded with Alexa488-labeled CLIP. 200 μl of this complex with a final concentration of 100 nm was incubated without a peptide or with 5 μm of each exchanging peptide in the absence or presence of 0.4 μm DM at 37 °C in pH 5.5 binding buffer. Fluorescence polarization was used to detect the dissociation of Alexa488-CLIP and association of each exchanging peptide. The sample was read 300 times with 4-min intervals with a Victor Multilabel plate reader.
DM Binding Studies
SPR experiments of DM binding to MHCII-peptide complexes were carried out on a BIAcore® 3000 instrument using CM5 chips (GE Healthcare) as described previously (23). Bio-layer interferometry experiments of DM binding to immobilized DO were carried out on a ForteBio Octet® instrument (Pall Life Sciences) as described previously (30).
Protein X-ray Crystallography
Crystals of the DR1-A1L9 complex obtained at 4 °C in 4% PEG 4000, 10% ethylene glycol, 100 mm sodium acetate (pH 5.0) were transferred to cryosolution containing 30% ethylene glycol and flash-cooled in liquid nitrogen. Diffraction data were collected from a single 100 × 100 × 300-μm crystal at the National Synchrotron Light Source X29 beamline. The structure was solved by molecular replacement with six copies per asymmetric unit using the known structure of DR1-A2(104–117) (31). Data processing, model building, and refinement were performed as described (30). Data processing and refinement statistics for the final model (PDB code 4OV5) are shown in Table 2.
TABLE 2.
Data collection and refinement statistics
| DR1-A1L9 (4OV5) | |
|---|---|
| Data collection | |
| Space group | P1 21 1 |
| Cell dimensions | |
| a, b, c (Å) | 95.96, 173.19, 96.48 |
| α, β, γ (°) | 90.00, 109.72, 90.00 |
| Resolution (Å) | 46.00-2.20 (2.24-2.20)a |
| Rmerge (%) | 4.8 (47.7) |
| I/σI | 16.09 (1.44) |
| Completeness (%) | 93.7 (81.8) |
| Redundancy | 2.5 (2.3) |
| Refinement | |
| Resolution (Å) | 46.00-2.20 (2.24-2.20) |
| No. of reflections | 140,577 |
| Rwork/Rfree | 20.52/23.95 |
| No. of atoms | 19,082 |
| Protein | 18,443 |
| Water | 639 |
| B-factors | 33.25 |
| Protein | 33.30 |
| Water | 31.77 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.773 |
| Ramachandran favored (%) | 98.08 |
| Allowed region | 1.88 |
| Outlier region | 0.04 |
a Values in parentheses are for highest resolution shell.
SDS Resistance Assay
SDS-PAGE experiments were performed for empty DR1 and DR1 loaded with each peptide. One-half of each sample (5 μg) was boiled for 3 min, and the other half was incubated at room temperature for 15 min in reduced Laemmli loading buffer. These samples were then applied to 12% polyacrylamide gels. After electrophoresis, the gel was stained with Coomassie Brilliant Blue R-250 (Bio-Rad) and destained with destaining buffer (10% acetic acid, 15% ethanol, and 75% water). The gel image was taken with VersaDoc 4000 MP (Bio-Rad).
Antibody Binding
A sandwich enzyme-linked immunosorbent assay (ELISA) was used to detect binding of LB3.1 and UL-5A1 antibodies to WT, A1, and A1L9 peptide-loaded DR1 as described previously (36). The 100% bound was normalized to the binding of DR1-WT at the highest 500-ng complexes. Half-maximal binding concentrations (EC50) were obtained by fitting the data to a four-parameter binding equation in GraphPad Prism 5.
Hydrodynamic Property Measurement
Gel filtration used a Superdex200 column (GE Healthcare), at a flow rate of 0.5 ml/min in PBS (pH 7.4), calibrated by γ-globulin (153 kDa), ovalbumin (44 kDa), and myoglobin (17 kDa). Five to 10 μg of each complex were injected, and the elution volume was derived from the chromatography traces. Dynamic light scattering experiments were read on Zetasizer Nano ZS (Malvern Instruments Ltd., Worcestershire, UK) at 25 °C. Samples were prepared in PBS at 0.5 mg/ml. 13–20 runs were taken for each sample, and the average diameter for each complex was obtained.
SAXS Analysis
SAXS data were collected at the SIBYLS beamline at the Lawrence Berkeley Laboratory (Berkeley, CA). Guinier analysis and P(r) (pair-distance distribution function) analyses were used to calculate the radius of gyration (Rg) for each complex, except for empty DR1, which had abnormal background subtraction and aggregation. To calculate Rg values from Guinier plot (lnI(q) versus q2), Equation 1 was used to get the slope of Guinier plot,
 |
where I(q) is the scattering intensity at scattering vector. To calculate Rg values from P(r) analysis, Equation 2 was used to get the P(r) function,
 |
where r is the allowable distance. Rg is obtained by integrating the P(r) function with r2 over all values of r. DR1-WT, DR1-A1L9, and DR1-A1 (at 2, 1 and 0.5 mg/ml) or peptide-free DR1 (at 0.92, 0.5, and 0.25 mg/ml) in PBS solution were shipped overnight at 4 °C in 96-well clear PCR microplates (AXYGEN Scientific, Union City, CA). A buffer well was included before and after each sample well.
Nuclear Magnetic Resonance (NMR) Spectroscopy and Analysis
NMR spectra were recorded on a Bruker AV 700 MHz magnet equipped with a 5-mm triple-resonance cryoprobe. Measurements of the individual protein complexes (200 μm) were performed at 310 K in PBS buffer (pH 5.8), containing 10% D2O. Spectra were processed with Topspin (Bruker) and analyzed with CCpNmr analysis (39). Chemical shift differences were calculated using the equation Δδ = (δH2 + (0.15δN)2)0.5 and were considered as significant if Δδ was larger than the sum of the average and standard deviations of all chemical shift differences. Signal-to-noise ratios were determined with Sparky (T.D. Goddard and D.G. Kneller, SPARKY 3, University of California, San Francisco) and were considered as significantly reduced if smaller than the mean value minus the standard deviation.
RESULTS
Identification of Peptides with Weakened Pocket 1 Interactions and Strengthened Interactions at Other Pockets
We focused on variants of the alloantigenic peptide HLA-A2(104–117) bound to the human class II MHC protein DR1 (HLA-DRA*01:01, DRB1*01:01). This MHC-peptide complex has been shown to be the target of graft rejection in solid organ transplant recipients (33, 34) and allospecific human monoclonal antibodies (40). HLA-A2(104–117) is the predominant endogenous peptide bound to DR1 isolated from a B lymphoid cell line (32), presumably reflecting its high binding affinity, resistance to DM-mediated exchange, and the abundance of the HLA-A2 protein in the endosomal subcellular compartment(s), where peptide binding and DM editing occur. The crystal structure of DR1-HLA-A2(104–117) has been solved previously defining the major and minor anchor residues (Fig. 1A) (31), which makes it an ideal example to study DM action.
FIGURE 1.

Identification of peptides with weakened pocket 1 interactions and strengthened interactions at other pockets. A, schematic view of peptide binding groove from previously solved crystal structure of peptide HLA-A2(104–117) bound to DR1 (PDB code 1AQD). Pockets 1, 4, 6, and 9 on DR1, which harbor the major anchor residues of HLA-A2(104–117), are indicated. B, competition binding with indicator peptide Alexa488-HA(306–318) to DR1 was measured for HLA-A2(104–117)-derived peptides harboring single mutation at pockets 1, 4, 6, and 9 anchor residues. The amino acid after substitution for each peptide was labeled at the x axis, with the open bar indicating wild-type sequence, and the star indicating peptides selected for following detailed studies. These data represent three independent experiments with two replicates each.
To address the role of pocket 1 in DM susceptibility, we designed variant peptides with nonoptimal pocket 1 side chains and combined these with optimal residues at other pockets. We reasoned that if we were able to find a pocket 4, pocket 6, or pocket 9 substitution that restored the wild-type (WT) affinity to a pocket 1-substituted variant, comparison of such peptides with the WT could illuminate the role of the pocket 1 residue in DM-mediated peptide exchange, because the WT peptide gains most of its binding affinity through the pocket 1 interactions, whereas the double-substituted peptides would derive most of their affinity elsewhere. We tested each A2 peptide variant using a fluorescence polarization inhibition assay with tight-binding indicator peptide HA(306–318) (KD ∼10 nm (41)). The WT (HLA-A2(104–117)) peptide bound to DR1 very tightly with IC50 values of 36 nm (Fig. 1B and Table 1), consistent with a previous report (32). As expected (42), single amino acid substitutions of the pocket 1 side chain generally reduced binding as indicated by increased IC50 values (Fig. 1B). We selected such four variants for continued work: L1, V1, T1, and A1. Substitution of pocket 4 and pocket 6 side chains with preferred residues (43) did not reduce IC50 values, and these positions were not investigated further. However, we found that leucine substitution at pocket 9 (L9) substantially enhanced the binding affinity as indicated by reduced IC50 values (Fig. 1B), consistent with previous reports that leucine is an optimal residue at this position (43, 44).
TABLE 1.
Binding and kinetic properties of HLA-DR1-peptide complexes formed with HLA-A2(104–117)-derived peptide mutants

a Peptides are named with the substituted anchor residues.
b The pockets 1, 4, 6, and 9 anchor residues in wild type peptides (WT) and corresponding substituted anchor residues are highlighted in bold and underlined. W1N-me represented peptide with N-methylated tryptophan in pocket 1.
c 50% inhibition concentration calculated from binding competition curves.
d Intrinsic dissociation half-life calculated from the dissociation curves.
e DM-mediated dissociation half-life measured in the presence of 0.5 μm DM calculated from the dissociation curves.
f DM susceptibility, which was calculated as the specific increase in dissociation rate in the presence of DM (koff, DM − koff, in)/[DM], where [DM] = 0.5 μm.
Leucine at Pocket 9 Counteracts the Low Binding Affinity, Low Kinetic Stability, and High DM Susceptibility of Peptides Having Nonoptimal Pocket 1 Anchor Residues or Broken Hydrogen Bonds
The L9 substitution that enhanced binding for the wild-type peptide also enhanced binding for the pocket 1 substituted variants L1, V1, T1, and A1. In each case, substitution of leucine at position 9 into a peptide carrying a substitution at position 1 (A1L9, T1L9, V1L9, and L1L9) reversed the effect of the pocket 1 substitution and returned the peptide binding affinity to nearly the wild-type level, as judged by a competition binding assay (Fig. 2, A and D, and Table 1). In the most extreme example, the A1 substitution showed more than 100-fold increase in IC50 relative to the wild-type peptide (Fig. 1B), as expected based on the major anchoring function of the pocket 1 residue (31), and consistent with a previously reported low affinity pocket 1 anchorless peptide derived from HA(306–318) (45). When the A1 peptide was reconstituted with leucine at pocket 9 (A1L9), it rescued the binding affinity to a similar level as WT (Fig. 2D and Table 1). Together, these data suggest that stronger interactions at the C-terminal end of the peptide (pocket 9) can compensate for the weaker interactions at the N-terminal end of peptide (pocket 1) in terms of binding to MHCII.
FIGURE 2.

Leucine at pocket 9 counteracts the low binding affinity, low kinetic stability, and high DM susceptibility of peptides having nonoptimal pocket 1 anchor residues or broken hydrogen bond bound to DR1. Binding affinity, intrinsic dissociation kinetics, DM-mediated dissociation kinetics, and DM susceptibility were shown for HLA-A2(104–117)-derived L1 series peptides (A), V1 series peptides (B), T1 series peptides (C), A1 series peptides (D), W1N-me series (E), and WLGQ (F). WT (wild-type HLA-A2(104–117)) was shown in each plot as a reference peptide. The sequence of each peptide is listed in Table 1. Binding affinity was measured by binding competition with the indicator peptide Alexa488-HA(306–318). Dissociation kinetics was measured in the absence or presence of 0.5 μm DM. DM susceptibility was calculated as (koff, DM − koff, in)/[DM]. These data represent at least two independent experiments with two replicates each.
We next looked at the effect of pocket 9 leucine reconstitution on MHCII-peptide kinetic stability and DM susceptibility. If the pocket 1 residue were very important in DM-mediated exchange, we would expect that L1L9, V1L9, T1L9, and A1L9 would exhibit similar DM susceptibilities as L1, V1, T1, and A1, respectively. We measured peptide dissociation kinetics in the absence or presence of DM (Fig. 2, A–D). DM accelerated the dissociation of peptides differentially. Following Weber et al. (16), we characterized the DM susceptibility of each peptide as the slope of the linear portion of the off-rate versus DM concentration curve ((koff, DM − koff, in)/[DM]). Another parameter has also been used to characterize the effect of DM (koff, DM/koff, in) (29). We prefer the DM susceptibility defined here because it emphasizes the specific rate enhancement of DM, and it can be interpreted in terms of a first-order rate constant. However, for the data shown below, either parameterization of DM action leads to the same conclusions. The DR1-WT complex (Fig. 2, A–D, green curves) showed high kinetic stability in the absence or presence of DM and low DM susceptibility. The L1, V1, T1, and A1 peptide complexes (red curves) all showed decreased intrinsic half-life (t½, in) and DM-mediated half-life (t½, DM), and an increase in DM susceptibility (Fig. 2, A–D), with the extreme A1 substitution decreasing the half-life over 200-fold and increasing the DM susceptibility by ∼400-fold (Fig. 2D and Table 1). Surprisingly, L1L9, V1L9, T1L9, and A1L9 (blue curves) all demonstrated intrinsic and DM-dependent half-lives, and DM susceptibility, similar to WT (Fig. 2, A–D). This indicates that a gain of interactions at the C-terminal end can compensate for loss of interactions at the N-terminal end of the peptide.
We also tested whether pocket 9 leucine reconstitution influences the DM susceptibility of peptides lacking the conserved hydrogen bonds between MHCII and backbone of peptides. Previously, the hydrogen bond between αSer-53 of DR1 and amide of pocket 1 residue has been shown to be important for DM-mediated kinetic stability (17). By breaking this particular hydrogen bond with N-methylated pocket 1 residue TrpN-me, we observed decreased binding affinity and half-lives and increased DM susceptibility, which were all counteracted by the reconstitution of leucine at pocket 9 (Fig. 2E). We observed decreased kinetic stability and increased DM susceptibility also for a peptide that retained the major anchor residues at positions 1, 4, 6, and 9 but had alanine substitutions elsewhere (Fig. 2F).
Collectively, these results show that multiple interactions along the entire peptide-binding site, not only the interactions near the N-terminal end of peptides, are responsible for kinetic stability and DM susceptibility.
One possible explanation for the unexpected role of L9 interaction could involve a potential shift to a different binding register with the A1L9 alanine not located in pocket 1. Peptide binding in a noncanonical reversed orientation has been observed, with DM able to accelerate the exchange of CLIP(106–120) either canonically or inversely bound to DR1 (37, 46). To confirm that A1L9 binds to DR1 with the same binding register as the WT peptide, we determined the x-ray crystal structure of DR1-AlL9 to a resolution of 2.2 Å (Fig. 3 and Table 2; PDB ID 4OV5). The structure shows A1L9 bound to DR1 in the canonical orientation with the expected pocket 1 and pocket 9 residues (Fig. 3, A and B), with no substantial differences in peptide conformation or MHCII side chain orientation relative to WT (Fig. 3C), including residues implicated in DM action (αTrp-43, αPhe-54, βAsn-82, and βPhe-89) (Fig. 3D).
FIGURE 3.

X-ray crystal structure of DR1-A1L9 confirms the expected peptide -binding motif and reveals similar conformation to that of DR1-WT. Electron density for peptide and DR1 residues around pocket 1 (A) and pocket 9 (B) with the peptide anchor residue and key contact residues on DR1 indicated in x-ray crystal structure of DR1-A1L9 complex (PDB code 4OV5). C, top view of DR1-A1L9 structure overlapped with DR1-WT structure. Four molecules in the asymmetric unit of DR1-WT and six molecules in the asymmetric unit of DR1-A1L9 are shown. DR1 in DR1-A1L9 is colored blue, A1L9 yellow, and DR1-WT green. D, a closer view at the pocket 1 with key residues is indicated. The pocket 1 alanine (A1) in DR1-A1L9 is highlighted as a yellow bulky residue, and the tryptophan in DR1-WT is highlighted as a transparent shaded residue. Statistics of x-ray data collection and refinement are listed in Table 2.
Peptides without an Optimal Hydrophobic Pocket 1 Anchor Residue Could Compete for the Binding to DR1 and Replace Other Peptides
One important prediction of a pocket 1-focused model for DM function is that peptides without a hydrophobic pocket 1 anchor residue would not be able to induce DR1 dissociation from DM and therefore could not compete effectively for the binding to DR1 in the peptide exchange reaction (24). We tested this idea directly by determining the ability of A1, A1L9, and WT peptides to displace CLIP that had been pre-associated with DR1. Surprisingly, we found that both A1 and A1L9 without a hydrophobic anchor residue at pocket 1 could displace labeled CLIP in the exchange reaction (Fig. 4). The equilibrium levels reached in the presence of A1, A1L9, and WT were different, as expected from their relative affinities, but each was able to efficiently displace CLIP. Equilibrium levels for each peptide were similar in the absence or presence of DM (Fig. 4). A1L9 behaved similarly to WT in terms of competition and equilibrium, consistent with their similar binding affinities and kinetic stabilities (Fig. 4 and Table 1). Therefore, a hydrophobic anchor residue at pocket 1 is not requisite for displacing bound peptide. Instead, the overall interactions determine the outcome of DM-catalyzed peptide exchange.
FIGURE 4.
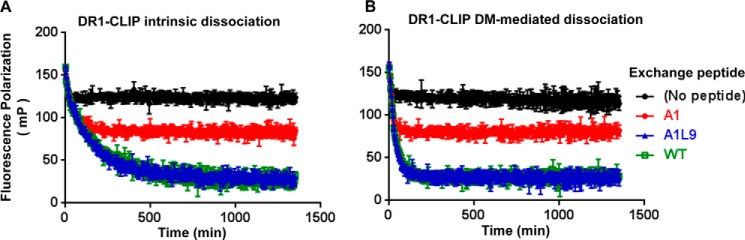
Peptides without an optimal hydrophobic pocket 1 anchor residue compete for the binding to DR1 and replace other peptides. DR1 loaded with Alexa488-labeled CLIP (DR1-CLIP, 0.1 μm) was incubated without a peptide or with 5 μm of exchanging A1, A1L9, and WT, respectively, in the absence of DM (A) and in the presence of 0.4 μm DM (B). These data represent at least two independent experiments with three or four replicates each.
DR1-A1 Shows Dose-dependent Binding to DM, although No Detectable Binding to DM Is Observed with Either DR1-WT or DR1-A1L9
We next sought to delineate features of the DR1-peptide complex that are required for DM association. We focused on the pocket 1 nonanchoring A1 peptide and its pocket 9 leucine reconstitution A1L9, because they showed the most dramatic changes in binding affinity, kinetic stability, and DM susceptibility. We first measured the direct binding of those complexes to DM by SPR. In previous studies, no detectable DM-DR binding was observed without manipulation of DR1 or bound peptide (22, 23), but direct binding to DM has been observed for DR1-CLIP with mutations in the 310 helix and extended strand region (23) and for DR1 bound with an N-terminally truncated HA(306–318) variant (22). Consistent with previous reports, we did not observe DM binding to DR1-WT at concentrations up to 16 μm (Fig. 5A). However, DR1-A1 showed clear dose-dependent binding to DM (Fig. 5B). Interestingly, DR1-AlL9 like DR1-WT had no detectable binding to DM (Fig. 5C). We verified that DM did not bind to empty DR1 (Fig. 5D). Collectively, these results suggest that DR1-A1 can adopt a conformation that is more accessible to DM, whereas leucine at pocket 9 could restore the conformation to that of DR1-WT, making it less susceptible to DM.
FIGURE 5.

DR1-A1 shows dose-dependent binding to DM, although no detectable binding to DM is observed with either DR1-WT or DR1-A1L9. Direct binding to DM was measured for DR1-WT (A), DR1-A1 (B), DR1-A1L9 (C), and empty DR1 (D) by SPR assay after blank flow cell subtraction. Each complex starting at 16 μm with a diluting factor of 2 flowed over the DM-coupled surface at 5 μl/min for 5 min and dissociated for 5 min. These results were repeated at least four times. RU, resonance unit.
DR1-A1 Shows SDS-sensitive Conformation and Decreased Recognition by a Conformation-specific Monoclonal Antibody UL-5A1, although DR1-A1L9 and DR1-WT Are Similar in Those Properties
A previous study (45) used the resistance of the DR1-peptide complex to SDS-induced denaturation and chain dissociation as an assay to identify SDS-sensitive “flexible” and SDS-resistant “compact” conformations of DR1-peptide complexes, and they found the hydrophobic residues in pocket 1 to be the determining factor of SDS resistance. A related study found that DM recognizes the flexible conformation of MHC II-peptide complexes (28). More recently, using a similar SDS-PAGE assay, another group identified a DM-labile conformer of the MHC II-peptide complex (47). We wanted to see whether this assay could distinguish the DM-sensitive DR1-A1 from the DM-resistant DR1-A1L9 and DR1-WT complex. Peptide-free DR1 and DR loaded with different A2(104–117) variant peptides were tested for their SDS resistance. Consistent with previous studies, empty DR1 and DR1 loaded with nonoptimal pocket 1 residues (A1 and T1) were sensitive to SDS, and DR1 loaded with WT or peptides with hydrophobic pocket 1 residues (V1, L1, and L9) were SDS-resistant (Fig. 6A). Interestingly, A1L9 and T1L9 showed SDS-resistant bands, indicating that leucine in pocket 9 could rescue the SDS-resistant conformation, and that hydrophobic residues in pocket 1 are not the sole determining factor of SDS resistance.
FIGURE 6.

DR1-A1 shows SDS-sensitive conformation and decreased recognition by a conformation-specific monoclonal antibody UL-5A1. A, SDS stability of empty DR1 or DR1 loaded with different peptides was determined. Each sample was split into two with one boiled (+) and the other one not (−). A protein marker was included in the 1st lane. The bands for α-subunit, β-subunit, and αβ complex were indicated. The multiple bands for α-subunit and β-subunit were due to glycosylation of DR1 produced in insect cells, as described previously (75). This gel is representative of three independent experiments. Detection of empty DR1, DR1 bound with HA(306–318), WT, A1, and A1L9 by UL-5A1 (B), LB3.1 (C), and streptavidin (D) was shown after BSA background subtraction. Each complex started at 500 ng with a diluting factor of 5. Each peptide was labeled with biotin. Percent bound (% Bound) was normalized to the absorbance of 500 ng of DR1-WT. Data are representative of at least two independent experiments with two replicates each.
To confirm that DR1-A1 adopted a different conformation compared with DR1-WT and DR1-A1L9, we tested the recognition of these complexes by UL-5A1, a human conformation-sensitive monoclonal antibody that recognizes the epitope-specific complex formed by HLA-A2(104–117) with DR1 (40). As a control, we used antibody LB3.1 that recognizes non-epitope-specific DR1-peptide complexes whose binding site is outside the peptide binding groove (48–50). Binding to streptavidin was also included as a control to verify peptide occupancy during the assay (in this experiment all of the peptides were biotin-labeled). We first mapped the binding site of UL-5A1 on the bound peptide. Substitutions of position 2 and 3 residues with alanine totally abrogated the recognition by UL-5A1, suggesting that residues at positions 2 and 3 were the major contact residues by UL-5A1 (data not shown), consistent with a T cell receptor-like recognition of this antibody. We then compared the recognition of DR1 loaded with WT, A1, and A1L9, reasoning that a difference would suggest a different conformation, because the direct contact residues are at positions 2 and 3 and not at pocket 1 or 9. Empty DR1 and DR1 loaded with HA(306–318) were not recognized by UL-5A1, confirming its specificity (Fig. 6B). Intriguingly, DR1-A1 showed a significant decrease of recognition by UL-5A1, with almost no detection of the 20-ng complex, whereas UL-5A1 detected the DR1-WT complex even at the lowest amount of 0.8 ng (Fig. 6B). The DR1-AlL9 complex demonstrated similar but slightly lower recognition by UL-5A1 compared with that of DR1-WT complex (Fig. 6B). As expected, DR1-WT, DR1-A1, and DR1-A1L9 complexes were recognized similarly by LB3.1 and streptavidin in a dose-dependent manner (Fig. 6, C and D). We confirmed the rescued recognition of A1L9 was not due to the direct recognition of leucine at pocket 9 itself, because DR1 loaded with L9 was recognized similarly as WT (data not shown). Instead, we interpret the L9 rescue of UL-5A1 binding to the A1 peptide to indicate that the L9 substitution has restored the conformation to that of DR1-WT.
DR1-A1 Shows Altered Hydrodynamic Behavior and Increased Radius of Gyration
To further investigate the conformational differences between DR1-WT, DR1-A1L9, and DR1-A1, we performed gel filtration and dynamic light scattering analyses, as described previously (51). Surprisingly, we observed a slight but consistent difference in the elution volume of DR1-A1 compared with DR1-WT, indicative of a slightly larger apparent size (Fig. 7A). DR1-A1L9 eluted similarly as DR1-WT (Fig. 7A). Because of the low affinity and short half-life of A1, one caveat is that peptide A1 may dissociate from DR1 during this assay. We have verified that A1 remains bound to DR1 after gel filtration by native gel analysis (data not shown), suggesting that peptide dissociation during gel filtration is not the cause of the difference in elution volume observed for DR1-A1. To confirm that the difference in gel filtration was due to actual difference in molecular sizes and not to differences in interactions with the gel filtration matrix, we used dynamic light scattering (DLS) to provide a direct measure of the effective hydrodynamic radius for the hydrated protein particles. DLS data indicated an effective hydrated diameter for DR1-A1 significantly larger than for DR1-WT (20% larger, Fig. 7B). DR1-A1L9 showed a similar diameter to DR1-WT (5% larger, Fig. 7B). Therefore, the hydrodynamic parameters obtained via two different methods indicate that DR1-A1 has a larger, less compact structure than DR1-WT, with an effective hydrodynamic diameter ∼20% greater.
FIGURE 7.

DR1-A1 shows altered hydrodynamic behavior and increased radius of gyration. A, gel filtration of DR1-WT, DR1-A1L9, and DR1-A1. The elution position and molecular mass of protein standards are shown above traces, with void volume (Vo) and total included volume (VT) indicated. These traces are representative of at least 20 independent experiments. B, dynamic light scattering measurement for DR1-WT, DR1-A1L9, and DR1-A1. The hydrodynamic radius (Rh) of each complex is indicated. Each sample was run for 13–20 times. C, SAXS profiles of DR1-WT, DR1-A1L9, and DR1-A1. The y axis is log of the scattering intensity, and the x axis is the scattering vector (q). The reciprocal of q can be interpreted as the resolution with which the sample is observed. D, Guinier plot of the SAXS profiles, with Rg indicated for each complex. E, P(r) (pair-distance distribution function) of the SAXS profiles, with Rg indicated. F, graphic presentation of the native DR1-A1L9 structure (blue), same structure with conformation of peptide adjusted at the P4 residue so that P-3-P3 are out of the binding site (red), and the same structure was adjusted at the P6 residue so that P-3-P5 are out of the binding site (green). G, P(r) function of the SAXS profiles theoretically calculated from the coordinates, with Rg indicated. H, Rg of each of 1800 conformations from molecular dynamic simulation trajectory is plotted. The molecular dynamic simulation was done previously for DR1 bound with a HIV-1 gag peptide (PDB code 1SJE). The Rg for the start model (trajectory 1, gray), model with largest Rg (trajectory index 15, red), and model with smallest Rg (trajectory index 1788, green) are indicated by arrows in the plot. I, overlapping view of the start model (gray), model with largest Rg (red), and model with smallest Rg (green).
We performed small angle x-ray scattering analysis to determine the radius of gyration (Rg) of each complex and to identify potential conformational alternation. SAXS has been widely used to give information on the size, shape, and orientation of biological macromolecules in solution (52–54). The SAXS profiles of the three complexes were almost identical; however, DR1-A1 differed from DR1-WT and DR1-A1L9 at high resolution (q < 0.2 Å−1) (Fig. 7C). We determined the Rg of each complex by Guinier plot analysis, which uses the linear points from the starting low-q region of the SAXS data (55). Consistent with gel filtration and DLS data, DR1-A1 had a larger size, with a Rg larger than that of DR1-WT or DR1-A1L9 by ∼1 Å (Fig. 7D). Alternatively, the Rg can be obtained from the pair-distance distribution function by calculating the mass distribution of the complex around its center of gravity, which uses all of the experimental data (56). Very similar Rg values were obtained, again with DR1-A1 having the largest radius of gyration (Fig. 7E).
We attempted to identify conformational changes that could contribute to the increased Rg. To examine the contribution from partial peptide release, we generated two coordinate sets based on the DR1-A1L9 x-ray crystal structure with the peptide flipped out of the site from the N terminus to pocket 4 or to pocket 6 (Fig. 7F). Theoretical SAXS data were calculated (57), and Rg was determined using the pair-distance distribution function. As expected, Rg increased with flipping the peptide from the binding groove (Fig. 7G). However, the difference in Rg even with peptide flipping until pocket 6 (0.4 Å) was not large enough for the observed ∼1 Å difference. To examine the contribution of MHCII conformational change to the Rg differences, we used a previously reported molecular dynamic simulation to identify dynamic conformational variations of MHCII-peptide complexes (50), calculating the Rg for >1800 representative conformational states (Fig. 7H). Rg differences of up to 1.1 Å, sufficient to explain the observed SAXS profiles, were observed (Fig. 7H), with conformational changes located in the 310 helix, extended strand region, peptide binding groove, and immunoglobulin domains (Fig. 7I). Overall, the native gel, gel filtration, DLS, and SAXS data demonstrate that DR1-A1 shows increased radius and altered conformation.
NMR Spectroscopy Reveals Conformational Differences between DR1-WT, DR1-A1L9, and DR1-A1
We used NMR spectroscopy to gain insight into possible conformational differences between DR1-WT, DR1-A1, and DR1-A1L9 that might be associated with DM susceptibility. The DR1 α-subunit was labeled with 15N and used to form DR1-peptide complexes with the WT, A1, and A1L9 peptides. 1H–15N heteronuclear single quantum coherence spectra were recorded for all complexes, and resonances were assigned based on previously published data (Fig. 8, A–C) (37, 46). Significant chemical shift differences between DR1-WT and DR1-A1 were observed for residues in the pocket 1 and surrounding areas, including the α 310 helix (Fig. 8D), consistent with the large environmental changes expected for removal of the buried tryptophan side chain. Chemical shift comparison of DR1-WT and DR1-A1L9 showed significant perturbations for residues in the vicinity of pockets 1 and 9 (Fig. 8E), again consistent with an altered environment due to the peptide substitutions. Substantial chemical shift differences between DR1-A1L9 and DR1-A1 were observed only for residues in close vicinity of pocket 9 (Fig. 8F). Thus the differential behavior of DR1-A1 and DR1-A1L9 complexes is not reflected by chemical shift differences, which also argues against peptide register shift or inversion of peptide orientation (37). Nevertheless, it is possible that DR1-A1L9 and DR1-A1 differ in conformational flexibility, which might affect signal intensities rather than chemical shifts. Therefore, we compared the signal intensity for all nonoverlapping peaks observed in both DR1-A1L9 and DR1-A1 spectra. In this analysis, DR1-A1 shows substantial loss in signal intensity near pocket 9 (αThr-74, αThr-75, αTyr-79 and αLys-80) but also at remote sites, including the peptide binding groove and the pocket 1 region (αGlu-11, αAsp-25, αAsp-29, αIle-31, αLeu-45, αGlu-55, αGln-57, αGly-58, αAsn-62, αCys-107, and αArg-146) (Fig. 8G). Of these, αLeu-45, αGlu-55, αGln-57, and αGly-58 are near sites of DM interaction observed in the crystal structure (24). Thus, despite the same pocket 1 alanine occupancy, DR1-A1 displays significantly more conformational flexibility compared with DR1-A1L9 that could be associated with its increased DM susceptibility and decreased kinetic stability.
FIGURE 8.

NMR spectroscopy reveals conformational differences between DR1-WT, DR1-A1L9, and DR1-A1. NMR spectra of DR1-WT (green) (A), DR1-A1 (red) (B), and DR1-A1L9 (blue) (C) with assigned residues indicated. Significant NMR chemical shift differences of DR1-WT versus DR1-A1 highlighted in green (Ala-10, Glu-11, Asp-25, Phe-26, Asp-27, Gly-28, Ile-31, Trp-43, Leu-45, Gly-49, and Lys-147) (D), DR1-WT versus DR1-A1L9 highlighted in red (Ala-10, Glu-11, Asp-25, Gly-28, Asp-29, Ile-31, Trp-43, Gly-49, Thr-74, Lys-75, Ser-77) (E), and DR1-A1 versus DR1-A1L9 highlighted in blue (Thr-74, Lys-75, Ser-77, Tyr-79, and Thr- 80) (F) are mapped onto the structure of DR1-A1L9. The 15N-labeled α-subunit is shown in light gray and the β-subunit in black in the schematic. G, significant signal-to-noise differences between DR1-A1 versus DR1-A19L are represented as blue spheres.
DR1 Substitutions Outside the DM-binding Site and Pocket 1 Alter DM Susceptibility
Based on the ability of peptide alterations in pocket 9 to reverse DM susceptibility induced by pocket 1 substitution, together with the conformational differences observed between DR1-A1 and DR1-A1L9 complexes, we suspected that DR1 peptide conformation rather than pocket 1 occupancy might be driving interaction with DM. To look at the effect of DR1 conformation independent of peptide sequence, we introduced mutations in the DR1 α-subunit outside the peptide binding region (Fig. 9A). In each case we substituted DR1 residues for those found in DO, a structural homolog that acts as a tight-binding competitive inhibitor (30). We constructed three mutants: H33A,A37K in the strand s3-s4 loop, a region that changes conformation in the DM complex (24, 30); P16Y,Q18K located at similar position but in the strand s1-s2 loop as a control; and T41A in the s4 strand where it contacts the DR1 extended strand region α51–57 implicated in DM modulation of DR1-peptide hydrogen-bonding interaction (17, 23). Both H33A,A37K and T41A mutations had dramatic effects in increasing DM binding, whereas the P16Y,Q18K mutation had no effect (Fig. 9, B–E). As with the A2 peptide mutants, the increased DM binding activity of H33A,A37K and T41A mutants was associated with increased DM susceptibility (Fig. 9F). Thus, DM association can be regulated by DR1 conformational alteration, irrespective of pocket 1 occupancy.
FIGURE 9.

DR1 substitutions away from the DM-binding site and pocket 1 alter DM susceptibility. A, mutated positions are indicated on ribbon diagram of DR1 bound to peptide A2(104–117) (gray, PDB code 1AQD) (31) or DR1 bound to DM (green, PDB code 4FQX) (24). Positions of P1 and P9 pockets in DR1-peptide complex and sites of DM interaction in DR1-DM complex are indicated. B–E, DM binding of CLIP-peptide complexes of wild-type DR1 (B), H33S,A37K (C), P16Y,Q18K (D), and T41A (E) as measured by SPR. F, DM susceptibility of CLIP bound to wild-type and mutant DR1 with wild-type and P95A DM. G, region around DR1 αThr-41, αTrp-43, and extended strand region shown for structure of DR1-A1L9. H, same region shown for structure of DR1 bound to DM (24). I, dissociation of CLIP peptide complexes of WT and T41A DR1 in the presence of various concentrations of WT and P95A DM. J, binding of WT and P95A DM to immobilized DO measured by bio-layer interferometry. K, dissociation kinetics were measured wild-type DR1 and T41A mutant bound to WT, A1, and A1L9 peptides, with DM susceptibility summarized in L.
We examined the T41A mutation in more detail. Residue αThr-41 interacts with the extended strand region α51–57, which participates in key MHCII-peptide hydrogen-bonding interactions in the vicinity of the pocket 1 (23), and also packs against αTrp-43, a residue crucial for efficient DM interaction (Fig. 9G) (22). In the DR1-DM complex observed crystallographically (24), DR1 αTrp-43 swings around to interact with DM, and DR1 αTrp-41 packs against DM αPro-95, reorienting away from the extended strand region (Fig. 9H). Mutation of DM αPro-95 to alanine blocked DM-facilitated peptide exchange (Fig. 9I, red). The DM αP95A mutation retains the high DO binding affinity of wild-type DM, ruling out substantial conformational or stability alteration induced by the mutation (Fig. 9J). The effect of the αP95A mutation on DM-mediated peptide release suggests a role for this residue in promoting conformational rearrangements associated with peptide release. In the context of the DR1 T41A mutation, where DM interaction was facilitated, DM P95A was effective in promoting peptide exchange, with DM susceptibility similar to that for wild-type DM interacting with DR1 (Fig. 9I, green). Thus, the effect of the DR1 T41A substitution is compensated by DM P95A, suggesting that these residues interact in the DM-mediated peptide exchange mechanism. The enhanced DM susceptibility of T41A can be observed for both A1 and A1L9 peptides, indicating that its effect is not dependent on pocket 1 occupancy (Fig. 9, K and L). These results provide further support for a mechanism in which MHCII-peptide conformational changes and not pocket 1 occupancy are key determinants of DM function.
DISCUSSION
In this study, we have evaluated the kinetic stability and DM susceptibility of DR1 complexes formed with HLA-A2(104–117) variant peptides with weakened interactions at pocket 1 and strengthened interactions at other positions. As expected from previous work, weakening interactions in the pocket 1 results in a decreased MHCII-peptide lifetime and increased susceptibility to DM. Unexpectedly, we found these effects could be completely compensated by substitutions elsewhere in the peptide. Judged by several criteria, the DR1-A1 complex with weakened pocket 1 interaction appears to adopt a conformation different from conventional DR1-peptide complexes, which is more susceptible to DM-mediated peptide exchange. Reconstitution with leucine at pocket 9 restores the conformation to the less susceptible form similar to that of DR1-WT.
Despite extensive investigation, peptide sequence determinants of DM susceptibility still have not been defined. Because the immunogenicity of epitopes after infection and vaccination is strongly linked to their relative DM susceptibility (8, 13, 58, 59), understanding these determinants is crucial to follow immune responses, improve vaccines, and understand the etiology of autoimmune disease. Previous models for predicting DM susceptibility have variously implicated particular conserved hydrogen bonds near pocket 1 (17, 18, 20, 21), spontaneous dissociation of the peptide N terminus (22, 60), conformational lability of the 310 helical region adjacent to pocket 1 (23), an SDS-sensitive flexible conformation determined by pocket 1 occupancy (28, 45), and an “compare-exchange-push-off” mechanism (25). Although these different approaches have generally implicated interactions around the pocket 1 region (61), some studies are consistent with more distributed effects (16, 25).
The idea that the pocket 1 region is crucial to DM susceptibility has been given additional prominence by a recent structure of DM bound to DR1 with a covalently trapped truncated peptide (24), in which DR1 conformational alterations around pocket 1 were observed and associated with DM interaction. Based on that structure, a deterministic role of pocket 1 interactions in DM susceptibility was proposed (24). This model for the peptide exchange reaction makes clear predictions that exchange peptides require hydrophobic pocket 1 residues and that the exchange activity will be dominated by pocket 1 occupancy. However, we have shown here that the A1 and A1L9 peptides, both with the same nonanchoring pocket 1 alanine, have dramatically different DM susceptibilities. Both of these peptides were able to efficiently displace bound peptides, in the absence or presence of DM, with the equilibrium level reached according their binding affinities and not their pocket 1 residues. Thus, DM susceptibility is not determined by interactions only in the pocket 1 region of the binding sites but rather by interactions throughout the peptide binding groove.
Conformational differences between DR1-A1 and DR1-A1L9 were revealed by SDS sensitivity, antibody binding, hydrodynamic measurements, SAXS, and NMR analyses. We suggest that the conformation of the MHCII-peptide complex, constrained by the interactions throughout the peptide-binding site, is the major determinant of DM susceptibility. Crystal structures of MHCII-peptide complexes show that they exhibit some degree of structural disorder, with flexibility observed in the α-subunit 310 helix and extended strand region, in the vicinity of a kink in the β-subunit helical region near residue β66, and in the orientation and loop of β2 Ig domain (62). Conformational changes observed here for the DR1-A1 complex could be due to the increased flexibility of 310 helix and extended region, as indicated by the NMR data. This is consistent with the effects of the T41A mutant, which appears to facilitate DM action by disrupting the stabilizing hydrogen bond between Thr-41 and the extended strand region.
Several studies indicating an important role for the conformation of MHCII-peptide complexes in determining DM susceptibility have appeared (23, 28, 47, 63–65). The largely decreased kinetic stability and increased DM susceptibility of A1, T1, and V1 bound to DR1 have confirmed the importance of interactions near pocket 1 (Fig. 2). However, we have demonstrated that other interactions, i.e. a single leucine reconstitution at pocket 9 far from the N terminus, could also influence the DM susceptibility by restoring the conformation of the MHCII bound to the original peptide with tryptophan in pocket 1. Notably, mutations in pocket 4 or pocket 6 of HLA-DR3 increase affinity for CLIP, make the mutant DR3-CLIP complexes resistant to DM and SDS-stable, and reduce their binding to DM (66). An HLA-DQ variant associated with autoimmunity (DQA1*0501) that is a poor substrate for DM (67) has residue α53 deleted with consequential structural alterations in the extended strand and 310 helical region (62, 68); DM susceptibility is restored by introduction of αGly-53 into the extended strand (69). All these data suggest that interactions along the entire peptide binding groove influence DM susceptibility by regulating the ability to adopt conformation(s) in which the 310 helix and extended strand regions can move into position for optimal interaction with DM.
The ability of an MHCII-peptide complex to interact productively with DM seems to involve a dynamic rather than a static conformation. The conformational differences between DR1-A1 and DR1-WT (or DR1-A1 and DR1-A1L9) observed by size exclusion chromatography, dynamic light scattering, and small angle x-ray scattering analyses were accompanied by changes in the intensity of heteronuclear single quantum coherence spectra without significant NMR chemical shift changes, suggesting an increase in conformational heterogeneity in the affected regions. Interestingly, the different P9 pocket occupations seem to translate into modulating the mobility of a network of residues that include regions remote from the P9 pocket and are prone to conformational editing by DM. As noted above, increased DM binding activity seems likely to be due to increased flexibility of residues in the DR1 α-subunit 310 helix and extended strand, which undergo substantial conformational alteration during DM complex formation. Previous work in other systems has emphasized the importance of dynamic conformational ensembles in biomolecular recognition (70). Moreover, MHCII-peptide interaction involves substantial conformational changes in both MHC and peptide components and has been characterized as a cooperative folding process (71). In general, such processes are characterized by funnel-shaped energy landscapes, with a large number of less stable conformers and fewer low energy forms. Viewed in this light, DM appears to be recognizing and editing out rare MHCII-peptide conformations that retain some mobility characteristic of intermediates in the overall multistep MHCII-peptide binding process (72).
These data provide the clearest evidence yet that DM edits conformations, not sequences, and helps to define a molecular basis for conformational editing. DM is known to promote exchange of peptides from unstable complexes, but how DM could detect those complexes well before peptide actually dissociates has not been clear. Peptide release from MHCII proteins requires substantial conformation change (51) as a result of the intricate arrangement of side-chain binding pockets (44), twisted peptide backbone conformation (73), and the cooperative nature of MHCII-peptide interactions (74). By taking advantage of transient conformational states along the pathway to full dissociation, DM is able to identify kinetically unstable MHCII-peptide complexes and promote full dissociation and exchange. This mechanism helps DM fulfill its role in quality control of MHCII-peptide complexes for antigen presentation during their relatively brief endosomal transit.
In conclusion, we have shown that the DM susceptibility of DR1-peptide complexes can be determined by peptide substitutions outside the P1 pocket and by MHC substitutions away from peptide and DM interaction sites. Our data support the idea that the dynamic conformation of MHCII-peptide complexes determined by interactions throughout the peptide binding groove is the key determinant of DM susceptibility and that DM acts as a conformational rather than sequence editor.
Acknowledgments
We thank Liying Lu and Loretta Lee for soluble DR1 and DM expression and purification; Zachary Maben for assistance in the SAXS experiment coordination and SAXS data discussion; Dr. Osman Bilsel for SAXS data discussion; Dr. Gang Han for assistance in DLS measurement; and Dr. Thomas Eiermann (Hamburg, Germany) for kind donation of the UL-5A1 antibody. Use of the National Synchrotron Light Source, beamline X29, Brookhaven National Laboratory, was supported by the United States Department of Energy, Office of Science. This work used resources from the University of Massachusetts X-ray Diffraction Core Facility (supported in part by National Institutes of Health Shared Instrumentation Grant S10 O012028-01).
This work was supported, in whole or in part, by National Institutes of Health Grants AI48833 and AI57319 (to L. J. S.), AI095813 (to E. D. M.), and UL1 RR025744 from NCRR (to W. J. and E. D. M.). This work was also supported by the Lucile Packard Foundation for Children's Health (to W. J. and E. D. M.) and Deutsche Forschungsgemeinschaft Grant FR 1325/11-1 (to C. F.).
The atomic coordinates and structure factors (code 4OV5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CLIP
- class II-associated invariant chain peptide
- DM
- HLA-DM
- SPR
- surface plasmon resonance
- DLS
- dynamic light scattering
- SAXS
- small-angle x-ray scattering
- PDB
- Protein Data Bank.
REFERENCES
- 1. Bakke O., Dobberstein B. (1990) MHC class II-associated invariant chain contains a sorting signal for endosomal compartments. Cell 63, 707–716 [DOI] [PubMed] [Google Scholar]
- 2. Roche P. A., Cresswell P. (1990) Invariant chain association with HLA-DR molecules inhibits immunogenic peptide binding. Nature 345, 615–618 [DOI] [PubMed] [Google Scholar]
- 3. Germain R. N. (1994) MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 76, 287–299 [DOI] [PubMed] [Google Scholar]
- 4. Sloan V. S., Cameron P., Porter G., Gammon M., Amaya M., Mellins E., Zaller D. M. (1995) Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature 375, 802–806 [DOI] [PubMed] [Google Scholar]
- 5. Kropshofer H., Vogt A. B., Moldenhauer G., Hammer J., Blum J. S., Hämmerling G. J. (1996) Editing of the HLA-DR-peptide repertoire by HLA-DM. EMBO J. 15, 6144–6154 [PMC free article] [PubMed] [Google Scholar]
- 6. Morris P., Shaman J., Attaya M., Amaya M., Goodman S., Bergman C., Monaco J. J., Mellins E. (1994) An essential role for HLA-DM in antigen presentation by class II major histocompatibility molecules. Nature 368, 551–554 [DOI] [PubMed] [Google Scholar]
- 7. Lovitch S. B., Petzold S. J., Unanue E. R. (2003) Cutting edge: H-2DM is responsible for the large differences in presentation among peptides selected by I-Ak during antigen processing. J. Immunol. 171, 2183–2186 [DOI] [PubMed] [Google Scholar]
- 8. Lazarski C. A., Chaves F. A., Sant A. J. (2006) The impact of DM on MHC class II-restricted antigen presentation can be altered by manipulation of MHC-peptide kinetic stability. J. Exp. Med. 203, 1319–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amria S., Hajiaghamohseni L. M., Harbeson C., Zhao D., Goldstein O., Blum J. S., Haque A. (2008) HLA-DM negatively regulates HLA-DR4-restricted collagen pathogenic peptide presentation and T cell recognition. Eur. J. Immunol. 38, 1961–1970 [DOI] [PubMed] [Google Scholar]
- 10. Lazarski C. A., Chaves F. A., Jenks S. A., Wu S., Richards K. A., Weaver J. M., Sant A. J. (2005) The kinetic stability of MHC class II:peptide complexes is a key parameter that dictates immunodominance. Immunity 23, 29–40 [DOI] [PubMed] [Google Scholar]
- 11. Sant A. J., Chaves F. A., Jenks S. A., Richards K. A., Menges P., Weaver J. M., Lazarski C. A. (2005) The relationship between immunodominance, DM editing, and the kinetic stability of MHC class II:peptide complexes. Immunol. Rev. 207, 261–278 [DOI] [PubMed] [Google Scholar]
- 12. Hartman I. Z., Kim A., Cotter R. J., Walter K., Dalai S. K., Boronina T., Griffith W., Lanar D. E., Schwenk R., Krzych U., Cole R. N., Sadegh-Nasseri S. (2010) A reductionist cell-free major histocompatibility complex class II antigen processing system identifies immunodominant epitopes. Nat. Med. 16, 1333–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin L., Calvo-Calle J. M., Dominguez-Amorocho O., Stern L. J. (2012) HLA-DM constrains epitope selection in the human CD4 T cell response to vaccinia virus by favoring the presentation of peptides with longer HLA-DM-mediated half-lives. J. Immunol. 189, 3983–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kremer A. N., van der Meijden E. D., Honders M. W., Goeman J. J., Wiertz E. J., Falkenburg J. H., Griffioen M. (2012) Endogenous HLA class II epitopes that are immunogenic in vivo show distinct behavior toward HLA-DM and its natural inhibitor HLA-DO. Blood 120, 3246–3255 [DOI] [PubMed] [Google Scholar]
- 15. Pu Z., Lovitch S. B., Bikoff E. K., Unanue E. R. (2004) T cells distinguish MHC-peptide complexes formed in separate vesicles and edited by H2-DM. Immunity 20, 467–476 [DOI] [PubMed] [Google Scholar]
- 16. Weber D. A., Evavold B. D., Jensen P. E. (1996) Enhanced dissociation of HLA-DR-bound peptides in the presence of HLA-DM. Science 274, 618–620 [DOI] [PubMed] [Google Scholar]
- 17. Stratikos E., Wiley D. C., Stern L. J. (2004) Enhanced catalytic action of HLA-DM on the exchange of peptides lacking backbone hydrogen bonds between their N-terminal region and the MHC class II α-chain. J. Immunol. 172, 1109–1117 [DOI] [PubMed] [Google Scholar]
- 18. Narayan K., Chou C. L., Kim A., Hartman I. Z., Dalai S., Khoruzhenko S., Sadegh-Nasseri S. (2007) HLA-DM targets the hydrogen bond between the histidine at position β81 and peptide to dissociate HLA-DR-peptide complexes. Nat. Immunol. 8, 92–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Narayan K., Su K. W., Chou C. L., Khoruzhenko S., Sadegh-Nasseri S. (2009) HLA-DM mediates peptide exchange by interacting transiently and repeatedly with HLA-DR1. Mol. Immunol. 46, 3157–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou Z., Callaway K. A., Weber D. A., Jensen P. E. (2009) Cutting edge: HLA-DM functions through a mechanism that does not require specific conserved hydrogen bonds in class II MHC-peptide complexes. J. Immunol. 183, 4187–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferrante A., Gorski J. (2010) Cutting edge: HLA-DM-mediated peptide exchange functions normally on MHC class II-peptide complexes that have been weakened by elimination of a conserved hydrogen bond. J. Immunol. 184, 1153–1158 [DOI] [PubMed] [Google Scholar]
- 22. Anders A. K., Call M. J., Schulze M. S., Fowler K. D., Schubert D. A., Seth N. P., Sundberg E. J., Wucherpfennig K. W. (2011) HLA-DM captures partially empty HLA-DR molecules for catalyzed removal of peptide. Nat. Immunol. 12, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Painter C. A., Negroni M. P., Kellersberger K. A., Zavala-Ruiz Z., Evans J. E., Stern L. J. (2011) Conformational lability in the class II MHC 310 helix and adjacent extended strand dictate HLA-DM susceptibility and peptide exchange. Proc. Natl. Acad. Sci. U.S.A. 108, 19329–19334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pos W., Sethi D. K., Call M. J., Schulze M. S., Anders A. K., Pyrdol J., Wucherpfennig K. W. (2012) Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell 151, 1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferrante A., Anderson M. W., Klug C. S., Gorski J. (2008) HLA-DM mediates epitope selection by a “compare-exchange” mechanism when a potential peptide pool is available. PLoS One 3, e3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mohan J. F., Unanue E. R. (2013) A novel pathway of presentation by class II-MHC molecules involving peptides or denatured proteins important in autoimmunity. Mol. Immunol. 55, 166–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schulze M. S., Anders A. K., Sethi D. K., Call M. J. (2013) Disruption of hydrogen bonds between major histocompatibility complex class II and the peptide N terminus is not sufficient to form a human leukocyte antigen-DM receptive state of major histocompatibility complex class II. PLoS One 8, e69228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chou C. L., Sadegh-Nasseri S. (2000) HLA-DM recognizes the flexible conformation of major histocompatibility complex class II. J. Exp. Med. 192, 1697–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Belmares M. P., Busch R., Wucherpfennig K. W., McConnell H. M., Mellins E. D. (2002) Structural factors contributing to DM susceptibility of MHC class II/peptide complexes. J. Immunol. 169, 5109–5117 [DOI] [PubMed] [Google Scholar]
- 30. Guce A. I., Mortimer S. E., Yoon T., Painter C. A., Jiang W., Mellins E. D., Stern L. J. (2013) HLA-DO acts as a substrate mimic to inhibit HLA-DM by a competitive mechanism. Nat. Struct. Mol. Biol. 20, 90–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murthy V. L., Stern L. J. (1997) The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implications for the structural basis of the specificity of peptide binding. Structure 5, 1385–1396 [DOI] [PubMed] [Google Scholar]
- 32. Chicz R. M., Urban R. G., Lane W. S., Gorga J. C., Stern L. J., Vignali D. A., Strominger J. L. (1992) Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature 358, 764–768 [DOI] [PubMed] [Google Scholar]
- 33. Hanvesakul R., Maillere B., Briggs D., Baker R., Larché M., Ball S. (2007) Indirect recognition of T-cell epitopes derived from the α3 and transmembrane domain of HLA-A2. Am. J. Transplant. 7, 1148–1157 [DOI] [PubMed] [Google Scholar]
- 34. Smith H. J., Hanvesakul R., Bentall A., Shabir S., Morgan M. D., Briggs D., Cockwell P., Borrows R., Larché M., Ball S. (2011) T lymphocyte responses to nonpolymorphic HLA-derived peptides are associated with chronic renal allograft dysfunction. Transplantation 91, 279–286 [DOI] [PubMed] [Google Scholar]
- 35. Busch R., Doebele R. C., von Scheven E., Fahrni J., Mellins E. D. (1998) Aberrant intermolecular disulfide bonding in a mutant HLA-DM molecule: implications for assembly, maturation, and function. J. Immunol. 160, 734–743 [PubMed] [Google Scholar]
- 36. Frayser M., Sato A. K., Xu L., Stern L. J. (1999) Empty and peptide-loaded class II major histocompatibility complex proteins produced by expression in Escherichia coli and folding in vitro. Protein Expr. Purif. 15, 105–114 [DOI] [PubMed] [Google Scholar]
- 37. Günther S., Schlundt A., Sticht J., Roske Y., Heinemann U., Wiesmüller K. H., Jung G., Falk K., Rötzschke O., Freund C. (2010) Bidirectional binding of invariant chain peptides to an MHC class II molecule. Proc. Natl. Acad. Sci. U.S.A. 107, 22219–22224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yin L., Stern L. J. (2014) A novel method to measure HLA-DM susceptibility of peptides bound to MHC class II molecules based on peptide binding competition assay and differential IC determination. J. Immunol. Methods 406, 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 40. Wölpl A., Halder T., Kalbacher H., Neumeyer H., Siemoneit K., Goldmann S. F., Eiermann T. H. (1998) Human monoclonal antibody with T-cell-like specificity recognizes MHC class I self-peptide presented by HLA-DR1 on activated cells. Tissue Antigens 51, 258–269 [DOI] [PubMed] [Google Scholar]
- 41. Roche P. A., Cresswell P. (1990) High-affinity binding of an influenza hemagglutinin-derived peptide to purified HLA-DR. J. Immunol. 144, 1849–1856 [PubMed] [Google Scholar]
- 42. Sato A. K., Zarutskie J. A., Rushe M. M., Lomakin A., Natarajan S. K., Sadegh-Nasseri S., Benedek G. B., Stern L. J. (2000) Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J. Biol. Chem. 275, 2165–2173 [DOI] [PubMed] [Google Scholar]
- 43. Hammer J., Valsasnini P., Tolba K., Bolin D., Higelin J., Takacs B., Sinigaglia F. (1993) Promiscuous and allele-specific anchors in HLA-DR-binding peptides. Cell 74, 197–203 [DOI] [PubMed] [Google Scholar]
- 44. Stern L. J., Brown J. H., Jardetzky T. S., Gorga J. C., Urban R. G., Strominger J. L., Wiley D. C. (1994) Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature 368, 215–221 [DOI] [PubMed] [Google Scholar]
- 45. Natarajan S. K., Stern L. J., Sadegh-Nasseri S. (1999) Sodium dodecyl sulfate stability of HLA-DR1 complexes correlates with burial of hydrophobic residues in pocket 1. J. Immunol. 162, 3463–3470 [PubMed] [Google Scholar]
- 46. Schlundt A., Günther S., Sticht J., Wieczorek M., Roske Y., Heinemann U., Freund C. (2012) Peptide linkage to the α-subunit of MHCII creates a stably inverted antigen presentation complex. J. Mol. Biol. 423, 294–302 [DOI] [PubMed] [Google Scholar]
- 47. Ferrante A., Gorski J. (2012) A Peptide/MHCII conformer generated in the presence of exchange peptide is substrate for HLA-DM editing. Sci. Rep. 2, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fu X. T., Karr R. W. (1994) HLA-DR α chain residues located on the outer loops are involved in nonpolymorphic and polymorphic antibody-binding epitopes. Hum. Immunol. 39, 253–260 [DOI] [PubMed] [Google Scholar]
- 49. Carven G. J., Chitta S., Hilgert I., Rushe M. M., Baggio R. F., Palmer M., Arenas J. E., Strominger J. L., Horejsi V., Santambrogio L., Stern L. J. (2004) Monoclonal antibodies specific for the empty conformation of HLA-DR1 reveal aspects of the conformational change associated with peptide binding. J. Biol. Chem. 279, 16561–16570 [DOI] [PubMed] [Google Scholar]
- 50. Painter C. A., Cruz A., López G. E., Stern L. J., Zavala-Ruiz Z. (2008) Model for the peptide-free conformation of class II MHC proteins. PLoS One 3, e2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zarutskie J. A., Sato A. K., Rushe M. M., Chan I. C., Lomakin A., Benedek G. B., Stern L. J. (1999) A conformational change in the human major histocompatibility complex protein HLA-DR1 induced by peptide binding. Biochemistry 38, 5878–5887 [DOI] [PubMed] [Google Scholar]
- 52. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 53. Svergun D. I., Koch M. H. (2003) Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 66, 1735–1782 [Google Scholar]
- 54. Vachette P., Koch M. H., Svergun D. I. (2003) Looking behind the beamstop: X-ray solution scattering studies of structure and conformational changes of biological macromolecules. Method Enzymol. 374, 584–615 [DOI] [PubMed] [Google Scholar]
- 55. Svergun D. I., Feigin L. A., Schedrin B. M. (1982) Direct method of interpretation of small-angle scattering by the solutions of biological macromolecules. Stud. Biophys. 87, 277–278 [Google Scholar]
- 56. Moore P. B. (1980) Small-angle scattering–information-content and error analysis. J. Appl. Crystallogr. 13, 168–175 [Google Scholar]
- 57. Schneidman-Duhovny D., Hammel M., Sali A. (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nanda N. K., Sant A. J. (2000) DM determines the cryptic and immunodominant fate of T cell epitopes. J. Exp. Med. 192, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hall F. C., Rabinowitz J. D., Busch R., Visconti K. C., Belmares M., Patil N. S., Cope A. P., Patel S., McConnell H. M., Mellins E. D., Sonderstrup G. (2002) Relationship between kinetic stability and immunogenicity of HLA-DR4/peptide complexes. Eur. J. Immunol. 32, 662–670 [DOI] [PubMed] [Google Scholar]
- 60. Schulze M. S., Wucherpfennig K. W. (2012) The mechanism of HLA-DM induced peptide exchange in the MHC class II antigen presentation pathway. Curr. Opin. Immunol. 24, 105–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yin L., Stern L. J. (2013) HLA-DM focuses on conformational flexibility around P1 pocket to catalyze peptide exchange. Front. Immunol. 4, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Painter C. A., Stern L. J. (2012) Conformational variation in structures of classical and non-classical MHCII proteins and functional implications. Immunol. Rev. 250, 144–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sadegh-Nasseri S., Natarajan S., Chou C. L., Hartman I. Z., Narayan K., Kim A. (2010) Conformational heterogeneity of MHC class II induced upon binding to different peptides is a key regulator in antigen presentation and epitope selection. Immunol. Res. 47, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ferrante A. (2013) HLA-DM: Arbiter conformationis. Immunology 138, 85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pu Z., Carrero J. A., Unanue E. R. (2002) Distinct recognition by two subsets of T cells of an MHC class II-peptide complex. Proc. Natl. Acad. Sci. U.S.A. 99, 8844–8849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Doebele R. C., Pashine A., Liu W., Zaller D. M., Belmares M., Busch R., Mellins E. D. (2003) Point mutations in or near the antigen-binding groove of HLA-DR3 implicate class II-associated invariant chain peptide affinity as a constraint on MHC class II polymorphism. J. Immunol. 170, 4683–4692 [DOI] [PubMed] [Google Scholar]
- 67. Fallang L. E., Roh S., Holm A., Bergseng E., Yoon T., Fleckenstein B., Bandyopadhyay A., Mellins E. D., Sollid L. M. (2008) Complexes of two cohorts of CLIP peptides and HLA-DQ2 of the autoimmune DR3-DQ2 haplotype are poor substrates for HLA-DM. J. Immunol. 181, 5451–5461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim C. Y., Quarsten H., Bergseng E., Khosla C., Sollid L. M. (2004) Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc. Natl. Acad. Sci. U.S.A. 101, 4175–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hou T., Macmillan H., Chen Z., Keech C. L., Jin X., Sidney J., Strohman M., Yoon T., Mellins E. D. (2011) An insertion mutant in DQA1*0501 restores susceptibility to HLA-DM: implications for disease associations. J. Immunol. 187, 2442–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Boehr D. D., Nussinov R., Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ferrante A., Gorski J. (2007) Cooperativity of hydrophobic anchor interactions: evidence for epitope selection by MHC class II as a folding process. J. Immunol. 178, 7181–7189 [DOI] [PubMed] [Google Scholar]
- 72. Joshi R. V., Zarutskie J. A., Stern L. J. (2000) A three-step kinetic mechanism for peptide binding to MHC class II proteins. Biochemistry 39, 3751–3762 [DOI] [PubMed] [Google Scholar]
- 73. Jardetzky T. S., Brown J. H., Gorga J. C., Stern L. J., Urban R. G., Strominger J. L., Wiley D. C. (1996) Crystallographic analysis of endogenous peptides associated with HLA-DR1 suggests a common, polyproline II-like conformation for bound peptides. Proc. Natl. Acad. Sci. U.S.A. 93, 734–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Anderson M. W., Gorski J. (2005) Cooperativity during the formation of peptide/MHC class II complexes. Biochemistry 44, 5617–5624 [DOI] [PubMed] [Google Scholar]
- 75. Stern L. J., Wiley D. C. (1992) The human class II MHC protein HLA-DR1 assembles as empty alpha beta heterodimers in the absence of antigenic peptide. Cell 68, 465–477 [DOI] [PubMed] [Google Scholar]