

Background: Whether ER to Golgi transport requires calcium, the source of calcium, and its mechanism is unknown.
Results: A requirement for luminal calcium is demonstrated, and evidence is presented for a molecular effector pathway.
Conclusion: Luminal calcium may regulate transport by activating these protein interactions.
Significance: The described calcium effector pathway may lead to greater insight into calcium action at multiple transport steps.
Keywords: Calcium-binding Protein, Endoplasmic Reticulum (ER), Golgi, Membrane Trafficking, Secretion, Vesicles, Endoplasmic Reticulum to Golgi Transport
Abstract
Luminal calcium released from secretory organelles has been suggested to play a regulatory role in vesicle transport at several steps in the secretory pathway; however, its functional roles and effector pathways have not been elucidated. Here we demonstrate for the first time that specific luminal calcium depletion leads to a significant decrease in endoplasmic reticulum (ER)-to-Golgi transport rates in intact cells. Ultrastructural analysis revealed that luminal calcium depletion is accompanied by increased accumulation of intermediate compartment proteins in COPII buds and clusters of unfused COPII vesicles at ER exit sites. Furthermore, we present several lines of evidence suggesting that luminal calcium affected transport at least in part through calcium-dependent interactions between apoptosis-linked gene-2 (ALG-2) and the Sec31A proline-rich region: 1) targeted disruption of ALG-2/Sec31A interactions caused severe defects in ER-to-Golgi transport in intact cells; 2) effects of luminal calcium and ALG-2/Sec31A interactions on transport mutually required each other; and 3) Sec31A function in transport required luminal calcium. Morphological phenotypes of disrupted ALG-2/Sec31A interactions were characterized. We found that ALG-2/Sec31A interactions were not required for the localization of Sec31A to ER exit sites per se but appeared to acutely regulate the stability and trafficking of the cargo receptor p24 and the distribution of the vesicle tether protein p115. These results represent the first outline of a mechanism that connects luminal calcium to specific protein interactions regulating vesicle trafficking machinery.
Introduction
The ER3-to-Golgi interface is the busiest vesicle trafficking step. Anterograde cargo is captured into a COPII prebudding complex with the activated GTPase Sar1 and the inner coat Sec23/24 heterodimer. Sar1 interacts directly with Sec23, whereas the cargo is bound in several distinct pockets on the membrane-proximal surface of Sec24 (1–4). Sec23 is a GAP for sar1, but its GAP activity is weak initially, preventing premature disassembly. Recruitment of the outer coat layer, composed of Sec13/31 heterotetramers, positions a flexible proline-rich region (PRR) loop of Sec31 across the membrane-distal surface of Sec23 and inserts residues into the Sar1 active site, potentiating the Sec23 GAP activity. Cyclical Sar1 GTPase activity is required for cargo concentration (5). Sec13/31 recruitment involves polymerization of at least 24 heterotetramers (4), triggering vesicle scission. Once free of the ER, the cargo-laden vesicle does not shed all of its coat; interactions with cargo retain ∼60% of the original Sec23/24 and ∼10% of the outer shell on the vesicle, and this residual coat possesses functions in targeting and the directionality of fusion (6–11). COPII vesicles fuse homotypically to produce vesicular tubular clusters (VTCs). This process involves multiple tethers and the ER/Golgi SNARE complex (12–15). Maturing VTCs are the primary site of cargo concentration (16). In the pericentriolar region, VTCs concentrate and fuse with Golgi cisternae (2).
Although Ca2+ is a required cofactor in evoked exocytosis, it is unclear what role Ca2+ plays in intracellular membrane fusions. Indications that Ca2+ is required for constitutive trafficking have come from in vitro studies of ER-to-Golgi (17), intra-Golgi (18), and endosome and lysosome (19–21) trafficking. These studies all point to a role for Ca2+ directly on the vesicle budding, docking, and/or fusion machinery. Chelator selectivities indicate that Ca2+ acts either through a short pulse or else a continuous gradient of leaking luminal Ca2+ very near the transport machinery (22, 23). In vivo evidence for a requirement for luminal Ca2+ in Golgi trafficking came with the recognition that mutations of the Golgi Ca2+ pump SPCA (secretory pathway Ca2+-ATPase) cause Hailey-Hailey disease (24) and that depletion of SPCA from cells arrests secretion at the Golgi (25, 26). In this case, the luminal Ca2+ may act in the lumen; SPCA-dependent luminal Ca2+ appears to be required in the TGN for sorting of a subset of secretory proteins into exocytic carriers via the luminal Ca2+-binding protein Cab45 (27, 28). Hence, Ca2+ does not play a universal or necessarily mechanistically consistent role; secretion is a mosaic of Ca2+-dependent and -independent transport steps (23). At the ER-to-Golgi stage, specific depletion of luminal Ca2+ using the reversible SERCA inhibitor cyclopiazonic acid (CPA) caused Rbet1, a rapidly recycling ER/Golgi SNARE, to accumulate in large peripheral punctate structures (10). Despite this morphological perturbation, potential functional effects of luminal Ca2+ depletion on anterograde ER/Golgi cargo transport remain to be established.
Effector mechanisms for luminal Ca2+ released from organelles are not understood. Calmodulin has been implicated in several transport steps (22), and Ca2+-dependent phospholipase A2 has been implicated in Golgi membrane dynamics (29, 30). Membrane-permeant BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) destabilizes COPI binding to membranes in vivo (31), but a Ca2+ sensor for this effect is not known. The PEF (penta-EF-hand-containing) protein apoptosis-linked gene-2 (ALG-2) acts as a Ca2+ sensor at ER exit sites and, only when Ca2+ is present, stabilizes association of Sec31 with the membrane (32–35). We previously demonstrated that ALG-2 stabilizes residual Sec31 on COPII fusion intermediates in a Ca2+-dependent manner and that excess ALG-2 inhibited COPII vesicle fusion in a luminal Ca2+-dependent manner (10). Recently, an in vitro budding reconstitution using mammalian components demonstrated that excess ALG-2 attenuated budding in a Ca2+-dependent manner and that ALG-2 binding to Sec31A directly promoted Sec31A/Sec23 interactions (36). Hence, in vitro studies have made significant progress in identifying the ALG-2/Sec31 axis as a potential effector of Ca2+. However, whether the ALG-2/Sec31 interaction is rate-limiting in the cell and at what step(s) has not been demonstrated.
Here we present evidence that luminal Ca2+ and ALG-2/Sec31A interactions regulate early steps in secretory traffic. This is the first functional demonstration that luminal Ca2+ plays a required role in pre-Golgi secretory trafficking. Furthermore, this work sheds light on a Ca2+ signaling mechanism involving ALG-2/Sec31A interactions that contributes to the requirement for Ca2+ in transport.
EXPERIMENTAL PROCEDURES
Antibodies and Expression Constructs
Rabbit polyconal anti-Sec31 (10) and anti-p24 (14) were produced in rabbits against synthetic peptides and affinity-purified as described previously. Monoclonal anti-Rbet1 antibodies were described before (37). Anti-vesicular stomatitis virus glycoprotein (VSV-G) trimer was obtained from tissue culture supernatant from the I-14 hybridoma (38) kindly provided by Dr. John Ngsee (University of Ottawa). Rabbit polyclonal anti-Sec24c was a kind gift from Dr. William Balch (Scripps Institute). Rabbit polyclonal anti-p58 was a kind gift from Dr. Jaakko Saraste (University of Bergen, Norway). Rabbit polyclonal anti-p115 was a kind gift from Dr. Elizabeth Sztul (University of Alabama, Birmingham). Rabbit polyclonal anti-ALG-2 was a kind gift from Dr. Masayuki Komada (Tokyo Institute of Technology). Mouse monoclonal anti-FLAG was purchased from Sigma (clone M2 product F3165). Rabbit anti-GPP130 antibody was purchased from Covance (product PRB144C). Rabbit anti-calnexin was from Stressgen (product SPA-865). Mouse monoclonal anti-mannosidase II antibody was purchased from Covance Research Products (product MMS-110R-200). Goat polyclonal anti-phosphoglycerate kinase and rabbit anti-TRAF2 were purchased from Santa Cruz Biotechnology, Inc. (products sc17943 and sc876, respectively). Secondary antibodies were FITC-, Cy3-, AMCA-, or Cy5-conjugated and purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
A mammalian expression construct encoding full-length, wild type human ALG-2 was provided by Dr. Masayuki Komada (Tokyo Institute of Technology) and was described before (35); point mutations/deletions to this construct and all other constructs were produced by PCR (QuikChange method, Agilent Technologies) and confirmed by sequencing. To produce RFP-ALG-2 for localization studies, tRFP was amplified by PCR from pCMV6-AC-RFP (Origene) with BamHI/EcoRI linker ends and ligated into mammalian expression vector pCDNA3(+). Next, full-length, wild type ALG-2 was amplified by PCR with EcoRV/XhoI linker ends and subcloned downstream and in-frame to tRFP in pCDNA3(+).
A mammalian construct in vector pME-FLAG containing residues 800–1113 of human SEC31A as described (35) was obtained from Dr. Masayuki Komada. Splicing of the human SEC31A gene is highly variable, currently comprising six full-length curated splicing variants with additional variants apparent among reported partial cDNAs. Our construct's exon structure corresponds closely to splicing variant 1 (NCBI reference sequence NM_014933.3) with the exception that there is a 13-amino acid insertion of ENQSIQDQAPMLE between Thr-989 and Gly-990 of the reference sequence. This exon is also expressed in a cDNA from cd34+ stem cells (GenBankTM AF161393.1) and is a predicted exon in the original analysis of the human gene structure (GenBankTM EAX05906.1), although it is not present in any of the currently curated full-length reference sequences. This exon is also expressed in the single curated murine transcript (NCBI reference sequence NM_026969.1). Most experiments in this work utilized the construct containing the additional exon; however, for clarity, we refer to amino acid positions relative to the that of human splicing variant 1; for example, when referring to mutation of Trp-995 in Fig. 7, this would actually correspond to amino acid 1008 in our cDNA because it expresses the 13-residue insert between Thr-989 and Gly-990. We did not notice a functional difference between SEC31A splicing variants within the PRR region. For example, the experiment in Fig. 5A employed the construct described above (35), whereas the experiment in Fig. 5B utilized PRR regions subcloned from full-length SEC31A expression constructs kindly provided by Dr. Hideki Shibata (Nagoya University, Japan) that had an exon structure identical to human splicing variant 1 (32).
FIGURE 7.

ALG-2/Sec31A interactions are required for p24 stability and targeting of p115. NRK cells were transfected with VSV-G-GFP along with ALG-2-RFP (A) or FLAG-sec31A (B) or with both (C–G). Cells were shifted to 32 °C for 12 min, fixed, and immunolabeled to detect FLAG-PRR and endogenous p24 (A–C), GPP130 (D), p58 (E), rbet1 (F), or p115 (G). Shown are single optical sections of deconvolved wide field image stacks. In each set of fields, green arrows are employed in the image plane that was merged as green and red arrows are used in the plane merged as red. White boxes indicate the area displayed in the merge if less than the entire field. Arrows in A, E, and F mark prominent cytoplasmic puncta decorated by ALG-2-RFP. Arrows in B–D, point out the Golgi area. The arrows in G mark regions of ER that co-localize with p115.
FIGURE 5.

ALG-2 targets the Sec31A PRR to a functionally critical site. A, NRK cells were transfected with VSV-G-GFP along with no additional construct (mock), β-galactosidase, or ALG-2 (wild type or indicated mutants) and/or a FLAG-Sec31-PRR construct consisting of residues 800–1113 and held at 40 °C. After ∼24 h, cells were shifted to 32 °C for 0 (left bar) or 12 min (bars 2–10). Cells were then fixed and immunolabeled to identify which cells expressed the constructs, and the arrival of total VSV-G-GFP in the Golgi area was quantified. Each bar represents the mean of ≥30 cells in one of three independent experiments with similar results. B, experiment as in A demonstrating that mutant FLAG-PRR lacking the ALG-2 binding site (ΔABS-PRR) is much less active as an inhibitor. C, NRK cells transfected with the indicated constructs were lysed and immunoblotted to detect FLAG-Sec31A-PRR, ALG-2, or phosphoglycerate kinase (PGK). Human ALG-2 migrates slightly slower on SDS-PAGE than the rat protein. D, NRK cells were mock-transfected or transfected with ALG-2 and the PRR constructs as indicated above. For lanes 1–9, cells were not pretreated and were extracted in CHAPS buffer containing ambient calcium. For lanes 10–13, cells were first pretreated with DMSO or dithiobis(succinimidyl propionate) (DSP) dissolved in DMSO and then extracted in Triton X-100-containing buffer including 2 mm EGTA and EDTA. The CHAPS extracts were immunoblotted to detect ALG-2 (lanes 1–5, top) or the FLAG constructs (bottom). Two nonspecific bands recognized by the FLAG antibody, one that co-migrates with FLAG-PRR 800–1113, are marked with asterisks. Extracts were then subjected to immunoprecipitation with anti-FLAG beads and washed extensively, and the precipitates were immunoblotted to detect ALG-2 (lanes 6–13). E, experiment as in A demonstrating that an ALG-2-RFP fusion construct had similar inhibitory activity. T-tests were performed between the 12-min control and each experimental condition. **, p < 0.001. Error bars, S.E.
Depletion of Luminal Calcium
Conditions were identical to our extensively characterized protocol (10). Briefly, Ca2+-free DMEM (U.S. Biological D9802-05B) was supplemented with 10% fetal bovine serum that had been dialyzed (twice for 2 h) in a 100-fold excess of Ca2+-free PBS. Cells to be depleted of luminal Ca2+ were washed twice with Ca2+-free DMEM, 10% FBS containing 10 μm CPA (Sigma C1530), 1 mm EGTA, and 25 mm Hepes, pH 7.2, and then incubated in that medium for 15 min. This was followed by a 15-min incubation in Ca2+-free DMEM, 10% FBS, no CPA, 0.3 mm EGTA, and 25 mm Hepes, pH 7.2. Some treatments (Figs. 1 and 2) included 10 μg/ml cycloheximide for a 30-min period prior to the addition of CPA; in these cases, cycloheximide was also maintained in the medium during the CPA treatment and subsequent Ca2+-free chase.
FIGURE 1.

Luminal calcium depletion inhibits ER-to-Golgi transport of VSV-G-GFP trimers. A, VSV-G-GFP-expressing NRK cells were CPA- or mock-treated. Cargo was then released from the ER by shifting cells from 40 to 32 °C. Cells were fixed after 0, 3, 10, 15, or 20 min; permeabilized; and labeled to detect VSV-G trimers using the I-14 antibody. The arrival of total VSV-G-GFP fluorescence (left plot) or VSV-G trimers (right panel) in the Golgi was quantified. Each value is a mean of ≥20 cells from a single experiment repeated at least three times with a similar outcome. Error bars, S.E. The transport index is much higher in the right panel because unfolded VSV-G-GFP in the ER is not detected. B, representative images from the experiment quantified in A. C, VSV-G trimerizes in 3 min. Cells expressing VSV-G-Myc or VSV-G-GFP were CPA- or mock-treated prior to temperature shifts and then chilled and lysed. Assembled VSV-G trimers were immunoprecipitated using the I-14 antibody and detected by immunoblot. The graph (top) was generated by averaging three experiments. Blots from one of the experiments are shown (bottom). D, CPA treatment inhibits ER-to-Golgi transport measured by acquisition of endoglycosidase H resistance (endo HR). NRK cells were transfected with VSV-G-Myc ts045 and grown for 24 h at 40 °C and then either left in regular medium (control) or else treated with CPA as in Fig. 1 at 40 °C (CPA). Cells were then shifted to 32 °C for 0 or 40 min to allow transport through the medial Golgi prior to standard procedures for determination of endo H resistance using endo H digestion followed by SDS-PAGE and anti-Myc immunoblotting. Plotted is the percentage transport after 40 min at 32 °C, quantitated from three replicates with the formula, percentage transport = endo H-resistant/(endo H-resistant + endo H-sensitive) × 100%. The 0-min background value of percentage transport for cells left at 40 °C was subtracted for each group of cells (∼20% for CPA-treated cells). **, p < 0.01 in a two-tailed t test. E, ATF-6 is not activated by our Ca2+ depletion conditions. NRK cells were transfected with FLAG-ATF-6, treated, lysed, and immunoblotted to detect FLAG. Lane 1, CPA as in A; lane 2, DMSO for 2 h; lane 3, 1 mm DTT for 2 h; lane 4, 2 μg/ml tunicamycin (Tun) for 2 h. Uncleaved ATF-6 is 100 kDa, whereas the cleaved ATF-6 is ∼60 kDa.
FIGURE 2.
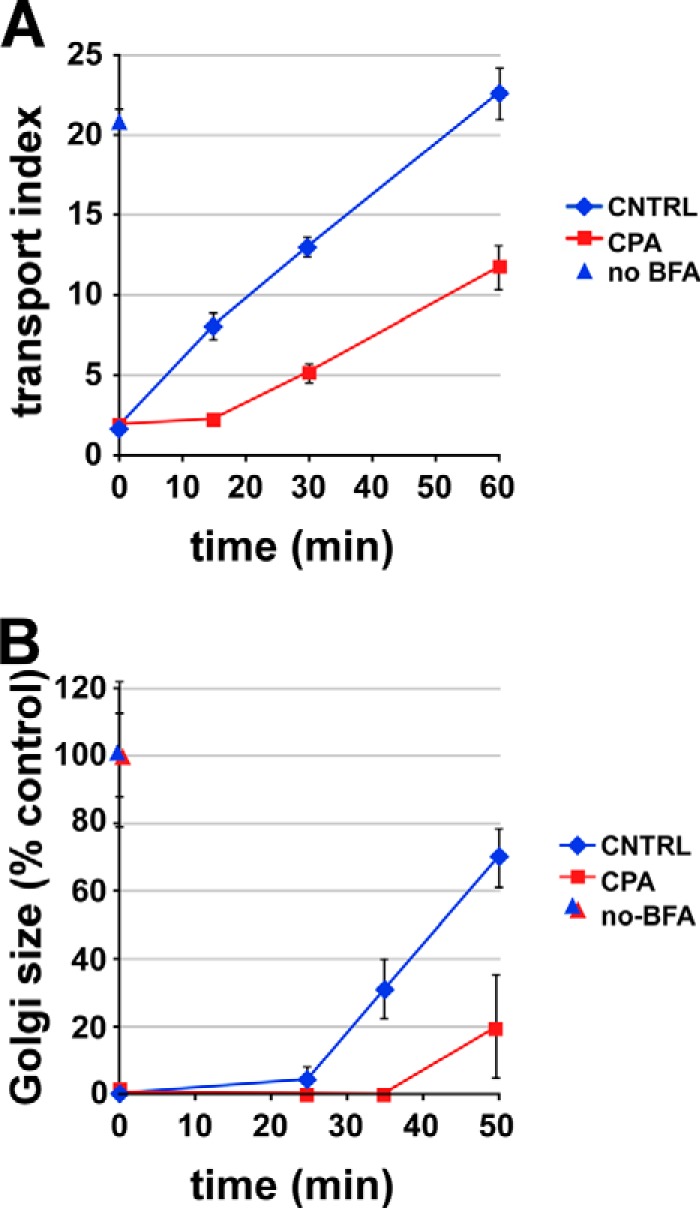
Luminal calcium depletion impairs ER-to-Golgi transport of mannosidase II. NRK cells were treated with BFA for 45 min, with CPA or DMSO present for the final 15 min. After BFA removal (t = 0 on plot), CPA-treated cells recovered in calcium-free medium, whereas control cells recovered in normal medium. At the indicated times, cells were fixed and labeled for mannosidase II. A, arrival of mannosidase II in the Golgi area was quantified using the transport index algorithm as in Fig. 1. no BFA, cells received neither BFA nor CPA (blue triangle). B, a similar experiment except that return to the Golgi was quantified by measuring the area of mannosidase II-positive puncta. Single experiments were representative of at least three similar experiments; each value is a mean of ≥20 cells. Error bars, S.E.
Immunofluorescence Microscopy
Coverslips were fixed with 4% paraformaldehyde containing 0.1 m sodium phosphate (pH 7) for 30 min at room temperature and quenched twice for 10 min with PBS containing 0.1 m glycine. Fixed cells were treated for 15 min at room temperature with permeabilization solution containing 0.4% saponin, 1% BSA, and 2% normal goat serum dissolved in PBS. The cells were then incubated with primary antibodies diluted in permeabilization solution for 1 h at room temperature. Next, coverslips were washed three times with permeabilization solution and incubated for 30 min at room temperature with different combinations of FITC-, Cy3-, AMCA-, and/or Cy5-conjugated anti-mouse or anti-rabbit secondary antibodies. After the secondary antibody incubation, coverslips were again washed three times using permeabilization solution and mounted on glass slides using Slow Fade Gold antifade reagent (Invitrogen), and the edges were sealed with nail polish. Slides were analyzed using a ×60 objective on a Nikon E800 microscope with excitation and emission filter wheels (Chroma Tech), a Hamamatsu Orca 2 camera, and a Nikon Z-drive, automated using OpenLab version 5.0 software (Improvision). For transport index assays (see below), following overexpression, typical images collected for each field of cells were VSV-G-GFP (FITC filters), anti-ALG-2 (cy3 filters), anti-FLAG (AMCA filters), and Golgi marker GPP130 (Cy5 filters).
For immunofluorescence co-localization experiments, such as those shown in Figs. 6 and 7, 21 images were captured from each color channel in 200-nm increments. These image stacks were deconvolved using Huygens Essential Widefield software (Scientific Volume Imaging, Hilversum, The Netherlands). Example images for display represent individual image planes near the center of the stack. For quantification of co-localization, deconvolved image planes from 10–12 transfected cells containing RFP-ALG-2 labeling in focus in the peripheral cytoplasm were randomly selected, and the background labeling was removed by defining a dark extracellular area of the image as zero. An RFP-ALG-2 object binary image mask was generated by thresholding the labeling such that 10–15 of the brightest RFP-ALG-2 spots were captured. Each of these objects was then manually tested for co-localization with a spot in the COPII marker plane, with partial overlap scored as positive co-localization. The percentage of bright RFP-ALG-2 objects that overlapped with ERES spots was directly calculated from this analysis for each cell, and the values reported in Fig. 6G represent means plus or minus S.E. of 10–12 cells/condition.
FIGURE 6.

Effects of ALG-2-RFP and Sec31A-PRR-FLAG on endogenous COPII components. NRK cells transfected with VSV-G-GFP along with no additional construct (A and B), with ALG-2-RFP (C and D), or with both ALG-2-RFP and FLAG-Sec31-PRR (E and F) were shifted to 32 °C for 0 (A) or 12 min (B–F) and then fixed and immunolabeled to detect FLAG-PRR and endogenous Sec31A (A–C and F) or endogenous Sec24C (D and E). Shown are single optical sections of deconvolved wide field image stacks. Arrows, prominent cytoplasmic puncta decorated by ALG-2-RFP that frequently co-localize with ERES and ERGIC markers. Merged images show superposition of the ALG-2-RFP (red) and COPII marker (green) in a magnified single cell from the field (marked with white boxes in the RFP-ALG-2 image). Insets within the merged planes show a smaller area with a higher magnification. G, quantification of the percentage of cytoplasmic ALG-2-RFP-positive structures that co-localize significantly with Sec24C and Sec31A. Error bars, S.E.
ER-to-Golgi Transport Assay
NRK cells were maintained in DMEM high glucose containing 10% fetal calf serum and penicillin-streptomycin. Suspensions of NRK cells were electroporated with 15 μg of VSV-G-GFP DNA or a mixture of that construct and constructs encoding β-galactosidase, ALG-2, and/or Sec31A-PRR and plated on polylysine-coated coverslips. Mock-transfected cells were electroporated with no DNA present. After 24 h of protein expression at 40 °C, the cells were either depleted of luminal Ca2+ (see above) and shifted to 32 °C or directly shifted to permissive temperature, depending upon the experiment. For the 0-min transport time point, coverslips were directly transferred from 40 °C to fixative. For other time points, coverslips were transferred from 40 °C into 6-well chambers containing pre-equilibrated 32 °C medium for various intervals and then transferred to fixative.
Morphological quantitation of ER-to-Golgi transport was accomplished as described before (39). Briefly, images were collected in a consistent manner with regard to cell morphology, protein expression levels, and exposure. After choosing a fixed exposure time for each color channel that would accommodate the vast majority of cells, we avoided any cell whose intensity values in any color exceeded the saturation value of our camera. We also avoided capturing cells lacking flat morphology and a single, well defined nucleus surrounded on all sides by an expanse of cytoplasm. A single wide field image plane was collected for each color channel for each field of cells randomly encountered; image deconvolution was not performed. Background in all quantification images was removed by defining a dark extracellular area of the image as zero. The Golgi region was defined by GPP130 immunofluorescence; a Golgi mask was created by thresholding the GPP130 image at 25% of its maximum value. This was used to identify the Golgi pixels in the associated VSV-G-GFP image. Transport index was calculated from the VSV-G-GFP image as the maximum pixel intensity within the Golgi region mask divided by the mean pixel intensity in the cell periphery. Maximum intensity was used for the Golgi instead of the mean to avoid having to calculate the precise cross-sectional area of the Golgi, which leads to greater variance. Line profiles across the maximum pixel confirmed that it fell within a peak of similar fluorescence intensity values. The peripheral fluorescence in the denominator of the transport index was derived from a sample of the ER taken by manually drawing an oval-shaped region of interest with the long dimension extending from the edge of the nucleus to roughly the edge of the cell; the surface of the nucleus on which the region of interest abuts was chosen so as to optimally avoid encompassing or being near any Golgi elements. The mean pixel intensity for this region of interest on the VSV-G-GFP image was the denominator of the transport index. Transport index was calculated for each cell separately.
For the brefeldin A (BFA) recovery ER-to-Golgi transport assay, NRK cells were grown at 37 °C on coverslips and treated for 45 min with 2.5 μg/ml BFA. Cells to be Ca2+-depleted were then switched to calcium-free DMEM, 10% FBS with 0.3 mm EGTA and 10 μm CPA plus 2.5 μg/ml BFA for 15 min, whereas control cells were incubated for an extra 15 min with 2.5 μg/ml BFA. Coverslips were then washed twice with either calcium-free DMEM, 10% FBS, 0.3 mm EGTA or control medium to remove the BFA and incubated for varying times in the wash medium. Throughout the experiment, all of the medium contained 10 μg/ml cycloheximide. Following the incubations, cells were then fixed as described above and labeled using anti-mannosidase II antibody and FITC-labeled secondary antibody.
Transport was then quantified using one of two methods; the first method employed the ER/Golgi transport index essentially as described above except that mannosidase II was employed as the cargo instead of VSV-G-GFP, and also the maximum ER intensity was used instead of the maximum Golgi intensity to generate a transport index at the early time points before a Golgi area was evident. Alternatively, a mannosidase II-positive object binary image mask was generated by thresholding the labeling at 4 times over its intensity in a peripheral area lacking punctate objects. The mean object size in this mask layer was calculated using Openlab and employed as an indicator of Golgi size; this value increased during the washout period as small puncta coalesced into larger puncta/Golgi fragments and then ribbons.
siRNA Knockdown of ALG-2 and Sec31A
NRK cells were electroporated with 0.5 μm custom-synthesized siRNA (Ambion, Invitrogen). After 2–3 days of normal growth, the cells were resuspended and re-electroporated, this time with a combination of the 0.5 μm siRNA and 15 μg of VSV-G-GFP, and the cells were allowed to recover and grow on coverslips at 40 °C. Twenty-four hours later, the cells were either lysed directly in SDS sample buffer for quantitative immunoblotting analysis or else processed for CPA treatments and transport assays as described above. Syntaxin 5 siRNA (40) and its effect on the transport index (39) were described before. Sec31A siRNA had the following sense strand sequence: 5′-GACCUUUGUUUACACGAUATT-3′ (containing Silencer Select chemical modifications). This target sequence is expressed in all annotated Sec31A splice variants. ALG-2 siRNAs had the following sense strand sequences: siRNA1, 5′-GGAGCGGAGUGAUUUCAGA-3′ (containing Silencer Select chemical modifications); siRNA2, 5′-GGACAACUCUGGGAUGAUU-3′ (Silencer Select); siRNA-3, 5′-GCAGAGGUUGACAGACAUAAA-3′ (lacking Silencer Select modifications). All of the siRNAs employed achieved >95% knockdown in the best experiments, with the primary determinant of success being the precise growth phase and density of the cells.
Immunoprecipitation Experiments
NRK cells were transfected with mammalian expression constructs encoding full-length, wild type human ALG-2 by itself or with one of three SEC31A pME-FLAG constructs: 1) residues 800–1113 of human SEC31A (PRR 800–1113); 2) a truncated PRR fragment containing residues 800–976 (PRR 800–976); or 3) a deletion mutant PRR fragment lacking the ALG-2 binding site (PRR ΔABS). 20–24 h after transfection, cells were washed with cold PBS and solubilized with 1.5 ml/10-cm plate CHAPS lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm KCl, 10 mm CHAPS, 2 μg/ml leupeptin, 4 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 mm PMSF) for 30 min in the cold. Extracts were collected and centrifuged at 12,000 × g for 15 min, and the supernatants were saved. To prepare extracts from cells cross-linked with DSP, cells were first washed with warm PBS and then incubated with DMSO in serum-free DMEM with or without 1 mg/ml dithiobis(succinimidyl propionate) (Pierce) for 15 min at 37 °C, transferred to ice, and quenched for at least 15 min with cold PBS (pH 7.4) containing 50 mm Tris and 0.1 m glycine. The cells were then washed with cold PBS prior to solubilization with 1.5 ml/10-cm plate of KCl buffer (20 mm Hepes, pH 7.2, 0.1 m KCl, 2 mm EGTA, 2 mm EDTA) containing 1% Triton X-100, 2 μg/ml leupeptin, 4 μg/ml aprotinin, 1 μg/ml pepstatin A, and 1 mm PMSF and centrifuged at 100,000 × g for 30 min, and the supernatants were saved. Lysates were used directly for SDS-PAGE and immunoblotting or immunoprecipitation followed by SDS-PAGE and immunoblotting. To perform immunoprecipitations, 500 μl of cell extract was incubated with 10 μl of 50% antibody beads (anti-FLAG® M2 Affinity Gel, Sigma-Aldrich) for 2 h at 4 °C with constant agitation. Beads were then washed three times with CHAPS lysis buffer or KCl buffer containing 0.1% Triton X-100, and proteins were eluted by incubation for 5 min at room temperature with non-reducing SDS-PAGE sample buffer. After removing the beads by centrifugation, the supernatant was adjusted to 5% β-mercaptoethanol and boiled, followed by SDS-PAGE and immunoblotting.
Correlative Light and Electron Microscopy
NRK cells treated with CPA under our previously characterized conditions (10) were fixed, and 70-nm ultrathin cryosections were prepared by the Tokuyasu method with modifications (41) and immobilized on copper grids containing alphanumeric markings for each sample quadrant. Because rabbit affinity-purified anti-Sec31 gave very strong labeling of the CPA-induced structures (Fig. 3), this primary antibody was used for the correlative protocol. Cryosections were labeled with anti-Sec31 followed by goat anti-rabbit Alexafluor-488 followed by 10-nm protein A-gold and then staining with Hoechst. Grids were then imaged by wide field fluorescence deconvolution microscopy using a Deltavision microscope. Alexafluor-488-labeled (green) cytoplasmic spots were similar to those observed previously in whole cell immunofluorescence experiments with anti-Sec31 and anti-Rbet1 antibodies. Spotty labeling of the ultrathin sections also occurred, apparently nonspecifically, over nuclei, although at a lower frequency than in cytoplasmic areas. Labeling of extracellular spaces was rarely observed. After recording z-stacks of wide field fluorescence, sections were stained with uranyl acetate and embedded in methylcellulose. Examination of the previously imaged quadrants by electron microscopy at low magnification allowed identification of landmark structures, such as nuclear material and extracellular spaces that could be aligned with the fluorescence images in EM fluorescence overlays performed using ACD Canvas graphics software. It was thus possible to establish an unambiguous correlation between major spots of interest decorated by Alexafluor-488 and 10-nm gold particles (Fig. 4A). However, the gold labeling was much less efficient, and there were occasional cases of fluorescence label without corresponding gold particle(s). The fluorescence label and gold particles correlated but were not always well aligned, probably due to the low resolution of light microscopy and/or section instability associated with two types of imaging. For annotation of EMs, vesicles (as opposed to buds) are defined when the membrane is clearly visible, encompassing at least 80% of a circular profile with no visible neck.
FIGURE 3.

Luminal calcium depletion causes COPII proteins to accumulate in enlarged, intensified peripheral puncta. NRK cells were CPA- or mock-treated and then fixed and immunolabeled for the endogenous VTC marker Rbet1 and the COPII subunit Sec31A (A) or Sec24C (B). Shown are single optical sections of deconvolved wide field fluorescence image stacks.
FIGURE 4.

Enlarged peripheral puncta induced by CPA contain abundant COPII buds and clusters of unfused vesicles. Correlative section light and electron microscopy of CPA structures in peripheral regions lacking Golgi. Anti-Sec31 primary antibody was followed by anti-rabbit Alexafluor-488 (green), 10-nm protein A-gold, and Hoechst (blue). Sections were imaged by fluorescence microscopy and then stained with uranyl acetate and embedded with methylcellulose prior to electron microscopy and fluorescence-transmission EM correlation. A and C, overlays of fluorescence with morphology visible by EM. Gold particles were enhanced with red. White arrowheads highlight swaths of fluorescence labeling over buds and vesicles not labeled by gold. B and D, magnified view of regions in red boxes in A and C, with buds (b), vesicles (v), VTCs, and ER labeled. Red arrow, vesicle cluster. Asterisks, gold particles not clearly associated with a membrane.
VSV-G Trimer Detection by Immmunoprecipitation
NRK cells were transfected with VSV-G-Myc or VSV-G-GFP and incubated for 24 h at 40 °C. Cells were depleted of luminal Ca2+ as described above and incubated for varying times at 32 °C to allow VSV-G trimerization, and then the medium was replaced with cold 25/125 buffer (25 mm HEPES, pH 7.2, 125 mm potassium acetate) containing 1% Triton X-100 and protease inhibitors (pepstatin, leupeptin, aprotinin) and incubated for 10 min at 4 °C. Next, the buffer and cell debris were scraped into a microtube and incubated with end-over-end rotation for at least an additional 10 min prior to removal of the insoluble fraction by centrifugation at 15,000 × g for 15 min. The soluble supernatant was then incubated with protein A beads that had been preincubated in I14 hybridoma culture medium and washed four times with 25/125. After 2 h of end-over-end rotation at 4 °C, the beads were pelleted and washed three times with 25/125. Samples of the washed beads were then analyzed using SDS-PAGE and Western blotting with either anti-GFP or anti-Myc antibodies. To prepare the graph in Fig. 1C from several experiments, 0-min values were subtracted from each data point, and then each point was expressed as a percentage of the maximum value.
ATF-6 Detection in NRK Cells
NRK cells were transfected with FLAG-ATF-6. Two days after electroporation, cells were washed once with PBS and once with swell buffer (10 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 0.5 mm EDTA, 0.5 mm EGTA, 1 mm DTT, and protease inhibitors ((N-(N-(N-acetyl-l-leucyl)-l-leucyl)-l-norleucine, PMSF, leupeptin, pepstatin, and aprotinin). Cells were then incubated on ice for 10 min in 1 ml of swell buffer and then scraped off of the dishes and passed through a syringe 15 times using a 22-gauge needle. After centrifugation at 1,000 × g for 10 min, the pellet containing total membranes was collected and analyzed using SDS-PAGE (8.5% acrylamide) and Western blotting using anti-FLAG and anti-calnexin antibodies.
RESULTS
Luminal Calcium Depletion Inhibits ER-to-Golgi Transport of VSV-G-GFP
The role of Ca2+ in ER-to-Golgi transport has been controversial (23, 42), and a specific role of luminal Ca2+ has not been addressed. We previously developed conditions for the specific depletion of luminal ER Ca2+ in living normal rat kidney (NRK) cells using CPA (43), a reversible inhibitor of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump. By employing calibrated Ca2+ sensors in living cells, we determined that luminal ionic ER Ca2+ could be reduced by more than 90% from 385 to 23 nm while maintaining resting cytosolic Ca2+ at 50 nm (10); this established the specificity of biological effects for luminal as opposed to cytosolic or total Ca2+. Furthermore, by 30 min, ER-derived pre-Golgi organelles should be depleted as well. Thapsigargin, a chemically unrelated irreversible SERCA inhibitor, produced similar phenotypes.
To test whether luminal Ca2+ was involved in ER-to-Golgi transport, we used the CPA protocol in conjunction with a morphological transport assay employing VSV-G-GFP ts045, which exhibits thermo-reversible assembly and transport (44). Cells were fixed at defined time points following the temperature shift, and a transport index was calculated by image analysis on dozens of cells. The transport index was a robust parameter unaffected by variations in Golgi architecture and cargo expression level (39). As shown in Fig. 1A (left plot), Ca2+ depletion slowed ER-to-Golgi transport of total VSV-G-GFP by 65–75%. Thus, luminal Ca2+ is required for efficient transport in the early secretory pathway. Example wide field fluorescence images are displayed in Fig. 1B. The transport defects observed did not result from effects on cargo folding or assembly because the effect was still pronounced after quantitation using a VSV-G trimer-specific monoclonal antibody (38) (Fig. 1A, right plot). As shown in Fig. 1C, VSV-G trimerization was very rapid and occurred within the first few min whether Ca2+ was present or not. In conjunction with the fact that cycloheximide was employed during transport experiments, these results indicate that the appearance of newly formed trimers did not complicate transport kinetics. As an alternative approach, the inhibition of ER/Golgi transport observed using the morphological assay described above was confirmed using a biochemical transport readout, the acquisition of resistance of VSV-G-Myc ts045 to endoglycosidase H cleavage (Fig. 1D).
A recent study found that activation of the unfolded protein response (UPR) using long term thapsigargin treatment inhibited ER export and led to Golgi dispersal (45). We avoided stress responses by minimizing the CPA treatment and by maintaining cytosolic Ca2+ at resting levels by using low Ca2+ medium to limit capacitative Ca2+ uptake. To confirm that our conditions did not lead to UPR activation, we immunoblotted control and treated cell lysates for the transcription factor ATF6. As shown in Fig. 1E, ATF6 was not cleaved as a result of our protocol. Hence, the effects we observed on transport reflect a direct requirement for luminal Ca2+ in transport.
To examine whether the requirement for luminal Ca2+ extended to an endogenous transmembrane cargo, we monitored the ER-to-Golgi transport of mature Golgi mannosidase II. This cargo was not newly synthesized but rather diverted to the ER by the addition of brefeldin A; its return to the Golgi was then monitored following brefeldin A washout. ER-to-Golgi transport of mannosidase II was initially severely impaired, whether monitored using the transport index method that measures the relative Golgi area intensity (Fig. 2A) or an algorithm that relies on the cross-sectional area of coalescing Golgi structures (Fig. 2B). These data expand our conclusions to an endogenous cargo whose folding and assembly preceded that of Ca2+ depletion. The data in Figs. 1 and 2 are the first functional demonstration that luminal Ca2+ plays a required role for efficient transport in pre-Golgi secretory trafficking.
Luminal Calcium Depletion Causes Accumulation of ERES and IC Markers in Buds and Clusters of Unfused Vesicles
We previously demonstrated that CPA treatment caused an enlargement of peripheral fluorescent spots labeling IC proteins, such as the SNARE Rbet1 and cargo receptor p24. On the other hand, the distribution of Sec16A, a COPII scaffold that does not enter vesicles (46), was not affected by luminal Ca2+ depletion (10). These findings were consistent with the enlarged spots representing aberrantly large early VTCs or abortive vesicle buds. To try to understand the architectural basis of this phenotype, we asked whether the COPII coat itself accumulates on the CPA-induced structures. As shown in Fig. 3A, CPA treatment resulted in a de-emphasis of pericentriolar ribbon-like labeling and an intensification and enlargement of peripheral COPII outer shell Sec31-positive puncta, where it co-localized with Rbet1. The inner shell component Sec24c displayed a similar behavior (Fig. 3B). Hence, the COPII coat accumulates on the CPA structures, in contrast to what had been observed for Sec16A. However, it is still not clear what precise structures the coat and IC markers accumulate on.
To identify the membrane structures that accumulate COPII and IC markers upon luminal Ca2+ depletion and to provide an architectural basis for the secretion defect, we performed correlative light and electron microscopy. CPA-treated cells were fixed, cryosectioned by the Tokayasu technique with modifications (41), placed on EM grids, and then labeled with an anti-Sec31 antibody followed by both anti-rabbit IgG-Alexafluor-488 and 10-nm protein A-gold to decorate the enlarged fluorescent spots and Hoechst to label nuclei. Sections were then imaged by wide field deconvolution microscopy, which revealed enlarged cytoplasmic spots very reminiscent of those observed in treated whole fixed cells as in Fig. 3. Subsequent imaging of the same quadrants of the grids by low magnification electron microscopy allowed identification and alignment of landmark structures, such as the unique shapes of nuclei and extracellular spaces when the two types of images were overlaid (data not shown). It was thus possible to establish an unambiguous correlation between the enlarged fluorescent spots decorated by Alexafluor-488, 10-nm gold particles, and the underlying membrane architecture. Fig. 4, A and C, shows fluorescence-overlaid electron micrographs, with Fig. 4, B and D, representing higher magnification electron micrographs of the boxed regions in A and C. In these representative peripheral locations lacking Golgi stacks, the labels were associated mainly with a preponderance of COPII buds as well as with clusters of unfused vesicles. Surprisingly, no enlarged or distended structures were observed, and VTCs, defined as apparent fusion products of two or more vesicles, were rare. The abundant buds emerging from the ER did not appear unusually large or elongated as would be expected for a membrane scission defect. Some vesicles near budding sites (marked with a red arrow in Fig. 4D) seemed tethered together into a cluster. Most gold particles were clearly associated with a visible membrane, whereas other gold particles were not (e.g. see Fig. 4D, red asterisks). Because these gold particles were nearly always found near an ERES, however, this label is either associated with membrane that is tangentially out of section or else represents Sec31 that has recently dissociated from a bud or vesicle. A highly useful feature of the immunofluorescence/EM correlation is the ability to identify the membranes comprising the CPA structures even when gold particles are sparse. For example, in Fig. 4C, gold particles are seen in only a subregion of a micrograph, but nevertheless the vesicles and membrane buds in a more extensive region correlated well with swaths of fluorescent labeling (Fig. 4C, white arrowheads). Altogether, we conclude that the CPA-induced, COPII-positive enlarged spots, as seen by immunofluorescence, are not, as previously speculated (10), enlarged VTCs or expanded/abortive budding structures but rather represent a collection of membrane buds and vesicles.
Based upon the ultrastructural analysis, CPA either caused an increase in buds and vesicles or alternatively redistributed coat and IC marker proteins from less and/or more mature structures to make them more concentrated on buds and vesicles. These possibilities are not mutually exclusive. The absence of luminal Ca2+ could have caused poor concentration of cargo in nascent vesicles, inefficient sorting of anterograde and retrograde cargo in VTCs, and/or increased vesicle back-fusion, leading to futile stimulation of the budding of vesicles with lower fusion competency due to an imbalance of docking/fusion machinery. Such phenomena would be predicted to both increase the density of active buds and clustered vesicles and cause excessive accumulation of COPII and IC markers at these sites.
ALG-2 Interactions with the Sec31A Proline-rich Region Are Functionally Critical for Transport
Studies of the role of Ca2+ in the COPII pathway have focused on interactions of Sec31A with the Ca2+-activated adaptor protein ALG-2 (10, 32, 33, 35, 47); however, a functional role for these interactions in intact cells has not been demonstrated. We tested the hypothesis that this interaction was functionally significant by co-overexpression studies in NRK cells combined with ER-to-Golgi transport assays. First, we expressed the isolated PRR of Sec31, residues 800–1113, containing the ALG-2 binding site (32), in NRK cells to potentially compete with endogenous Sec31A for its binding partners, including that for ALG-2. Overexpression of PRR-FLAG itself mildly inhibited transport (Fig. 5A, bars 2 and 3 versus bar 7). Likewise, overexpression of wild type ALG-2 did not consistently inhibit transport (Fig. 5A, bar 4). Surprisingly, however, simultaneous overexpression of both proteins created a strongly synergistic inhibition of transport of ∼60% (Fig. 5A, bar 8). The two overexpressed proteins together create a uniquely interpretable dominant negative inhibitor; whereas inhibition by either protein alone could be a consequence of titrating out endogenous, functional Sec31 and/or ALG-2, the apparent requirement for overexpressed ALG-2 to bind and place or activate the PRR inhibitor illustrates their specific interaction at a functional, saturable site. That the two must interact is reinforced by the functional inactivity of the ALG-2 F60A mutant (Fig. 5A, bar 9), which lacks interactions with Sec31 but not other ALG-2 effectors (48). Reciprocally, a PRR peptide with a deletion of the 13-amino acid ALG-2 binding site (PRR ΔABS) was also much less active when co-expressed (Fig. 5B). Hence, although ALG-2 inevitably has multiple binding targets in the cell, Sec31A binding must be involved in the observed effects. In addition, both constructs and the mutants are well expressed (Fig. 5C). Interestingly, the inactivity of the ALG-2 ΔGF mutant (Fig. 5A, bar 10), which reportedly exhibits Sec31 binding but fails to bind several other ALG-2 targets (48), implies that a distinct ALG-2 target may be involved, as predicted by current models of PEF protein function, wherein an ALG-2 dimer cross-links and stabilizes the otherwise weak association of two effectors (47). This scenario is also consistent with the in vitro activity of ALG-2 to kinetically stabilize Sec31A at or near ERES and on COPII vesicles (10, 32, 33, 35), which implies that ALG-2 cross-links Sec31A to an “anchor” site.
To further demonstrate that the two overexpressed proteins functioned as an inhibitory complex, we performed immunoprecipitations from NRK cell extracts. As shown in Fig. 5D, the transfected human ALG-2 construct ran slightly slower than endogenous rat ALG-2 on SDS gels of CHAPS extracts containing ambient calcium (lane 1 versus lanes 2–5). When the extracts were immunoprecipitated with anti-FLAG antibodies, ALG-2 was specifically co-precipitated, but only in extracts co-transfected with both constructs (lane 6 versus lane 7). Furthermore, co-expression and immunoprecipitation of FLAG-PRR 800–976, a minimal inhibitory fragment containing the ALG-2 binding site but lacking the binding sites for Sec16 and Sec23 (see below; Fig. 9), also robustly interacted with ALG-2 in cell extracts (Fig. 5D, lane 8). Finally, the much less inhibitory FLAG-PRR ΔABS lacking the major 13-amino acid ALG-2 binding site interacted much less efficiently (lane 9). The small residual co-precipitation and slight inhibitory activity (Fig. 5B, last bar) may be attributable to a second ALG-2 binding site (residues 869–884) that may contribute to minor binding in the absence of the 839–851 site (32). That the constructs exist as a calcium-dependent complex in living cells, as opposed to forming in cell extracts, is definitively demonstrated by the complete dependence of co-immunoprecipitation upon prior treatment of living cells with the reversible cross-linker dithiobis(succinimidyl propionate), when the extracts are produced using chelator-containing extraction buffer (lane 12 versus lane 13).
FIGURE 9.
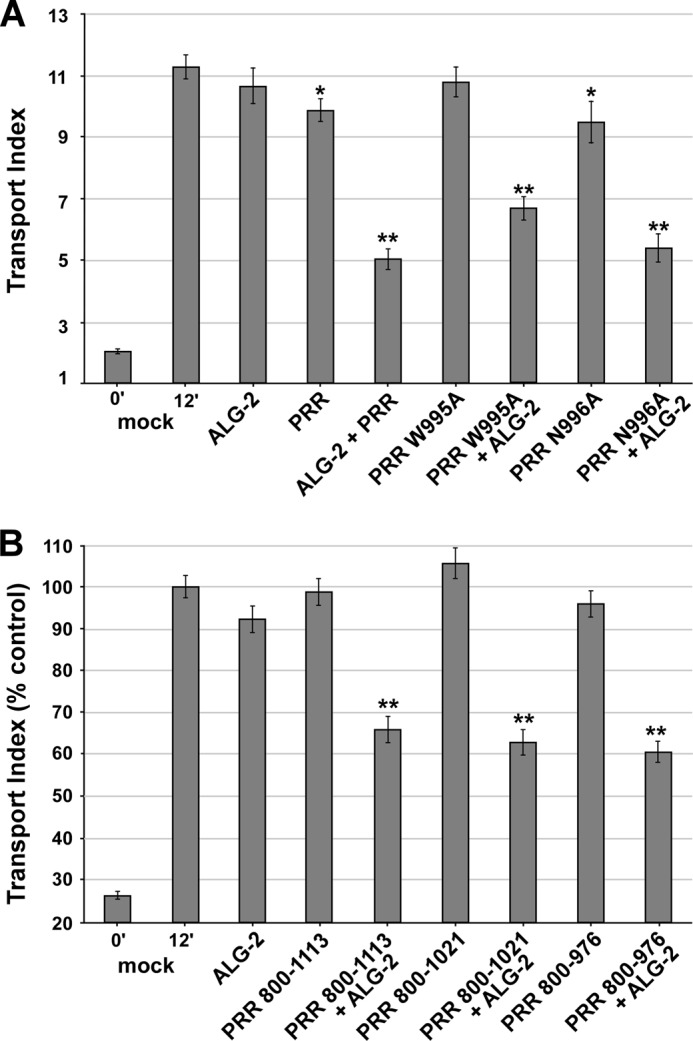
ALG-2/Sec31-PRR inhibits transport independently of known interactions with Sec16, Sec23, or Sar1. A, as in Fig. 5A but employing FLAG-Sec31-PRR residues 800–1113 (PRR) or the indicated PRR construct mutants. B, as in A but employing PRR deletion constructs as indicated rather than point mutations. Asterisks, p values for two-tailed t tests for the indicated value versus the 12-min mock control; *, p < 0.05; **, p < 0.01. Shown are averages of three independent replicates of each experiment. Each bar represents ≥90 cells. Error bars, S.E.
The observed inhibitory complex displays a preference for the exogenous ALG-2 (Fig. 5D, lanes 7, 8, and 13; note the prominence of the upper ALG-2 band compared with the lower one). This preference seemed stronger than that expected from the ∼4:1 exogenous/endogenous ALG-2 cellular concentration ratio (calculated from the roughly equal abundance of the two bands in extracts and our ∼25% electroporation efficiency of NRK cells). Thus, the functional synergy produced by co-overexpressing ALG-2 with PRR is probably not merely a result of mass action driving more ALG-2 into inhibitory ALG-2·PRR complexes. Rather, it may arise from the timing of exogenous versus endogenous ALG-2 expression because endogenous ALG-2 may be less available to form complexes with the newly expressed PRR due to established interactions at multiple other sites in the cell. Alternatively, it may arise from slight structural differences in the human ALG-2 that either increase its binding affinity for the PRR or decrease its affinity for other competing ALG-2 binding sites in the rat cells. In either case, the preference for interaction with exogenous ALG-2 precisely correlates with the functional inhibition, further indicating that the complex functions as the inhibitor, and provides a molecular explanation for why PRR does not inhibit efficiently when expressed by itself, despite the presence of endogenous ALG-2. Taken together, the data of Fig. 5, A–D, represent the first evidence in living cells that the interaction of ALG-2 with Sec31 can regulate ER-to-Golgi transport.
ALG-2/Sec31A-PRR Interactions Acutely Regulate Cargo Receptors and Tethers but Exert Minor Effects on Coat Localization
To better understand the functional significance of disrupted endogenous ALG-2/Sec31-PRR interactions, we needed to examine the ALG-2·PRR inhibitor targeting sites relative to endogenous cellular markers. An RFP-ALG-2 construct was created. RFP-ALG-2 also produced a synergistic inhibition with Sec31-PRR similarly to wild type ALG-2 (Fig. 5D). Fig. 6, A and B, illustrates the morphological change in VSV-G-GFP localization during transport and demonstrates that Sec31A is unperturbed by VSV-G-GFP transport. We next asked whether the RFP-ALG-2 construct localized to ERES, as indicated by COPII markers Sec31A (outer shell) and Sec24C (inner shell). As shown in Fig. 6, C and D, the RFP-ALG-2 construct was present in cytoplasmic puncta, a high percentage of which co-localized precisely with ERES marked by both Sec31A (Fig. 6C, Merge) and Sec24C (Fig. 6D, Merge). However, the vast majority of ERES lacked concentrated ALG-2-RFP, indicating that a small subset of ERES were much more heavily targeted. We then asked what happens to the coat when RFP-ALG-2 and FLAG-PRR are co-expressed. As shown in Fig. 6, E and F, RFP-ALG-2 in these triply transfected cells had a similar localization as before; however, VSV-G-GFP was retained in the ER and/or other peripheral sites. The FLAG-PRR construct was highly expressed and displayed a nonselective localization. Cytoplasmic puncta were visible in some cells, but these never co-localized with secretory pathway markers, indicating that the fraction of FLAG-PRR targeted to ERES may have been very small. We carefully examined ERES markers in the triply transfected cells. Endogenous Sec31A decorated with an N-terminal antibody that does not recognize the PRR region was surprisingly unperturbed; the ratio of cytosolic to particulate Sec31A remained about the same, and the number and size of ERES spots were not noticeably altered (Fig. 6F, middle, compare cell with arrows with surrounding cells). However, upon close inspection, the particular subset of ERES highly enriched in RFP-ALG-2 were usually depleted of Sec31A, resulting in less co-localization of RFP-ALG-2 and Sec31A (Fig. 6, contrast C (Merge) with F (Merge), arrows). A similar result was found for the inner shell component Sec24c (Fig. 6, D and E). The co-localization results for RFP-ALG-2 and COPII subunits Sec24c and Sec31A are quantified from multiple cells in Fig. 6G.
So far, these results support two conclusions. First, despite the transport block, the targeting of COPII to membranes and ERES per se was not generally affected by the inhibitors. Hence, displacement of the coat from the membrane and ERES was not necessary to produce the inhibitory effect because the majority of VSV-G-GFP transport was blocked, but only a small subset of ERES displayed bright RFP-ALG-2 spots and coat displacement (whereas 30–50% of bright RFP-ALG-2 spots co-localized with COPII markers, a much smaller fraction of COPII spots co-localized with RFP-ALG-2). Rather, it seems more likely that the inhibitory effect was due to selective disruption of ALG-2/Sec31A interactions at relatively low concentrations of inhibitor that are not apparent as accumulations by immunofluorescence and that the functionally disrupted ERES contain a normal amount of coat. This conclusion is consistent with the continued presence of COPII proteins on buds and vesicles upon luminal Ca2+ depletion (Figs. 3 and 4), another manipulation that should disrupt ALG-2/Sec31A interactions. However, because we did not directly examine ALG-2/Sec31 interactions at these sites, this conclusion is tentative. The second conclusion is that at the highest concentration of inhibitor present at a relatively small percentage of ERES, a fundamental disruption of ERES structure, surprisingly including both inner and outer coat displacement, seems to have occurred. At these sites, the inhibitor present in many copies may have nonselectively interfered with scaffolding and/or linking interactions mediated through the PRR that are necessary to nucleate multiple ERES components.
The effects of the inhibitors on COPII prompted us to examine other ERES, VTC, and Golgi components. p24 is a type I integral membrane protein required for ER/Golgi transport most likely as a cargo receptor and/or as a coat recruitment accessory protein (49–51); at steady state, it localizes to the early Golgi, VTCs, and ERES. As shown in Fig. 7, A and B, p24 displayed its normal pericentriolar localization in cells transfected with either RFP-ALG-2 (Fig. 7A) or FLAG-PRR (Fig. 7B). In addition to the strong Golgi area labeling, p24 was also present in peripheral spots that co-localized with the RFP-ALG-2 construct (Fig. 7A, merge). Much to our surprise, when the two inhibitor constructs were co-expressed, p24 labeling was strongly reduced or ablated. This is demonstrated in Fig. 7C (Merge), where the cell on the left lacks transfection with RFP-ALG-2, transported VSV-G-GFP efficiently and displays a yellow-containing Golgi area, whereas the triply transfected cell on the right displays a cytoplasmic build-up of cargo and a green-only Golgi area. The p24 labeling pattern in ∼50 cells from each condition was quantified in Fig. 8 (left two groups of columns), further demonstrating a dramatic shift toward ablation and/or dispersion of p24 upon ALG-2·RFP-PRR expression. Furthermore, this effect did not require the RFP moiety because it was observed using untagged ALG-2 as well (Fig. 8, far right group of columns). One justification for the ablation of p24 could be ablation of the Golgi itself. However, Fig. 7D illustrates that GPP130, a cis-Golgi integral membrane protein (52), is localized normally in transport-inhibited cells. One possibility is that p24, which requires a phenylalanine motif for binding to Sec23 (53, 54) directly and/or indirectly interacts with the PRR region of Sec31A near the ALG-2 binding site and/or with ALG-2 itself and that this interaction is required for the proper localization and physical stability of p24. Other manipulations that block ER-to-Golgi cargo transport, such as BFA treatment, cause dispersal but not ablation of p24 (data not shown). That p24 was degraded in an acidic compartment as opposed to otherwise masked from immunofluorescence was confirmed by the addition of the lysosomal protease inhibitors leupeptin and aprotinin or by neutralization of acidic compartments with chloroquine, which largely prevented p24 ablation in cells co-expressing the RFP-ALG-2 and PRR (Fig. 8, third and fourth groups of columns).
FIGURE 8.
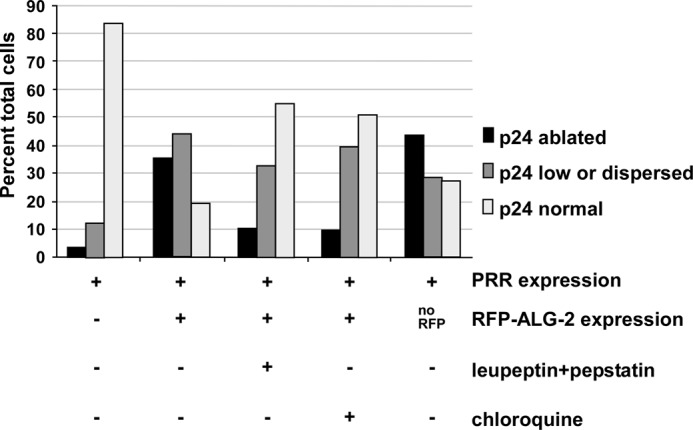
ALG-2·Sec31-PRR causes ablation of p24 by proteolytic degradation in an acidic compartment. Cells were transfected as indicated and then incubated for 8 h in regular medium or medium containing 100 μm leupeptin + 20 μm pepstatin or containing 100 μm chloroquine prior to fixation and immunofluorescence microscopy. At least 50 cells from each experimental condition were imaged, and each cell's p24 was scored (conditions were blinded, and images were randomly interspersed) as either normal, partially disrupted, or ablated. Data shown are from a single experiment but are qualitatively similar to several other experiments obtained under similar conditions. The condition for the fifth group of bars (no RFP) employed a wild type untagged ALG-2 construct, demonstrating that the p24 phenotype was not dependent upon the RFP tag.
We wondered whether the structure of the IC and stability of other vesicle machinery were similarly affected. We examined p58, another cargo receptor that rapidly recycles between the ER and Golgi (Fig. 7E), and Rbet1, an ER/Golgi SNARE with similar dynamics (Fig. 7F). In triply transfected cells displaying the expected transport delay, both of these markers did not change significantly compared with non-transfected neighbor cells. However, there may have been a slight intensification of peripheral spots reminiscent of CPA treatment (10), and there appears to have been a displacement of these proteins from the particular ERES most loaded with inhibitor (particularly for Rbet1; arrows in Fig. 7F, merge). The results with p58 and Rbet1 demonstrate that the overall structure and molecular composition of the IC was normal, further emphasizing the specificity of the dependence of p24 on Sec31/ALG-2 interactions.
We also examined the localization of a peripheral membrane tether protein, p115. p115 appears to be packaged in budding COPII vesicles (15), is essential for their fusion (14), and rapidly cycles on and off the membrane (55) presumably as a function of SNARE availability. In triply transfected cells displaying delayed transport, p115 distribution was noticeably altered (Fig. 7G). Rather than displaying mostly an intense Golgi area concentration, p115 in these cells was visible throughout the cytoplasm. Much of the peripheral p115 labeling had a hazy appearance, suggesting cytosol. However, there were a few instances where overlap could be seen between the arrested VSV-G-GFP and p115, suggesting that at least some of the p115 became concentrated at inhibited ERES (Fig. 7G, Merge). The displacement of p115 by the inhibitor constructs suggests that the on/off membrane dynamics of this protein was highly dependent upon Sec31A/ALG-2 interactions.
To summarize, the morphological results suggest unexpectedly that targeting of Sec31A on the membrane per se is not the primary functional locus of ALG-2/Sec31A interactions; rather, these interactions may selectively regulate specific transport machinery, such as cargo receptors and tethers, at ERES.
Sar1-GAP Potentiation and Sar1, Sec23, or Sec16 Binding Are Not Required by the ALG-2·PRR Inhibitor
The Sec31A PRR domain (residues 800–1113) can be visualized as an unstructured chain connecting and perhaps nucleating a number of important ERES components. ALG-2 binding (residues 839–851) (32) is the most N-terminal known interaction, followed by a region (residues 981–1015) that contacts both inner shell components Sec23 and Sar1 (1), followed by a region (not precisely known but C-terminal to the others) that binds the coat scaffold Sec16 (56), which also independently interacts with both inner and outer shell coat layers (56). ALG-2/Sec31A interactions might regulate Sec31 interactions with its binding partners along this chain. For example, one possibility was that ALG-2 helped position the Sec31-PRR onto the Sar1·Sec23 complex, where PRR residues Trp-995 and Asn-996 insert into the Sar1 active site and potentiate Sec23 GAP activity. If this were the case, the inhibitor peptide could work by “falsely” stimulating GAP activity of Sar1. We mutated Sec31A-PRR residues Trp-995 and Asn-996 individually to Ala; both mutations have been shown to inactivate GAP potentiating activity (1). When combined with ALG-2 overexpression, both peptides produced strongly synergistic inhibitions (Fig. 9A), indicating that the GAP potentiating activity was not required for the disruption of transport.
To systematically test whether PRR interactions along the chain are required for the functional inhibition, we truncated the FLAG-PRR 800–1113 from the C-terminal end. The PRR 800–1021 construct eliminates known interactions with Sec16. The PRR 800–976 construct eliminates known interactions with Sec16, Sec23, and Sar1. As shown in Fig. 9B, both PRR 800–1021 and PRR 800–976 retained full inhibitory potential compared with the full-length PRR 800–1113 construct. Note that in the presence of excess PRR 800–976, endogenous Sec31A, Sec23, Sar1, and Sec16 can, in principle, still interact through all of their known binding sites, implying that the inhibitor functionally acted by disrupting interactions between Sec31A and ALG-2 itself rather than by directly competing with other binding partners of the PRR. One possibility consistent with the data is that ALG-2/PRR interactions activate PRR interactions with its downstream binding partners by virtue of ALG-2-induced PRR structural changes. Indeed, recent in vitro studies demonstrated that the purified Sec31A·Sec13 complex bound to Sec23 only in the presence of ALG-2 and Ca2+, yet ALG-2 itself did not interact with Sec23 (36). A structural mechanism of this effect mediated in the context of a proline-rich loop domain remains to be elucidated. However, our experiments demonstrate the regulatory significance for the ALG-2/Sec31A interaction in vivo. Furthermore, our mutagenesis experiments rule out the less incisive interpretation that the targeted PRR construct merely competes with, displaces, and/or directly dysregulates the other binding partners of Sec31A. Thus, it appears that proper ALG-2/PRR interactions per se are required for transport. Although the functionally critical step(s) affected by this interaction remains unknown, several possibilities suggested by in vitro studies include the timed attenuation of budding to allow proper vesicle filling (36), proper on/off dynamics of Sec31A at ERES (32), regulation of cargo receptor p24 stability and trafficking (Fig. 7), regulation of p115 distribution (Fig. 7), or the timing of vesicle uncoating (10) and fusion.
ALG-2/Sec31A Interactions and Sec31A Function in Transport Require Luminal Calcium
To strengthen the link between the luminal calcium requirement for transport (Figs. 1–4) and the effects of ALG-2/Sec31A interactions on transport (Figs. 5–9), we examined the luminal calcium dependence of ALG-2 and Sec31A function using gene silencing and overexpression combined with CPA treatments. First, we examined the functional effects of ALG-2 depletion from NRK cells using siRNA. Using three equally efficacious non-overlapping siRNAs, the best experiments achieved greater than 95% depletion of ALG-2, as judged from immunoblots of NRK cell extracts (data not shown). In these experiments, we observed a statistically significant ∼13% inhibition of VSV-G-GFP transport (background-subtracted mock siRNA transport value, 8.21 ± 0.32; ALG-2 siRNA 1 transport value 7.12 ± 0.32; p = 0.0192; n = 90 cells/condition; background transport value without incubation at 32 °C = 2). Similar effects were observed with all three siRNAs. Although suggestive, these results fall short of demonstrating more than an ancillary role for ALG-2 under steady-state conditions. In addition, quantitative immunoblotting of a parallel set of knockdown experiments revealed that ALG-2 depleted cells displayed a ∼50% increase in Sec31A abundance relative to other constitutively expressed cytosolic proteins (Fig. 10A; quantified in Fig. 10B), suggesting a potential compensatory mechanism that could help account for the mild transport phenotype. Compensatory Sec31A overexpression would also support a positive functional role for ALG-2 in trafficking but does not provide quantitative functional information. Therefore, other components of the proposed effector pathway were targeted.
FIGURE 10.

Sec31A function in transport and modulation of transport by ALG-2/Sec31A interactions require luminal Ca2+. A, knockdown of ALG-2 and Sec31A by siRNA. Western blotting of treated cell extracts with the indicated antibodies demonstrates specificity of the depletions. B, quantitation of replicate samples and Western blots as in A, displaying relative amounts of the five indicated proteins in mock- or siRNA-treated samples. To compare and combine band intensities from multiple samples, each protein amount was normalized to the amount of the constitutively expressed cytosolic protein, TRAF2, in the same lane and then expressed relative to the mean normalized value obtained in mock-treated samples. C, ER-to-Golgi transport of VSV-G-GFP following siRNA and/or CPA treatment as indicated. siRNA-treated or mock-treated cells were transfected with VSV-G-GFP and incubated overnight at 40 °C. After ∼24 h, cells were either left in control medium (+Ca2+) or treated with CPA at 40 °C as in Fig. 1 (−Ca2+/CPA) and then shifted to 32 °C for the indicated times. Cells were then fixed, and the arrival of VSV-G-GFP in the Golgi area was quantified using the transport index algorithm. Each bar represents the mean of ≥90 cells from three independent experiments that were averaged for the graph. The inset shows the 12- and 3-min groups' transport indices, normalized such that their own mock-treated value equals 100%. D, ER-to-Golgi transport of VSV-G-GFP upon overexpression of the ALG-2 and Sec31A-PRR inhibitor constructs and/or CPA treatment. NRK cells were transfected with VSV-G-GFP along with no additional construct, ALG-2 wild type, and/or a FLAG-Sec31-PRR construct consisting of residues 800–1113, as indicated, and held at 40 °C. After ∼24 h, cells were either left in control medium or treated with CPA at 40 °C as in Fig. 1 and then shifted to 32 °C for the indicated times. Cells were then fixed and immunolabeled to identify which cells expressed the constructs, and the arrival of total VSV-G-GFP in the Golgi area was quantified using the transport index algorithm. Each bar represents ≥90 cells from three independent experiments that were averaged for the graph. Asterisks, p values for two-tailed t tests for the indicated value versus its corresponding mock-treated value. *, p < 0.05; **, p < 0.01 (B) or p < 10−8 (C and D); ns, p > 0.05. Error bars, S.E.
siRNA-mediated depletion of Sec31A by over 85% caused an ∼50% inhibition of VSV-G-GFP transport, using the transport index assay (Fig. 10C, bar 1 versus bar 2), similar to the inhibition caused by depletion of the ER/Golgi SNARE syntaxin 5 (Fig. 10C, bar 2 versus bar 3) (39). It was somewhat surprising that significant VSV-G-GFP transport occurred in the virtual absence of Sec31A because the outer shell is often described as an essential element of COPII vesicle formation. One possibility would be that another gene provides some minimal outer shell function. The Sec31B gene product has not been demonstrated to function in ER/Golgi transport. If Sec31B were expressed and provided some outer shell function, it might involve a very different mechanism of coordination with inner shell components because Sec31B exhibits its lowest sequence similarity to Sec31A in the PRR region and contains no consensus ALG-2 binding sites (not shown). Another possibility to explain the residual transport in the absence of Sec31A is that some cargo transport is possible using primarily or entirely inner shell components, at least for the very efficient cargo VSV-G. Indeed, the inner shell component Sec23 was significantly elevated in Sec31A knockdown cells (Fig. 10, A and B), suggesting an artificially enhanced role for the inner shell in the absence of Sec31A. Upon luminal Ca2+ depletion using the CPA protocol, transport was severely inhibited in control NRK cells (Fig. 10C, bar 1 versus bar 4). Interestingly, however, transport was not further inhibited by Sec31A depletion, apparently indicating that Sec31A does not contribute to transport in the absence of luminal Ca2+ (Fig. 10C, bar 4 versus bar 5). This was not due to the transport index algorithm being less sensitive at small values or a non-physiological transport mechanism because syntaxin 5 depletion caused an easily detectable inhibition in the absence of luminal Ca2+ (Fig. 10C, bar 4 versus bar 6) and because Sec31A depletion caused an easily detectable inhibition in the presence of Ca2+ but at time-limited increments of transport (Fig. 10C, bar 7 versus bar 8). In fact, if the background-subtracted transport indices for the normal, CPA-depleted, and time-limited groups are each normalized to their own positive control, we found that the knockdowns inhibited to a similar degree under all conditions except Sec31A depletion after CPA treatment (Fig. 10C, inset). Ca2+ dependence of Sec31A function has not been observed before; this novel finding provides a potential basis for the observed requirement for luminal Ca2+ in ER-to-Golgi transport (Figs. 1–4) and is strongly suggestive of an important role for ALG-2/Sec31A interactions because ALG-2 is the only known Ca2+ binding protein with which Sec31A interacts.
If indeed ALG-2/Sec31A interactions contribute to Sec31- and Ca2+-dependent traffic, then luminal Ca2+ depletion should also negate the transport inhibition caused by dominant-negative disruption of ALG-2/Sec31A interactions. Indeed, as shown in Fig. 10D, the inhibition caused by ALG-2·PRR overexpression was eliminated following CPA treatment. Likewise and perhaps more importantly, in the presence of ALG-2·PRR overexpression, the further effect of CPA addition was very minimal (Fig. 10D, last bar in the first group versus the last bar in the second group). Hence, luminal Ca2+ and the ALG-2·PRR disruption depend upon each other to effect transport, implying that the protein interactions disrupted by the inhibitors at least in part account for the luminal Ca2+ requirement. This experiment, when combined with the above results, strongly implies an important role for ALG-2/Sec31A interactions in luminal Ca2+-dependent trafficking from the ER to Golgi. However, more direct experiments will be required to elucidate the precise mechanisms.
DISCUSSION
A Model for Luminal Calcium and ALG-2/Sec31A Interactions in ER-to-Golgi Transport
Here we demonstrate that luminal Ca2+ is required post-cargo folding for ER-to-Golgi transport. Previous studies employing chelators and/or in vitro reconstitutions did not address whether luminal Ca2+ pools, as opposed to resting cytosolic Ca2+, was involved (17, 23, 42). Likewise, previous studies using SERCA inhibitors would have resulted in activation of UPR. Although UPR may additionally regulate ER/Golgi transport machinery (45, 57, 58), our results identify a more direct involvement of luminal Ca2+.
A leading candidate mechanism of action of Ca2+ in ER-to-Golgi transport is regulation of coat dynamics by the Ca2+ adaptor protein ALG-2 (32, 33, 35, 47). However, the functional role of ALG-2 has been difficult to assess, perhaps due to functional redundancy of PEF proteins (59) or cellular compensatory mechanisms (this work). Here we present evidence that ALG-2 targets the Sec31 PRR to a functionally critical, saturable site. Overexpression of reagents designed to disrupt ALG-2/Sec31A interactions blocks transport, implying a required role. Furthermore, luminal Ca2+ is required for this interaction to modulate transport and for Sec31A function in transport, strongly implying that Ca2+-dependent ALG-2/Sec31A interactions contribute to the Ca2+ dependence of ER/Golgi transport. Our data are compatible with the model shown in Fig. 11, wherein escaping luminal Ca2+ regulates ALG-2/Sec31A interactions with each other and an unknown membrane anchor site (“M”). This model conforms to the current paradigm that PEF proteins act as Ca2+-dependent adaptors to stabilize otherwise weak complexes (47). The ALG-2/Sec31/M interaction could directly regulate Sec31A interactions with other components, such as inner shell components (36), cargo receptors, and tethers, and/or directly regulate Sec31 on/off dynamics, including uncoating (10). Our data argue that the localization of Sec31A to ERES, per se, is not a consequence of ALG-2 interactions because a very significant transport block was usually accompanied by very limited effects on coat localization. However, our data do support a role for the Sec31A PRR region as an integrator of ERES components because, at sites where the ALG-2·PRR inhibitor targeted with the highest copy number, several ERES components, including both the inner and outer COPII coat and Rbet1, were displaced.
FIGURE 11.
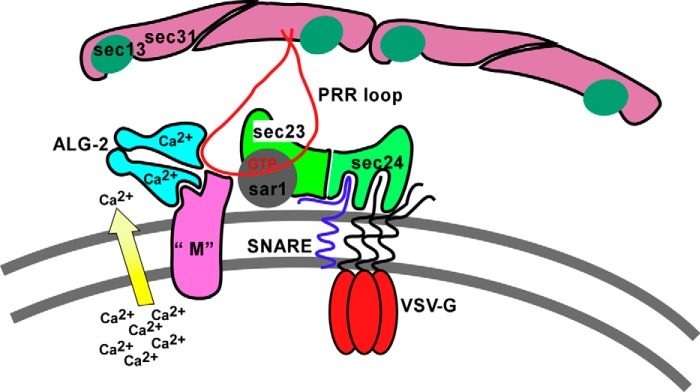
Model for action of luminal Ca2+, ALG-2, and COPII components. Component “M” represents an unknown membranous site to which ALG-2 targets the PRR loop (shown in red extending downward from Sec31A) in response to escaping luminal Ca2+.
In an earlier work (10), we reported that Ca2+-dependent ALG-2 interactions stabilized Sec31A on isolated vesicles in vitro and inhibited their homotypic fusion. This finding highlighted the question of whether Ca2+-dependent mechanisms were required for as opposed to inhibiting the overall transport process. Likewise, recent in vitro budding studies found that Ca2+-dependent ALG-2/Sec31A interactions attenuated vesicle budding with purified components (36). Once again, the in vitro model did not illuminate whether this regulatory action of ALG-2 at an isolated step was required for proper transport in cells (e.g. for correct timing of vesicle scission). The present work helps to clarify the physiological role of Ca2+ and ALG-2/Sec31A interactions. First, it demonstrates a required role for luminal Ca2+ in membrane cargo transport, with Ca2+ depletion leading to slowed cargo transport accompanied by dense accumulations of both COPII buds and unfused vesicles. Second, it demonstrates that abrogation of Ca2+-dependent ALG-2/Sec31A interactions inhibits transport, implying a required role for these Ca2+-dependent interactions. Third, it demonstrates that Sec31A function in transport requires luminal Ca2+, again implying a required, overall positive role for Ca2+ and implicating ALG-2, as the only known Ca2+-binding partner of Sec31A, in that positive role. However, because our results did not pinpoint the precise functional step affected by ALG-2/Sec31A interactions, several potential mechanisms of action, including attenuation-based mechanisms, remain possible.
The potential functional consequences of ALG-2/Sec31A interactions for ER-to-Golgi transport are severalfold. The interactions may facilitate vesicle budding and cargo loading. Cargo loading is only beginning to be understood and appears to involve subtly timed interactions by multiple ERES components to properly regulate vesicle size and content (60, 61). The functional locus of the ALG-2/Sec31A interaction could also be shortly after vesicle formation. Stabilization of the outer coat on budded vesicles by ALG-2 can restrict their fusion (10), which could limit back-fusion with the Ca2+-rich ER and/or coordinate fusion with lower-Ca2+ mature VTCs. Alternatively, by delaying complete uncoating and contributing to the immobilization of Sec23/24, postbudding ALG-2/Sec31A interactions could create anterograde cargo microdomains on the VTC surface that could facilitate efficient cargo sorting. Any of the potential mechanisms outlined above could result in an overall requirement for luminal Ca2+ and ALG-2/Sec31A interactions, although some involve attenuation mechanisms consistent with the observed inhibition of in vitro budding and COPII vesicle fusion by excess ALG-2 (10, 36). The phenotypes we observed upon luminal Ca2+ depletion and targeting of ALG-2/Sec31A in intact cells are all consistent with but do not distinguish between the mechanisms above, which are not mutually exclusive.
ALG-2/Sec31a interactions may not be the sole functional pathway for luminal Ca2+ in ER-to-Golgi transport. COPI stabilization on membranes in vitro is also Ca2+-regulated, but it appears to be independent of ALG-2 (10, 31). Also, Ca2+-dependent phospholipase A2 has been implicated in Golgi membrane dynamics (29, 30) where a luminal Ca2+ requirement for transport is also evident (24–26). Last, Ca2+-dependent cargo receptors, such as ERGIC-53/p58/LMAN1, bind cargo in the ER and release it in the lower Ca2+ environment of the VTC (62, 63). Hence, there is ample precedent for multiple calcium sensors that could, in principle, affect ER-to-Golgi transport.
Although most ERES remained intact in the presence of excess ALG-2·PRR, those with the highest targeted inhibitor appeared to conspicuously lack several components, including both inner and outer COPII shells and rbet1. Our best interpretation is that the inhibitor had multiple effects: a functional block of most ERES that remained molecularly intact and, at higher concentrations, a breakdown in ERES structure. Formally, however, we cannot exclude the counterintuitive possibility that the relatively few obviously disrupted ERES were in fact responsible for the block in transport and that the vast majority of ERES that remained intact can never transport VSV-G anyway.
ALG-2 and the Cargo Receptor p24
One of the most striking phenotypes of disrupted ALG-2/Sec31A interactions was the ablation of p24 expression (Fig. 7C). p24 cycles between the ER and Golgi, is a major component of both COPII and COPI vesicles (50), serves as a primer for COPI vesicle formation (51, 64), and, in isoform-specific fashion, appears to serve as a cargo receptor for glycosylphosphatidylinositol-anchored proteins (65, 66), G-protein coupled receptors (67), and the Wnt ligand wingless (68, 69). Proper p24 trafficking and interaction with other binding partners requires hetero-oligomerization among several closely related p24 family isoforms (70–72). The monomeric form appears to be intrinsically unstable (73), such that knocking down one isoform will deplete other members of the hetero-oligomer (74). p24 interacts directly with COPII (54); perhaps this binding is assisted by ALG-2 and promotes or proofreads correct oligomer formation of p24. The binding of ALG-2 to Sec31A in proximity to p24 may therefore promote p24 expression. p24 expression levels appear to be regulated in sync with cargo needs (75). It is thus possible that p24 stability is linked homeostatically to the cargo folding and export environment of the ER through ALG-2/Sec31A interactions and the availability of free luminal Ca2+.
Acknowledgments
Many investigators kindly provided reagents (see “Experimental Procedures”); however, we especially thank Dr. Masayuki Komada (Tokyo Institute of Technology) and Dr. Hideki Shibata (Nagoya University, Japan) for providing the ALG-2 reagents.
This work was supported, in whole or in part, by National Institutes of Health Grant GM106323 (to J. C. H.) and Fellowship NS067834 (to J. R. H.). This work was also supported by the University of Montana Center for Structural and Functional Neuroscience.
- ER
- endoplasmic reticulum
- ERES
- ER exit site(s)
- ALG-2
- apoptosis-linked gene-2
- BFA
- brefeldin A
- CPA
- cyclopiazonic acid
- IC
- intermediate compartment
- GAP
- GTPase-activating protein
- NRK
- normal rat kidney
- SERCA
- sarco-endoplasmic reticulum Ca2+ ATPase
- PRR
- Sec31A proline-rich region
- VSV-G
- vesicular stomatitis virus glycoprotein
- VTC
- vesicular tubular cluster
- UPR
- unfolded protein response
- endo H
- endoglycosidase H
- AMCA
- 7-amino-4-methylcoumarin-3-acetic acid.
REFERENCES
- 1. Bi X., Mancias J. D., Goldberg J. (2007) Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Dev. Cell 13, 635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hughes H., Stephens D. J. (2008) Assembly, organization, and function of the COPII coat. Histochem. Cell Biol. 129, 129–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller E. A., Barlowe C. (2010) Regulation of coat assembly: sorting things out at the ER. Curr. Opin. Cell Biol. 22, 447–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stagg S. M., LaPointe P., Razvi A., Gürkan C., Potter C. S., Carragher B., Balch W. E. (2008) Structural basis for cargo regulation of COPII coat assembly. Cell 134, 474–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tabata K. V., Sato K., Ide T., Nishizaka T., Nakano A., Noji H. (2009) Visualization of cargo concentration by COPII minimal machinery in a planar lipid membrane. EMBO J. 28, 3279–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cai H., Yu S., Menon S., Cai Y., Lazarova D., Fu C., Reinisch K., Hay J. C., Ferro-Novick S. (2007) TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature 445, 941–944 [DOI] [PubMed] [Google Scholar]
- 7. Lord C., Bhandari D., Menon S., Ghassemian M., Nycz D., Hay J., Ghosh P., Ferro-Novick S. (2011) Sequential interactions with Sec23 control the direction of vesicle traffic. Nature 473, 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trahey M., Hay J. C. (2010) Transport vesicle uncoating: it's later than you think. F1000 Biol. Rep. 2, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barlowe C., Orci L., Yeung T., Hosobuchi M., Hamamoto S., Salama N., Rexach M. F., Ravazzola M., Amherdt M., Schekman R. (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77, 895–907 [DOI] [PubMed] [Google Scholar]
- 10. Bentley M., Nycz D. C., Joglekar A., Fertschai I., Malli R., Graier W. F., Hay J. C. (2010) Vesicular calcium regulates coat retention, fusogenicity, and size of pre-Golgi intermediates. Mol. Biol. Cell 21, 1033–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Angers C. G., Merz A. J. (2011) New links between vesicle coats and Rab-mediated vesicle targeting. Semin. Cell Dev. Biol. 22, 18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu D., Hay J. C. (2004) Reconstitution of COPII vesicle fusion to generate a pre-Golgi intermediate compartment. J. Cell Biol. 167, 997–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu S., Satoh A., Pypaert M., Mullen K., Hay J. C., Ferro-Novick S. (2006) mBet3p is required for homotypic COPII vesicle tethering in mammalian cells. J. Cell Biol. 174, 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bentley M., Liang Y., Mullen K., Xu D., Sztul E., Hay J. C. (2006) SNARE status regulates tether recruitment and function in homotypic COPII vesicle fusion. J. Biol. Chem. 281, 38825–38833 [DOI] [PubMed] [Google Scholar]
- 15. Allan B. B., Moyer B. D., Balch W. E. (2000) Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science 289, 444–448 [DOI] [PubMed] [Google Scholar]
- 16. Martínez-Menárguez J. A., Geuze H. J., Slot J. W., Klumperman J. (1999) Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell 98, 81–90 [DOI] [PubMed] [Google Scholar]
- 17. Beckers C. J., Balch W. E. (1989) Calcium and GTP: essential components in vesicular trafficking between the endoplasmic reticulum and Golgi apparatus. J. Cell Biol. 108, 1245–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Porat A., Elazar Z. (2000) Regulation of intra-Golgi membrane transport by calcium. J. Biol. Chem. 275, 29233–29237 [DOI] [PubMed] [Google Scholar]
- 19. Colombo M. I., Beron W., Stahl P. D. (1997) Calmodulin regulates endosome fusion. J. Biol. Chem. 272, 7707–7712 [DOI] [PubMed] [Google Scholar]
- 20. Peters C., Mayer A. (1998) Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature 396, 575–580 [DOI] [PubMed] [Google Scholar]
- 21. Pryor P. R., Mullock B. M., Bright N. A., Gray S. R., Luzio J. P. (2000) The role of intraorganellar Ca2+ in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J. Cell Biol. 149, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burgoyne R. D., Clague M. J. (2003) Calcium and calmodulin in membrane fusion. Biochim. Biophys. Acta 1641, 137–143 [DOI] [PubMed] [Google Scholar]
- 23. Chen J.-L., Ahluwalia J. P., Stamnes M. (2002) Selective effects of calcium chelators on anterograde and retrograde protein transport in the cell. J. Biol. Chem. 277, 35682–35687 [DOI] [PubMed] [Google Scholar]
- 24. Hu Z., Bonifas J. M., Beech J., Bench G., Shigihara T., Ogawa H., Ikeda S., Mauro T., Epstein E. H. (2000) Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat. Genet. 24, 61–65 [DOI] [PubMed] [Google Scholar]
- 25. Sepúlveda M. R., Vanoevelen J., Raeymaekers L., Mata A. M., Wuytack F. (2009) Silencing the SPCA1 (secretory pathway Ca2+-ATPase isoform 1) impairs Ca2+ homeostasis in the Golgi and disturbs neural polarity. J. Neurosci. 29, 12174–12182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Micaroni M., Perinetti G., Berrie C. P., Mironov A. A. (2010) The SPCA1 Ca2+ pump and intracellular membrane trafficking. Traffic 11, 1315–1333 [DOI] [PubMed] [Google Scholar]
- 27. von Blume J., Alleaume A.-M., Cantero-Recasens G., Curwin A., Carreras-Sureda A., Zimmermann T., van Galen J., Wakana Y., Valverde M. A., Malhotra V. (2011) ADF/cofilin regulates secretory cargo sorting at the TGN via the Ca2+ ATPase SPCA1. Dev. Cell 20, 652–662 [DOI] [PubMed] [Google Scholar]
- 28. von Blume J., Alleaume A.-M., Kienzle C., Carreras-Sureda A., Valverde M., Malhotra V. (2012) Cab45 is required for Ca2+-dependent secretory cargo sorting at the trans-Golgi network. J. Cell Biol. 199, 1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Micaroni M. (2012) Calcium around the Golgi apparatus: implications for intracellular membrane trafficking. Adv. Exp. Med. Biol. 740, 439–460 [DOI] [PubMed] [Google Scholar]
- 30. Brown W. J., Chambers K., Doody A. (2003) Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic 4, 214–221 [DOI] [PubMed] [Google Scholar]
- 31. Ahluwalia J. P., Topp J. D., Weirather K., Zimmerman M., Stamnes M. (2001) A role for calcium in stabilizing transport vesicle coats. J. Biol. Chem. 276, 34148–34155 [DOI] [PubMed] [Google Scholar]
- 32. Shibata H., Inuzuka T., Yoshida H., Sugiura H., Wada I., Maki M. (2010) The ALG-2 binding site in Sec31A influences the retention kinetics of Sec31A at the endoplasmic reticulum exit sites as revealed by live-cell time-lapse imaging. Biosci. Biotechnol. Biochem. 74, 1819–1826 [DOI] [PubMed] [Google Scholar]
- 33. Shibata H., Suzuki H., Yoshida H., Maki M. (2007) ALG-2 directly binds Sec31A and localizes at endoplasmic reticulum exit sites in a Ca2+-dependent manner. Biochem. Biophys. Res. Commun. 353, 756–763 [DOI] [PubMed] [Google Scholar]
- 34. la Cour J. M., Mollerup J., Berchtold M. W. (2007) ALG-2 oscillates in subcellular localization, unitemporally with calcium oscillations. Biochem. Biophys. Res. Commun. 353, 1063–1067 [DOI] [PubMed] [Google Scholar]
- 35. Yamasaki A., Tani K., Yamamoto A., Kitamura N., Komada M. (2006) The Ca2+-binding protein ALG-2 is recruited to endoplasmic reticulum exit sites by Sec31A and stabilizes the localization of Sec31A. Mol. Biol. Cell 17, 4876–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. la Cour J. M., Schindler A. J., Berchtold M. W., Schekman R. (2013) ALG-2 attenuates COPII budding in vitro and stabilizes the Sec23/Sec31A complex. PLoS One 8, e75309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hay J. C., Klumperman J., Oorschot V., Steegmaier M., Kuo C. S., Scheller R. H. (1998) Localization, dynamics, and protein interactions reveal distinct roles for ER and Golgi SNAREs. J. Cell Biol. 141, 1489–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lefrancois L., Lyles D. S. (1982) The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 121, 157–167 [PubMed] [Google Scholar]
- 39. Thayanidhi N., Helm J. R., Nycz D. C., Bentley M., Liang Y., Hay J. C. (2010) α-Synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 21, 1850–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gordon D. E., Bond L. M., Sahlender D. A., Peden A. A. (2010) A targeted siRNA screen to identify SNAREs required for constitutive secretion in mammalian cells. Traffic 11, 1191–1204 [DOI] [PubMed] [Google Scholar]
- 41. Slot J. W., Geuze H. J. (2007) Cryosectioning and immunolabeling. Nat. Protoc. 2, 2480–2491 [DOI] [PubMed] [Google Scholar]
- 42. Flanagan J. J., Barlowe C. (2006) Cysteine-disulfide cross-linking to monitor SNARE complex assembly during endoplasmic reticulum-Golgi transport. J. Biol. Chem. 281, 2281–2288 [DOI] [PubMed] [Google Scholar]
- 43. Mason M. J., Garcia-Rodriguez C., Grinstein S. (1991) Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Comparison of the effects of thapsigargin, 2,5-di-(tert-butyl)-1,4-hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. J. Biol. Chem. 266, 20856–20862 [PubMed] [Google Scholar]
- 44. Gallione C. J., Rose J. K. (1985) A single amino acid substitution in a hydrophobic domain causes temperature-sensitive cell-surface transport of a mutant viral glycoprotein. J. Virol. 54, 374–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Amodio G., Renna M., Paladino S., Venturi C., Tacchetti C., Moltedo O., Franceschelli S., Mallardo M., Bonatti S., Remondelli P. (2009) Endoplasmic reticulum stress reduces the export from the ER and alters the architecture of post-ER compartments. Int. J. Biochem. Cell Biol. 41, 2511–2521 [DOI] [PubMed] [Google Scholar]
- 46. Hughes H., Budnik A., Schmidt K., Palmer K. J., Mantell J., Noakes C., Johnson A., Carter D. A., Verkade P., Watson P., Stephens D. J. (2009) Organisation of human ER-exit sites: requirements for the localisation of Sec16 to transitional ER. J. Cell Sci. 122, 2924–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maki M., Shibata H. (2007) The penta-EF-hand protein ALG-2 and its interacting proteins. Calcium Binding Proteins 2, 4–10 [Google Scholar]
- 48. Shibata H., Suzuki H., Kakiuchi T., Inuzuka T., Yoshida H., Mizuno T., Maki M. (2008) Identification of Alix-type and non-Alix-type ALG-2-binding sites in human phospholipid scramblase 3: differential binding to an alternatively spliced isoform and amino acid-substituted mutants. J. Biol. Chem. 283, 9623–9632 [DOI] [PubMed] [Google Scholar]
- 49. Bonnon C., Wendeler M. W., Paccaud J.-P., Hauri H.-P. (2010) Selective export of human GPI-anchored proteins from the endoplasmic reticulum. J. Cell Sci. 123, 1705–1715 [DOI] [PubMed] [Google Scholar]
- 50. Strating J. R. P. M., Martens G. J. M. (2009) The p24 family and selective transport processes at the ER-Golgi interface. Biol. Cell 101, 495–509 [DOI] [PubMed] [Google Scholar]
- 51. Aguilera-Romero A., Kaminska J., Spang A., Riezman H., Muñiz M. (2008) The yeast p24 complex is required for the formation of COPI retrograde transport vesicles from the Golgi apparatus. J. Cell Biol. 180, 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mukhopadhyay S., Bachert C., Smith D. R., Linstedt A. D. (2010) Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Mol. Biol. Cell 21, 1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Contreras I., Yang Y., Robinson D. G., Aniento F. (2004) Sorting signals in the cytosolic tail of plant p24 proteins involved in the interaction with the COPII coat. Plant Cell Physiol. 45, 1779–1786 [DOI] [PubMed] [Google Scholar]
- 54. Dominguez M., Dejgaard K., Füllekrug J., Dahan S., Fazel A., Paccaud J. P., Thomas D. Y., Bergeron J. J., Nilsson T. (1998) gp25L/emp24/p24 protein family members of the cis-Golgi network bind both COP I and II coatomer. J. Cell Biol. 140, 751–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brandon E., Szul T., Alvarez C., Grabski R., Benjamin R., Kawai R., Sztul E. (2006) On and off membrane dynamics of the endoplasmic reticulum-Golgi tethering factor p115 in vivo. Mol. Biol. Cell 17, 2996–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shaywitz D. A., Espenshade P. J., Gimeno R. E., Kaiser C. A. (1997) COPII subunit interactions in the assembly of the vesicle coat. J. Biol. Chem. 272, 25413–25416 [DOI] [PubMed] [Google Scholar]
- 57. Higashio H., Kohno K. (2002) A genetic link between the unfolded protein response and vesicle formation from the endoplasmic reticulum. Biochem. Biophys. Res. Commun. 296, 568–574 [DOI] [PubMed] [Google Scholar]
- 58. Sato M., Sato K., Nakano A. (2002) Evidence for the intimate relationship between vesicle budding from the ER and the unfolded protein response. Biochem. Biophys. Res. Commun. 296, 560–567 [DOI] [PubMed] [Google Scholar]
- 59. Kitaura Y., Matsumoto S., Satoh H., Hitomi K., Maki M. (2001) Peflin and ALG-2, members of the penta-EF-hand protein family, form a heterodimer that dissociates in a Ca2+-dependent manner. J. Biol. Chem. 276, 14053–14058 [DOI] [PubMed] [Google Scholar]
- 60. Kung L. F., Pagant S., Futai E., D'Arcangelo J. G., Buchanan R., Dittmar J. C., Reid R. J. D., Rothstein R., Hamamoto S., Snapp E. L., Schekman R., Miller E. A. (2012) Sec24p and Sec16p cooperate to regulate the GTP cycle of the COPII coat. EMBO J. 31, 1014–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gillon A. D., Latham C. F., Miller E. A. (2012) Vesicle-mediated ER export of proteins and lipids. Biochim. Biophys. Acta 1821, 1040–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Appenzeller-Herzog C., Roche A.-C., Nufer O., Hauri H.-P. (2004) pH-induced conversion of the transport lectin ERGIC-53 triggers glycoprotein release. J. Biol. Chem. 279, 12943–12950 [DOI] [PubMed] [Google Scholar]
- 63. Zhang B., Kaufman R. J., Ginsburg D. (2005) LMAN1 and MCFD2 form a cargo receptor complex and interact with coagulation factor VIII in the early secretory pathway. J. Biol. Chem. 280, 25881–25886 [DOI] [PubMed] [Google Scholar]
- 64. Mitrovic S., Ben-Tekaya H., Koegler E., Gruenberg J., Hauri H.-P. (2008) The cargo receptors Surf4, endoplasmic reticulum-Golgi intermediate compartment (ERGIC)-53, and p25 are required to maintain the architecture of ERGIC and Golgi. Mol. Biol. Cell 19, 1976–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Takida S., Maeda Y., Kinoshita T. (2008) Mammalian GPI-anchored proteins require p24 proteins for their efficient transport from the ER to the plasma membrane. Biochem. J. 409, 555–562 [DOI] [PubMed] [Google Scholar]
- 66. Muñiz M., Nuoffer C., Hauri H. P., Riezman H. (2000) The Emp24 complex recruits a specific cargo molecule into endoplasmic reticulum-derived vesicles. J. Cell Biol. 148, 925–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bartoszewski S., Luschnig S., Desjeux I., Grosshans J., Nüsslein-Volhard C. (2004) Drosophila p24 homologues eclair and baiser are necessary for the activity of the maternally expressed Tkv receptor during early embryogenesis. Mech. Dev. 121, 1259–1273 [DOI] [PubMed] [Google Scholar]
- 68. Buechling T., Chaudhary V., Spirohn K., Weiss M., Boutros M. (2011) p24 proteins are required for secretion of Wnt ligands. EMBO Rep. 12, 1265–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Port F., Hausmann G., Basler K. (2011) A genome-wide RNA interference screen uncovers two p24 proteins as regulators of Wingless secretion. EMBO Rep. 12, 1144–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Béthune J., Kol M., Hoffmann J., Reckmann I., Brügger B., Wieland F. (2006) Coatomer, the coat protein of COPI transport vesicles, discriminates endoplasmic reticulum residents from p24 proteins. Mol. Cell. Biol. 26, 8011–8021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Barr F. A., Preisinger C., Kopajtich R., Körner R. (2001) Golgi matrix proteins interact with p24 cargo receptors and aid their efficient retention in the Golgi apparatus. J. Cell Biol. 155, 885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Füllekrug J., Suganuma T., Tang B. L., Hong W., Storrie B., Nilsson T. (1999) Localization and recycling of gp27 (hp24γ3): complex formation with other p24 family members. Mol. Biol. Cell 10, 1939–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Belden W. J., Barlowe C. (1996) Erv25p, a component of COPII-coated vesicles, forms a complex with Emp24p that is required for efficient endoplasmic reticulum to Golgi transport. J. Biol. Chem. 271, 26939–26946 [DOI] [PubMed] [Google Scholar]
- 74. Denzel A., Otto F., Girod A., Pepperkok R., Watson R., Rosewell I., Bergeron J. J., Solari R. C., Owen M. J. (2000) The p24 family member p23 is required for early embryonic development. Curr. Biol. 10, 55–58 [DOI] [PubMed] [Google Scholar]
- 75. Rötter J., Kuiper R. P., Bouw G., Martens G. J. M. (2002) Cell-type-specific and selectively induced expression of members of the p24 family of putative cargo receptors. J. Cell Sci. 115, 1049–1058 [DOI] [PubMed] [Google Scholar]