

Abstract
A pure culture of an actinobacterium previously described as ‘Candidatus Rhodoluna lacicola’ strain MWH-Ta8 was established and deposited in two public culture collections. Strain MWH-Ta8T represents a free-living planktonic freshwater bacterium obtained from hypertrophic Meiliang Bay, Lake Taihu, PR China. The strain was characterized by phylogenetic and taxonomic investigations, as well as by determination of its complete genome sequence. Strain MWH-Ta8T is noticeable due to its unusually low values of cell size (0.05 µm3), genome size (1.43 Mbp), and DNA G+C content (51.5 mol%). Phylogenetic analyses based on 16S rRNA gene and RpoB sequences suggested that strain MWH-Ta8T is affiliated with the family Microbacteriaceae with Pontimonas salivibrio being its closest relative among the currently described species within this family. Strain MWH-Ta8T and the type strain of Pontimonas salivibrio shared a 16S rRNA gene sequence similarity of 94.3 %. The cell-wall peptidoglycan of strain MWH-Ta8T was of type B2β (B10), containing 2,4-diaminobutyric acid as the diamino acid. The predominant cellular fatty acids were anteiso-C15 : 0 (36.5 %), iso-C16 : 0 (16.5 %), iso-C15 : 0 (15.6 %) and iso-C14 : 0 (8.9 %), and the major (>10 %) menaquinones were MK-11 and MK-12. The major polar lipids were diphosphatidylglycerol, phosphatidylglycerol and two unknown glycolipids. The combined phylogenetic, phenotypic and chemotaxonomic data clearly suggest that strain MWH-Ta8T represents a novel species of a new genus in the family Microbacteriaceae, for which the name Rhodoluna lacicola gen. nov., sp. nov. is proposed. The type strain of the type species is MWH-Ta8T ( = DSM 23834T = LMG 26932T).
Members of the phylum Actinobacteria frequently contribute large proportions to freshwater bacterioplankton of lakes, ponds and rivers (Glöckner et al., 2000; Hahn, 2006; Newton et al., 2011). Shares of 10–60 % of bacterioplankton cell numbers were frequently reported (Allgaier & Grossart, 2006; Glöckner et al., 2000; Newton et al., 2011; Salcher et al., 2010); however, none of the abundant lineages of freshwater Actinobacteria is represented by a species with a validly published name (Newton et al., 2011). Only eight Candidatus species representing six Candidatus genera have been described so far (Hahn, 2009; Jezbera et al., 2009). Here we present the first species description of an abundant freshwater bacterium representing a lineage of indigenous freshwater bacteria in the phylum Actinobacteria.
Strain MWH-Ta8T was obtained from the hypertrophic Meiliang (Mailing) Bay (31° 23′ 55″ N 120° 13′ 50″ E) Lake Taihu, People’s Republic of China (Hahn, 2009) by using the filtration-acclimatization method (Hahn et al., 2004). Lake Taihu is a large and shallow subtropical lake characterized by a surface area of 2338 km2, a mean depth of 1.9 m and a maximum depth of only 2.6 m (Sun & Mao, 2008). The lake is the third largest freshwater lake in China and plays an important role as a freshwater source for a number of cities.
Initially only a mixed culture consisting of strain MWH-Ta8T and uncharacterized members of the class Betaproteobacteria could be established, which only enabled the description of a Candidatus species, i.e. ‘Candidatus Rhodoluna lacicola’ MWH-Ta8 (Hahn, 2009). Further purification efforts finally resulted in establishment of a pure culture growing in liquid medium and on NSY agar plates. Strain MWH-Ta8T shares with typical freshwater members of the phylum Actinobacteria characterized by fluorescent in situ hybridization (FISH) in water samples from freshwater systems, cell sizes of <0.1 µm3 (Salcher et al., 2010). Bacteria with cell sizes below this threshold comprise the majority of cells in freshwater and marine bacterioplankton, and are termed ultramicrobacteria (UMB) (Duda et al., 2012). Interestingly, strain MWH-Ta8T represents an obligate UMB (Duda et al., 2012), which maintains its small cell sizes even when grown in rich complex media.
Strain MWH-Ta8T strongly differed in cell size from all previously described species of the family Microbacteriaceae, and most likely differed from them all in growth potential when cultivated and tested by using standard bacteriological methods. Previous experiments on phenotypic characterization of various freshwater actinobacteria by using API 50 CH tests (bioMérieux) did not result in any positive response; therefore, a non-standard test method for subfstrate utilization (see below) previously used for other strains with only weak growth potential was used for the characterization of strain MWH-Ta8T. Another problem was encountered when we tried to evaluate the results of Gram staining tests by microscopy at a magnification of ×1000. Due to the tininess of the stained cells, it was impossible to recognize any colours, which made it impossible to conclude which Gram stain reaction occurs. Several of the performed phenotypic tests (see below) resulted only in weak reactions that were difficult to interpret. Repetition of such experiments often did not improve the quality of the test result. One consequence of the unusual traits of strain MWH-Ta8T is that it was not possible to fulfil the minimal standards for the description of new taxa of the suborder Micrococcineae (Schumann et al., 2009). We compensated for the limited phenotypic characterization of strain MWH-Ta8T by a characterization of the genome of this strain and by an extended phylogenetic analysis.
Phylogeny.
The 16S rRNA gene sequence of strain MWH-Ta8T used for phylogenetic analyses was established as described previously (Hahn, 2009). This sequence was obtained from the previous mixed culture; however, the sequence is identical to the 16S rRNA gene sequence determined by genome sequencing (see below) of the later established pure culture of the strain. For phylogenetic inference, aligned ribosomal sequences of reference strains were downloaded from the Greengenes database (DeSantis et al., 2006), while sequences not available from this source were retrieved from the GenBank database. The latter sequences were aligned by using already aligned sequences as a template, and the whole alignment was examined for errors, which were then manually corrected. The final alignment consisted of 1405 positions, and the length of the aligned sequence of strain MWH-Ta8T was 1365 bp (without alignment gaps). Maximum-likelihood (ML), neighbour-joining (NJ) and maximum-parsimony (MP) trees were calculated with the mega software version 5.2 (Tamura et al., 2011). The best substitution model for phylogeny inference by the ML method was searched by the model test option of mega 5.2. Accordingly, the General Time Reversible model with non-uniformity of evolutionary rates among sites by using a discrete Gamma distribution (+G) with five rate categories and by assuming that a certain fraction of sites are evolutionarily invariable (+I) was used for calculation of a ML tree. A NJ tree was calculated by using the Tamura-3-parameter model with a gamma distribution with five categories, a heterogeneous pattern among lineages, and pairwise deletion of gaps. All alignment positions (all sites) were used in the calculation of a MP tree. The number of bootstrap replications in calculation of the ML, NJ and MP trees was 1000, 1000 and 100, respectively. In all trees based on a 16S rRNA gene sequence set consisting of the type species of all described genera within the family Microbacteriaceae and some closely related Candidatus species, the sequence of strain MWH-Ta8T clustered within this family (Fig. 1). As already revealed by previous analyses, strain MWH-Ta8T formed, together with other freshwater actinobacteria, a tight cluster (Hahn, 2009) previously designated Luna-1 cluster (Hahn et al., 2003). This cluster was supported by all three phylogenetic methods (Fig. 1). This freshwater cluster including strain MWH-Ta8T appeared in all trees calculated by the three methods together with Pontimonas salivibrio CL-TW6T (Jang et al., 2013). The close relationship of these taxa was supported with high bootstrap values by at least two methods, but the phylogenetic relationship of the Pontimonas/Luna-1 cluster to other genera of the family Microbacteriaceae could not be resolved. The Pontimonas/Luna-1 cluster appeared in all three trees together with the type species of six other genera, but this relationship was not supported by high bootstrap values in any tree, and the branching order of these taxa markedly differed between trees calculated by using different methods. Furthermore, Pontimonas salivibrio clustered in the trees presented here (Fig. 1) with other taxa than in a previous phylogenetic reconstruction (Jang et al., 2013).
Fig. 1.

NJ tree based on partial 16S rRNA gene sequences reconstructing the phylogenetic position of strain MWH-Ta8T within the family Microbacteriaceae. Results from analyses by the ML and MP methods are also indicated. Bootstrap values (percentage of replicates) above the threshold of ≥60 % are shown for those nodes supported in at least one of the three methods; these bootstrap values are depicted in the order NJ/ML/MP. Filled diamonds indicate nodes reconstructed by all three methods independent of their respective bootstrap values; filled circles indicate those nodes only present in NJ and ML trees; filled square indicates one node only reconstructed by the NJ and MP methods. Brevibacterium linens DSM 20425T, which is not affiliated with the family Microbacteriaceae was used as an out-group. GenBank accession numbers of 16S rRNA gene sequences are depicted in parentheses. Bar, 0.02 substitutions per nucleotide position.
Because strain MWH-Ta8T greatly differed in a couple of important taxonomic traits from all previously described taxa affiliated with the family Microbacteriaceae, we additionally evaluated the phylogenetic position of the strain by using amino acid (aa) sequences of the beta subunit of the RNA polymerase (RpoB) retrieved from genome databases. Forty-three genomes of strains classified as Microbacteriaceae were found in the integrated microbial genomes (IMG) database (Markowitz et al., 2012) and one additional genome assigned to this family but not contained in the IMG database was found in the GenBank database. Because only 20 of those 44 genomes represented taxonomically evaluated type species (Table S1, available in the online Supplementary Material), their putative affiliation with the family Microbacteriaceae was first checked by a phylogenetic analysis of their 16S rRNA gene sequences. Tree reconstruction and other analyses confirmed for 42 strains, a putative affiliation with the family Microbacteriaceae (Fig. S1). One strain (Microbacterium sp. KROCY2) clustered outside the family Microbacteriaceae, and one strain could not be tested because its draft genome completely lacked a gene annotated as 16S or SSU rRNA gene (Table S1).
Nucleotide sequences of the rpoB gene of reference strains were retrieved, translated to aa sequences and aligned by the muscle aligner provided by the mega software package. The complete RpoB aa sequence of strain MWH-Ta8T had a length of 1204 aa, while 1103 aa were contained in the aligned and trimmed sequence set used for the phylogenetic analyses. NJ, ML and MP trees were reconstructed by using mega software version 6.05. The ML tree was calculated, as suggested by model test, by using the LG model +G +I. The NJ tree was calculated by using the Poisson model and a gamma distribution (+G) with a parameter of five. For calculation of ML, NJ and MP trees all alignment columns containing gaps were completely omitted, and 1000, 1000 and 100 bootstrap replications were performed in ML, NJ and MP analyses, respectively. The obtained RpoB trees support the affiliation of strain MWH-Ta8T with the family Microbacteriaceae (Fig. 2) but in contrast to the phylogenetic reconstruction based on 16S rRNA gene sequences (Fig. 1), strain MWH-Ta8T did not appear in a nested position within the family. The difference in position within the family could have resulted from the different sets of taxa used in the two phylogenetic reconstructions. Interestingly, strain MWH-Ta8T clustered together with ‘Candidatus Aquiluna’ sp. IMCC13023 in both the 16S rRNA gene tree and the RpoB tree, but the relationship to Pontimonas salivibrio and the type species of six other genera appearing close to strain MWH-Ta8T in the 16S rRNA gene trees could not be evaluated since RpoB sequences of these taxa were not available.
Fig. 2.

NJ tree based on partial RpoB (DNA-directed RNA polymerase subunit beta) amino acid sequences reconstructing the phylogenetic position of strain MWH-Ta8T. Amino acid sequences were used for phylogenetic analysis in order to omit phylogenetic noise potentially cause by the pronounced differences in DNA G+C content of rpoB genes (range 51 to 68 mol%). Results from analyses by the ML and MP methods are also indicated. Bootstrap values (percentage of replicates) above the threshold of ≥60 % are shown for those nodes supported at least in inference by one of the three methods; values are presented in the order NJ/ML/MP. Filled diamonds indicate nodes reconstructed by all three methods independent of their respective bootstrap values; filled circles indicate those nodes only present in NJ and ML trees; filled squares indicate nodes only reconstructed by the NJ and MP methods. If available, IMG Gene ID numbers of genes encoding the RpoB proteins are given in parentheses; for other proteins, the RefSeq or GenBank accession numbers are provided. Microbacterium sp. KROCY2 represents a strain classified by the IMG database as a member of the family Microbacteriaceae, and acI actinobacteria SCGC AAA027-L06 represents a freshwater actinobacterium affiliated with the acI lineage (Garcia et al., 2013). Ten type strains of species not affiliated with the family Microbacteriaceae were used as an out-group. Bar, 0.05 substitutions per amino acid position.
16S rRNA gene sequence similarities were calculated by using the mega software. Based on the sequence alignment, a matrix of pairwise nucleotide differences was calculated, and the numbers of nucleotide differences were converted to pairwise sequence similarity values. Strain MWH-Ta8T did not share sequence similarity values >97 % with any of the 43 type species of the currently described genera affiliated with the family Microbacteriaceae. The highest similarities were observed with sequences of Alpinimonas psychrophila Cr8-25T (94.5 %), Pontimonas salivibrio CL-TW6T (94.3 %) and Compostimonas suwonensis SMC46T (94.2 %).
Phenotypic and chemotaxonomic traits.
Strain MWH-Ta8T formed small, circular, convex colonies with a shiny surface and a red pigmentation on NSY agar plates (Hahn, 2009); however, unpigmented mutants were also observed and one such strain (MWH-Ta8n) was deposited at the BCCM/LMG culture collection under the accession number LMG 27024. Cell morphology and size (Table 1) were examined by epifluorescence microscopy (Axioplan, Zeiss, Germany) after staining with the fluorochrome 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) at a magnification of ×1250 as described previously (Hahn et al., 2003). This method enabled a suitable microscopic examination of this ultramicrobacterium. Due to tininess of cells, observation of motility by phase-contrast microscopy (×1000) did not yield reliable results. Therefore, tests by culturing strain MWH-Ta8T on soft agar (NSY medium, 0.4 % agar) were performed. Repeated experiments suggested a weak potential for spreading on the agar surface; however, the absence of genes required for flagellum synthesis in the genome sequence of the strain (see below) suggested that the strain was negative for motility by flagella (Table 1).
Table 1. Morphological and phenotypic traits that characterize strain MWH-Ta8T.
−, Negative; +, positive; w, weakly positive.
| Characteristic | MWH-Ta8T |
| Cell morphology | Curved (selenoid) rods |
| Cell length (µm) | 0.85 |
| Cell width (µm) | 0.30 |
| Cell volume (µm3) | 0.053 |
| Pigmentation | Red |
| Motility | − |
| Temperature range of growth (°C) | 9–36 (w at 37 °C) |
| NaCl tolerance (%, w/v) | 0–0.4 (w at 0.5 %) |
| Anaerobic growth | − |
| Catalase | + |
| Oxidase | − |
| Assimilation of: | |
| Formic acid | − |
| Glyoxylic acid | − |
| Glycolic acid | − |
| Propionic acid | − |
| Acetic acid | w |
| Fumaric acid | − |
| Malonic acid | − |
| Oxaloacetic acid | w |
| Citric acid | − |
| d-Fucose | − |
| d-Glucose | − |
| d-Mannose | + |
| d-Sorbitol | + |
| l-Glutamate | w |
| l-Alanine | w |
| Betaine | − |
The temperature range of growth and salinity tolerance were tested by cultivation on NSY agar plates (Hahn et al., 2004). Inocula from cultures grown at room temperature in liquid NSY medium were spread on standard NSY plates and incubated at different temperatures, or were spread on NSY plates supplemented with 0, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 1.0, 1.5 or 2.0 % (w/v) NaCl and incubated at room temperature. Temperature experiments were performed at 4, 8, 9, 15, 17, 25, 30, 34, 36, 37 and 39 °C. These experiments suggested a mesophilic thermal adaptation and a low salinity tolerance (Table 1). Growth under anoxic conditions on standard NSY agar and NSY agar supplemented with nitrate (0.8 mM) was examined by using an anaerobic chamber [anoxic conditions generated by Anerocult A (Merck)]. Catalase activity was determined by bubble production in 3 % (v/v) H2O2, and cytochrome oxidase activity was examined by using Bactident Oxidase test stripes (Merck). The latter test was repeated several times and resulted in comparison to oxidase-positive reference strains only in very faint bluish reactions, which were evaluated as negative test results. Due to difficulties in the examination of substrate utilization of strain MWH-Ta8T by standard methods, a method previously successfully applied for characterization of novel members of the genus Polynucleobacter (Hahn et al., 2009) was employed. Briefly, growth enabled by utilization of a specific substrate was determined by comparison of the optical density established in liquid one-tenth-strength NSY medium (0.3 g) with and without 0.5 g test substance. OD 575 differences of 10 %, 10–50 % and 50 % of the OD 575 established in the medium without test substance were scored after 10 days of growth as no utilization, weak utilization and good utilization, respectively. Good utilization was only observed for d-mannose and d-sorbitol (Table 1).
The analyses of peptidoglycan structure, respiratory quinones and polar lipids were carried out by the Identification Service of the Leibniz-Institut DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen), Braunschweig, Germany. For these analyses and for fatty acid analysis, biomass of strain MWH-Ta8T was harvested by centrifugation from cultures grown in liquid NSY medium on a shaker (50 r.p.m.) at 22–24 °C. Peptidoglycan analyses were performed as described previously (Schumann, 2011; Schumann et al., 2012). The total hydrolysate (4 M HCl, 100 °C, 16 h) of the peptidoglycan of strain MWH-Ta8T contained the amino acids 2,4-diaminobutyric acid (Dab), alanine (Ala), glycine (Gly), homoserine (Hse) and glutamic acid (Glu) in an approximate molar ratio of 0.8 : 0.5 : 0.9 : 0.4 : 1.0, respectively. The partial hydrolysate (4 M HCl, 100 °C, 0.75 h) contained the peptides Mur–Gly, d-Ala–d-Dab, Gly–d-Glu. The latter peptide is characteristic of B-type peptidoglycan, and it was concluded that the peptidoglycan of strain MWH-Ta8T represented the type B2β (type B10) (Schleifer & Kandler, 1972; Schumann, 2011). The respiratory quinones of strain MWH-Ta8T were menaquinones MK-11 (56 %), MK-12 (24 %), MK-10 (9 %), MK-9 (5 %), MK-13 (2 %) and some minor components. The major polar lipids were diphosphatidylglycerol, phosphatidylglycerol and two unknown glycolipids.
Cellular fatty acids from whole bacteria samples were extracted with a 4 : 2 : 1 (by vol.) chloroform/methanol/water mixture (Parrish, 1999). These samples were then sonicated and vortexed twice and the organic phases were removed and pooled. Fatty acids were transmethylated at 50 °C overnight using 1 % sulfuric acid as a catalyst. Fatty acid methyl esters were analysed with a gas chromatograph (Agilent 6890N) with mass spectrometric detection (Agilent 5973N). An Agilent DB-23 column (30 m×0.25 mm×0.15 µm) was used with the following temperature program: 60 °C was maintained for 1.5 min, then the temperature was increased at 10 °C min−1 to 100 °C, followed by 2 °C min−1 to 140 °C, and 1 °C min−1 to 180 °C, and finally heated up at 2 °C min−1 to 210 °C and then held for 6 min. Helium was used as carrier gas with a mean velocity of 34 cm s−1. The predominant cellular fatty acids of strain MWH-Ta8T were anteiso-C15 : 0, iso-C16 : 0, iso-C15 : 0 and iso-C14 : 0 (Table 2). Strain MWH-Ta8T differed only slightly in cellular fatty acid composition from the phylogenetically most closely related characterized species, Pontimonas salivibrio CL-TW6T.
Table 2. Comparison of cellular fatty acids contents of strain MWH-Ta8T and the phylogenetically most closely related strain Pontimonas salivibrio CL-TW6T.
tr, Traces; nd, not detected.
| Fatty acid | MWH-Ta8T | Pontimonas salivibrio CL-TW6T |
| Saturated | ||
| C14 : 0 | 2.2 | 2.2 |
| C15 : 0 | 1.4 | 2.6 |
| C16 : 0 | 7.0 | 7.0 |
| C17 : 0 | tr | tr |
| C18 : 0 | 2.5 | 1.1 |
| Total saturated | 13.1 | 12.9 |
| Branched | ||
| iso-C14 : 0 | 8.9 | 11.8 |
| iso-C15 : 0 | 15.6 | 13.2 |
| anteiso-C15 : 0 | 36.5 | 32.6 |
| iso-C16 : 0 | 16.5 | 20.4 |
| iso-C17 : 0 | 2.4 | 1.9 |
| anteiso-C17 : 0 | 4.1 | 2.8 |
| Total branched | 75.1 | 70.9 |
| Monounsaturated | ||
| C16 : 1ω7 | nd | nd |
| C18 : 1ω7 | tr | tr |
| Total monounsaturated | 0.0 | 0.0 |
Genomic traits.
DNA used for genome sequencing was extracted from biomass of strain MWH-Ta8T grown in liquid NSY medium. The extraction was performed as described previously (Meincke et al., 2012). Construction of a paired-end library and sequencing by using a Roche GS FLX system and Titanium chemistry was performed at McGill University and Génome Québec Innovation Centre (Montréal, Canada). The paired-end library had an mean insert length of approximately 7000 kb, and sequencing of the library resulted in 239 000 filtered reads with median length of 323 bp. Sequence assembly by using the GS De novo Assembler software (Newbler software, Roche) resulted in four large contigs, which were ordered and oriented into one scaffold. Gap closure was performed by primer design, PCR amplification and Sanger sequencing of obtained amplicons. The closed genome consists of 1 430 433 bp with a G+C content of 51.53 mol%. The closed genome sequence was annotated by using the IMG/ER annotation pipeline (Markowitz et al., 2012). The genome of strain MWH-Ta8T differed strongly in genome size and G+C content from the vast majority of genome sequences of members of the phylum Actinobacteria (1296 genomes; excluding draft genomes obtained from single cells) currently (February 2014) available in the IMG database (Markowitz et al., 2012) (Fig. 3). Only five of the 1296 sequences possessed smaller genome sizes than strain MWH-Ta8T. Four of these five genomes represent obligately host-associated genera of the phylum Actinobacteria (Tropheryma, Atopobium and ‘Candidatus Ancillula’), while the only free-living strain with a smaller genome size was ‘Ca. Aquiluna’ sp. IMCC13023 (Kang et al., 2012), which represented a close relative of strain MWH-Ta8T (Figs 1 and 2). A very small genome size of about 1.2 Mbp was suggested recently for a member of the so-called acI lineage of freshwater actinobacteria (Garcia et al., 2013). This estimation of genome size is based on a draft genome sequence obtained from a single cell and consists of 75 contigs. The phylogenetic position and taxonomic affiliation of the acI lineage, which is represented by ‘Candidatus Planktophila limnetica’, could not be determined with certainty so far (Jezbera et al., 2009); however, it is obvious that this lineage of freshwater Actinobacteria has a different phylogenetic origin than strain MWH-Ta8T (Fig. 2).
Fig. 3.
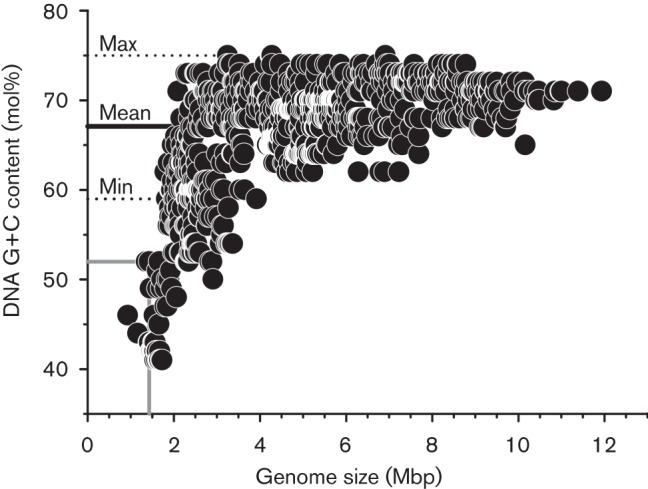
Genome size and DNA G+C content of all genomes of actinobacteria currently contained in the IMG database. Only genomes of strains established as pure cultures were considered. Values of strain MWH-Ta8T are indicated by solid grey lines, and minimum, mean and maximum DNA G+C values of type strains of members of the family Microbacteriaceae (Table S1) are indicated by dotted and solid black lines.
Apart from the genomes of strain MWH-Ta8T and ‘Ca. Aquiluna’, the DNA G+C content of genomes putatively affiliated with the family Microbacteriaceae (Table S1) ranged from 59 to 73 mol% (mean 68.9 mol%; n = 42). These data are quite similar to the DNA G+C content of type strains of previously described species of the family Microbacteriaceae (Table 3). Values for these type strains ranged from 59 mol% [Alpinimonas (Schumann et al., 2012) and Schumannella (An et al., 2008)] to 76 mol% [Agrococcus (Zhang et al., 2010)] and the mean DNA G+C content was 67.1 mol%. Thus, strains MWH-Ta8T and ‘Ca. Aquiluna’ sp. IMCC13023 strongly differ in both traits, genome size and DNA G+C content, from previously described species of the family Microbacteriaceae.
Table 3. Genome characteristics of strain MWH-Ta8T and other strains affiliated with the family Microbacteriaceae.
The column presenting data for other members of the family Microbacteriaceae summarizes in the first three lines characteristics of 41 genome-sequenced strains (Table S1) and in the last line data of type species determined by a different method. Phylogenetic analyses with 16S rRNA gene sequences of the genome-sequenced strains confirmed their affiliation with the family Microbacteriaceae (Fig. S1). The data presented in the last column represent minimum–maximum (mean) values. The genome sequence of MWH-Ta8T represents a closed genome organized in one contig, while the genome sequence strain IMCC13023 is organized in six scaffolds (Kang et al., 2012). nd, Not determined.
| Characteristic | MWH-Ta8T | ‘Ca. Aquiluna’ sp. IMCC13023 | Other Microbacteriaceae |
| Genome Size (Mbp) | 1.43 | 1.36 | 2.2–4.8 (3.3) |
| Putative ORFs | 1408 | 1410 | 2110–4766 (3298) |
| DNA G+C content (mol%)* | 51.5 | 51.7 | 59–73 (68.9) |
| DNA G+C content (mol%)† | nd | nd | 59–76 (67.1) |
Determined by genome sequencing.
Determined by HPLC analyses.
The genome of strain MWH-Ta8T encoded a putative phosphotransferase system (PTS) for fructose but a PTS for glucose was lacking. A complete gene set for gluconeogenesis was found but a gene encoding an enzyme for phosphorylation of fructose 6-phosphate to fructose 1,6-bisphosphate [e.g. a gene for phosphofructokinase-1 (EC 2.7.1.11)], which represents a crucial step of the glycolysis pathway, was missing in the genome annotations. A complete citrate cycle is encoded and all genes required for a respiratory electron transport chain were present in the annotated genome. On the other hand, the genome lacked genes encoding a cytochrome bd complex, which suggests that the strain is not adapted to low oxygen concentrations. Putative ABC transporters for phosphate, iron (III) ions, spermidine/putrescine, sorbitol/mannitol, d-xylose, and branched-chain amino acids were found in the genome. Two ammonium transporter genes of the amt family and genes encoding ammonia assimilation were found. Assimilatory and respiratory nitrite and nitrate reductases were lacking, which suggested that strain MWH-Ta8T is able to assimilate ammonia but not nitrate or nitrite. Furthermore, the genome of the strain completely lacked genes of flagellum synthesis.
The genome of strain MWH-Ta8T also encoded a putative actinorhodopsin/proteorhodopsin (Sharma et al., 2009). These proteins represent putative light-driven proton pumps enabling conservation of light energy. Only two of the 42 investigated reference genomes of members of the family Microbacteriaceae also encoded such genes. One of these two genomes (‘Ca. Aquiluna’ sp. IMCC13023) is also affiliated with the Luna-1 cluster of freshwater actinobacteria (Figs 1 and 2). As demonstrated previously (Sharma et al., 2009) the majority of investigated members of this cluster encoded actinorhodopsin genes. Interestingly, the second strain among the reference genome strains that possessed such a gene, Leifsonia rubra CMS 76rT, was also isolated from an aquatic habitat, i.e. from a cyanobacterial mat of a pond in McMurdo dry valleys, Antarctica (Kumar Pinnaka et al., 2013).
The genome of strain MWH-Ta8T represented a strongly streamlined genome sharing an equally small genome size with ‘Candidatus Pelagibacter ubique’ (Giovannoni et al., 2005b) a member of the marine SAR11 cluster (Alphaproteobacteria). Interestingly, both organisms also share the presence of proteorhodopsin/actinorhodopsin genes (Giovannoni et al., 2005a), but they differ strongly in their ability to grow in artificial media at high substrate concentrations or on the surface of agar plates (Carini et al., 2013).
Ecology and biogeography.
Strain MWH-Ta8T was isolated from the water column of a freshwater lake, suggesting that the strain possessed a planktonic lifestyle. This assumption is supported by the revealed genome characteristics, which are similar to those observed for other planktonic strains (Giovannoni et al., 2005b), as well as by detection of closely related ribotypes, which shared >99 % 16S rRNA gene sequence similarity and the presence of two diagnostic sequences defining ‘Ca. Rhodoluna lacicola’ (Hahn, 2009) with strain MWH-Ta8T, in the water column of various freshwater systems. Three related strains were obtained as mixed cultures from freshwater systems located in Australia (GenBank accession number AM999979), China (AM999980) and Austria (FJ545223). These bacteria also share with strain MWH-Ta8T small cell sizes and red pigmentation. In total, nine ribosomal sequences retrieved by cultivation-independent methods, which also share high sequence similarity and the presence of the two diagnostic sequences, originate from freshwater and estuary systems located in North America (HQ530784, EU800868, AY947900, AY947943, AF289150 and EF471688) and lakes located in China (JN232906, JF697478 and JF697420). These data indicate that the narrow phylogenetic taxon including strain MWH-Ta8T consists of planktonic freshwater bacteria with a worldwide distribution, at least in freshwater systems of the temperate and subtropical climate zone.
Proposal of a novel genus and species.
The results on phylogeny, phenotypic and chemotaxonomic characteristics of strain MWH-Ta8T suggest that this strain represents a novel species of a new genus in the family Microbacteriaceae. This strain can be well distinguished from Pontimonas salivibrio strain CL-TW6T (Table 4), which represented the closest related described species. In fact, strain MWH-Ta8T can be easily distinguished from all previously described species of the family due to its unusually small cell size and the unusually low DNA G+C content (Table 3). According to the previous description of strain MWH-Ta8T as ‘Ca. Rhodoluna lacicola’, we propose for this new taxon the name Rhodoluna lacicola gen. nov., sp. nov.
Table 4. Selected differential characteristics of strain MWH-Ta8T and the type strain of the most closely related described species.
| Characteristic | MWH-Ta8T | Pontimonas salivibrio CL-TW6T* |
| Isolation source | Freshwater, lake | Seawater, solar saltern |
| Mean cell length >1 µm | − | + |
| Growth at <15 °C | + | − |
| Salt tolerance (%) | 0–0.6 | 1–9 |
| Utilization of d-glucose | − | + |
| Major respiratory quinones (MK) | 11, 12 | 9, 10 |
| Homoserine present in peptidoglycan | + | − |
| DNA G+C content (mol%) | 51.5 | 60.0 |
Data from Jang et al. (2013).
Description of Rhodoluna gen. nov.
Rhodoluna [Rho.do.lu′na. Gr. n. rhodon the rose; L. fem. n. luna the moon; N.L. fem. n. Rhodoluna red-coloured moon, referring to the red pigmentation and the seemingly selenoid (crescent shape) morphology of the strain].
Aerobic chemo-organoheterotrophs. Cells are tiny, curved (selenoid) rods with cell volumes <0.1 µm3. The predominant cellular fatty acids are anteiso-C15 : 0, iso-C16 : 0, iso-C15 : 0 and iso-C14 : 0. The polar lipid profile contains diphosphatidylglycerol, phosphatidylglycerol and unknown glycolipids. The major menaquinones are MK-11 and MK12. The cell-wall peptidoglycan is of the B-type and contains 2,4-diaminobutyric acid as the diamino acid. The DNA G+C content is circa 52 mol%. The genome size is circa 1.4 Mbp. The type species is Rhodoluna lacicola.
Description of Rhodoluna lacicola sp. nov.
Rhodoluna lacicola [la.ci′co.la. L. masc. n. lacus lake; L. suffix n. -cola (from incola the inhabitant); N.L. masc. n. lacicola inhabitant of lakes].
General descriptions of morphological and chemotaxonomic features are as given in the genus description. Cells grown in liquid NSY medium are tiny, curved (selenoid) rods with a size of about 0.3×0.9 µm and cell volume of 0.04–0.06 µm3. Colonies on NSY agar are small, circular and convex with a shiny surface and red pigmentation. Unpigmented mutants may occur. Grows on NSY medium aerobically but not under anaerobic conditions. The temperature range for growth on NSY is 9 to 36 °C, and weak growth may occur at 37 °C. NaCl concentrations of 0–0.4 % in NSY medium are well tolerated. Weak growth may occur at concentrations of 0.5 % NaCl but growth is absent at higher salinities. Positive result in tests for catalase activity, but only very weak signals in tests for oxidase activity (scored as oxidase-negative). Assimilates d-mannose and d-sorbitol, but not d-glucose, d-fucose, propionic acid, fumaric acid, malonic acid, citric acid or betaine. Lacks genes for flagella synthesis, phosphofructokinase and cytochrome bd complex, but encodes an actinorhodopsin gene (GenBank accession no. FJ545221). Furthermore, characterized by the combined presence of two oligonucleotide sequences, 5′-CTTGCTCCGGTGGATTAGTGG-3′ (Escherichia coli positions 83–105) and 5′-ACGACACCTTGGGGCATCCCAGGGTGTGGAA-3′ (E. coli positions 181–196), within the 16S rRNA gene.
The type strain, MWH-Ta8T ( = DSM 23834T = LMG 26932T), was isolated from the water column of hypertrophic Mailing Bay, Lake Taihu, China. The G+C content of the genomic DNA of the type strain is 51.5 mol% and the genome size is 1.43 Mbp.
Acknowledgements
We are grateful to Maria Gadermaier (University of Innsbruck, Mondsee, Austria) and Katrin Sommerfeld (Dalhousie University, Halifax, Canada) for excellent technical assistance. This project was supported by a Canadian Institutes for Health Research grant (MOP-4467) to W. F. D., an Academy of Finland grant (251665) to S. T., and an Austrian Science Fund grant (I482-B09) to M. W. H.
Abbreviations:
- IMG
integrated microbial genomes
- ML
maximum-likelihood
- MP
maximum-parsimony
- NJ
neighbour-joining
- UMB
ultramicrobacteria.
Footnotes
A supplementary table and a supplementary figure are available with the online version of this paper.
References
- Allgaier M., Grossart H. P. (2006). Diversity and seasonal dynamics of Actinobacteria populations in four lakes in northeastern Germany. Appl Environ Microbiol 72, 3489–3497 10.1128/AEM.72.5.3489-3497.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S.-Y., Xiao T., Yokota A. (2008). Schumannella luteola gen. nov., sp. nov., a novel genus of the family Microbacteriaceae. J Gen Appl Microbiol 54, 253–258 10.2323/jgam.54.253 [DOI] [PubMed] [Google Scholar]
- Carini P., Steindler L., Beszteri S., Giovannoni S. J. (2013). Nutrient requirements for growth of the extreme oligotroph ‘Candidatus Pelagibacter ubique’ HTCC1062 on a defined medium. ISME J 7, 592–602 10.1038/ismej.2012.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis T. Z., Hugenholtz P., Larsen N., Rojas M., Brodie E. L., Keller K., Huber T., Dalevi D., Hu P., Andersen G. L. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with arb. Appl Environ Microbiol 72, 5069–5072 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda V. I., Suzina N. E., Polivtseva V. N., Boronin A. M. (2012). [Ultramicrobacteria: Formation of the concept and contribution of ultramicrobacteria to biology]. Mikrobiologiia 81, 415–427 (in Russian). [PubMed] [Google Scholar]
- Garcia S. L., McMahon K. D., Martinez-Garcia M., Srivastava A., Sczyrba A., Stepanauskas R., Grossart H. P., Woyke T., Warnecke F. (2013). Metabolic potential of a single cell belonging to one of the most abundant lineages in freshwater bacterioplankton. ISME J 7, 137–147 10.1038/ismej.2012.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni S. J., Bibbs L., Cho J. C., Stapels M. D., Desiderio R., Vergin K. L., Rappé M. S., Laney S., Wilhelm L. J. & other authors (2005a). Proteorhodopsin in the ubiquitous marine bacterium SAR11. Nature 438, 82–85 10.1038/nature04032 [DOI] [PubMed] [Google Scholar]
- Giovannoni S. J., Tripp H. J., Givan S., Podar M., Vergin K. L., Baptista D., Bibbs L., Eads J., Richardson T. H. & other authors (2005b). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245 10.1126/science.1114057 [DOI] [PubMed] [Google Scholar]
- Glöckner F. O., Zaichikov E., Belkova N., Denissova L., Pernthaler J., Pernthaler A., Amann R. (2000). Comparative 16S rRNA analysis of lake bacterioplankton reveals globally distributed phylogenetic clusters including an abundant group of actinobacteria. Appl Environ Microbiol 66, 5053–5065 10.1128/AEM.66.11.5053-5065.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M. W. (2006). The microbial diversity of inland waters. Curr Opin Biotechnol 17, 256–261 10.1016/j.copbio.2006.05.006 [DOI] [PubMed] [Google Scholar]
- Hahn M. W. (2009). Description of seven candidate species affiliated with the phylum Actinobacteria, representing planktonic freshwater bacteria. Int J Syst Evol Microbiol 59, 112–117 10.1099/ijs.0.001743-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M. W., Lünsdorf H., Wu Q. L., Schauer M., Höfle M. G., Boenigk J., Stadler P. (2003). Isolation of novel ultramicrobacteria classified as Actinobacteria from five freshwater habitats in Europe and Asia. Appl Environ Microbiol 69, 1442–1451 10.1128/AEM.69.3.1442-1451.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M. W., Stadler P., Wu Q. L., Pöckl M. (2004). The filtration-acclimatization method for isolation of an important fraction of the not readily cultivable bacteria. J Microbiol Methods 57, 379–390 10.1016/j.mimet.2004.02.004 [DOI] [PubMed] [Google Scholar]
- Hahn M. W., Lang E., Brandt U., Wu Q. L., Scheuerl T. (2009). Emended description of the genus Polynucleobacter and the species Polynucleobacter necessarius and proposal of two subspecies, P. necessarius subsp. necessarius subsp. nov. and P. necessarius subsp. asymbioticus subsp. nov. Int J Syst Evol Microbiol 59, 2002–2009 10.1099/ijs.0.005801-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang G. I., Cho Y., Cho B. C. (2013). Pontimonas salivibrio gen. nov., sp. nov., a new member of the family Microbacteriaceae isolated from a seawater reservoir of a solar saltern. Int J Syst Evol Microbiol 63, 2124–2131 10.1099/ijs.0.043661-0 [DOI] [PubMed] [Google Scholar]
- Jezbera J., Sharma A. K., Brandt U., Doolittle W. F., Hahn M. W. (2009). ‘Candidatus Planktophila limnetica’, an actinobacterium representing one of the most numerically important taxa in freshwater bacterioplankton. Int J Syst Evol Microbiol 59, 2864–2869 10.1099/ijs.0.010199-0 [DOI] [PubMed] [Google Scholar]
- Kang I., Lee K., Yang S.-J., Choi A., Kang D., Lee Y. K., Cho J.-C. (2012). Genome sequence of “Candidatus Aquiluna” sp. strain IMCC13023, a marine member of the Actinobacteria isolated from an arctic fjord. J Bacteriol 194, 3550–3551 10.1128/JB.00586-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Pinnaka A., Singh A., Ara S., Begum Z., Reddy G. S., Shivaji S. (2013). Draft genome sequence of Leifsonia rubra strain CMS 76RT, isolated from a cyanobacterial mat sample from a pond in Wright Valley, McMurdo, Antarctica. Genome announcements. 10.1128/genomeA.00633-13 [DOI] [PMC free article] [PubMed]
- Markowitz V. M., Chen I. M. A., Palaniappan K., Chu K., Szeto E., Grechkin Y., Ratner A., Jacob B., Huang J. H. & other authors (2012). IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res 40 (Database issue), D115–D122 10.1093/nar/gkr1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meincke L., Copeland A., Lapidus A., Lucas S., Berry K. W., Del Rio T. G., Hammon N., Dalin E., Tice H. & other authors (2012). Complete genome sequence of Polynucleobacter necessarius subsp. asymbioticus type strain (QLW-P1DMWA-1T). Stand Genomic Sci 6, 74–83 10.4056/sigs.2395367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R. J., Jones S. E., Eiler A., McMahon K. D., Bertilsson S. (2011). A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75, 14–49 10.1128/MMBR.00028-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish C. C. (1999). Determination of total lipid, lipid classes, and fatty acids in aquatic samples. In Lipids in Freshwater Ecosystems, pp. 4–20 Edited by Arts M. T., Wainman B. C. New York: Springer; 10.1007/978-1-4612-0547-0_2 [DOI] [Google Scholar]
- Salcher M. M., Pernthaler J., Posch T. (2010). Spatiotemporal distribution and activity patterns of bacteria from three phylogenetic groups in an oligomesotrophic lake. Limnol Oceanogr 55, 846–856 10.4319/lo.2009.55.2.0846 [DOI] [Google Scholar]
- Schleifer K.-H., Kandler O. (1972). Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 36, 407–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann P. (2011). Peptidoglycan structure. Methods Microbiol 38, 101–129 10.1016/B978-0-12-387730-7.00005-X [DOI] [Google Scholar]
- Schumann P., Kämpfer P., Busse H.-J., Evtushenko L. I., Subcommittee on the Taxonomy of the Suborder Micrococcineae of the International Committee on Systematics of Prokaryotes (2009). Proposed minimal standards for describing new genera and species of the suborder Micrococcineae. Int J Syst Evol Microbiol 59, 1823–1849 10.1099/ijs.0.012971-0 [DOI] [PubMed] [Google Scholar]
- Schumann P., Zhang D.-C., Redzic M., Margesin R. (2012). Alpinimonas psychrophila gen. nov., sp. nov., an actinobacterium of the family Microbacteriaceae isolated from alpine glacier cryoconite. Int J Syst Evol Microbiol 62, 2724–2730 10.1099/ijs.0.036160-0 [DOI] [PubMed] [Google Scholar]
- Sharma A. K., Sommerfeld K., Bullerjahn G. S., Matteson A. R., Wilhelm S. W., Jezbera J., Brandt U., Doolittle W. F., Hahn M. W. (2009). Actinorhodopsin genes discovered in diverse freshwater habitats and among cultivated freshwater Actinobacteria. ISME J 3, 726–737 10.1038/ismej.2009.13 [DOI] [PubMed] [Google Scholar]
- Sun S., Mao R. (2008). Hydrography and drainage basin. In Lake Taihu, China: Dynamics and Environmental Change, pp. 1–10 Edited by Qin B. Springer: Berlin; 10.1007/978-1-4020-8555-0_1 [DOI] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28, 2731–2739 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-Y., Liu X.-Y., Liu S.-J. (2010). Agrococcus terreus sp. nov. and Micrococcus terreus sp. nov., isolated from forest soil. Int J Syst Evol Microbiol 60, 1897–1903 10.1099/ijs.0.013235-0 [DOI] [PubMed] [Google Scholar]