

Abstract
We recently demonstrated direct evidence of increased monoamine oxidase (MAO) activity in the brain of a simian immunodeficiency virus (SIV) model of human immunodeficiency virus (HIV)-associated central nervous system (CNS) disease, consistent with previously reported dopamine deficits in both SIV and HIV infection. In this study, we explored potential mechanisms behind this elevated activity. MAO B messenger RNA was highest in macaques with the most severe SIV-associated CNS lesions and was positively correlated with levels of CD68 and GFAP transcripts in the striatum. MAO B messenger RNA also correlated with viral loads in the CNS of SIV-infected macaques and with oxidative stress. Furthermore, in humans, striatal MAO activity was elevated in individuals with HIV encephalitis, compared with activity in HIV-seronegative controls. These data suggest that the neuroinflammation and oxidative stress caused by SIV infection in the CNS may provide the impetus for increased transcription of MAO B and that MAO, and more broadly, oxidative stress, have significant potential as therapeutic targets in CNS disease due to HIV.
Keywords: HIV, SIV, monoamine oxidase, oxidative stress, neuroinflammation, reactive oxygen species
The neuropathogenesis of human immunodeficiency virus (HIV) infection is mediated by complex interactions between the virus and the host. While HIV does not directly infect neurons, the indirect effects of central nervous system (CNS) viral infection cause neuronal dysfunction. In the CNS, macrophages and microglia are the primary producers of virus [1]. Infected cells produce virus and secrete neurotoxic viral proteins (eg, Tat, gp120, and Nef), cytokines (such as tumor necrosis factor α and interleukin 1β), and reactive oxygen species (ROS), all of which have deleterious effects on neurons [2–6]. Many of these products induce oxidative stress, which can facilitate viral replication [7–9] and can disrupt neuronal function via lipid peroxidation, protein carbonylation, protein nitrosylation, and DNA damage [4, 5, 10]. Virus, viral proteins, cytokines, and ROS can additionally activate surrounding uninfected macrophages, microglia, and astrocytes, perpetuating neuroinflammation and neuronal damage.
While HIV may affect any part of the brain, the basal ganglia region is particularly vulnerable. This area is a hot spot of viral replication and neuropathology and is atrophied in HIV-infected individuals [11–14]. Dopaminergic neurons, which send many of their projections to the basal ganglia, are particularly susceptible to ROS and many virologic and immunologic insults [15]. Levels of dopamine and its metabolites are lower in both HIV-infected individuals and simian immunodeficiency virus (SIV)–infected macaques in the basal ganglia [16–20], and this decrease has been shown to correlate with neurological deficits in humans [20]. Furthermore, SIV studies have shown increased ratios of dopamine metabolites relative to dopamine, indicating increased dopamine catabolism or decreased production [16, 17].
One of the primary enzymes responsible for dopamine catabolism is monoamine oxidase (MAO). There are 2 isoforms of MAO, A and B, each a product of separate genes on the X chromosome. MAO A is primarily found in catecholaminergic neurons, whereas MAO B is primarily found in serotonergic neurons and glia [21]. The 2 isoforms were originally distinguished biochemically by their substrate and inhibitor specificities, and both are important for dopamine metabolism in the primate CNS [22]. In the oxidation of monoamines, MAO produces H2O2, a damaging ROS, as a byproduct. Experimentally elevating MAO activity in rodents decreases levels of dopamine and increases levels of H2O2, glutathione oxidation, astrocytosis, and neuronal damage [23–25]. Therefore, MAO activity together with autoxidation of dopamine and its metabolites create a delicate redox environment surrounding dopaminergic neurons in homeostasis.
We recently demonstrated increased MAO activity in SIV-infected macaques in late-stage disease [17]. Transcription of the MAO B isoform, but not MAO A, is increased in a p38-dependent manner in vitro [26]. Interestingly, the p38/MAPK signaling pathway is a point of convergence for HIV/SIV virions, viral proteins, oxidative stress, and neuroinflammatory processes [27–29]. While p38 activation is dependent on cell type and context, we have previously shown p38 activation in astrocytes and neurons of SIV-infected macaques [30]. We therefore hypothesized that MAO B transcription could be induced by the neuroinflammatory environment generated in SIV/HIV infection of the CNS.
In this study, we demonstrated that elevated MAO activity in the basal ganglia of SIV-infected macaques was due to increased transcription of the MAO B isoform. MAO activity positively correlated with several neuroinflammatory measures in the basal ganglia of SIV-infected macaques, with viral loads in the CNS, and with oxidative stress. Furthermore, MAO activity was increased in the striatum of HIV-infected individuals with encephalitis. Together, these data suggest that the neuroinflammatory environment of HIV/SIV-associated CNS disease may promote MAO activation, which can contribute to neuropathogenesis via monoaminergic dysregulation and ROS production.
MATERIALS AND METHODS
Animal Infection and Treatment
Experiments were performed using archived samples from juvenile pigtailed macaques (Macaca nemestrina) that were either mock inoculated or inoculated intravenously with the neurovirulent clone SIV/17E-Fr and the immunosuppressive swarm SIV/DeltaB670 [31]. Blood and CSF specimens were collected periodically after inoculation and at euthanasia to monitor viral loads and inflammatory markers. Macaques were scheduled to undergo euthanasia 3 months after inoculation, the time at which all macaques have AIDS and most have developed encephalitis [31, 32]. Occasionally, animals were euthanized before the scheduled euthanasia date if the animal presented with clinical symptoms, as described previously [33]. At the time of euthanasia, macaques were perfused with phosphate-buffered saline to remove blood from tissue vasculature. The brain was sliced into 0.5 cm coronal sections. Sections were snap frozen and stored at −80°C until use, or they were fixed and embedded in paraffin. The Johns Hopkins Animal Care and Use Committee approved all animal studies. Animals were treated humanely in accordance with federal guidelines.
Human Samples Obtained From Tissue Banks
Samples of fresh frozen caudate or putamen (striatum) from human autopsy specimens were obtained from tissue banks that are part of the National NeuroAIDS Tissue Consortium (Manhattan HIV Brain Bank, Texas NeuroAIDS Research Center, National Neurological AIDS Bank, and California NeuroAIDS Tissue Network) or other independent tissue banks (Harvard Brain Tissue Resource Center and Human Brain and Spinal Fluid Resource Center). Samples were acquired from 14 HIV-seronegative controls and 14 HIV-positive individuals with encephalitis (HIVE). We requested specimens from individuals without history of drug abuse, psychiatric illness and with no known use of MAO inhibitors. However, this information was often not available. While the utmost care was taken to ensure the groups were demographically matched as well as possible, the seronegative group was significantly older than the HIVE group (median ages, 52.2 and 42.0 years, respectively; P = .003, by the Mann–Whitney test). All specimens were male, except for 1 specimen from the HIVE group. All specimens were from the caudate nucleus, except for 2 cases from the HIVE group, which were from the putamen.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Measurement of MAO A, MAO B, and SIV Messenger RNA (mRNA)
RNA was isolated from macaque basal ganglia and CSF as previously described [31]. For measurement of MAO A and MAO B mRNA, 100–200 ng of RNA was used for complementary DNA synthesis using Superscript III reverse transcriptase (Invitrogen). qPCR was performed with NoROX Multiplex Mix (Qiagen), using iQ5 (BioRad) or Rotorgene (Qiagen) thermocyclers. MAO A and MAO B were normalized to 18S and were expressed as fold-change from the average of uninfected controls, using the ΔΔCt method. PCR conditions were 95°C for 12 minutes, followed by 45–55 cycles at 95°C for 15 seconds, 55°C for 15 seconds, and 62°C for 15 seconds. Primers and probes were designed on the basis of rhesus macaque (Macaca mulatta) sequences in GenBank. Primer sequences were as follows:
for MAO A: 5′-TGAAGCTGAACCAACCTGTC-3′ (forward), 5′-AATGCGATCCCTCCGACCTTGAC-3′ (probe), and 5′-AAGCTCTGGTCTGAAGTGAATC-3′ (reverse); and for MAO B: 5′-ACTCTGGACTGAATGTGGTTG-3′ (forward), 5′-TCCTAAGAGTGTAAGTCCTGCCTCCC-3′ (probe), and 5′-GATACGATTCTGGGTTGGTCC -3′ (reverse). 18S primer/probe sequences were previously published [34].
The SIV viral loads in the CSF and basal ganglia of macaques were determined by qRT-PCR, as previously described [17]. Viral loads were expressed as SIV copy equivalents per milliliter of CSF or per 1 µg of RNA isolated from basal ganglia. Statistical analyses involving viral loads were performed using log-transformed values.
MAO Activity Measurement
MAO activity was measured in macaque basal ganglia (75 µg of protein) or human striatum (15 µg of protein), using the luminescence-based MAO-Glo Assay (Promega), as previously described [17].
Nanostring nCounter Gene Expression Analysis
A custom Nanostring CodeSet (Nanostring Technologies) was designed to measure 249 transcripts of interest and 11 putative housekeeping transcripts. For the purposes of this study, we were only interested in MAO A, MAO B, genes involved in ROS detoxification (CAT, GPX1, GPX3, GPX4, GPX7, GSR, GSS, PRDX1, PRDX2, PRDX3, PRDX4, PRDX6, SRXN1, TXN, TXNRD1, TXNRD2, SOD1, SOD2, SOD3, and CCS), CD68 (macrophage/microglial marker), and GFAP (astrocytic marker), in addition to putative housekeeping genes (ACTB, B2M, GAPDH, HMBS, HPRT1, RPL13A, rpS9, SDHA, TPB, UBC, and YWHAZ). Transcript target sequences can be found in Supplementary Table 1.
Total RNA was isolated from macaque striatum by using RNA STAT-60 (Tel-Test), treated with DNase, and purified using the mirVana total RNA purification protocol (Life Technologies). RNA concentration and purity was determined using a NanoDrop Spectrophotometer (Thermo Fisher Scientific). A total of 100 ng of RNA per sample was used for the custom nCounter Gene Expression Assay according to the manufacturer's protocol. The assay was performed by the Johns Hopkins University Microarray Core Facility. To assess the probe-specific background, 100 ng of MS2 phage RNA (Roche) was run in triplicate as a negative control.
Nanostring data were analyzed by adding a value of 1 to all counts, normalizing to the geometric mean of manufacturer spike-in controls, and normalizing to the geometric mean of the most stably expressed housekeeping genes (GAPDH, RPL13A, SDHA, and TBP). Data were expressed as normalized counts. The probe-specific limit of detection was set to the mean value + 2 SDs for the MS2 phage RNA negative control. More-detailed methods can be found in the Supplementary Materials.
Semiquantitative Measurement of Macaque CNS Lesion Severity
SIV and HIV encephalitis are characterized by infiltrating macrophages, perivascular cuffing, microglial nodules, and multinucleated giant cells. CNS lesion severity (defined as none, mild, moderate, or severe) was quantified for each macaque, as previously described [31]. We have previously shown that CNS lesion severity correlates with several markers of neuroinflammation and CNS viral loads [31].
Glutathione Measurement
The small molecule glutathione is an important redox regulator that detoxifies the ROS H2O2, which is produced by multiple cellular enzymes, such as superoxide dismutase during the inactivation of the ROS superoxide and monoamine oxidase in the catabolism of monoamines [35]. Reduced and oxidized forms of glutathione (GSH and GSSG, respectively) were measured using a GSH/GSSG Ratio Detection Assay Kit (Abcam) according to the manufacturer's protocol. Three-millimeter-punch specimens were taken from macaque striatum and homogenized by sonication in 0.1% ascorbic acid (ratio of weight to volume, 1:20) while on ice. Samples were aliquoted and stored at −80°C until use. Insoluble debris was removed by centrifugation, and 10 µL of homogenate was used, in duplicate, for detection of GSH or GSSG. Results are the average of data from 2 separate experiments.
RESULTS
Increased MAO B Transcription and MAO Activity in the Brains of SIV-Infected Macaques
We recently showed that MAO activity is elevated in late-stage SIV infection [17], and we hypothesized that the increase in MAO activity might be due to enhanced transcription of MAO, particularly the MAO B isoform. To test this hypothesis, we used qRT-PCR and Nanostring gene expression analysis to measure mRNA of the MAO A and MAO B isoforms in the basal ganglia of uninfected and SIV-infected pigtailed macaques. The MAO B mRNA level measured by qRT-PCR was significantly increased in the striatum of SIV-infected macaques, compared with the level for uninfected controls (P = .002; Figure 1A), which was confirmed by Nanostring analysis (2107 counts vs 1431 counts; P = .030, by the Mann–Whitney test). The increase in MAO B mRNA level positively correlated with the previously published levels of MAO activity (P = .014; Figure 1B [17]).
Figure 1.
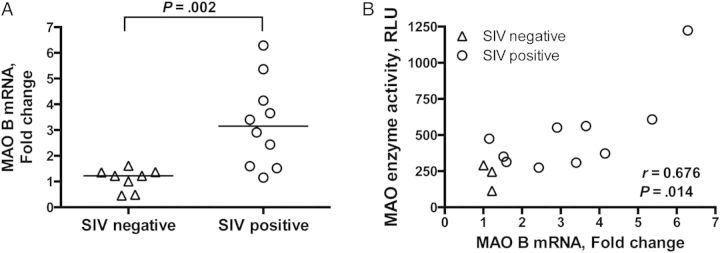
Monoamine oxidase (MAO) B transcription level was elevated in the basal ganglia of simian immunodeficiency virus (SIV)–infected macaques and correlated with MAO activity. A, MAO B messenger RNA (mRNA) level was measured by quantitative real-time polymerase chain reaction analysis and was increased in SIV-infected macaques, compared with uninfected controls (P = .002). MAO B mRNA level was normalized to 18S and is expressed as fold-change relative to the average of uninfected controls, using the ΔΔCt method. B, MAO B mRNA level positively correlated with MAO enzyme activity (P = .014), as measured by a luminescent method, indicating that increased enzyme activity was at least partly due to increased transcription of MAO B. Bars in panel A represent medians. Statistical analyses used the Mann–Whitney test (A) or Spearman correlation (B). Abbreviation: RLU, relative light units.
The MAO A mRNA level measured by qRT-PCR was near the limit of detection, making replicates too variable to report. However, there was no difference in MAO A mRNA levels between uninfected and SIV-infected macaques as measured by Nanostring analysis (data not shown).
MAO Activity and MAO B mRNA Level Positively Correlated With Neuroinflammation
MAO B transcription can be elevated by the p38/MAPK signaling pathway [26], which can be triggered by inflammatory cytokines, virus, viral proteins, and oxidative stress [27–29]. Therefore, we hypothesized that the elevations in MAO activity and MAO B mRNA level would correlate with markers of neuroinflammation. SIV-induced CNS lesions, characterized by infiltrating macrophages, perivascular cuffing, microglial nodules, and multinucleated giant cells, are a pathological indication of neuroinflammation. The severity of CNS lesions was scored as none, mild, moderate, or severe. The MAO activity and MAO B mRNA level, as measured by Nanostring analysis, were higher in macaques with moderate/severe encephalitis than those with none/mild encephalitis (P = .067 and P = .002, respectively; Figure 2A and 2B).
Figure 2.

Monoamine oxidase (MAO) was highest in the basal ganglia of macaques with more neuroinflammation. A, Simian immunodeficiency virus (SIV)–infected macaques with moderate or severe encephalitis tended to have higher MAO activity in the basal ganglia than those with no or mild encephalitis (P = .067). B, SIV-infected macaques with moderate or severe encephalitis had higher levels of MAO B messenger RNA (mRNA) in the striatum, as measured by Nanostring analysis, than those with no or mild encephalitis (P = .002). MAO B mRNA levels positively correlated with CD68 (C) and GFAP (D) transcripts in the striatum, as measured by Nanostring analysis (P < .001 for both). Bars in panels A and B represent medians. Statistical analyses used the Mann–Whitney test (A and B) or Spearman correlation (C and D). Points falling within the gray shading are below the transcript-specific limit of detection. Abbreviations: CNS, central nervous system; RLU, relative light units.
Since measurement of CNS lesion severity is semiquantitative, we also used Nanostring analysis to quantitate mRNA levels of CD68 (a marker of macrophages and microglia and their activation) and GFAP (a marker of astrocytes and their activation). The MAO B mRNA level positively correlated with CD68 and GFAP mRNA levels (P < .001 for both comparisons; Figure 2C and 2D). These data indicate that MAO mRNA levels are increased in the proinflammatory environment of the brain during SIV infection.
MAO B mRNA Levels Correlated With CNS Viral Loads
We also hypothesized that MAO B mRNA levels would correlate with levels of virus in the CNS because activated macrophages/microglia are a major source of viral production. Viral loads were quantified by qRT-PCR in the basal ganglia and CSF. MAO B mRNA levels positively correlated with terminal viral loads in the basal ganglia and CSF of SIV-infected macaques (P = .030 and P = .001, respectively; Figure 3).
Figure 3.
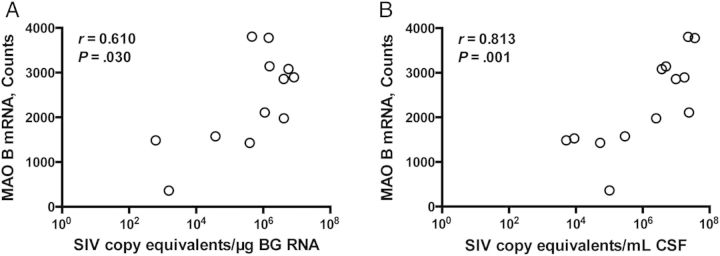
Monoamine oxidase (MAO) B messenger RNA (mRNA) levels in the striatum correlated with central nervous system viral loads. A, MAO B mRNA level, as measured by Nanostring analysis, positively correlated with viral load in the basal ganglia (BG, P = .030). B, MAO B mRNA level also positively correlated with viral load in the cerebrospinal fluid (CSF; P = .001). Viral loads were expressed as simian immunodeficiency virus (SIV) copy equivalents per microgram of RNA (A) or per milliliter of CSF (B). Statistical analyses used Spearman correlation.
Oxidative Stress Levels Were Elevated in the Brains of SIV-Infected Macaques and Correlated With MAO B mRNA Levels
Whereas induction of inflammatory cytokines, neuroinflammation, and high viral loads have been well established in this SIV/macaque model, oxidative stress has only been demonstrated by the dual oxidative/nitrosative stress product nitrotyrosine [17, 36]. To further examine the hypothesis that the level of oxidative stress is elevated in the CNS of SIV-infected macaques, we measured the ratio of reduced to oxidized glutathione (GSH/GSSG), a decrease in which is representative of an increased level of oxidative stress. The GSH/GSSG ratio in the striatum of SIV-infected macaques was significantly lower than that in uninfected controls (P < .001; Figure 4A).
Figure 4.

Oxidative stress level was elevated in the striatum of simian immunodeficiency virus (SIV)–infected macaques and correlated with monoamine oxidase (MAO) B messenger RNA (mRNA) level. A, Reduced and oxidized glutathione (GSH and GSSG, respectively) were measured by a commercially available fluorimetric method. SIV-infected macaques had lower GSH/GSSG ratios than uninfected macaques (P < .001), which is indicative of oxidative stress. B, mRNA levels of genes involved in reactive oxygen species (ROS) detoxification (CAT, GPX1, GPX3, GPX4, GPX7, GSR, GSS, PRDX1, PRDX2, PRDX3, PRDX4, PRDX6, SRXN1, TXN, TXNRD1, TXNRD1, SOD1, SOD2, SOD3, and CCS) were measured using a custom Nanostring gene expression assay. To assess the cumulative impact on genes involved in ROS detoxification, the geometric mean of the counts of these transcripts was calculated and compared between SIV-negative and SIV-positive macaques. Transcripts of genes involved in ROS detoxification were elevated in SIV-infected macaques, compared with those in uninfected controls (P = .007). C, MAO B mRNA level, as measured by Nanostring analysis, negatively correlated with the ratio of GSH/GSSG (P = .044). D, MAO B mRNA positively correlated with the geometric mean counts of transcripts involved in ROS detoxification (P < .001). Bars represent medians. Statistical analyses used the Mann–Whitney test (A and B) or Spearman correlation (C and D).
We also quantitated the mRNA expression of the following genes involved in ROS detoxification, using Nanostring gene expression analysis: CAT, GPX1, GPX3, GPX4, GPX7, GSR, GSS, PRDX1, PRDX2, PRDX3, PRDX4, PRDX6, SRXN1, TXN, TXNRD1, TXNRD2, SOD1, SOD2, SOD3, and CCS. To assess the cumulative level of ROS detoxification transcripts, the geometric mean of the counts of these transcripts was calculated for each macaque, and this geometric mean was compared between uninfected and SIV-infected macaques. The geometric mean of ROS detoxification transcripts was increased in the striatum of SIV-infected macaques, compared with uninfected controls (P = .007; Figure 4B). Together, the decrease in GSH/GSSG ratios and the elevated ROS detoxification transcript levels support the hypothesis of elevated levels of oxidative stress in SIV infection of the CNS.
We then tested the hypothesis that the MAO B mRNA level would correlate with these two measures of oxidative stress. The MAO B mRNA level negatively correlated with GSH/GSSG ratios and positively correlated with levels of ROS detoxification transcripts (P = .044 and P < .001, respectively; Figure 4C and 4D). A caveat to these data is that the uninfected controls anchor the low MAO B/high GSH/GSSG relationship, thereby significantly impacting this correlation. Indeed, separation of the SIV-negative and SIV-positive groups eliminates the MAO B/GSSG reciprocity, indicating that MAO B is unlikely the only contributor to glutathione dysregulation in SIV infection. In contrast, the correlation of the MAO B mRNA level with ROS detoxification is maintained for the SIV-negative and SIV-positive groups. These data demonstrate that as the level oxidative stress increases, so does the level MAO B transcription.
MAO Activity Was Increased in the Striatum of HIV-Infected Individuals With Encephalitis
Since we determined that MAO activity was upregulated in the SIV/macaque model, we tested whether this phenomenon was recapitulated in HIV-infected individuals with encephalitis. We found that MAO activity was increased in the striatum of HIV-infected individuals with encephalitis, compared with activity in seronegative controls (P = .009; Figure 5). These findings are likely even more meaningful since the seronegative control group was significantly older than the HIVE group and MAO activity increases with age [37]. There was not a corresponding increase in MAO B transcription in human brain, as was found in the SIV-infected macaques (data not shown).
Figure 5.

Monoamine oxidase (MAO) activity was increased in the striatum of individuals with human immunodeficiency virus infection encephalitis (HIVE), compared with uninfected controls. MAO activity was measured in human caudate nucleus or putamen homogenates by a luminescent method. MAO activity was higher in individuals with HIVE, compared with seronegative controls (P = .009). Bars represent medians. Statistical comparisons were made using the Mann–Whitney test. Abbreviation: RLU, relative light unit.
DISCUSSION
To our knowledge, this is the first report showing increased MAO activity in the brains of HIV-infected individuals with encephalitis. In our SIV model of HIV-associated CNS disease, we determined that increased MAO activity was at least partly due to an increase in transcription of the MAO B isoform. Elevated levels of MAO B mRNA correlated with more severe neuroinflammation (as measured by CNS lesion severity and activation markers of macrophages/microglia and astrocytes), SIV viral loads in the basal ganglia and CSF, as well as higher levels of oxidative stress. Since MAO can contribute to oxidative stress via generation of H2O2, these data suggest that elevated MAO levels may be both a consequence of and a contributor to the neuropathogenesis of SIV and HIV infection.
Taking a comprehensive view of this correlative study together with current knowledge of HIV/SIV neuropathogenesis, we propose a chain of events whereby (1) HIV/SIV infects the CNS; (2) infection produces virus, toxic viral proteins, neuroinflammatory cytokines, and oxidative stress, which activate p38/MAPK signaling; (3) p38/MAPK activation increases MAO B transcription and thus MAO activity; and (4) elevated MAO levels increase monoaminergic neurotransmitter turnover and H2O2 production, thus further contributing to oxidative stress and neuroinflammation. This model suggests potential for a self-perpetuating positive feedback mechanism whereby the neuroinflammatory environment promoted by HIV/SIV infection of the CNS leads to overactive MAO and H2O2 generation, which stimulates further neuroinflammation and more MAO activity.
Whereas p38/MAPK signaling promotes MAO B transcription, it has the opposite action on MAO A, decreasing enzyme activity via inhibitory phosphorylation of serine 209 [38], meaning that p38/MAPK signaling could have complex effects on overall MAO activity. However, levels of MAO B are higher than levels of MAO A in the primate striatum [22], so any inhibitory effect on MAO A activity due to p38/MAPK signaling is likely more than counterbalanced by increases in MAO B transcription. Similarly, Chaudhuri et al recently identified a microRNA, the level of which is increased in the brains of HIV-infected humans and SIV-infected macaques, that is capable of downregulating MAO A transcription via SIRT1 [39]. However, in our studies, the MAO A mRNA level in the striatum was not different between SIV-infected macaques and uninfected controls, and the MAO B mRNA level correlated with MAO activity, indicating that an increase in the level of MAO B isoform mRNA is the major contributor to increased enzyme activity in the basal ganglia of SIV-infected macaques in late-stage disease in this model.
While we found an increase in MAO activity in the brains of HIV-infected individuals with encephalitis, we did not find a parallel increase in MAO B transcription. One reason for the divergence could be differences in disease stages, with human subjects likely having more variation in disease history and progression than the macaques in our tightly controlled, consistent model of infection. Whereas the macaques in this study were not given any drugs (eg, antiretrovirals or drugs of abuse), the patients' complete drug histories are unknown and could confound results. Furthermore, an increase in MAO activity without an increase in mRNA level could indicate heretofore-unidentified posttranscriptional or posttranslational regulatory mechanisms of MAO.
Although encephalitis remains an important clinical problem when treatment is unavailable, the relevance of our findings must be tested directly in the setting of combination antiretroviral therapy (cART). While successful cART greatly reduces neuroinflammation, levels of many neuroinflammatory markers often remain above the levels observed in uninfected subjects [40, 41]. If our hypothesis that neuroinflammation drives the elevation in MAO activity is correct, then cART would decrease MAO activity, but the activity may remain above homeostatic levels in some individuals, potentially causing dopamine dysregulation and further neuroinflammation via generation of H2O2.
Oxidative stress and MAO have both been targets for clinical trials to treat HIV-associated neurocognitive disorders. Antioxidants such as OPC-14117, thioctic acid, and minocycline have all been tested in HIV-positive patients with neurocognitive impairment, and unfortunately none have improved neurocognition as measured in those trials [42–45]. Selegiline, a MAO B–selective inhibitor with ROS-scavenging and neurotrophic properties, has been tested in several clinical trials for HIV-infected patients with cognitive impairment and showed some limited benefit [43, 46–49]. In contrast, selegiline administered at peak acute viremia in an SIV model of CNS disease protected dopamine levels but also exacerbated neuropathology and virus production [16].
The contradiction between the HIV clinical trials and SIV experiments using selegiline could be due to differences in drug doses, routes of administration, and, perhaps more importantly, timing of treatment initiation with respect to disease progression. In the human trials, the drug was administered to patients experiencing neurological complications, indicating an advanced stage of neurological disease. However, in the SIV experiments, selegiline was administered during peak viremia, 2 weeks after inoculation, a time when, perhaps, dopamine levels were not yet impacted by SIV, potentially leading to pathologically high levels of dopamine in the early stages of infection.
Previous studies in our macaque model indicate that there may be a therapeutic window for intervention with antioxidants, such as minocycline [17]. Unfortunately, given the variable times since infection onset for most individuals in clinical trials, the question of treatment initiation is, as of yet, unaddressed. Therapies that improve neurocognition in patients with established neurocognitive deficits are needed, and it is also important to identify therapies that prevent the onset of neurocognitive decline. In light of these studies, a reevaluation of the therapeutic potential of MAO inhibitors and, more broadly, antioxidant therapy for the treatment of HIV-associated neurocognitive disorders, with careful attention to the timing of treatment initiation with respect to disease progression, may be warranted.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Bob Adams for overseeing the health and care of the animals. We thank Elizabeth Engle, Ming Li, and Suzanne Queen, for technical assistance; Patrick Tarwater, for statistical advice; staff at the Johns Hopkins Microarray Core Facility, for running the Nanostring assays; and staff at the Johns Hopkins Brain Science Institute, for partial financial support of the Nanostring assays.
Human tissue samples were provided by the following repositories, which obtain funding from multiple institutions: Manhattan HIV Brain Bank (New York, NY: U01MH083501 and R24MH59724), Texas NeuroAIDS Research Center (Galveston, TX: U01MH083507 and R24NS45491), National Neurological AIDS Bank (Los Angeles, CA: 5U01MH083500 and NS38841), California NeuroAIDS Tissue Network (San Diego, CA: U01MH083506 and R24MH59745), National NeuroAIDS Tissue Consortium Statistics and Data Coordinating Center (Rockville, MD, and Indianapolis, IN: U01MH083545 and N01MH32002), Harvard Brain Tissue Resource Center (Belmont, MA: R24MH068855), and Human Brain and Spinal Fluid Resource Center, VA West Los Angeles Healthcare Center (Los Angeles, CA: Sponsored by NINDS/NIMH, National Multiple Sclerosis Society, and the Department of Veterans Affairs).
Disclaimer. The contents of these studies are solely the responsibility of the authors and do not necessarily represent the official view of the funding agencies or repositories.
Financial support. This work was supported by the National Institutes of Health (grants R01 MH069116, R01 MH087233, R01 MH085554, P01 MH070306 and P40 OD013117).
Potential conflicts of interest. All authors: No potential conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 2.Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4:307–18. doi: 10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- 3.Rumbaugh JA, Nath A. Developments in HIV neuropathogenesis. Curr Pharm Des. 2006;12:1023–44. doi: 10.2174/138161206776055877. [DOI] [PubMed] [Google Scholar]
- 4.Steiner J, Haughey N, Li W, et al. Oxidative stress and therapeutic approaches in HIV dementia. Antioxid Redox Signal. 2006;8:2089–100. doi: 10.1089/ars.2006.8.2089. [DOI] [PubMed] [Google Scholar]
- 5.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–23. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 6.Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci. 2002;25:537–62. doi: 10.1146/annurev.neuro.25.112701.142822. [DOI] [PubMed] [Google Scholar]
- 7.Israel N, Gougerot-Pocidalo MA, Aillet F, Virelizier JL. Redox status of cells influences constitutive or induced NF-kappa B translocation and HIV long terminal repeat activity in human T and monocytic cell lines. J Immunol. 1992;149:3386–93. [PubMed] [Google Scholar]
- 8.Piette J, Legrand-Poels S. HIV-1 reactivation after an oxidative stress mediated by different reactive oxygen species. Chem Biol Interact. 1994;91:79–89. doi: 10.1016/0009-2797(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 9.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–58. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pocernich CB, Sultana R, Mohmmad-Abdul H, Nath A, Butterfield DA. HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. Brain Res Brain Res Rev. 2005;50:14–26. doi: 10.1016/j.brainresrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Becker JT, Sanders J, Madsen SK, et al. Subcortical brain atrophy persists even in HAART-regulated HIV disease. Brain Imaging Behav. 2011;5:77–85. doi: 10.1007/s11682-011-9113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar AM, Borodowsky I, Fernandez B, Gonzalez L, Kumar M. Human immunodeficiency virus type 1 RNA Levels in different regions of human brain: quantification using real-time reverse transcriptase-polymerase chain reaction. J Neurovirol. 2007;13:210–24. doi: 10.1080/13550280701327038. [DOI] [PubMed] [Google Scholar]
- 13.Wiley CA, Soontornniyomkij V, Radhakrishnan L, et al. Distribution of brain HIV load in AIDS. Brain Pathol. 1998;8:277–84. doi: 10.1111/j.1750-3639.1998.tb00153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19:525–35. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- 15.Agrawal L, Louboutin JP, Marusich E, Reyes BA, Van Bockstaele EJ, Strayer DS. Dopaminergic neurotoxicity of HIV-1 gp120: reactive oxygen species as signaling intermediates. Brain Res. 2010;1306:116–30. doi: 10.1016/j.brainres.2009.09.113. [DOI] [PubMed] [Google Scholar]
- 16.Czub S, Koutsilieri E, Sopper S, et al. Enhancement of central nervous system pathology in early simian immunodeficiency virus infection by dopaminergic drugs. Acta Neuropathol. 2001;101:85–91. doi: 10.1007/s004010000313. [DOI] [PubMed] [Google Scholar]
- 17.Meulendyke KA, Pletnikov MV, Engle EL, Tarwater PM, Graham DR, Zink MC. Early minocycline treatment prevents a decrease in striatal dopamine in an SIV model of HIV-associated neurological disease. J Neuroimmune Pharmacol. 2012;7:454–64. doi: 10.1007/s11481-011-9332-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sardar AM, Czudek C, Reynolds GP. Dopamine deficits in the brain: the neurochemical basis of parkinsonian symptoms in AIDS. Neuroreport. 1996;7:910–2. doi: 10.1097/00001756-199603220-00015. [DOI] [PubMed] [Google Scholar]
- 19.Scheller C, Sopper S, Jenuwein M, et al. Early impairment in dopaminergic neurotransmission in brains of SIV-infected rhesus monkeys due to microglia activation. J Neurochem. 2005;95:377–87. doi: 10.1111/j.1471-4159.2005.03373.x. [DOI] [PubMed] [Google Scholar]
- 20.Kumar AM, Ownby RL, Waldrop-Valverde D, Fernandez B, Kumar M. Human immunodeficiency virus infection in the CNS and decreased dopamine availability: relationship with neuropsychological performance. J Neurovirol. 2011;17:26–40. doi: 10.1007/s13365-010-0003-4. [DOI] [PubMed] [Google Scholar]
- 21.Westlund KN, Denney RM, Kochersperger LM, Rose RM, Abell CW. Distinct monoamine oxidase A and B populations in primate brain. Science. 1985;230:181–3. doi: 10.1126/science.3875898. [DOI] [PubMed] [Google Scholar]
- 22.Youdim MB, Riederer P. Dopamine metabolism and neurotransmission in primate brain in relationship to monoamine oxidase A and B inhibition. J Neural Transm Gen Sect. 1993;91:181–95. doi: 10.1007/BF01245231. [DOI] [PubMed] [Google Scholar]
- 23.Mallajosyula JK, Kaur D, Chinta SJ, et al. MAO-B elevation in mouse brain astrocytes results in Parkinson's pathology. PLoS One. 2008;3:e1616. doi: 10.1371/journal.pone.0001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siddiqui A, Mallajosyula JK, Rane A, Andersen JK. Ability to delay neuropathological events associated with astrocytic MAO-B increase in a Parkinsonian mouse model: implications for early intervention on disease progression. Neurobiol Dis. 2010;40:444–8. doi: 10.1016/j.nbd.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spina MB, Cohen G. Dopamine turnover and glutathione oxidation: implications for Parkinson disease. Proc Natl Acad Sci U S A. 1989;86:1398–400. doi: 10.1073/pnas.86.4.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong WK, Ou XM, Chen K, Shih JC. Activation of human monoamine oxidase B gene expression by a protein kinase C MAPK signal transduction pathway involves c-Jun and Egr-1. J Biol Chem. 2002;277:22222–30. doi: 10.1074/jbc.M202844200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–26. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 28.Medders KE, Kaul M. Mitogen-activated protein kinase p38 in HIV infection and associated brain injury. J Neuroimmune Pharmacol. 2011;6:202–15. doi: 10.1007/s11481-011-9260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiizaki S, Naguro I, Ichijo H. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Adv Biol Regul. 2013;53:135–44. doi: 10.1016/j.jbior.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC. Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol. 2004;164:355–62. doi: 10.1016/S0002-9440(10)63125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zink MC, Suryanarayana K, Mankowski JL, et al. High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J Virol. 1999;73:10480–8. doi: 10.1128/jvi.73.12.10480-10488.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clements JE, Mankowski JL, Gama L, Zink MC. The accelerated simian immunodeficiency virus macaque model of human immunodeficiency virus-associated neurological disease: from mechanism to treatment. J Neurovirol. 2008;14:309–17. doi: 10.1080/13550280802132832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weed MR, Hienz RD, Brady JV, et al. Central nervous system correlates of behavioral deficits following simian immunodeficiency virus infection. J Neurovirol. 2003;9:452–64. doi: 10.1080/13550280390218751. [DOI] [PubMed] [Google Scholar]
- 34.Witwer KW, Gama L, Li M, et al. Coordinated regulation of SIV replication and immune responses in the CNS. PLoS One. 2009;4:e8129. doi: 10.1371/journal.pone.0008129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–71. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 36.Follstaedt SC, Barber SA, Zink MC. Mechanisms of minocycline-induced suppression of simian immunodeficiency virus encephalitis: inhibition of apoptosis signal-regulating kinase 1. J Neurovirol. 2008;14:376–88. doi: 10.1080/13550280802199898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fowler CJ, Wiberg A, Oreland L, Marcusson J, Winblad B. The effect of age on the activity and molecular properties of human brain monoamine oxidase. J Neural Transm. 1980;49:1–20. doi: 10.1007/BF01249185. [DOI] [PubMed] [Google Scholar]
- 38.Cao X, Rui L, Pennington PR, et al. Serine 209 resides within a putative p38(MAPK) consensus motif and regulates monoamine oxidase-A activity. J Neurochem. 2009;111:101–10. doi: 10.1111/j.1471-4159.2009.06300.x. [DOI] [PubMed] [Google Scholar]
- 39.Chaudhuri AD, Yelamanchili SV, Fox HS. MicroRNA-142 reduces monoamine oxidase A expression and activity in neuronal cells by downregulating SIRT1. PLoS One. 2013;8:e79579. doi: 10.1371/journal.pone.0079579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64:529–36. doi: 10.1093/jnen/64.6.529. [DOI] [PubMed] [Google Scholar]
- 41.Hagberg L, Cinque P, Gisslen M, et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res Ther. 2010;7:15. doi: 10.1186/1742-6405-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Safety and tolerability of the antioxidant OPC-14117 in HIV-associated cognitive impairment. The Dana Consortium on the Therapy of HIV Dementia and Related Cognitive Disorders. Neurology. 1997;49:142–6. doi: 10.1212/wnl.49.1.142. [DOI] [PubMed] [Google Scholar]
- 43.A randomized, double-blind, placebo-controlled trial of deprenyl and thioctic acid in human immunodeficiency virus-associated cognitive impairment. Dana Consortium on the Therapy of HIV Dementia and Related Cognitive Disorders. Neurology. 1998;50:645–51. doi: 10.1212/wnl.50.3.645. [DOI] [PubMed] [Google Scholar]
- 44.Nakasujja N, Miyahara S, Evans S, et al. Randomized trial of minocycline in the treatment of HIV-associated cognitive impairment. Neurology. 2013;80:196–202. doi: 10.1212/WNL.0b013e31827b9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sacktor N, Miyahara S, Deng L, et al. Minocycline treatment for HIV-associated cognitive impairment: results from a randomized trial. Neurology. 2011;77:1135–42. doi: 10.1212/WNL.0b013e31822f0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans SR, Yeh TM, Sacktor N, et al. Selegiline transdermal system (STS) for HIV-associated cognitive impairment: open-label report of ACTG 5090. HIV Clin Trials. 2007;8:437–46. doi: 10.1310/hct0806-437. [DOI] [PubMed] [Google Scholar]
- 47.Sacktor N, Schifitto G, McDermott MP, Marder K, McArthur JC, Kieburtz K. Transdermal selegiline in HIV-associated cognitive impairment: pilot, placebo-controlled study. Neurology. 2000;54:233–5. doi: 10.1212/wnl.54.1.233. [DOI] [PubMed] [Google Scholar]
- 48.Schifitto G, Yiannoutsos CT, Ernst T, et al. Selegiline and oxidative stress in HIV-associated cognitive impairment. Neurology. 2009;73:1975–81. doi: 10.1212/WNL.0b013e3181c51a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schifitto G, Zhang J, Evans SR, et al. A multicenter trial of selegiline transdermal system for HIV-associated cognitive impairment. Neurology. 2007;69:1314–21. doi: 10.1212/01.wnl.0000268487.78753.0f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.