

Abstract
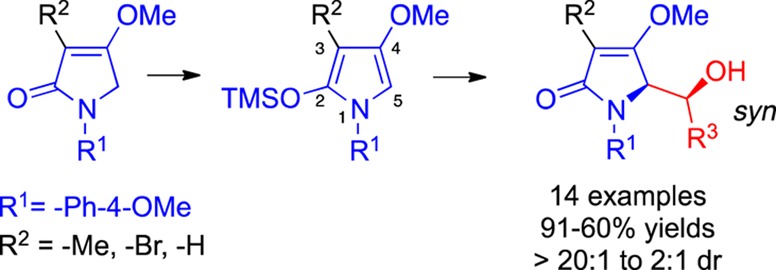
A catalytic diastereoselective aldol reaction has been developed for N1-arylated/C2-O-silylated/C3-methylated and brominated/C4-O-methylated pyrroles in its reactions with various aldehydes. Syn adducts emerge with regard to the vicinal nitrogen and oxygen heteroatom substituents. The N1-aryl residue undergoes oxidative cleavage, and the C3-bromine atom undergoes palladium-mediated coupling reactions, both without disturbing the newly created stereocenters.
Tetramic acid aldolate adducts are known with both syn and anti stereochemical arrangements. Natural product examples include tetrapetalone B (1),1 cylindramide A (2),2 militarinone B (3),3 paecilosetin (4),4 and cryptocin (5)5 (Figure 1).
Figure 1.
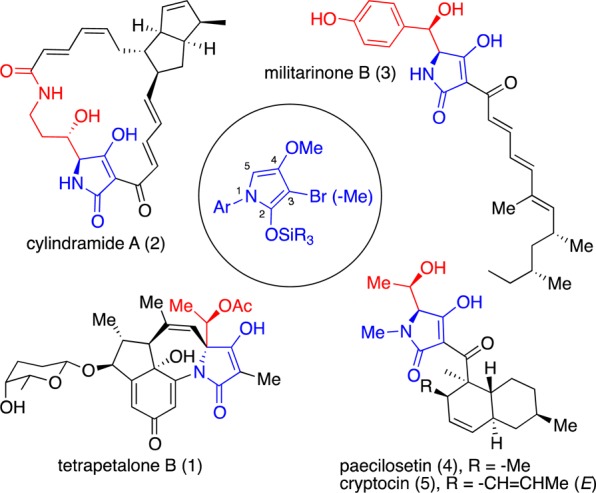
Some natural products containing tetramic acid aldolate motifs; varying tautomeric forms including those depicted.
Given their occurrence, it was surprising for us to learn that a general diastereoselective aldol method leading to the construction of this motif had not been reported. A chief problem appears to have been selection of the protecting group for the nitrogen atom. This residue affects the acidity, sterics, selectivity, and chemistry surrounding the neighboring C5 atoms far more so than for their corresponding des-C4-methoxy counterparts.6 Our analysis of the chemical literature revealed only two reported examples of aldol reactions involving tetramic acid derivatives. Neither was comprehensively studied nor appeared general. Hunter employed an N1-benzylated/C2-O-silylated/C4-O-methylated pyrrole in combination with a chiral α-alkoxy aldehyde and observed an anti-aldolate,7 whereas Stachel employed an N1-benzylated/C2-O-lithiated/C4-O-methylated pyrrole with an α-alkoxy aldehyde and observed a syn-aldolate.8 We speculated that an N1-aryl residue might prove sterically smaller than the customary N1-benzyl and -Boc derivatives and lessen the acidity of the C5 proton(s) of the tetramic acid and thereby fortify any emerging stereochemistry by thwarting potential epimerization processes or retroaldol events that are notorious among these systems.9
We prepared the desired starting compounds by employing the Jones’ protocol (Figure 2).10 The known γ-bromo unsaturated ethyl esters 6a–b11 were independently coupled with p-methoxy aniline in a single pot to afford the corresponding tetramic acid derivatives 7a–b in good yields. The adduct 7b was then converted to the corresponding C3-bromo derivative 7c by exposure to bromine.
Figure 2.
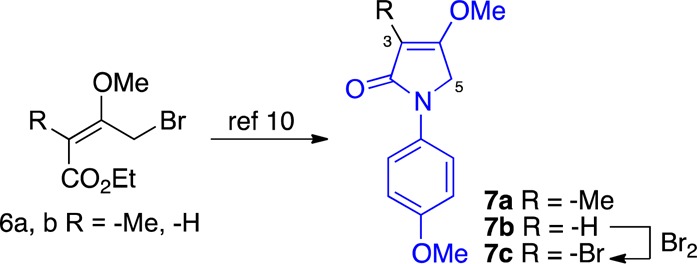
Jones’ 1986 procedure for preparation of tetramic acid derivatives adapted to prepare 7a–c.
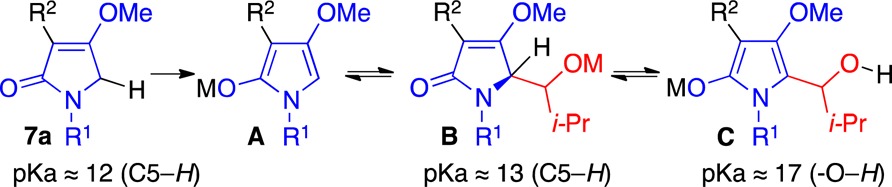
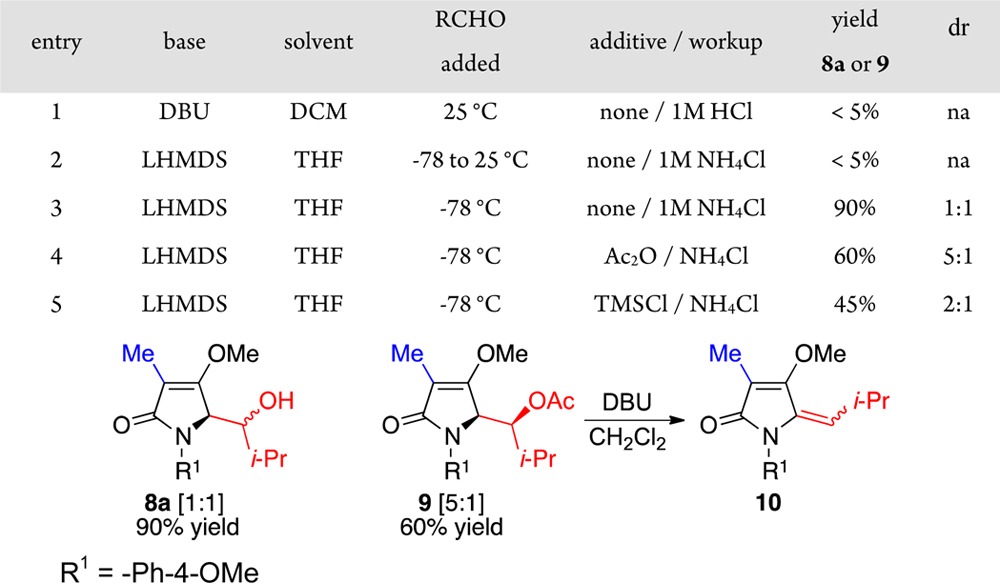
With these in hand, we set out to determine their reactivity as their respective enolate A. We found that treatment of the tetramic acid derivative 7a in dichloromethane (DCM) at 50 °C with triethylamine (Et3N) and deuterium oxide (D2O) yielded very little deuterium incorporation at the C5 position. However, treatment with either 1,8-diazabicycloundec-7-ene (DBU) and D2O, or lithium bis-(trimethylsilyl)amide (LHMDS) and subsequent D2O workup, led to complete bis-deuteration at the C5 position. From these observations, we estimated the pKa of the C5 hydrogen in derivative 7a to be between 12 and 16. Deprotonation of adduct 7a with DBU followed by addition of isobutyraldehyde at ambient temperature failed to yield appreciable amounts of product (Table 1, entry 1). Similarly, deprotonation at −78 °C with LHMDS (1.3 equiv, 0.7 M in toluene) followed by the addition of isobutyraldehyde and subsequent quenching with 1 M ammonium chloride (NH4Cl) at ambient temperature also failed to yield product (entry 2). However, upon quenching aldol reactions initiated with LHMDS at −78 °C with 1 M NH4Cl the desired aldolate 8a formed (90% yield, 1:1 mixture; entry 3). These observations, when taken together, implied to us that a facile retroaldol had returned the starting material at higher temperatures owing to the stability of the enolate A as compared to the alkoxide B. This assumption was further supported by the observation that, when the product 8a was retreated with LHMDS, as in entry 2, the starting material 7a was returned. However, we attributed the poor diastereoselection to an intramolecular deprotonation and enolization to C followed by stereoindiscriminate reprotonation, as this was supported by the observation that, when the alcohol 8a(12) was treated with DBU at 50 °C, the diastereomeric ratio was nearly fully degraded. We subsequently found that if the addition of the aldehyde was rapidly followed by the addition of acetic anhydride (Ac2O), then the selectivity improved to 5:1 (entry 4). None of the elimination product 10 was observed. However, it could be formed upon exposing the acetate 9 to DBU. Application of trimethylsilyl chloride (TMSCl) as the quenching additive failed to provide gains in selectivity (entry 5).
Table 1. Enolate Outcomes of the N1-Ar Tetramic Acid Derivative 7a.
We next investigated the 2-siloxypyrroles 11a–c (Table 2). These materials were formed from three different protocols from the corresponding tetramic acid derivatives 7a–c. In the first, the material was deprotonated with LHMDS followed by the addition of TMSCl and then used in crude form at −78 °C. Thus, the effects of lithium chloride (LiCl), hexamethyldisilazane (HMDS), and tetrahydrofuran (THF) had to be considered. In the second, the starting compound was deprotonated with potassium bis(trimethylsilyl)amide (KHMDS), followed by the addition of TMSCl and then evaporated, whereupon the residue was suspended into toluene and used at −78 °C. Thus, the effects of potassium chloride (KCl), HMDS, and THF were eliminated. In the third, the starting compound was individually subjected in DCM to an admixture of Et3N and trimethylsilyl trifluoromethanesulfonate (TMSOTf) and then used at −78 °C. Thus, the effects of Et3NH+–OTf had to be contemplated. For example in our hands spectroscopic characterization of the pyrroles 11a–c prepared from protocol-III proved to be fruitless, as they fell apart upon removal of the solvent. However, they could be characterized if obtained from protocol-II.
Table 2. Aldol Reactions of Pyrroles 11a–c with Isobutyraldehyde.
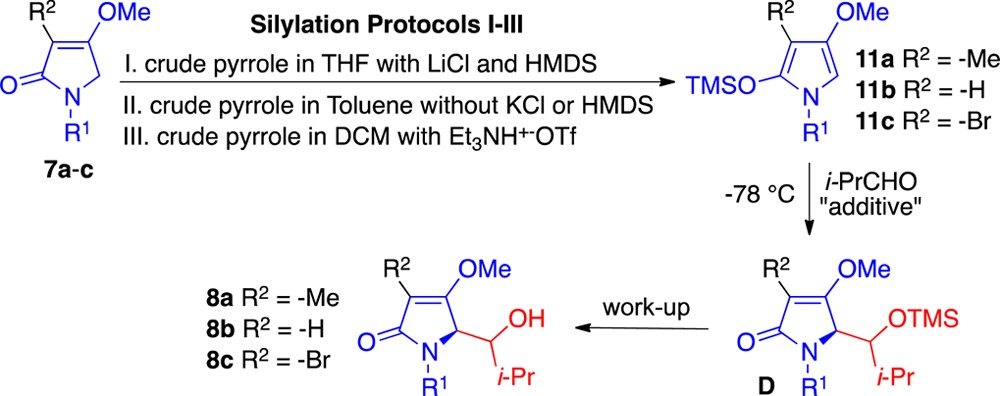
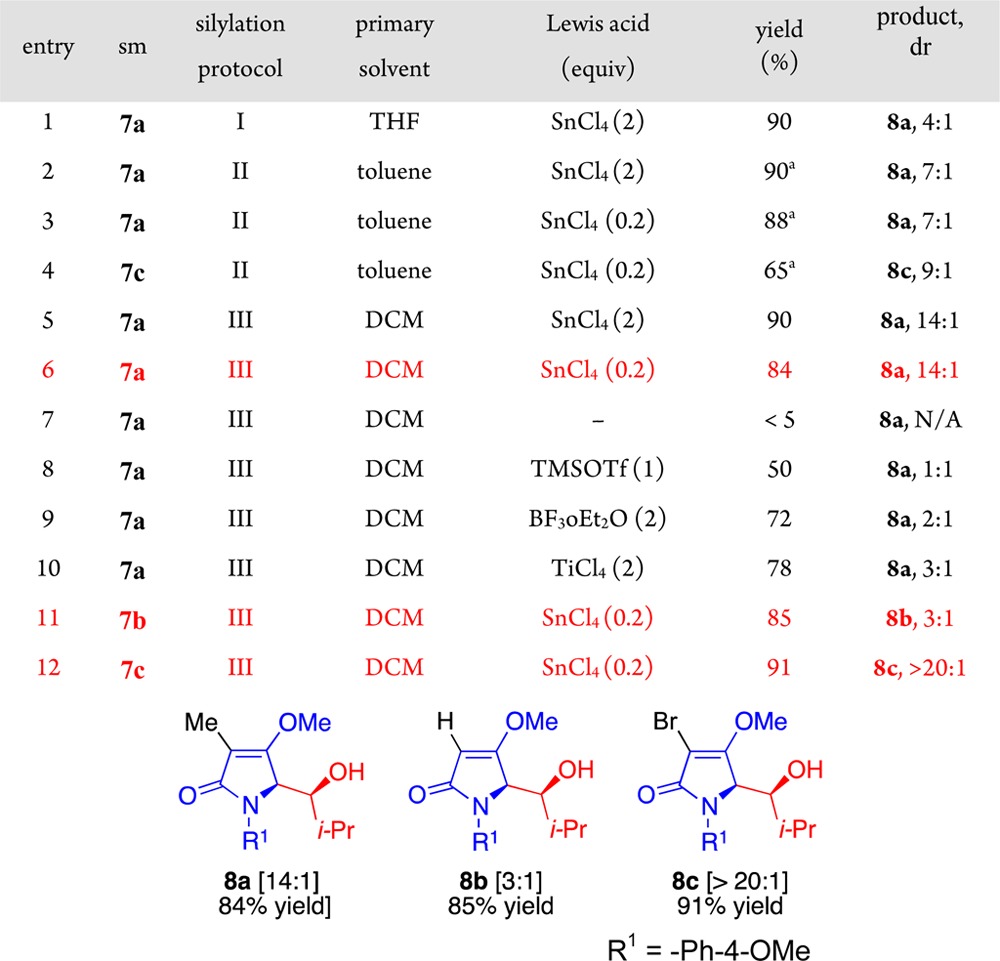
Isolated yield based on recovered starting material.
When the pyrrole 11a (protocol-I) was successively treated with isobutyraldehyde, tin tetrachloride (SnCl4) and aqueous NH4Cl, compound 8a emerged in a 4:1 ratio of diastereomers (Table 2, entry 1). On the other hand, when pyrrole 11a (protocol-II/THF-free) was treated in an identical fashion, then an improved 7:1 ratio was realized (entry 2). Application of catalytic SnCl4 (0.2 equiv) afforded comparable results albeit with a slightly reduced yield (entry 3). The bromopyrrole 11c prepared in similar fashion also underwent reaction mediated by catalytic SnCl4 to afford compound 8c in a 9:1 ratio (entry 4).
Treatment of pyrrole 11a (protocol-III) with SnCl4 in DCM provided the adduct 8a with a further improved ratio and yield (entry 5). Use of catalytic SnCl4 (0.2 equiv) afforded comparable selectivity albeit at a slightly reduced yield (entry 6). However, if SnCl4 was not added at all, then the reaction failed (entry 7). Use of TMSOTf resulted in no selectivity for the reaction (entry 8). Boron trifluoride diethyl etherate (BF3·Et2O) and titanium tetrachloride (TiCl4) led to some improvement (entry 9–10). The C3 substituent had a profound consequence upon the diastereoselectivity [8b < C3-methyl 8a < C3-bromo 8c].
We next explored the substrate scope for our catalytic conditions by combining various aldehydes with the starting pyrroles 11a–c. Our results are summarized in Figure 3. In general, the C3 pyrrole 11b proved significantly less diastereoselective than its C3-methyl and -bromo counterparts. The exception was its reaction with pivalaldehyde, which afforded outstanding selectivity among adducts 12–14 for all three pyrrole cores. Both acyclic and cyclic aliphatic aldehydes afforded excellent yields and selectivity for pyrroles 11a and 11c. In general, branched aldehydes expectedly led to higher selectivities as seen for 2-ethylbutyraldehyde and cyclohexanecarboxaldehyde in adducts 15–16. Aryl aldehydes, which are prone toward reversible reactions, provided slightly lower selectivity as seen in compounds 17–18. The linear n-butyl aldehyde led to the poor outcome we observed in products 19–20. It should be noted that the reactions with chiral 2-phenylpropionaldehyde, which is well-known to undergo diastereoselective reactions owing to internal allylic strain,13 led to almost a single adduct for both the methyl- and bromo- products 21–22, a further testament to the diastereoselectivity and asymmetric synthetic potential for this new protocol.
Figure 3.
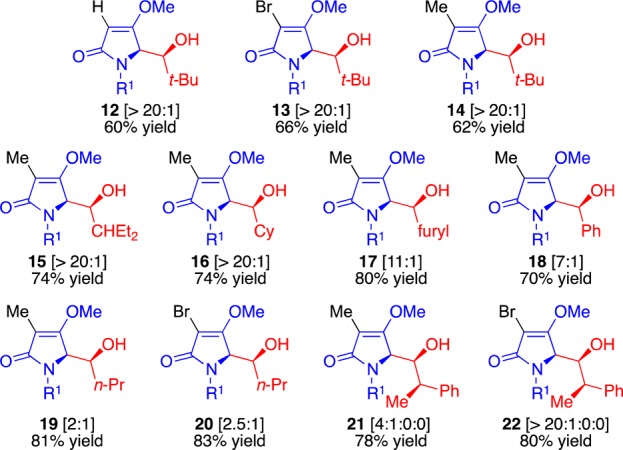
Range and scope examined for the reaction via protocol-III.
Since both of the diastereomers gave similar 1H NMR signals for their respective C5-methine in adducts 8a–c and 12–22, we obtained an X-ray structure of 8a and learned that the preferred isomer displays a syn relationship between the vicinal nitrogen and oxygen atoms (Figure 4).14 We then assumed that further syn and anti assignments could be determined empirically from the 1H NMR, as the C5 methine signal appears further downfield for the syn diastereomers as compared with the anti diastereomer in adducts arising from aliphatic aldehydes.
Figure 4.
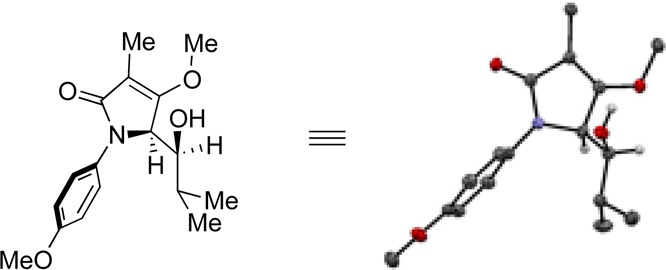
X-ray structure determined for major diastereomer of compound 8a and found to be syn.
Equipped with this knowledge, we can imagine three transition states, open, closed, or [4 + 2]-exo arrangement, as leading to the syn adduct we have observed (Figure 5). The exo [4 + 2] transition state seems unlikely because an unfavorable steric interaction exists between the Lewis acid and the R2 substituent. Moreover, chelation of the metal atom with the siloxy oxygen atom (RO) seems unlikely. The closed transition state can be excluded because of an unfavorable interaction between the R3 aldehyde substituent and the pyrrole R2 substituents, poor alignment between the lone pairs and the coordinating metal atom, as well as the unlikely circumstance that the metal atom and R3 substituent would be on the same side of the carbonyl. We therefore attribute the selectivity to an open transition state; as the R1 substituent (-N1-aryl) is sterically small, it can adopt coplanarity and increase the pyrrole’s nucleophilicity and provide room for the coordinated metal atom (M).
Figure 5.
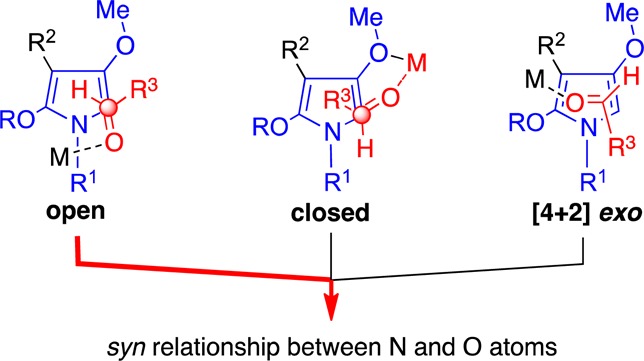
Three transition states leading to syn products.
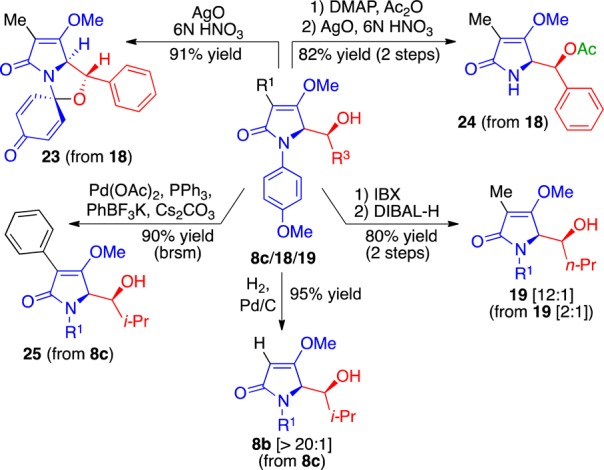
These novel adducts participate in a number of other notable transformations (Scheme 1). For example, exposure of the compound 18 to argentic oxide15 (6 equiv 6 N HNO3) afforded the unusual spirocyclic aminal 23 in 91% yield. On the other hand, the alcohol 18, once protected as its corresponding acetate, when similarly treated smoothly afforded the unprotected tetramic acid derivative 24 in 82% yield over two steps. Cerium ammonium nitrate could also be used. The brominated compound 8c underwent palladium-mediated coupling under Molander’s conditions to afford the C3-aryl derivative 25.16 This outcome suggests potential late-stage stitching strategies for a number of C3-substituted tetramic acid natural products rather than concluding their syntheses with its construction as has been customarily done.2c The bromo derivative 8c can also be converted into the aldolate 8b by hydrogenolysis, thereby enabling access to these aldol products with better stereocontrol. In addition, aldol products with lower diastereoselectivities from linear aldehydes, such as adduct 19 (2:1) are readily improved to >12:1 by oxidation of the secondary alcohol and subsequent reduction with DIBAL-H.
Scheme 1. Some Reactivities of Aldol Adducts 8c, 18, and 19.
In summary, we have developed the first general diastereoselective method for accessing tetramic acid aldolate derivatives from their corresponding siloxy pyrroles. SnCl4 was found to be an outstanding Lewis acid among those examined, and the reaction proved to be catalytic. The C3–Br-substituted tetramic acid derivative afforded superior results. The reactions with chiral aldehydes, which resulted in compounds 21 and 22 with three contiguous stereocenters, demonstrate that this procedure can be used to access nonracemic substrates in an enantioselective fashion. Lower diastereoselectivities emanating from adducts such as 8b or linear aldehydes such as 19 can be respectively improved by hydrogenolysis of the corresponding bromo adduct 8c or by an oxidation–reduction sequence. The bromo derivative 8c participates in palladium-mediated coupling reactions, indicating access to an even greater range of tetramic acid derivatives. It therefore appears that this new method, which employs tetramic acid-derived pyrroles bearing an N1-aryl residue, may be amenable to catalysis with chiral Lewis acids in future asymmetric regimes.
Acknowledgments
T.R.R.P is grateful that the National Institute of General Medical Sciences has supported this work (GM064831) and other studies related to the synthesis of tetrapetalones. W.-J.B. is appreciative of the UCSB Dean’s Fellowship.
Supporting Information Available
Experimental procedures, characterization data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ J.G.D. and W.-J.B. equally contributed.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Isolation:; a Komoda T.; Kishi M.; Abe N.; Sugiyama Y.; Hirota A. Biosci. Biotechnol. Biochem. 2004, 68, 903. [DOI] [PubMed] [Google Scholar]; Synthetic studies; b Marcus A. P.; Sarpong R. Org. Lett. 2010, 12, 4560. [DOI] [PubMed] [Google Scholar]; c Carlsen P. N.; Mann T. J.; Hoveyda A. M.; Frontier A. J. Angew. Chem., Int. Ed. 2014, 10.1002/anie.201404410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isolation:; a Kanazawa S.; Fusetani N.; Matsunaga S. Tetrahedron Lett. 1993, 34, 1065. [Google Scholar]; Synthetic studies:; b Cramer N.; Laschat S.; Baro A.; Schwalbe H.; Richter C. Angew. Chem., Int. Ed. 2004, 44, 820. [DOI] [PubMed] [Google Scholar]; c Hart A. C.; Phillips A. J. J. Am. Chem. Soc. 2006, 128, 1094. [DOI] [PubMed] [Google Scholar]
- Schmidt K.; Riese U.; Li Z.; Hamburger M. J. Nat. Prod. 2003, 66, 378. [DOI] [PubMed] [Google Scholar]
- Lang G.; Blunt J. W.; Cummings N. J.; Cole A. L. J.; Munro M. H. G. J. Nat. Prod. 2005, 68, 810. [DOI] [PubMed] [Google Scholar]
- Li J. Y.; Strobel G.; Harper J.; Lobkovsky E.; Clardy J. Org. Lett. 2000, 2, 767. [DOI] [PubMed] [Google Scholar]
- a Casiraghi G.; Rassu G.; Spanu P.; Pinna L. J. Org. Chem. 1992, 57, 3760. [DOI] [PubMed] [Google Scholar]; b Bella M.; Piancatelli G.; Squarcia A.; Trolli C. Tetrahedron Lett. 2000, 41, 3669. [Google Scholar]
- a Hunter R.; Rees-Jones S. C. M.; Su H. Tetrahedron Lett. 2007, 48, 2819. [Google Scholar]; b Hunter R.; Rees-Jones S. C. M.; Su H. Beilstein. J. Org. Chem. 2007, 3, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachel H.-D.; Zeitler K.; Lotter H. Liebigs Ann. Chem. 1994, 1129. [Google Scholar]
- The chiral C5 position of the tetramic acid derivatives may undergo racemization quickly, see:; a Hosseini M.; Kringelum H.; Murray A.; Tønder J. E. Org. Lett. 2006, 8, 2103. [DOI] [PubMed] [Google Scholar]; b Bai W.-J.; Jackson S. K.; Pettus T. R. R. Org. Lett. 2012, 14, 3862. [DOI] [PubMed] [Google Scholar]
- Jones R. C. F.; Bates A. D. Tetrahedron Lett. 1986, 27, 5285. [Google Scholar]
- a Zhou Y.; Xu Q.; Zhai H. Tetrahedron Lett. 2008, 49, 5271. [Google Scholar]; b Welch S. C.; Gruber J. M. J. Org. Chem. 1982, 47, 385. [Google Scholar]
- The highly diastereoselective alcohol 8a (14:1 dr) used here was obtained from a later protocol, see Table 2, entry 5.
- a Heathcock C. H.; Flippin L. A. J. Am. Chem. Soc. 1983, 105, 1667. [Google Scholar]; b Mori I.; Ishihara K.; Heathcock C. H. J. Org. Chem. 1990, 55, 1114. [Google Scholar]; c Tomo Y.; Yamamoto K. Tetrahedron Lett. 1985, 26, 1061. [Google Scholar]
- Submitted to the Cambridge Crystal Database, CCDC 1000459.
- a Snyder C. D.; Rapoport H. J. Am. Chem. Soc. 1972, 94, 227. [Google Scholar]; b Bai W.-J.; Green J. C.; Pettus T. R. R. J. Org. Chem. 2012, 77, 379. [DOI] [PubMed] [Google Scholar]
- Molander G. A.; Felix L. A. J. Org. Chem. 2005, 70, 3950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.