

Abstract
Recently, we used in vitro selection to identify a new class of naturally occurring GTP aptamer called the G motif. Here we report the discovery and characterization of a second class of naturally occurring GTP aptamer, the “CA motif.” The primary sequence of this aptamer is unusual in that it consists entirely of tandem repeats of CA-rich motifs as short as three nucleotides. Several active variants of the CA motif aptamer lack the ability to form consecutive Watson-Crick base pairs in any register, while others consist of repeats containing only cytidine and adenosine residues, indicating that noncanonical interactions play important roles in its structure. The circular dichroism spectrum of the CA motif aptamer is distinct from that of A-form RNA and other major classes of nucleic acid structures. Bioinformatic searches indicate that the CA motif is absent from most archaeal and bacterial genomes, but occurs in at least 70 percent of approximately 400 eukaryotic genomes examined. These searches also uncovered several phylogenetically conserved examples of the CA motif in rodent (mouse and rat) genomes. Together, these results reveal the existence of a second class of naturally occurring GTP aptamer whose sequence requirements, like that of the G motif, are not consistent with those of a canonical secondary structure. They also indicate a new and unexpected potential biochemical activity of certain naturally occurring tandem repeats.
Keywords: naturally occurring aptamer, tandem repeats, in vitro selection, GTP, RNA, microsatellite
Introduction
GTP plays a central role in diverse cellular processes,1,2 and at least three percent of the proteins in the human proteome bind GTP.3 The fundamental role played by GTP in cells, as well as the emerging realization that the functions of biological proteins and RNAs may not be as distinct as once thought, led us to hypothesize that RNA molecules that bind GTP might also be widespread in nature. To test this hypothesis, we used in vitro selection to search a pool of phylogenetically diverse genome-derived RNA fragments for new examples of naturally occurring GTP aptamers.4 This study revealed that such aptamers are abundant, especially in the genomes of eukaryotes. The majority of the aptamers identified had the potential to form G-quadruplex structures, and further analysis confirmed that most G-quadruplexes exhibit an intrinsic GTP-binding activity.4
Here, we report the discovery of a second class of naturally occurring GTP-binding RNA aptamer, which we have designated the “CA motif.” Members of this aptamer class contain multiple copies of CA-rich sequence motifs such as CAACA arranged in tandem repeats. Although recent studies indicate that RNA tandem repeats can play biological roles, including roles in human disease,5-7 the functional properties of such repeats are still not well understood. A second intriguing feature of the CA motif aptamer is that its sequence requirements are not consistent with those of a canonical nucleic acid structure. For example, CA motif variants with limited potential for Watson-Crick base pairing, including those made of repeats containing only cytidine and adenosine nucleotides, can still bind GTP. Circular dichroism (CD) analysis of the CA motif aptamer also supports the idea of an unusual structure: the characteristic CD spectrum of the CA motif aptamer is distinct from that of several common types of nucleic acid structures, including A-form RNA. Although much less widespread in nature than the previously described G motif,4 the CA motif was detected in 70 percent of the approximately 400 eukaryotic genomes analyzed, and several examples were identified whose ability to bind GTP has been conserved in evolution. Taken together, these results reveal the existence of a second type of naturally occurring GTP aptamer with a noncanonical structure, and reveal a new potential biochemical activity of naturally occurring RNA tandem repeats.
Results
GTP aptamers made of tandem repeats
Previously, we used in vitro selection to isolate GTP aptamers from a pool of phylogenetically diverse genome-derived RNA fragments (Fig. 1A).4,8-11 The majority of these aptamers had the potential to form G-quadruplex structures, and further analysis demonstrated that both RNA and DNA G-quadruplexes possess an intrinsic GTP-binding activity. Most of the remaining sequences isolated in the selection contained short CA-rich motifs such as CAACA that often occurred in the context of tandem repeats (Fig. 1B). To determine whether members of this second family of sequences were also GTP aptamers, three examples were synthesized and tested for the ability to bind GTP. Each example bound GTP-agarose at least 20-fold better than a random sequence control RNA, and none bound agarose lacking GTP at significant levels (Fig. 1C).

Figure 1. GTP aptamers made of CA-rich tandem repeats. (A) Isolation of naturally occurring GTP aptamers using in vitro selection. (B) Examples of tandem repeat GTP aptamers isolated in the selection. (C) Binding of radiolabeled tandem repeat aptamers shown in B to control agarose (Con) and GTP-agarose (GTP). The blue, orange, and white bars correspond to the tandem repeat aptamer sequences in B. Guanosines were added to the 5′ end of each aptamer to facilitate T7 transcription (Table S1). Binding of a random sequence control with the sequence GG(N)48 is also shown as the leftmost set of bars. Percent bound values reflect the average of three independent experiments, and error bars indicate one standard deviation.
The most effective GTP binder among these aptamers, GG(GCAACA)6, was chosen for further characterization. This aptamer could be eluted from GTP-agarose columns with GTP, indicating that it binds free as well as immobilized GTP (Fig. 2A). Based on the concentration dependence of elution,12,13 we estimate that this aptamer binds free GTP with a dissociation constant (Kd) of 1.8 mM (Fig. 2B). Elution profiles were consistent with a single binding site for GTP (as was assumed in Fig. 2B), but could be described equally well by more complex models in which multiple, independent binding sites are considered. The aptamer could also be efficiently eluted from GTP-agarose columns with ddGTP, and somewhat efficiently eluted with GDP, but not with 7-methyl-GTP, 6-methylthio-GTP, ITP, ATP, CTP, or UTP (Fig. 2C). These observations indicate that the CA motif recognizes GTP primarily through interactions with its nucleobase (Fig. 2D). Taken together, these results demonstrate that naturally occurring simple tandem repeats can form binding sites specific for biological small-molecule ligands such as GTP.

Figure 2. Characterization of the affinity and specificity of the CA motif aptamer using competitive column elution. (A) Elution of the CA motif aptamer from a GTP-agarose column with free GTP. (B) Elution of the CA motif aptamer from a GTP-agarose column as a function of free GTP concentration. (C) Elution of the CA motif aptamer from a GTP-agarose column in the presence of various GTP analogs. (D) Competitive column elution data mapped onto the chemical structure of GTP. The CA motif construct GG(GCAACA)6 was used for these experiments. Values reflect the average of three independent experiments, and error bars indicate one standard deviation.
Unusual sequence requirements of the CA motif aptamer
To better understand the types of tandem repeats that can bind GTP, we used site-directed mutagenesis to characterize the sequence requirements of this aptamer in greater detail. We focused our efforts on the (GCAACA)n aptamer, both because its sequence was the simplest among the tandem repeat aptamers identified and because it bound GTP the most efficiently (Fig. 1C). To determine the minimum number of repeats needed for efficient binding as well as the number required for maximal binding, a series of aptamer variants containing between two and 16 repeats were synthesized and tested for the ability to bind GTP-agarose. No significant binding was observed for variants containing two or four repeats (Fig. 3A). The ability to bind GTP increased dramatically for a variant containing six repeats, and continued to increase as repeat number increased to ten, before gradually decreasing (Fig. 3A). We hypothesize that at higher repeat numbers the expected increase in GTP-binding activity from additional binding sites is offset by less efficient aptamer folding.14
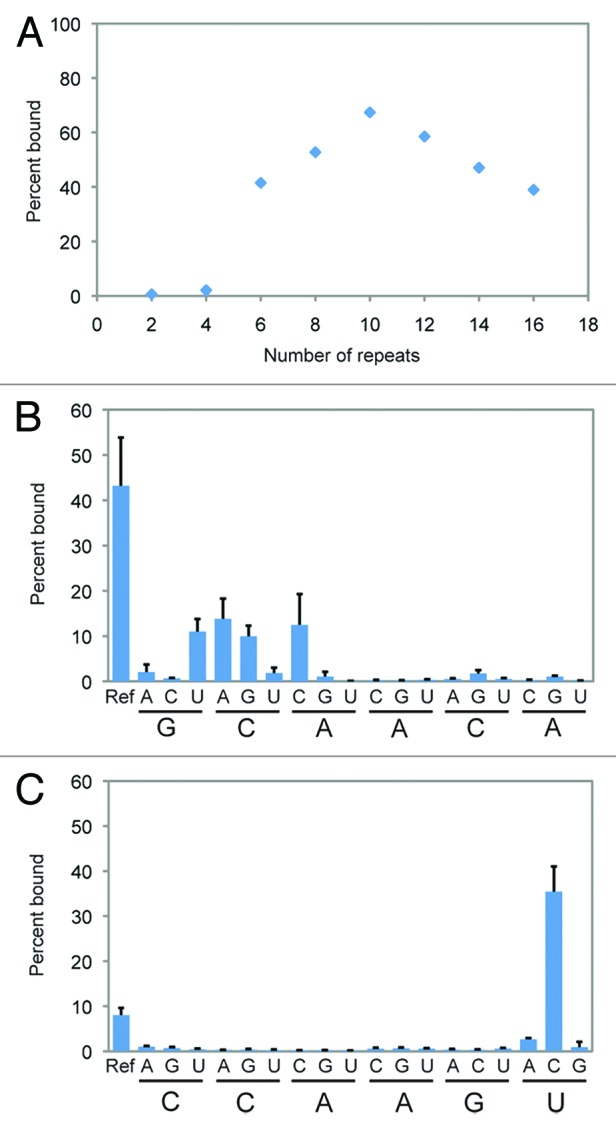
Figure 3. Sequence requirements of the CA motif aptamer. (A) Binding of the GG(GCAACA)n aptamer to GTP-agarose as a function of repeat number. (B) Ability of all single-mutation variants of the GCAACA motif in the GG(GCAACA)6 aptamer to bind GTP-agarose. (C) Ability of all single-mutation variants of the CCAAGU motif in the G(GCAUCCCAAGUGAUGUA)3 aptamer to bind GTP-agarose. Percent bound values reflect the average of three independent experiments, and error bars indicate one standard deviation.
To identify nucleotides in the aptamer most important for its GTP-binding activity, variants containing each of the 18 possible single-mutation changes in the GCAACA motif were synthesized and tested for the ability to bind GTP in the context of an aptamer containing six tandem repeats. These results confirmed the importance of the CA-rich portion of the sequence. While in some cases mutational changes at the first, second, or third positions had only small effects, all single-mutation changes at the fourth, fifth, and six positions either strongly decreased or abolished the GTP-binding activity of the aptamer (Fig. 3B). This analysis also revealed that, at least in the context of single-mutation changes, the optimal nucleotide at five out of six positions in the GCAACA repeat is either a cytidine or adenosine (Fig. 3B). Similar analysis of the GCAUCCCAAG UGAUGUA aptamer showed that the CA-rich motif CCAA is important for its activity, and that variants containing the CCAAGC motif bind GTP more effectively than the original isolate (Fig. 3C).
To further probe the sequence requirements of the CA motif, we analyzed possible secondary structures formed by this aptamer. As a consequence of its CA-rich sequence, the potential of this aptamer to form canonical base pairs is limited: only two of the 15 possible pairs of positions in each GCAACA repeat, G1-C2 and G1-C5, have the potential to form a Watson-Crick pair (Fig. 4). Furthermore, only the G1-C2 pairing can generate secondary structures containing consecutive base pairs (Fig. 4A). Two observations, however, suggest that the potential G1-C2 interaction is unlikely to be important for aptamer activity. First, of the five different single-mutation variants of this aptamer we generated in which this putative base pair was disrupted, the GTP-binding activity of only two were significantly lower than that of the reference sequence (Figs. 3B and 4A). Second, none of the three potential compensatory mutations tested at positions 1 and 2 rescued aptamer activity (Fig. 4A). In contrast, mutations that disrupted the putative G1-C5 pairing typically reduced aptamer activity as expected (Figs. 3B and 4B). Furthermore, in one case this loss of GTP-binding activity could be rescued by compensatory mutations consistent with a standard Watson-Crick base pair, although in two other cases such compensatory mutations did not restore activity (Fig. 4B). These results suggest that positions 1 and 5 interact in some way, although not necessarily in the context of a canonical Watson-Crick base pair. For example, the observed rescue patterns at these positions are more consistent with either trans-Watson-Crick/Watson-Crick or trans-Hoogsteen/Hoogsteen base pairing geometries (in which G-C to C-G changes are isosteric but G-C to either U-G or U-A are not) than with a standard cis-Watson-Crick/Watson-Crick pairing.15

Figure 4. Evaluation of potential Watson-Crick base pairs in the CA motif aptamer. (A) Binding of single and double mutants at positions 1 and 2 in each repeat of the GG(GCAACA)6 aptamer to GTP-agarose. (B) Binding of single and double mutants at positions 1 and 5 in each repeat of the GG(GCAACA)6 aptamer to GTP-agarose. The antiparallel-strand model being tested is shown above each graph, with potential Watson-Crick base pairs indicated by dashes. For simplicity, only two copies of the GCAACA motif are shown. Percent bound values reflect the average of three independent experiments, and error bars indicate one standard deviation.
CA motif aptamer repeats only require cytidine and adenosine
Taken together, these results suggest that the CA motif aptamer may not require canonical base pairs for its activity. To further explore this hypothesis, we investigated the extent to which aptamer variants with little or no ability to form canonical base pairs could bind GTP. Previous results suggested that the GG(ACAACA)6 aptamer was a good starting point for these experiments because, although its ACAACA repeats cannot form canonical base pairs in any register, this aptamer still binds GTP at low but detectable levels (Fig. 3B). As was the case for the reference GCAACA construct (Fig. 3A), the ability of the ACAACA aptamer to bind GTP increased with repeat number, and constructs containing 18 or 24 copies of this motif bound GTP almost as well as the reference GG(GCAACA)6 sequence (Fig. 5A). Like other aptamer variants tested, GG(ACAACA)24 did not bind control agarose lacking GTP at significant levels under these conditions (Fig. 5B). These results show that noncanonical interactions involving cytidine and/or adenosine nucleotides play critical roles in the structure of the CA motif aptamer.

Figure 5. CA motif variants with repeats that contain only cytidine and adenosine. (A) Binding of the GG(ACAACA)n aptamer to GTP-agarose as a function of repeat number. (B) Binding of GG(ACAACA)24 to control agarose (Con) and GTP-agarose (GTP). Binding of a random sequence control with the sequence GG(N)48 is also shown as the leftmost set of bars. (C) Compensatory CA to AC mutations at positions 2 and 6 in each repeat of the aptamer. Mutations were tested in the context of the 3C background, which has the sequence GG(GCCACA)6. (D) Compensatory CA to AC mutations at positions 2 and 6 in each repeat of the aptamer. Mutations were tested in the context of the reference background, which has the sequence GG(GCAACA)6. (E) Parallel-strand model with the 1–5 and 2–6 constraints indicated by dashes. For simplicity only two copies of the GCAACA motif are shown. For A–D, percent bound values reflect the average of three independent experiments, and error bars indicate one standard deviation.
To more clearly elucidate the nature of these interactions, we used site-directed mutagenesis to search for compensatory mutational effects among positions 2, 3, 4, and 6 in the GG(GCAACA)6 aptamer. Fifteen of the 16 possible variants containing either a cytidine or adenosine at these positions were synthesized and tested for the ability to bind GTP. Although the majority of these mutants exhibited little or no GTP-binding activity (Table S1), several containing the 2C6A to 2A6C change bound GTP up to 50-fold more efficiently than expected based on the separate effects of 2C to 2A and 6A to 6C mutations (Fig. 5C and D). When considered together, the 2–6 constraint and the previously identified 1–5 constraint are consistent with a structure containing parallel strands (Fig. 5E; compare with antiparallel models in Fig. 4). Although uncommon, parallel strand nucleic acid structures have been previously described,16 and they occur frequently in certain noncanonical folds such as G-quadruplexes.17,18
Circular dichroism spectrum of the GCAACA aptamer
Different classes of nucleic acid structures often exhibit characteristic circular dichroism (CD) spectra.19 To investigate the relationship between the structure of the CA motif and those of other types of nucleic acid motifs, we obtained the CD spectrum of the GG(GCAACA)6 reference sequence and four variants of this aptamer. These variants differed from the reference sequence at six positions, and each bound GTP-agarose with an efficiency comparable to that of the GG(GCAACA)6 aptamer (Fig. 3B). The CD spectra of these five sequences were similar: each contained a negative peak at ~210 nm and ~250 nm, a positive peak at ~270 nm, and a peak that was usually positive at ~220 nm (Fig. 6A). In the presence of 1 mM GTP, spectra were similar to those measured in the absence of GTP, although some differences were observed for the 1U construct (Fig. 6A). In contrast, spectra of CA motif variants differed significantly from that of tRNA (A-form RNA), which contains a negative peak at ~220 nm and a positive peak at ~260 nm (Fig. 6B). They also differed from typical spectra of several other important types of nucleic acid structures, including the B-form helix, the Z-form helix, the parallel strand G-quadruplex, the antiparallel strand G-quadruplex, and the i-motif.19

Figure 6. Unusual CD spectrum of the CA motif aptamer. (A) CD spectra of five sequence variants of the CA motif aptamer in the absence or presence of GTP. (B) CD spectra of five sequence variants of the CA motif aptamer compared with the CD spectra of tRNA. Ref = GG(GCAACA)6; 1U = GG(UCAACA)6; 2A = GG(GAAACA)6; 2G = GG(GGAACA)6; 3C = GG(GCCACA)6.
The spectrum of the CA motif most closely resembles those of several sequences that adopt triple-stranded architectures,20 although we note that considerable heterogeneity in such spectra has been reported.19 Like the CD spectrum of the CA motif, the spectra of these triplex sequences contain negative peaks at ~210 nm and ~250 nm and positive peaks at ~220 nm and ~270 nm.21-24 Taken together, these results provide additional evidence that the CA motif does not form a canonical A-form duplex, and are consistent with an unusual structure such as a triple-stranded helix.
Phylogenetic distribution and evolutionary conservation of the CA motif aptamer
Our original selection revealed that the CA motif occurs in both the human and chicken genomes,4 but provided little information about other species in which this aptamer is found. To better characterize the phylogenetic distribution of the CA motif, we searched genomic sequence databases for additional examples using a sequence model derived from the site-directed mutagenesis experiments described in Figures 3A and B, 4B, and 5A. This model required sequences to contain at least six copies of the GCAACA motif or at least ten copies of the ACAACA motif, allowed for five mutational changes (1G to U, 2C to A, 2C to G, 3A to C, and 1G5C to 1C5G) observed in active variants of the GCAACA aptamer, and required that tandem repeats be perfect (model summarized in Fig. 7A).

Figure 7. Evolutionary conservation of the CA motif aptamer. (A) Sequence model used to search genomic sequence databases for the CA motif aptamer. (B) Evolutionary conservation of the CA motif aptamer. Above: conservation of the primary sequence of the CA motif. Mouse aptamers and their rat orthologs were identified using the UCSC Genome Browser. For each pair, differences between the mouse and rat sequences are indicated in orange. Below: conservation of the GTP-binding activity of the CA motif. One or two guanosine residues were added to the 5′ end of each aptamer to facilitate T7 transcription (Table S1). Percent bound values reflect the average of three independent experiments, and error bars indicate one standard deviation.
A survey of approximately 430 phylogenetically diverse genomes using this sequence model failed to detect the CA motif aptamer in either bacterial or archaeal genomes. In contrast, this aptamer was identified in about 70 percent of the eukaryotic genomes examined. Approximately, 20 percent of these genomes contained a single example of the CA motif, while 5 percent contained 35 or more examples. Examination of the species in which the CA motif was most abundant (Table S2) revealed several taxonomic groups in which this aptamer occurred frequently in all available genomes, including family Muridae (Mus musculus and Rattus norvegicus) and family Culicoidea (Anopheles gambiae, Aedes aegypti, and Culex quinquefasciatus). Analysis of gene expression databases such as the Gene Expression Omnibus25 indicated that at least some of these examples are also expressed. For example, of the 29 species in which the CA motif occurs 25 or more times, 19 contain at least one expressed example supported by EST evidence, and six more contain at least one example predicted to be expressed (Table S2). Because we have likely not identified all possible variants of the CA motif that can bind GTP, the results of these searches represent lower limits on the true number of CA motif aptamers in the genomes analyzed.
In addition to examining the overall abundance of the CA motif in different species, we also investigated the extent to which specific examples have been conserved in evolution. Because the CA motif is abundant in both mouse and rat, and the comparative genomics of this pair is well studied, we focused most of our efforts to identify conserved variants on these two species. Rat orthologs of mouse CA motif variants were identified using whole-genome alignments, manually examined to determine their similarity to the CA motif consensus sequence, and in some cases tested for the ability to bind GTP. Although conserved orthologs were not detected for most of the approximately 120 CA motif variants analyzed, two mouse/rat pairs were identified in which the CA motif was potentially conserved (Fig. 7B). For both pairs, the mouse sequence was an exact match to the CA motif consensus, while the rat ortholog was similar but not identical (Fig. 7B). One of these examples (mouse/rat 1) mapped to an intergenic region on chromosome 5, while the other (mouse/rat 2) occurred antisense to an intron in the Hid1 gene. Both mouse sequences as well as their rat orthologs bound GTP-agarose (Fig. 7B), indicating that in some cases the ability of the CA motif to bind GTP has been conserved in evolution.
Discussion
Although antiparallel, double-stranded, right-handed helices formed by canonical Watson-Crick base pairs are the most common structural elements in both RNA and DNA, numerous variations on this theme have been identified.26 In the most commonly observed case, noncanonical base pairs occur in the context of otherwise standard helical elements.27,28 Unusual helical architectures such as Z-DNA26,29 and triplex DNA20,30 have likewise been reported. Structures in which canonical base pairs are entirely absent are also known. Examples include the G-quadruplex, which contains guanosine tetrads rather than standard base pairs,17,18 and the i-motif, which contains hemiprotonated C-C base pairs organized into a four-stranded structure.31
The sequence requirements of the CA motif suggest that, like the G-quadruplex and the i-motif, the structure of this aptamer does not require canonical base pairs. Although our experiments do not provide direct information about the three-dimensional fold of this structure, they significantly constrain the possibilities. Variants of the CA motif made up of repeats containing only cytidine and adenosine can still bind GTP (Fig. 5A and B), suggesting that the essential interactions made by the aptamer only require these two nucleotides. Compensatory mutations involving CA to AC changes (Fig. 5C and D) suggest that in some cases these interactions occur between cytidine and adenosine. The CD spectrum of the CA motif suggests that it does not form a standard A-form helix (Fig. 6B). Finally, if the 1–5 (Fig. 4B) and 2–6 interactions (Fig. 5C and D) reflect physical contacts, they are consistent with a structure containing parallel strands (Fig. 5E).
The structure of the CA motif may be related to that of a viral translational enhancer element called the omega sequence.32 This enhancer consists almost entirely of imperfect CAA repeats,33 although the number of repeats is not high enough to be expected to bind GTP under the conditions used in our study. Analysis of the omega sequence by ultracentrifugation, thermal melting, and chemical probing indicates that, despite the inability to form canonical Watson-Crick base pairs, it adopts a compact and stable structure.34-36 The CA-rich region of this enhancer was proposed to form a triple helix containing both parallel and antiparallel strand topologies,37 and some details of this model are consistent with our data. For example, our site-directed mutagenesis experiments suggest that the CA motif aptamer could form a structure containing parallel strands (Figs. 4B and 5C–E). The CD spectrum of the CA motif (Fig. 6) is also consistent with a triple helix,21-24 although a high-resolution structure will be needed to confirm this hypothesis. A second known example of a stable structure formed by an RNA containing only cytidine and adenosine is a dimer formed by two molecules of CA and an intercalated proflavine molecule.38 Like the proposed fold of the omega sequence, the structure of this dimer contains parallel strands.
The results of our selection using a pool of genome-derived RNA fragments differ significantly from those of most previous studies in which functional RNAs were isolated from random sequence pools.39 The majority of the aptamers and ribozymes isolated in these studies form secondary structures containing standard helical elements. For example, 14 GTP aptamers previously identified using in vitro selection, including at least six distinct motifs isolated from random sequence pools, form secondary structures containing canonical Watson-Crick base pairs.40-45 In contrast, neither of the GTP-binding motifs we isolated from a pool of genome-derived RNA fragments adopts such a structure. This observation is consistent with the idea that nucleic acid motifs that form noncanonical structures may be more abundant in eukaryotic genomes than they are in random sequence pools. It also suggests that efforts to identify naturally occurring functional RNA elements should include methods capable of identifying noncanonical motifs, such as the selection-based approach used here, in addition to those designed to identify phylogenetically conserved RNA secondary structures containing Watson-Crick base pairs. Recent studies suggest that such motifs can bind a wide range of biological cofactors, providing further evidence of the functional diversity of noncanonical nucleic acid structures.46,47
RNA aptamers that bind small molecules are widespread in bacteria, and play important roles in the regulation of gene expression.48 The results described in this and a previous study4 are consist with the possibility that such aptamers also occur more frequently in eukaryotes than is currently appreciated.49 We anticipate that direct selection-based approaches will continue to serve as a powerful approach to further explore this possibility.
Materials and Methods
In vitro selection
Pools were generated by fragmentation of genomic DNA using DNase I followed by gel purification of ~100–600 bp fragments on agarose gels. Fragments with 3′ adenosine overhangs were generated by incubating first with DNA polymerase I, and then with dATP and Taq DNA polymerase. These fragments were ligated into pGEM-T vectors and amplified by PCR using primers flanking the insertion site, one of which contained a T7 promoter at its 5′ end. Templates were transcribed using T7 RNA polymerase to generate starting pools for in vitro selection experiments.
GTP aptamers were isolated by incubating pool RNA with GTP-agarose, washing away unbound molecules with selection buffer, and eluting bound RNAs with EDTA. Eluted molecules were subjected to RT-PCR and transcribed to generate RNA for the next round of selection. After four rounds of selection, the pool was cloned using the TOPO TA kit (Invitrogen) and sequenced. See our previous study for detailed descriptions of these protocols.4
GTP-agarose binding assays
Assays were performed as previously described4 using GTP-agarose from Innova Biosciences and micro-spin columns from Pierce (catalog number 89879). Following the addition of RNA (100 nM final concentrations with a trace amount of body labeled RNA in a volume of 170 µL), columns were incubated for 15 min and then washed four times with 170 µL 1 × binding buffer (20 mM MgCl2, 200 mM KCl, 20 mM HEPES pH 7.1). Bound RNA was eluted by washing twice with 170 µL of 5 mM EDTA, pH 9.
To determine the percent of RNA bound to GTP-agarose, equivalent percentages of wash and elution fractions were ethanol precipitated, resuspended, and spotted on TLC plates. Plates were scanned using a Typhoon phosphorimager, and counts determined using ImageQuant software. For constructs for which the fraction bound to control agarose was also determined, this value was never significantly different from the fraction bound by a random sequence control pool (Fig. 1C).
Competitive column elution
The GG(GCAACA)6 construct of the CA motif aptamer was bound to GTP-agarose using the protocol described above. After washing twice with 170 µL of 1 × binding buffer to remove non-specifically bound RNA, 50 μL of elution buffer (20 mM MgCl2, 200 mM KCl, 20 mM HEPES pH 7.1 and 5 mM GTP analog) was added to the column. After incubating for 5 min, the column was briefly spun in a benchtop centrifuge, and the flowthrough was transferred to a clean tube. After performing this elution protocol zero (Fig. 2C) or four (Fig. 2A) more times, the remaining RNA was removed from the column by washing twice with 170 µL 5 mM EDTA. Fractions were precipitated and analyzed as described above. The total number of bound counts was defined as the sum of the counts in all elution fractions plus the sum of the counts in all EDTA washes.
A modified elution buffer (20 mM MgCl2, 200 mM KCl, 20 mM HEPES pH 7.1, 50 mM NaCl and varying concentrations of GTP) was used for the experiments described in Figure 2B. Elution volumes were either 50 μL (10 μM to 3 mM GTP) or 20 μL (5 mM to 10 mM GTP), and three elutions were performed for each GTP concentration. The solubility of GTP in the elution buffer prevented us from using concentrations greater than 10 mM. Elution profiles were fit to the equation F = e-kvwhere F is the fraction of aptamer remaining on the column, k is the decay constant, and v is the elution volume. Similar approaches have previously been used to model elution profiles in affinity chromatography experiments.50,51 This formula was used to calculate the volume required to elute 50 percent of the aptamer from the column at each GTP concentration. These volumes were then plotted as a function of GTP concentration and fit to the equation VeL = ((Kd × Ve) + (L × Vn)) / (L + Kd) where VeL is the median elution volume at the indicated free GTP concentration, Kd is the dissociation constant for free GTP, Ve is the median elution volume in the absence of free GTP in the elution buffer, L is the concentration of free GTP in the elution buffer, and Vn is the median elution volume in the presence of control agarose lacking immobilized GTP.12,13 VeL and Ve were measured as described above, L was known, and the best fit was determined using a value of Vn that never exceeded 32 μL (the median elution volume at 10 mM GTP, the highest GTP concentration at which the rate of elution was determined).
Circular dichroism
In a typical experiment, a 200 μM nucleic acid solution in a volume of 100 μL was heated at 65 °C for 5 min and cooled at room temperature for 5 min. After addition of 100 μL of 2 × aptamer buffer (40 mM MgCl2, 400 mM KCl, 40 mM HEPES pH 7.1), the solution was incubated for 30 min at room temperature and the circular dichroism spectrum was measured using a JASCO J-715 Spectropolarimeter.
Bioinformatics
CA motif variants were typically identified by BLAT52 (run from the DOE Joint Genome Institute website53,54) and BLAST55 (run from the NCBI website56). BLAT searches were performed on unmasked genomes, while BLAST searches were performed in the absence of filters and masking. Results obtained from BLAT were virtually identical to those obtained by “replace all” searches in Microsoft Word files of downloaded genomes (R2 = 0.99), while results from BLAST searches were similar (R2 = 0.86) but underestimated the number of aptamers in some genomes. In some cases, results of BLAST searches were also compared with those of BLAT searches of masked genomes (implemented using the UCSC Genome Browser57). These two approaches typically gave similar results, although BLAT could not reliably detect the (ACAACA)10 aptamer in most masked genomes. Expressed examples of the CA motif were identified by BLAT and BLAST (as described above). Mouse/rat whole-genome alignments were analyzed using the UCSC Genome Browser.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Mike Lawrence as well as John Guilinger and other members of the group for helpful discussions. This work was supported by HHMI and the NIH/NIGMS (R01 GM065865).
References
- 1.Dever TE, Merrick WC. The Guanine-Nucleotide Binding Proteins. New York: Plenum Publishing Corp; 1989. [Google Scholar]
- 2.Alberts B, Johnson A, Lewis J, Raff M, Roberts P. Molecular Biology of the Cell. New York: Garland; 2007. [Google Scholar]
- 3.Murray JM, Bussiere DE. Targeting the purinome. Methods Mol Biol. 2009;575:47–92. doi: 10.1007/978-1-60761-274-2_3. [DOI] [PubMed] [Google Scholar]
- 4.Curtis EA, Liu DR. Discovery of widespread GTP-binding motifs in genomic DNA and RNA. Chem Biol. 2013;20:521–32. doi: 10.1016/j.chembiol.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hui J, Stangl K, Lane WS, Bindereif A. HnRNP L stimulates splicing of the eNOS gene by binding to variable-length CA repeats. Nat Struct Biol. 2003;10:33–7. doi: 10.1038/nsb875. [DOI] [PubMed] [Google Scholar]
- 6.Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 2010;44:445–77. doi: 10.1146/annurev-genet-072610-155046. [DOI] [PubMed] [Google Scholar]
- 7.Galka-Marciniak P, Urbanek MO, Krzyzosiak WJ. Triplet repeats in transcripts: structural insights into RNA toxicity. Biol Chem. 2012;393:1299–315. doi: 10.1515/hsz-2012-0218. [DOI] [PubMed] [Google Scholar]
- 8.Gold L, Brown D, He Y, Shtatland T, Singer BS, Wu Y. From oligonucleotide shapes to genomic SELEX: novel biological regulatory loops. Proc Natl Acad Sci U S A. 1997;94:59–64. doi: 10.1073/pnas.94.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salehi-Ashtiani K, Lupták A, Litovchick A, Szostak JW. A genomewide search for ribozymes reveals an HDV-like sequence in the human CPEB3 gene. Science. 2006;313:1788–92. doi: 10.1126/science.1129308. [DOI] [PubMed] [Google Scholar]
- 10.Zimmermann B, Bilusic I, Lorenz C, Schroeder R. Genomic SELEX: a discovery tool for genomic aptamers. Methods. 2010;52:125–32. doi: 10.1016/j.ymeth.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vu MM, Jameson NE, Masuda SJ, Lin D, Larralde-Ridaura R, Lupták A. Convergent evolution of adenosine aptamers spanning bacterial, human, and random sequences revealed by structure-based bioinformatics and genomic SELEX. Chem Biol. 2012;19:1247–54. doi: 10.1016/j.chembiol.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunn BM, Chaiken IM. Quantitative affinity chromatography. Determination of binding constants by elution with competitive inhibitors. Proc Natl Acad Sci U S A. 1974;71:2382–5. doi: 10.1073/pnas.71.6.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connell GJ, Illangesekare M, Yarus M. Three small ribooligonucleotides with specific arginine sites. Biochemistry. 1993;32:5497–502. doi: 10.1021/bi00072a002. [DOI] [PubMed] [Google Scholar]
- 14.Sabeti PC, Unrau PJ, Bartel DP. Accessing rare activities from random RNA sequences: the importance of the length of molecules in the starting pool. Chem Biol. 1997;4:767–74. doi: 10.1016/S1074-5521(97)90315-X. [DOI] [PubMed] [Google Scholar]
- 15.Leontis NB, Stombaugh J, Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rippe K, Jovin TM. Parallel-stranded duplex DNA. Methods Enzymol. 1992;211:199–220. doi: 10.1016/0076-6879(92)11013-9. [DOI] [PubMed] [Google Scholar]
- 17.Gellert M, Lipsett MN, Davies DR. Helix formation by guanylic acid. Proc Natl Acad Sci U S A. 1962;48:2013–8. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis JT. G-quartets 40 years later: from 5′-GMP to molecular biology and supramolecular chemistry. Angew Chem Int Ed Engl. 2004;43:668–98. doi: 10.1002/anie.200300589. [DOI] [PubMed] [Google Scholar]
- 19.Kypr J, Kejnovská I, Renciuk D, Vorlícková M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009;37:1713–25. doi: 10.1093/nar/gkp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 2008;36:5123–38. doi: 10.1093/nar/gkn493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JS, Johnson DA, Morgan AR. Complexes formed by (pyrimidine)n. (purine)n DNAs on lowering the pH are three-stranded. Nucleic Acids Res. 1979;6:3073–91. doi: 10.1093/nar/6.9.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manzini G, Xodo LE, Gasparotto D, Quadrifoglio F, van der Marel GA, van Boom JH. Triple helix formation by oligopurine-oligopyrimidine DNA fragments. Electrophoretic and thermodynamic behavior. J Mol Biol. 1990;213:833–43. doi: 10.1016/S0022-2836(05)80267-0. [DOI] [PubMed] [Google Scholar]
- 23.Xodo LE, Manzini G, Quadrifoglio F. Spectroscopic and calorimetric investigation on the DNA triplex formed by d(CTCTTCTTTCTTTTCTTTCTTCTC) and d(GAGAAGAAAGA) at acidic pH. Nucleic Acids Res. 1990;18:3557–64. doi: 10.1093/nar/18.12.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray DM, Hung SH, Johnson KH. Absorption and circular dichroism spectroscopy of nucleic acid duplexes and triplexes. Methods Enzymol. 1995;246:19–34. doi: 10.1016/0076-6879(95)46005-5. [DOI] [PubMed] [Google Scholar]
- 25.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rich A. DNA comes in many forms. Gene. 1993;135:99–109. doi: 10.1016/0378-1119(93)90054-7. [DOI] [PubMed] [Google Scholar]
- 27.Turner DH. Thermodynamics of base pairing. Curr Opin Struct Biol. 1996;6:299–304. doi: 10.1016/S0959-440X(96)80047-9. [DOI] [PubMed] [Google Scholar]
- 28.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–20. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 29.Wang AH, Quigley GJ, Kolpak FJ, Crawford JL, van Boom JH, van der Marel G, Rich A. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature. 1979;282:680–6. doi: 10.1038/282680a0. [DOI] [PubMed] [Google Scholar]
- 30.Felsenfeld G, Rich A. Studies on the formation of two- and three-stranded polyribonucleotides. Biochim Biophys Acta. 1957;26:457–68. doi: 10.1016/0006-3002(57)90091-4. [DOI] [PubMed] [Google Scholar]
- 31.Gehring K, Leroy JL, Guéron M. A tetrameric DNA structure with protonated cytosine.cytosine base pairs. Nature. 1993;363:561–5. doi: 10.1038/363561a0. [DOI] [PubMed] [Google Scholar]
- 32.Sleat DE, Gallie DR, Jefferson RA, Bevan MW, Turner PC, Wilson TM. Characterisation of the 5′-leader sequence of tobacco mosaic virus RNA as a general enhancer of translation in vitro. Gene. 1987;60:217–25. doi: 10.1016/0378-1119(87)90230-7. [DOI] [PubMed] [Google Scholar]
- 33.Gallie DR, Walbot V. Identification of the motifs within the tobacco mosaic virus 5′-leader responsible for enhancing translation. Nucleic Acids Res. 1992;20:4631–8. doi: 10.1093/nar/20.17.4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovtun AA, Shirokikh NE, Gudkov AT, Spirin AS. The leader sequence of tobacco mosaic virus RNA devoid of Watson-Crick secondary structure possesses a cooperatively melted, compact conformation. Biochem Biophys Res Commun. 2007;358:368–72. doi: 10.1016/j.bbrc.2007.04.152. [DOI] [PubMed] [Google Scholar]
- 35.Shirokikh NE, Agalarov SCh, Spirin AS. Chemical and enzymatic probing of spatial structure of the omega leader of tobacco mosaic virus RNA. Biochemistry (Mosc) 2010;75:405–11. doi: 10.1134/S0006297910040024. [DOI] [PubMed] [Google Scholar]
- 36.Agalarov SC, Sogorin EA, Shirokikh NE, Spirin AS. Insight into the structural organization of the omega leader of TMV RNA: the role of various regions of the sequence in the formation of a compact structure of the omega RNA. Biochem Biophys Res Commun. 2011;404:250–3. doi: 10.1016/j.bbrc.2010.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Efimov AV, Spirin AS. Intramolecular triple helix as a model for regular polyribonucleotide (CAA)(n) Biochem Biophys Res Commun. 2009;388:127–30. doi: 10.1016/j.bbrc.2009.07.133. [DOI] [PubMed] [Google Scholar]
- 38.Westhof E, Sundaralingam M. X-ray-structure of a cytidylyl-3′,5′-adenosine-proflavine complex: a self-paired parallel-chain double helical dimer with an intercalated acridine dye. Proc Natl Acad Sci U S A. 1980;77:1852–6. doi: 10.1073/pnas.77.4.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartel DP, Unrau PJ. Constructing an RNA world. Trends Cell Biol. 1999;9:M9–13. doi: 10.1016/S0962-8924(99)01669-4. [DOI] [PubMed] [Google Scholar]
- 40.Connell GJ, Yarus M. RNAs with dual specificity and dual RNAs with similar specificity. Science. 1994;264:1137–41. doi: 10.1126/science.7513905. [DOI] [PubMed] [Google Scholar]
- 41.Davis JH, Szostak JW. Isolation of high-affinity GTP aptamers from partially structured RNA libraries. Proc Natl Acad Sci U S A. 2002;99:11616–21. doi: 10.1073/pnas.182095699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Z, Szostak JW. Evolution of aptamers with a new specificity and new secondary structures from an ATP aptamer. RNA. 2003;9:1456–63. doi: 10.1261/rna.5990203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carothers JM, Oestreich SC, Davis JH, Szostak JW. Informational complexity and functional activity of RNA structures. J Am Chem Soc. 2004;126:5130–7. doi: 10.1021/ja031504a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carothers JM, Oestreich SC, Szostak JW. Aptamers selected for higher-affinity binding are not more specific for the target ligand. J Am Chem Soc. 2006;128:7929–37. doi: 10.1021/ja060952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carothers JM, Davis JH, Chou JJ, Szostak JW. Solution structure of an informationally complex high-affinity RNA aptamer to GTP. RNA. 2006;12:567–79. doi: 10.1261/rna.2251306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kröner C, Röthlingshöfer M, Richert C. Designed nucleotide binding motifs. J Org Chem. 2011;76:2933–6. doi: 10.1021/jo2003067. [DOI] [PubMed] [Google Scholar]
- 47.Kröner C, Göckel A, Liu W, Richert C. Binding cofactors with triplex-based DNA motifs. Chemistry. 2013;19:15879–87. doi: 10.1002/chem.201303098. [DOI] [PubMed] [Google Scholar]
- 48.Tucker BJ, Breaker RR. Riboswitches as versatile gene control elements. Curr Opin Struct Biol. 2005;15:342–8. doi: 10.1016/j.sbi.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Cheah MT, Wachter A, Sudarsan N, Breaker RR. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447:497–500. doi: 10.1038/nature05769. [DOI] [PubMed] [Google Scholar]
- 50.Hage DS, Xuan H, Nelson MA. Application and elution in affinity chromatography. In: Hage DS, editor. Handbook of affinity chromatography. Boca Raton, FL: Taylor & Francis Group; 2006. page 944. [Google Scholar]
- 51.Nelson MA, Papastavros E, Dodlinger M, Hage DS. Environmental analysis by on-line immunoextraction and reversed-phase liquid chromatography: optimization of the immunoextraction/RPLC interface. J Agric Food Chem. 2007;55:3788–97. doi: 10.1021/jf063286l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, et al. The genome portal of the department of energy joint genome institute. Nucleic Acids Res. 2012;40:D26–32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42:D699–704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 56.NCBI Resource Coordinators Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2014;42:D7–17. doi: 10.1093/nar/gkt1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.