

Abstract
BACKGROUND
Honokiol (HNK), derived from the bark of an oriental medicinal plant (Magnolia officinalis), is a promising anticancer agent with in vitro (PC-3 and LNCaP cells) and in vivo (PC-3 xenografts) efficacy against prostate cancer cells. However, the mechanisms affecting anticancer response to HNK are not fully understood.
METHODS
Human (androgen-independent PC-3 and androgen-responsive LNCaP) and murine (Myc-CaP) prostate cancer cells, and PC-3 tumor xenografts were used for various assays. Autophagy was assessed by transmission electron microscopy, immunofluorescence (LC3 puncta), and immunoblotting (LC3BII detection). Cell viability was determined by trypan blue assay. Apoptosis was quantitated by DNA fragmentation detection and Annexin V/propidium iodide assay. Reactive oxygen species were detected by electron paramagnetic resonance spectrometry and flow cytometric/microscopic analysis of MitoSOX red fluorescence.
RESULTS
Exposure of PC-3, LNCaP, and Myc-CaP cells to pharmacologic doses of HNK resulted in autophagy induction. The PC-3 tumor xenografts from HNK-treated mice contained higher levels of LC3BII protein compared with control tumors. Cell viability inhibition and apoptosis induction resulting from HNK exposure were significantly augmented by pharmacological inhibition of autophagy using 3-methyladenine as well as RNA interference of autophagy regulator ATG5. HNK-mediated increase in levels of LC3BII protein was partially but markedly diminished in the presence of antioxidants, including N-acetylcysteine, polyethylene glycol-conjugated (PEG)-superoxide dismutase, and PEG-catalase. On the other hand, antioxidants had no impact on HNK-induced apoptosis.
CONCLUSIONS
In conclusion, the present study demonstrates, for the first time, that HNK induces reactive oxygen species-mediated cytoprotective autophagy in prostate cancer cells.
Keywords: honokiol, prostate cancer, autophagy, chemoprevention
INTRODUCTION
Honokiol, a neolignan present in parts (e.g., stem bark) of the oriental medicine plant Magnolia officinalis, is an interesting small-molecule exhibiting a variety of pharmacological effects in preclinical experimental models [1, 2]. For example, honokiol (HNK) not only protected myocardial damage from ischemic injury but also inhibited ventricular arrhythmia during ischemia and reperfusion [3]. Some of the other notable pharmacological effects of HNK include activity against Gram-positive and Gram-negative bacteria as well as fungi [4], blockade of leukotriene synthesis via inhibition of 5-lipoxygenase activity [5], inhibition of platelet aggregation [6], and anticancer effects [reviewed in 2, 7]. Anticancer and proapoptotic effects of HNK were initially documented in leukemic cells [8, 9]. Antineoplastic activity of HNK was subsequently extended to several different solid tumor types such as breast, prostate, gastric, and ovarian cancer [2, 7, 10–14].
While most studies on honokiol have utilized cultured cells to delineate the mechanism of its antineoplastic effect, experimental evidence for the in vivo anticancer efficacy of this natural product continues to accumulate. For example, daily intraperitoneal injection of 3 mg HNK/mouse resulted in nearly 50% growth inhibition of SVR angiosarcoma cells subcutaneously implanted in athymic mice; the antiangiogenic effect of HNK was also shown in this study [15]. Administration of HNK (2 mg/mouse) was shown to retard the growth of RKO colorectal cancer cells implanted in nude mice [16]. The lifespan of RKO tumor bearing animals was also prolonged by HNK therapy [16]. We have also shown previously that oral administration of 2 mg HNK/mouse (three times/week) inhibits the growth of PC-3 human prostate cancer xenografts in association with in vivo apoptosis induction [10]. Another study showed in vivo inhibition of bone metastatic growth of androgen-independent C4-2 prostate cancer cells by intraperitoneal administration of 2.5 mg HNK [13]. HNK-mediated chemoprevention of UVB-induced skin carcinogenesis and in vivo potentiation of the antitumor effects of ionizing radiation as well as epidermal growth factor receptor inhibitors were also documented [17–19].
Analogous to other naturally-occurring agents [20], HNK exhibits multifaceted effects likely contributing to its growth inhibitory activity against cancer [1, 2, 10, 21, 22]. Notable mechanisms implicated in anticancer effect of HNK include cell cycle arrest [21], apoptosis induction [8, 10–12], and inhibition of angiogenesis [15]. HNK treatment also suppresses oncogenic signaling pathways mediated by Notch [19], epidermal growth factor receptor [18], nuclear factor-κB [22], c-Src [23], mammalian target of rapamycin (mTOR) [24], hypoxia inducible factor-1 [25], signal transducer and activator of transcription 3 [26], and Wnt/β-catenin [27] leading to inhibition of cancer cells growth and invasion. Inhibition of cancer stem-like cells upon treatment with HNK was shown very recently for colon and oral cancers [19, 27]. HNK inhibits activity of androgen receptor in prostate cancer cells [28].
Previous mechanistic studies, including those from our own laboratory, indicated that the cell cycle arrest and apoptosis induction by HNK in cancer cells was associated with production of reactive oxygen species (ROS) [21, 29]. Because ROS are implicated in regulation of autophagy [30], which can either contribute to the overall cell death or serve to protect against apoptosis [31–35], it was of interest to determine if HNK induced autophagy. The present study addresses this question using prostate cancer cells as a model.
MATERIALS AND METHODS
Ethics Statement
PC-3 xenografts from our published study evaluating the in vivo efficacy of HNK [10] were used for western blot analysis of autophagy markers. Use and care of mice were consistent with the Institutional Animal Care and Use Committee guidelines.
Reagents and Cell Lines
Stock solution (50 mM) of HNK (purity ≥ 98%), which was purchased from LKT Laboratories (St. Paul, MN), was prepared in dimethyl sulfoxide (DMSO) and diluted with cell culture medium prior to use in the experiments. Final concentration of DMSO did not exceed 0.1%. Other reagents including 3-methyladenine (3-MA), N-acetylcysteine (NAC), polyethylene glycol-conjugated catalase (PEG-catalase), polyethylene glycol-conjugated superoxide dismutase (PEG-SOD), gentamycin, anti-tubulin antibody, and anti-actin antibody were purchased from Sigma-Aldrich (St. Louis, MO). Anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was from GeneTex (Irvine, CA). Cell culture reagents, OligoFECTAMINE, Alexa Fluor 488-conjugated goat anti-rabbit antibody, MitoSOX Red, and MitoTracker Green were purchased from Invitrogen-Life Technologies (Grand Island, NY). 1-Hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH) for electron paramagnetic resonance spectrometry (EPR) was bought from Noxygen Science Transfer and Diagnostics (Elzach, Germany). Antibodies against cleaved poly-(ADP-ribose)-polymerase (PARP), LC3B, ATG5 (for detection of ATG5-ATG12), phospho-(Ser473)-AKT and total AKT, phospho-(Ser2448)-mTOR, total mTOR, phospho-(Ser65)-eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), and total 4E-BP1 were from Cell Signaling (Danvers, MA). Anti-LC3 antibody used for immunofluorescence microscopy was purchased from MBL International (Woburn, MA). Control small-interfering RNA (siRNA) and ATG5-targeted siRNA were purchased from Qiagen (Valencia, CA) and Santa Cruz Biotechnology (Dallas, TX), respectively. Human prostate cancer cells (PC-3 and LNCaP) were obtained from the American Type Culture Collection (Manassas, VA) and maintained as described previously [10, 21]. The MDA-MB-231 cells expressing mRuby/LC3 fusion protein as an autophagosome marker were obtained from the University of Pittsburgh Cancer Institute Lentiviral Core Facility, and maintained in RPMI1640 supplemented with 10% fetal bovine serum, 10 μg/mL of gentamycin, and 0.5 μg/mL of puromycin. The Myc-CaP cell line established from prostate tumor of a Hi-Myc transgenic mouse was a generous gift from Dr. Charles L. Sawyers (Howard Hughes Medical Institute, Memorial Sloan Kettering Cancer Center, New York, NY) [36]. Myc-CaP cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 4.5 g/L glucose, 10% fetal bovine serum, and 1% antibiotics.
Microscopic Visualization of Autophagic Vacuoles
Transmission electron microscopy in PC-3 cells after 24 h treatment with DMSO (control) or 40 μM HNK was performed for visualization and quantitation of autophagic vacuoles as previously described [31, 35].
Western Blot Analysis
Lysates from cultured cells and PC-3 xenograft supernatants were prepared as previously described [37, 38]. Details of immunoblotting can be found in our published studies [37, 38]. In some experiments, cells were pretreated with 4 mM of 3-MA or 4 mM of NAC for 2 h or 100 U/mL of PEG-SOD or PEG-catalase for 1 h prior to 24 h treatment with 40 μM HNK.
Immunocytochemical Analysis of LC3 Puncta
Cells were seeded on coverslips (in 12-well plates in triplicate), allowed to attach by overnight incubation, and then exposed to DMSO or 40 μM HNK for 24 h. Cells were then fixed with 2% paraformaldehyde for 1 h at room temperature, permeabilized with 0.5% Triton X-100 for 10 min, blocked with buffer containing 0.5% bovine serum albumin and 0.15% glycine in phosphate-buffered saline (PBS) for 1 h, followed by incubation with anti-LC3 antibody at 4°C. After overnight incubation, the cells were incubated with 2 μg/mL Alexa Fluor 488-conjugated secondary antibody for 1 h at room temperature. After washing with PBS, cells were mounted and observed under a Leica DC300F fluorescence microscope at 100× objective lens magnification. MDA-MB-231/RFP-LC3 cells (1×105 cells per well) were plated onto coverslips in 12-well plates and then treated with DMSO or 40 μM HNK for 24 h. After treatment, cells were fixed with 2% paraformaldehyde at room temperature for 1 h and then mounted with anti-fading mounting medium. RFP-LC3 puncta images were captured using a fluorescence microscope at 100× objective magnification.
Cell Viability Assay
Trypan blue dye exclusion assay was performed to determine the effect of HNK treatment on cell viability essentially as described by us previously [39]. Cells were seeded at a density of 5×104 per well in 12-well plates in triplicate, allowed to attach overnight, pretreated with 4 mM of 3-MA for 2 h, and then exposed to 40 μM of HNK for 24 h in the absence or presence of 3-MA.
DNA Fragmentation Assay
Cells (5~7.5×104 cells per well in 12-well plates) were plated in triplicate, allowed to attach overnight, pretreated with 4 mM of 3-MA for 2 h and then treated with 40 μM HNK in the absence or presence of 3-MA for 24 h. Apoptosis induction was assessed by quantitation of histone-associated DNA fragment release into the cytosol using a Cell Death Detection ELISAPLUS kit from Roche Diagnostics (Indianapolis, IN).
RNA Interference of ATG5
Cells were seeded in six-well plates and transfected at 50% confluency with a control (nonspecific) siRNA or ATG5 siRNA. Twenty-four hours after transfection, cells were treated with DMSO (control) or 40 μM of HNK for 24 h. Cells were then collected and processed for immunoblotting, cell viability, and DNA fragmentation assay.
Measurement of Reactive Oxygen Species Generation
Several methods were employed for detection of ROS in HNK-treated prostate cancer cells. First, a cell-permeable spin probe (CMH) was used to detect ROS by EPR. In brief, 1×106 cells were plated in triplicate in 10 cm dish and exposed to DMSO or 40 μM of HNK for 4 h. Cells were collected by scraping, centrifuged, and the pellet was resuspended in 100 μL of Krebs HEPES buffer (pH 7.4). EPR was performed as described by us previously [40]. Second, flow cytometry using MitoSOX Red was performed for ROS detection. For these experiments, cells were plated in triplicate, treated with DMSO (control) or 40 μM of HNK for 2 and 4 h, and then incubated with 5 μM of MitoSOX Red for 30 min at 37°C. Cells were collected by trypsinization, washed with PBS, and processed for flow cytometry using BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). Finally, ROS generation upon treatment with HNK was visualized by MitoSOX Red fluorescence microscopy essentially as described by us previously [40, 41].
Apoptosis Detection by Annexin V/Propidium Iodide Flow Cytometry
Early- and late-stage apoptotic cell death was determined using Annexin V/propidium iodide Apoptosis Detection kit (BD Biosciences) according to the manufacturer’s instructions. Briefly, cells (2×105 cells per well) were seeded in 6-well plates in triplicate, incubated overnight, pretreated with 100 U/mL of PEG-SOD or PEG-catalase, and then exposed to DMSO or 40 μM HNK in the absence or presence of PEG-SOD or PEG-catalase for an additional 24 h. After treatment, harvested cells were resuspended in 100 μL of binding buffer and then stained with 4 μL of Annexin V and 2 μL of propidium iodide solution for 15 min at room temperature in the dark, and analyzed using a flow cytometer.
Statistical Analysis
All data were analyzed using the Prism 4 (version 4.03) (GraphPad Software, Inc., La Jolla, CA). Statistical significance was determined by analysis of variance (ANOVA) followed by Dunnett’s test or Bonferroni’s multiple comparison test or Student t test. P < 0.05 was considered statistically significant.
RESULTS
Treatment with HNK Resulted in Autophagy Induction in Prostate Cancer Cells
Initially, transmission electron microscopy was performed to ascertain autophagy induction by HNK using PC-3 cell line, which is a widely studied cellular model of human prostate cancer. The HNK concentrations used in the present study (20 and 40 μM) are pharmacologically relevant based on rodent pharmacokinetic studies [16, 42]. Fig. 1A shows representative transmission electron micrographs from PC-3 cells following 24 h treatment with DMSO or 40 μM HNK. Because autophagy is a normal physiological process for bulk degradation of macromolecules, some autophagic vacuoles were visible in the DMSO-treated controls (identified by arrows in Fig. 1A). In addition, the vehicle-treated control cells were enriched with healthy looking mitochondria with normal cristae structure (Fig. 1A). On the other hand, HNK-treated PC-3 cells displayed a nearly 3.5-fold increase in number of autophagic vacuoles in comparison with DMSO-treated control (Fig. 1B). Some of the autophagic vacuoles in HNK-treated PC-3 cells seem to contain fragments of organelles including mitochondria. The number of healthy looking mitochondria was reduced after treatment of PC-3 cells with HNK. Increase in levels of 14 kDa lipidated form of LC3 (LC3BII) is considered a biochemical hallmark of autophagy [43]. Western blotting was performed to test whether HNK treatment affected levels of LC3BII. As can be seen in Fig. 1C, HNK treatment caused a dose-dependent increase in protein levels of LC3BII (LC3BII band is identified by an arrow in Fig. 1C) in both PC-3 cells and androgen-responsive LNCaP cells. The increase in protein levels of LC3BII after treatment with HNK was evident as early as 8 h post-treatment especially at the 40 μM concentration.
Fig. 1.

HNK induces autophagy in prostate cancer cells. A: Representative transmission electron microscopic images (25,000×magnification; scale bars = 500 nm) showing autophagic vacuoles (indicated by arrows) in PC-3 cells treated for 24 h with DMSO or 40 μM HNK. B: Quantitation of number of autophagic vacuoles from experiment in (A). Quantitation from images acquired from six different sections of each group is shown as mean ± SD (n = 6). Statistical significance was determined by Student t test. C: Immunoblotting for LC3BII (pointed by arrows) in PC-3 and LNCaP cells treated with DMSO or HNK. Numbers on top of bands are fold change in LC3BII protein level relative to corresponding DMSO-treated control.
We used a cell line (Myc-CaP) established from prostate tumor of a transgenic mouse (Hi-Myc) to confirm autophagy induction by HNK. Viability of Myc-CaP cells was decreased significantly after 24 h treatment with 40 μM HNK (Fig. 2A). In cells undergoing autophagy, localization of LC3 is characterized by a punctate arrangement [43]. Fig. 2B shows localization of LC3 in Myc-CaP cells after 24 h treatment with 40 μM HNK (the LC3 puncta is identified by an arrow in Fig. 2B). The number of LC3 puncta/cell in Myc-CaP cells was increased significantly upon HNK treatment compared with control (Fig. 2C). In agreement with these results, HNK exposure resulted in an increase in levels of LC3BII protein in the Myc-CaP cell line (Fig. 2D).
Fig. 2.
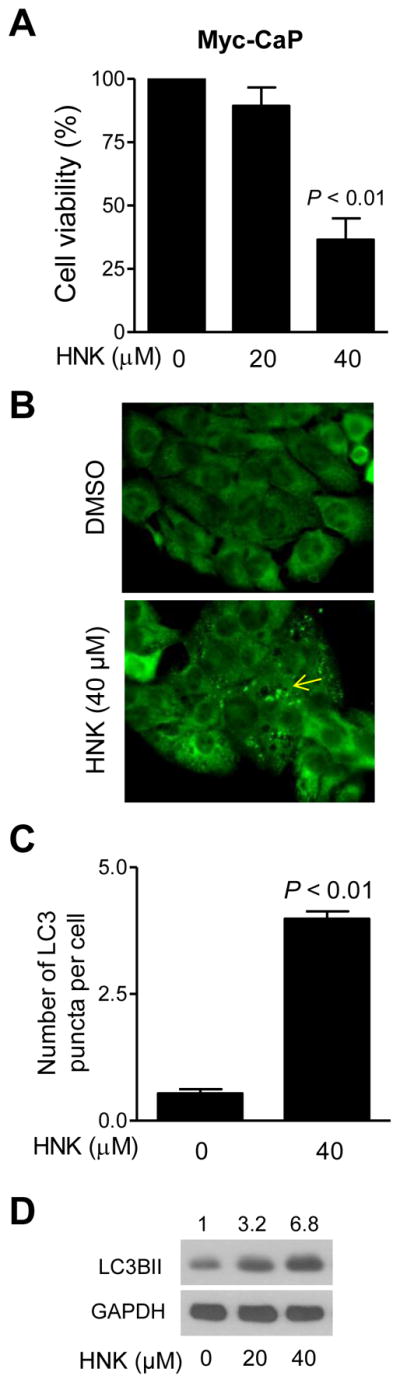
HNK induces autophagy in Myc-CaP murine prostate cancer cells. Effect of HNK treatment (24 h treatment) on A: Myc-CaP cell viability (mean ± SD; n = 3; statistical significance was determined by one-way ANOVA followed by Dunnett’s test), B: LC3 puncta (100× objective magnification), C: Quantitation of LC3 puncta (mean ± SD; n = 3; statistical significance was determined by Student t test), and D: LC3BII level.
Fig. 3A depicts LC3 puncta in PC-3 and LNCaP cells after 24 h treatment with 40 μM HNK. The number of LC3 puncta/cell was increased by about 2.5-fold and 3.2-fold in HNK-treated PC-3 and LNCaP cells, respectively, when compared with DMSO-treated control (Fig. 3B). We used a breast cancer cell line with stable overexpression of RFP-LC3 to further test autophagic response to HNK. The LC3 puncta were rare in controls but clearly visible after 24 h treatment with 40 μM HNK (Fig. 3C). Collectively, these results indicated that HNK treatment resulted in autophagy induction in prostate and breast cancer cells.
Fig. 3.

HNK enriches LC3 puncta in cancer cells. A: Representative immunofluorescence images (100× objective magnification) showing HNK-induced LC3 puncta in PC-3 and LNCaP cells after 24 h treatment with DMSO or 40 μM HNK. B: Quantitation of LC3 puncta per cell in PC-3 and LNCaP cells. Results combined from two independent experiments are presented as mean ± SD (n = 6). Statistical significance was determined by Student t test. C: Representative immunofluorescence images (100× objective magnification) showing puncta (identified by arrows) in MDA-MB-231/RFP-LC3 cells treated with DMSO or 40 μM of HNK for 24 h. D: Immunoblot for LC3BII using PC-3 xenograft supernatants from control or HNK-treated mice. Tumor tissues from two different mice of each group were used. Number on top of the band reflects quantification of protein expression level (mean ± SD). E: Western blotting for LC3BII using lysates from LNCaP cells after 2 h pretreatment with 4 mM of 3-MA followed by 24 h exposure to DMSO or 40 μM of HNK in the absence or presence of 3-MA.
PC-3 Tumor Xenografts Exhibited Increased Levels of LC3BII Protein
We used PC-3 xenografts from our previously published study [10] to determine the effect of HNK on levels of LC3BII in vivo. The level of LC3BII protein was relatively higher in the tumors from HNK-treated mice compared with control (Fig. 3D). These results indicated that HNK treatment promoted processing of LC3 protein in vivo.
Autophagy Induction by HNK Was Cytoprotective
Because autophagy can either promote or inhibit apoptosis [31–35], it was of interest to study the functional significance of autophagy induction by HNK. Initially, we utilized a chemical inhibitor of autophagy (3-MA) to address this question. Cells were first pretreated for 2 h with 4 mM 3-MA then treated with 40 μM HNK for an additional 24 h in the presence of the inhibitor. As shown in Fig. 3E, HNK-induced increase in protein level of LC3BII was nearly fully inhibited in the presence of 4 mM 3-MA in LNCaP cells. Treatment with 3-MA alone did not increase LC3BII protein level (Fig. 3E). There are several examples in the published literature indicating inhibition of experimentally-induced autophagy in the presence of 1-5 mM 3-MA in PC-3 cells [44, 45]. For example, increase in LC3BII protein level after treatment of PC-3 cells with another phytochemical (benzyl isothiocyanate) was markedly suppressed in the presence of 1 mM 3-MA (1 h pretreatment with 3-MA followed by 24 h treatment with benzyl isothiocyanate in the presence of 3-MA) [45]. In agreement with our prior published observations [21], viability of PC-3 and LNCaP cells was decreased significantly after 24 h treatment with HNK (Fig. 4A). The cell viability inhibition resulting from HNK treatment was significantly augmented in the presence of 3-MA in both PC3 and LNCaP cells; although the inhibitor alone exhibited some cytotoxicity in LNCaP cells (Fig. 4A). Apoptosis induction by HNK was assessed by measuring the release of histone-associated DNA fragments into the cytosol and cleavage of PARP, which are commonly employed techniques for apoptosis detection. As shown in Fig. 4B, the DNA fragmentation resulting from HNK treatment was significantly intensified in the presence of 3-MA in both cells. In agreement with these results, the HNK+3-MA combination was more efficacious in causing cleavage of PARP in comparison with HNK alone (Fig. 4C). Because DNA fragmentation or cleavage of PARP was minimal or not seen at all with 3-MA alone, it is possible that the cytotoxicity observed with the inhibitor in the LNCaP cell line is due to necrosis. Nevertheless, these results indicated that autophagy was protective against HNK-induced apoptosis in both PC-3 and LNCaP cells.
Fig. 4.

Pharmacological inhibition of autophagy by 3-MA enhances HNK-mediated apoptosis. Effects of 3-MA and/or HNK treatments on A: Cell viability, B: DNA fragmentation, and C: PARP cleavage in PC-3 and LNCaP cells after 2 h pretreatment with 4 mM of 3-MA and then 24 h exposure to DMSO or 40 μM of HNK in the absence or presence of 3-MA. Results shown are mean ± SD (n = 3). *Statistically significant (panels A and B) between the indicated groups by one-way ANOVA followed by Bonferroni’s multiple comparison test.
HNK-Induced Apoptosis Was Augmented by Knockdown of ATG5 Protein
We performed knockdown of ATG5, a protein critical for autophagy process, to obtain additional evidence for the cytoprotective nature of HNK-induced autophagy using PC-3 cells. The level of ATG5 protein was decreased by about 50% after transfection of PC-3 cells with 100 nM ATG5-targeting siRNA (Fig. 5A). RNA interference of ATG5 was also inhibitory against HNK-mediated increase in LC3BII protein level (Fig. 5A). HNK treatment alone for 24 h resulted in modest (30%) increase in the level of ATG5 protein (Fig. 5A). HNK-induced decrease in cell viability (Fig. 5B) as well as apoptosis induction (Fig. 5C) was augmented by knockdown of ATG5 protein. These observations confirmed that HNK-induced autophagy was cytoprotective in prostate cancer cells.
Fig. 5.
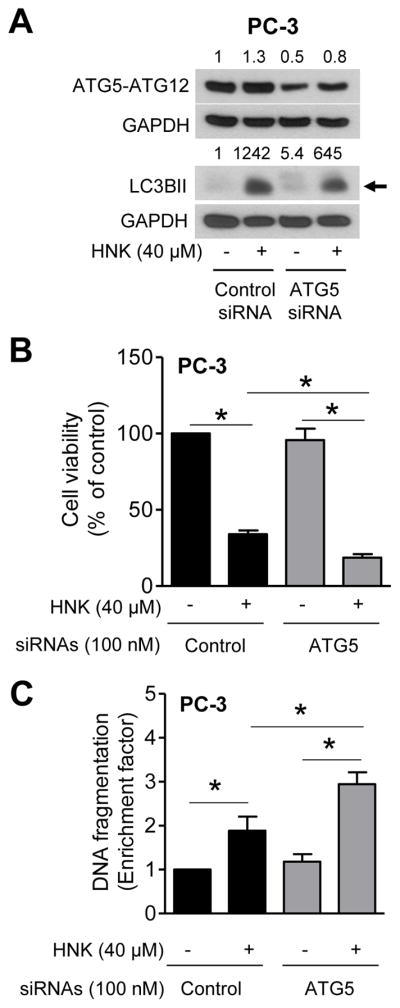
ATG5 knockdown enhances HNK-induced apoptosis. A: Immunoblotting for ATG5-ATG12 and LC3BII in PC-3 cells. Numbers on top of bands are fold change in ATG5-ATG12 protein expression relative to DMSO-treated control siRNA-transfected cells (first lane). B: Viability of PC-3 cells. C: Detection of apoptosis in PC-3 cells. For data shown in panels AC, PC-3 cells were transiently transfected with control siRNA or ATG5 siRNA followed by 24 h treatment with DMSO or 40 μM HNK. Results shown are mean ± SD (n = 3). *Statistically significant between the indicated groups by one-way ANOVA followed by Bonferroni’s multiple comparison test.
Effect of HNK Treatment on Phospho-mTOR
Inhibition of mTOR induces autophagy in cancer cells including PC-3 and LNCaP cells [31, 46]. We explored the possibility whether autophagy induction by HNK was associated with suppression of mTOR. As shown in Fig. 6, the levels of phospho-mTOR as well as its upstream regulator (phospho-AKT) and downstream target phospho-4E-BP1 were decreased after treatment with HNK in PC-3 cells especially at the 24 h time point. In LNCaP cells, level of phospho-4E-BP-1 protein was decreased in a dose- and time-dependent manner upon HNK treatment (Fig. 6). The level of phospho-mTOR was reduced by 30% after 8 and 16 h treatment of LNCaP cells with 40 μM HNK (Fig. 6). These results indicated that suppression of mTOR alone may not fully explain autophagy induction by HNK especially in LNCaP cells.
Fig. 6.

HNK treatment alters mTOR pathway in prostate cancer cells. Immunoblotting for mTOR signaling-related proteins using whole lysates from PC-3 and LNCaP cells treated with DMSO or various doses of HNK. Numbers on top of bands are fold change of level relative to corresponding DMSO-treated control.
HNK Treatment Resulted in ROS Generation
ROS are implicated in autophagy induction by different stimuli [30, 31]. We proceeded to experimentally test whether autophagy induction by HNK was dependent on ROS generation. Several methods were employed to determine ROS generation by HNK in PC-3 cells. EPR signals in PC-3 cells after 4 h treatment with DMSO (control) or 40 μM HNK are shown in Fig. 7A. The intensity of EPR signal was significantly higher in HNK-treated cells than in DMSO-treated control (Fig. 7A, bar graph). ROS generation by HNK was confirmed by analysis of MitoSOX Red fluorescence. Fig. 7B shows representative flow histograms for MitoSOX Red fluorescence in PC-3 cells after 2 or 4 h treatment with 40 μM HNK or DMSO (control). HNK-mediated increase in MitoSOX Red fluorescence in comparison with DMSO control was statistically significant at both time points (Fig. 7B, bar graph). MitoSOX Red fluorescence can be visualized in Fig. 7C, which was minimal in DMSO treated control cells. On the other hand, MitoSOX Red fluorescence was increased dramatically after treatment of PC-3 cells with HNK. Moreover, the MitoSOX Red fluorescence was co-localized with MitoTracker Green signal suggesting mitochondrial origin of ROS in HNK-treated cells.
Fig. 7.

HNK causes ROS production in prostate cancer cells. A: Representative EPR spectra using CMH (1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine) as a spin probe, and quantitation of EPR signal intensity from PC-3 cells treated with DMSO or 40 μM HNK for 4 h. Combined results from two independent experiments are shown as mean ± SD (n = 6). Statistical significance was determined by Student t test. B: Representative flow histograms and quantitation of MitoSOX Red fluorescence in PC-3 cells treated with DMSO or 40 μM HNK for 2 h or 4 h. Results shown are mean ± SD (n = 3). Statistical significance was determined by Student t test. C: Representative microscopic images showing MitoSOX Red fluorescence in PC-3 cells after 2 h or 4 h treatment with DMSO or 40 μM HNK (100× objective magnification).
HNK-Induced Increase in LC3BII Protein Level Was Partially Abolished in the Presence of Antioxidants
To test whether autophagy induction by HNK was dependent on ROS generation, we performed western blotting for LC3BII using lysates from PC-3 and LNCaP cells treated for 24 h with HNK and/or NAC (2 h pretreatment). HNK-induced increase in levels of LC3BII protein was partially but markedly suppressed in the presence of NAC (Fig. 8A). The role of ROS in autophagy induction by HNK was confirmed using cell-permeable antioxidants, including PEG-SOD (Fig. 8B) and PEG-catalase (Fig. 8C). Similar to NAC, HNK-mediated processing of LC3 was partially blocked in the presence of these antioxidants. These results indicated that autophagy induction by HNK was partly dependent on ROS generation.
Fig. 8.
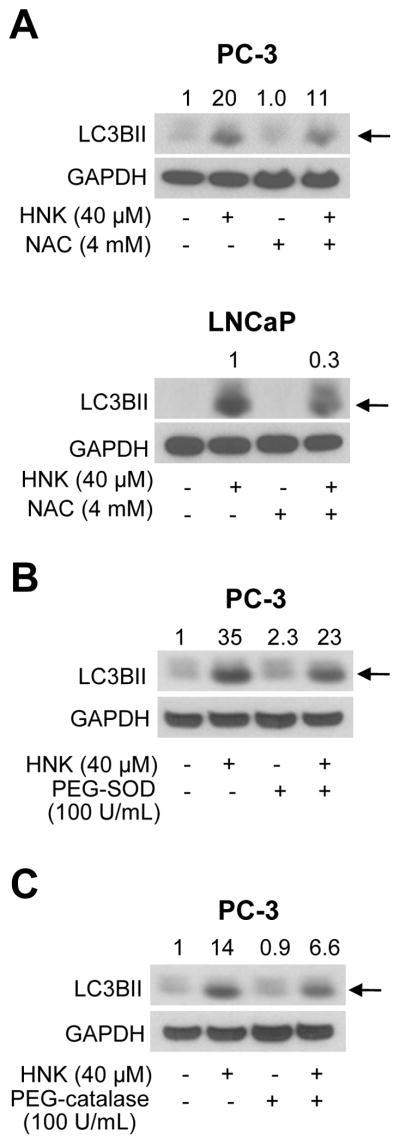
Antioxidants inhibit HNK-induced increase in LC3BII protein level. A: Immunoblotting for LC3BII (pointed by arrows) in PC-3 and LNCaP cells after 2 h pretreatment with 4 mM NAC and then 24 h exposure to DMSO or 40 μM HNK in the absence or presence of NAC. B: Immunoblotting for LC3BII (pointed by an arrow) from PC-3 cells after 1 h pretreatment with 100 U/mL of PEG-SOD and then 24 h exposure to DMSO or 40 μM HNK in the absence or presence of PEG-SOD. C: Immunoblotting for LC3BII (pointed by an arrow) in PC-3 cells after 1 h pretreatment with 100 U/mL of PEG-catalase and then 24 h exposure to DMSO or 40 μM HNK in the absence or presence of PEG-catalase. Quantitation relative to DMSO-treated control (first lane) is shown on top of bands.
Effect of Antioxidants on HNK-Induced Apoptosis
Next, we determined the effect of PEG-SOD and PEG-catalase on HNK-induced apoptosis by using Annexin V-propidium iodide method that measures both early and late apoptotic fraction. HNK treatment resulted in enrichment of early + late apoptotic fraction in comparison with DMSO control but the HNK-induced apoptosis was not significantly altered in the presence of PEG-SOD or PEG-catalase (data not shown).
DISCUSSION
A few previously published studies have documented the effect of HNK treatment on mitochondrial morphology or lipidation of LC3 using different cells [47–49]. Notably, these studies have relied on a single marker or technique to assess autophagy after HNK treatment. Furthermore none of these studies provided any evidence for functional relevance of HNK-induced autophagy [47–49]. For example, Steinmann et al. [49] observed vacuoles in the cytoplasm and mitochondrial structural changes (transmission electron microscopy) in LM8-LacZ mouse osteosarcoma cells after 1.5–6 h treatment with 15 μg/mL of HNK. The ultrastructure of these vacuoles increased in size and number after HNK treatment exhibiting enlarged and swollen endoplasmic lumen and changes in morphology of mitochondrial cristae [49]. Based solely on these morphological observations, these investigators concluded that autophagy was not triggered after treatment with HNK in LM8-LacZ cells [49]. On the other hand, Chang et al. [47] noted lipidated form of LC3 (immunoblotting for LC3BII) in DBTRG-05MG glioblastoma multiforme cell line after 72 h treatment with 50 μM HNK. Immunoblotting for LC3BII alone is not sufficient to conclude induction of autophagy. Transmission electron microscopy alone was utilized to show appearance of autophagic vacuoles in B16-F10 melanoma cells after 24 h treatment with 30 μM HNK [48]. Another limitation of these studies was lack of time-course kinetics for morphological changes or LC3 lipidation [47–49]. The present study utilizes various methods (transmission electron microscopy to visualize autophagic vacuoles containing remnants of organelles such as mitochondria, western blotting for LC3BII, and microscopic visualization of LC3 puncta) to clearly establish autophagy induction by HNK in prostate and breast cancer cells. Because HNK-induced autophagy is observed in both PC-3 and LNCaP cells, we also conclude that this phenomenon is not affected by the expression of androgen receptor or androgen-responsiveness.
Autophagy induction with different functional consequences has been described for a number of structurally divergent naturally-occurring anticancer agents, including watercress constituent phenethyl isothiocyanate [31], D,L-sulforaphane [33, 34], and a chemical (benzyl isothiocyanate) isolated from garden cress [35]. Autophagy induction contributed to overall cell death after treatment with phenethyl isothiocyanate and benzyl isothiocyanate [31, 35]. On the other hand, autophagy induction served to protect against D,L-sulforaphane-induced apoptosis by delaying release of cytochrome c [33]. The present study clearly shows that autophagy is cytoprotective against apoptosis induction by HNK at least in prostate cancer cells. This conclusion is based on the following observations: (a) inhibition of autophagy in the presence of 3-MA potentiates HNK-induced apoptosis in PC-3 and LNCaP cells and (b) RNA interference of an autophagy regulator (ATG5) has similar effect as 3-MA on HNK-induced apoptosis. These results suggest that autophagy inhibition can be exploited to increase anticancer effect of HNK.
Previous studies have implicated mTOR as a negative regulator of autophagy. We observed suppression of mTOR and AKT phosphorylation by HNK treatment, but these effects were relatively more pronounced in the PC-3 cells than in LNCaP (Fig. 6). Thus mTOR inhibition alone can’t explain autophagy induction by HNK. Results described herein indicate that autophagy induction by HNK is partly dependent on ROS generation as evidenced by rescue of LC3BI conversion to LC3BII. Our expectation was that inhibition of autophagy in the presence of antioxidants would lead to an increase in HNK-induced apoptosis. However, the proapoptotic response to HNK was not affected by the antioxidants (data not shown). Our interpretation of these seemingly negative results is that increased apoptosis arising from inhibition of autophagy in the presence of antioxidants is likely masked by a ROS-dependent apoptotic mechanism for HNK (which would be inhibited in the presence of antioxidants). We have shown previously that apoptosis induction by certain other natural agents (e.g., phenethyl isothiocyanate) is significantly attenuated by antioxidants because of reduced activation of multidomain Bcl-2 family member Bax[41]. In this context, it is important to mention that the HNK-induced apoptosis in PC-3 and LNCaP cells is associated with induction of Bax and Bak [10]. We have also shown previously that Bax and Bak knockdown confers significant protection against HNK-induced apoptotic cell death in PC-3 cells [10]. It is possible that ROS generation by HNK leads to activation and Bax and/or Bak, but further work is necessary to explore this possibility.
In conclusion, the present study demonstrates, for the first time, that HNK treatment triggers cytoprotective autophagy in prostate cancer cells, which can be exploited to increase anticancer effect of this promising natural agent
Acknowledgments
This work was supported by the grants RO1 CA101753-10, RO1 CA115498-08, and RO1 CA113363-10 awarded by the National Cancer Institute. This research used the Cell and Tissue Imaging Facility, Flow Cytometry Facility, and the Lentiviral Facility supported in part by a grant from the National Cancer Institute at the National Institutes of Health (P30 CA047904).
Abbreviations
- ANOVA
analysis of variance
- CMH
1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine
- DMSO
dimethyl sulfoxide
- 4E-BP-1
eukaryotic translation initiation factor 4E-binding protein 1
- EPR
electron paramagnetic resonance
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HNK
honokiol
- LC3
microtubule-associated protein 1 light chain 3
- LC3BII
a lipidated form of microtubule-associated protein 1 light chain 3 beta
- 3-MA
3-methyladenine
- mTOR
mammalian target of rapamycin
- NAC
N-acetylcysteine
- PBS
phosphate-buffered saline
- PARP
poly-(ADP-ribose)-polymerase
- PEG-catalase
polyethylene glycol-conjugated catalase
- PEG-SOD
polyethylene glycol-conjugated superoxide dismutase
- ROS
reactive oxygen species
- siRNA
small interfering RNA
Footnotes
Disclosure Statement: None.
AUTHOR DISCLOSURE STATEMENT
The authors declare no competing financial interest.
References
- 1.Maruyama Y, Kuribara H. Overview of the pharmacological features of honokiol. CNS Drug Rev. 2000;6(1):35–44. [Google Scholar]
- 2.Arora S, Singh S, Piazza GA, Contreras CM, Panyam J, Singh AP. Honokiol: a novel natural agent for cancer prevention and therapy. Curr Mol Med. 2012;12(10):1244–1252. doi: 10.2174/156652412803833508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai SK, Huang SS, Hong CY. Myocardial protective effect of honokiol: An active component in Magnolia officinalis. Planta Med. 1996;62(6):503–506. doi: 10.1055/s-2006-957957. [DOI] [PubMed] [Google Scholar]
- 4.Clark AM, El-Feraly FS, Li WS. Antimicrobial activity of phenolic constituents of Magnolia grandiflora L. J Pharm Sci. 1981;70(8):951–952. doi: 10.1002/jps.2600700833. [DOI] [PubMed] [Google Scholar]
- 5.Hamasaki Y, Muro E, Miyanji S, Yamamoto S, Kobayashi I, Sato R, Zaitu M, Matsuo M, Ichimaru T, Tasaki H, Miyazaki S. Inhibition of leukotriene synthesis by honokiol in rat basophilic leukemia cells. Int Arch Allergy Immunol. 1996;110(3):278–281. doi: 10.1159/000237299. [DOI] [PubMed] [Google Scholar]
- 6.Teng CM, Chen CC, Ko FN, Lee LG, Huang TF, Chen YP, Hsu HY. Two antiplatelet agents from Magnolia officinalis. Thromb Res. 1988;50(6):757–765. doi: 10.1016/0049-3848(88)90336-2. [DOI] [PubMed] [Google Scholar]
- 7.Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009;11(5):1139–1148. doi: 10.1089/ars.2009.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hibasami H, Achiwa Y, Katsuzaki H, Imai K, Yoshioka K, Nakanishi K, Ishii Y, Hasegawa M, Komiya T. Honokiol induces apoptosis in human lymphoid leukemia Molt 4B cells. Int J Mol Med. 1998;2(6):671–673. doi: 10.3892/ijmm.2.6.671. [DOI] [PubMed] [Google Scholar]
- 9.Hirano T, Gotoh M, Oka K. Natural flavonoids and lignans are potent cytostatic agents against human leukemic HL-60 cells. Life Sci. 1994;55(13):1061–1069. doi: 10.1016/0024-3205(94)00641-5. [DOI] [PubMed] [Google Scholar]
- 10.Hahm ER, Arlotti JA, Marynowski SW, Singh SV. Honokiol, a constituent of oriental medicinal herb Magnolia officinalis, inhibits growth of PC-3 xenografts in vivo in association with apoptosis induction. Clin Cancer Res. 2008;14(4):1248–1257. doi: 10.1158/1078-0432.CCR-07-1926. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Liu Y, Zhao X, Pan X, Yin R, Huang C, Chen L, Wei Y. Honokiol, a natural therapeutic candidate, induces apoptosis and inhibits angiogenesis of ovarian tumor cells. Eur J Obstet Gynecol Reprod Biol. 2008;140(1):95–102. doi: 10.1016/j.ejogrb.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 12.Sheu ML, Liu SH, Lan KH. Honokiol induces calpain-mediated glucose-regulated protein-94 cleavage and apoptosis in human gastric cancer cells and reduces tumor growth. PLoS ONE. 2007;2(10):e1096. doi: 10.1371/journal.pone.0001096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shigemura K, Arbiser JL, Sun SY, Zayzafoon M, Johnstone PA, Fujisawa M, Gotoh A, Weksler B, Zhau HE, Chung LW. Honokiol, a natural plant product, inhibits the bone metastatic growth of human prostate cancer cells. Cancer. 2007;109(7):1279–1289. doi: 10.1002/cncr.22551. [DOI] [PubMed] [Google Scholar]
- 14.Nagalingam A, Arbiser JL, Bonner MY, Saxena NK, Sharma D. Honokiol activates AMP-activated protein kinase in breast cancer cells via an LKB1-dependent pathway and inhibits breast carcinogenesis. Breast Cancer Res. 2012;14(1):R35. doi: 10.1186/bcr3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, Der CJ, Battle T, Frank DA, Ye K, Murad E, Dubiel W, Soff G, Arbiser JL. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278(37):35501–35507. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- 16.Chen F, Wang T, Wu YF, Gu Y, Xu XL, Zheng S, Hu X. Honokiol: A potent chemotherapy candidate for human colorectal carcinoma. World J Gastroenterol. 2004;10(23):3459–3463. doi: 10.3748/wjg.v10.i23.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guillermo RF, Chilampalli C, Zhang X, Zeman D, Fahmy H, Dwivedi C. Time and dose-response effects of honokiol on UVB-induced skin cancer development. Drug Discov Ther. 2012;6(3):140–146. [PubMed] [Google Scholar]
- 18.Leeman-Neill RJ, Cai Q, Joyce SC, Thomas SM, Bhola NE, Neill DB, Arbiser JL, Grandis JR. Honokiol inhibits epidermal growth factor receptor signaling and enhances the antitumor effects of epidermal growth factor receptor inhibitors. Clin Cancer Res. 2010;16(9):2571–2579. doi: 10.1158/1078-0432.CCR-10-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ponnurangam S, Mammen JM, Ramalingam S, He Z, Zhang Y, Umar S, Subramaniam D, Anant S. Honokiol in combination with radiation targets notch signaling to inhibit colon cancer stem cells. Mol Cancer Ther. 2012;11(4):963–972. doi: 10.1158/1535-7163.MCT-11-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3(10):768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 21.Hahm ER, Singh SV. Honokiol causes G0-G1 phase cell cycle arrest in human prostate cancer cells in association with suppression of retinoblastoma protein level/phosphorylation and inhibition of E2F1 transcriptional activity. Mol Cancer Ther. 2007;6(10):2686–2695. doi: 10.1158/1535-7163.MCT-07-0217. [DOI] [PubMed] [Google Scholar]
- 22.Ahn KS, Sethi G, Shishodia S, Sung B, Arbiser JL, Aggarwal BB. Honokiol potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through modulation of nuclear factor-κB activation pathway. Mol Cancer Res. 2006;4(9):621–633. doi: 10.1158/1541-7786.MCR-06-0076. [DOI] [PubMed] [Google Scholar]
- 23.Park EJ, Min HY, Chung HJ, Hong JY, Kang YJ, Hung TM, Youn UJ, Kim YS, Bae K, Kang SS, Lee SK. Down-regulation of c-Src/EGFR-mediated signaling activation is involved in the honokiol-induced cell cycle arrest and apoptosis in MDA-MB-231 human breast cancer cells. Cancer Lett. 2009;277(2):133–140. doi: 10.1016/j.canlet.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 24.Crane C, Panner A, Pieper RO, Arbiser J, Parsa AT. Honokiol-mediated inhibition of PI3K/mTOR pathway: A potential strategy to overcome immunoresistance in glioma, breast, and prostate carcinoma without impacting T cell function. J Immunother. 2009;32(6):585–592. doi: 10.1097/CJI.0b013e3181a8efe6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lan KL, Lan KH, Sheu ML, Chen MY, Shih YS, Hsu FC, Wang HM, Liu RS, Yen SH. Honokiol inhibits hypoxia-inducible factor-1 pathway. Int J Radiat Biol. 2011;87(6):579–590. doi: 10.3109/09553002.2011.568572. [DOI] [PubMed] [Google Scholar]
- 26.Rajendran P, Li F, Shanmugam MK, Vali S, Abbasi T, Kapoor S, Ahn KS, Kumar AP, Sethi G. Honokiol inhibits signal transducer and activator of transcription-3 signaling, proliferation, and survival of hepatocellular carcinoma cells via the protein tyrosine phosphatase SHP-1. J Cell Physiol. 2012;227(5):2184–2195. doi: 10.1002/jcp.22954. [DOI] [PubMed] [Google Scholar]
- 27.Yao CJ, Lai GM, Yeh CT, Lai MT, Shih PH, Chao WJ, Whang-Peng J, Chuang SE, Lai TY. Honokiol eliminates human oral cancer stem-like cells accompanied with suppression of Wnt/β-catenin signaling and apoptosis induction. Evid Based Complement Alternat Med. 2013;2013:146136. doi: 10.1155/2013/146136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hahm ER, Karlsson AI, Bonner MY, Arbiser JL, Singh SV. Honokiol inhibits androgen receptor activity in prostate cancer cells. Prostate. 2014;74(4):408–420. doi: 10.1002/pros.22762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han LL, Xie LP, Li LH, Zhang XW, Zhang RQ, Wang HZ. Reactive oxygen species production and Bax/Bcl-2 regulation in honokiol-induced apoptosis in human hepatocellular carcinoma SMMC-7721 cells. Environ Toxicol Pharmacol. 2009;28(1):97–103. doi: 10.1016/j.etap.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Ishdorj G, Gibson SB. Reactive oxygen species regulation of autophagy in cancer: Implications for cancer treatment. Free Radic Biol Med. 2012;53(7):1399–1410. doi: 10.1016/j.freeradbiomed.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 31.Bommareddy A, Hahm ER, Xiao D, Powolny AA, Fisher AL, Jiang Y, Singh SV. Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 2009;69(8):3704–3712. doi: 10.1158/0008-5472.CAN-08-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gewirtz DA. Cytoprotective and nonprotective autophagy in cancer therapy. Autophagy. 2013;9(9):1263–1265. doi: 10.4161/auto.25233. [DOI] [PubMed] [Google Scholar]
- 33.Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome c and apoptosis in human prostate cancer cells. Cancer Res. 2006;66(11):5828–5835. doi: 10.1158/0008-5472.CAN-06-0139. [DOI] [PubMed] [Google Scholar]
- 34.Vyas AR, Hahm ER, Arlotti JA, Watkins S, Stolz DB, Desai D, Amin S, Singh SV. Chemoprevention of prostate cancer by D,L-sulforaphane is augmented by pharmacological inhibition of autophagy. Cancer Res. 2013;73(19):5985–5995. doi: 10.1158/0008-5472.CAN-13-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao D, Bommareddy A, Kim SH, Sehrawat A, Hahm ER, Singh SV. Benzyl isothiocyanate causes FoxO1-mediated autophagic death in human breast cancer cells. PLoS ONE. 2012;7(3):e32597. doi: 10.1371/journal.pone.0032597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watson PA, Ellwood-Yen K, King JC, Wongvipat J, Lebeau MM, Sawyers CL. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65(24):11565–11571. doi: 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 37.Xiao D, Srivastava SK, Lew KL, Zeng Y, Hershberger P, Johnson CS, Trump DL, Singh SV. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. 2003;24(5):891–897. doi: 10.1093/carcin/bgg023. [DOI] [PubMed] [Google Scholar]
- 38.Powolny AA, Bommareddy A, Hahm ER, Normolle DP, Beumer JH, Nelson JB, Singh SV. Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. J Natl Cancer Inst. 2011;103(7):571–584. doi: 10.1093/jnci/djr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao D, Choi S, Johnson DE, Vogel VG, Johnson CS, Trump DL, Lee YJ, Singh SV. Diallyl trisulfide-induced apoptosis in human prostate cancer cells involves c-Jun N-terminal kinase and extracellular-signal regulated kinase-mediated phosphorylation of Bcl-2. Oncogene. 2004;23(33):5594–5606. doi: 10.1038/sj.onc.1207747. [DOI] [PubMed] [Google Scholar]
- 40.Hahm ER, Moura MB, Kelley EE, Van Houten B, Shiva S, Singh SV. Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS ONE. 2011;6(8):e23354. doi: 10.1371/journal.pone.0023354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao D, Powolny AA, Moura MB, Kelley EE, Bommareddy A, Kim SH, Hahm ER, Normolle D, Van Houten B, Singh SV. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem. 2010;285(34):26558–26569. doi: 10.1074/jbc.M109.063255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai TH, Chou CJ, Cheng FC, Chen CF. Pharmacokinetics of honokiol after intravenous administration in rats assessed using high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1994;655(1):41–45. doi: 10.1016/0378-4347(94)00031-x. [DOI] [PubMed] [Google Scholar]
- 43.Hönscheid P, Datta K, Muders MH. Autophay: Detection, regulation and its role in cancer and therapy response. Int J Radiat Biol. 2014 doi: 10.3109/09553002.2014.907932. in press. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Li X, Wang J, Ye Z, Li JC. Oridonin up-regulates expression of P21 and induces autophagy and apoptosis in human prostate cancer cells. Int J Biol Sci. 2012;8(6):901–912. doi: 10.7150/ijbs.4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin JF, Tsai TF, Liao PC, Lin YH, Lin YC, Chen HE, Chou KY, Hwang TIS. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis. 2013;34(2):406–414. doi: 10.1093/carcin/bgs359. [DOI] [PubMed] [Google Scholar]
- 46.Lorin S, Hamaï A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Semin Cancer Biol. 2013;23(5):361–379. doi: 10.1016/j.semcancer.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 47.Chang KH, Yan MD, Yao CJ, Lin PC, Lai GM. Honokiol-induced apoptosis and autophagy in glioblastoma multiforme cells. Oncol Lett. 2013;6(5):1435–1438. doi: 10.3892/ol.2013.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaushik G, Ramalingam S, Subramaniam D, Rangarajan P, Protti P, Rammamoorthy P, Anant S, Mammen JM. Honokiol induces cytotoxic and cytostatic effects in malignant melanoma cancer cells. Am J Surg. 2012;204(6):868–873. doi: 10.1016/j.amjsurg.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinmann P, Walters DK, Arlt MJ, Banke IJ, Ziegler U, Langsam B, Arbiser JL, Muff R, Born W, Fuchs B. Antimetastatic activity of honokiol in osteosarcoma. Cancer. 2012;118(8):2117–2127. doi: 10.1002/cncr.26434. [DOI] [PubMed] [Google Scholar]