

Abstract
Gastrointestinal (GI) illnesses are a significant health burden for the US population, with 40 million office visits each year for gastrointestinal complaints and nearly 250,000 deaths. Acute and chronic inflammation are a common element of many GI diseases. Inflammatory processes may be initiated by a chemical injury (acid reflux in the esophagus), an infectious agent (Helicobacter pylori infection in the stomach), autoimmune processes (graft versus host disease after bone marrow transplantation), or idiopathic (as in the case of inflammatory bowel diseases). Inflammation in these settings can contribute to acute complaints (pain, bleeding, obstruction, diarrhea) as well as chronic sequelae including strictures and cancer. Research into the pathophysiology of these conditions has been limited by the availability of primary human tissues or appropriate animal models that attempt to physiologically model the human disease. With the many recent advances in tissue engineering and primary human cell culture systems, it is conceivable that these approaches can be adapted to develop novel human ex vivo systems that incorporate many human cell types to recapitulate in vivo growth and differentiation in inflammatory microphysiological environments. Such an advance in technology would improve our understanding of human disease progression and enhance our ability to test for disease prevention strategies and novel therapeutics. We will review current models for the inflammatory and immunological aspects of Barrett’s esophagus, acute graft versus host disease, and inflammatory bowel disease and explore recent advances in culture methodologies that make these novel microphysiological research systems possible.
Keywords: inflammation, oxidative stress, autophagy, gastrointestinal (GI) disease, gastroesophageal reflux disease (GERD), Barrett’s esophagus (BE), esophageal adenocarcinoma (EAC), graft-versus-host disease (GvHD), inflammatory bowel disease (IBD), human 3D organotypic model systems (OTC)
Introduction
Gastrointestinal inflammatory conditions in humans, both acute and chronic, are a significant source for disease-related morbidity and mortality, both in the U.S. and worldwide. In addition to the human costs in pain, disability, and premature death, these disorders carry a considerable financial impact as well. Productivity loses and the enormous expense of medical care strain both familial and societal budgets. Indeed, the costs for care of U.S. patients with inflammatory bowel disease (IBD), a single chronic inflammatory condition, were estimated to be over $6 billion in 2008 (1). Altogether, this severe impact on patient well-being and the enormous resulting financial burden confirms the importance of research efforts to better understand and treat these disorders.
Microphysiological culture systems hold great promise to revolutionize research into these important disease conditions. Although tissue and cell culture methodologies as laboratory techniques have been established for well over a century, and have significantly contributed to the advancement of biomedical research and therapeutics, it has become increasingly clear that current standard culture techniques remain highly artificial (2,3). Limitations include an over-reliance on transformed and neoplastic cell lines, utilization of stiff plastic and glass culture plates, and a dependence upon undefined serums and matrices to support and sustain cell growth. Microphysiological culture systems, in contrast, seek to better replicate in vivo microenvironments in an in vitro setting suitable for research laboratories.
Microphysiological culture systems, in contrast, attempt to better model human tissues through a combination of innovative bioengineered platforms, novel materials, and the use of primary or immortalized human cells rather than transformed cell lines. This edition of Experimental Biology and Medicine explores novel bioengineered devices and 3-dimensional culture conditions that are attempting to replicate every major organ system of the human body. Each utilizes novel approaches to study the biology and physiology of these engineered tissue representations. Moreover, many of these systems are being explored as platforms for toxicology testing and drug screening, two very powerful applications of this technology. However, no single platform currently recapitulates all the cell types and biology inherent in the endogenous tissues. In this review we propose another innovative use of these novel human culture systems: to model the effects of acute and chronic inflammatory microenvironments on human gastrointestinal epithelium. We will explore several inflammatory diseases of the esophagus and intestine, briefly discuss current cell culture and animal models employed to study them, and propose how innovative modifications to microphysiological culture conditions can endow them with new functionalities such as improved modeling of human inflammatory diseases in the laboratory.
Gastroesophageal Reflux Disease and Barrett’s Esophagus
Acute and chronic inflammatory conditions of the esophagus including gastroesophageal reflux disease (GERD), reflux esophagitis, eosinophilic esophagitis (EOE), Barrett’s esophagus (BE), and esophageal adenocarcinoma (EAC), cumulatively afflict a significant portion of the US population each year (4). These diseases progress sequentially, with gastric acid and bile refluxed to the esophagus in GERD inflicting tissue injury and inflammation that leads first to esophagitis (Figure 1) (5). An estimated 10–15% of these patients then proceed to develop BE, a metaplasia characterized by replacement of normal esophageal stratified squamous epithelium with an intestinal columnar epithelium (Figure 2) (6). These metaplastic regions then progress to low-grade dysplasia (LGD), followed by high-grade dysplasia (HGD), culminating in esophageal adenocarcinoma (EAC). EAC is a deadly cancer with an increasing incidence rate in the US, and the risk of EAC development for patients with BE is approximately 0.5% per year (7). Despite its prevalence, its association with EAC, and the increasing incidence of EAC, the biology of BE and its progression to cancer are poorly understood and are therefore critically important clinical and basic research imperatives.
Figure 1.
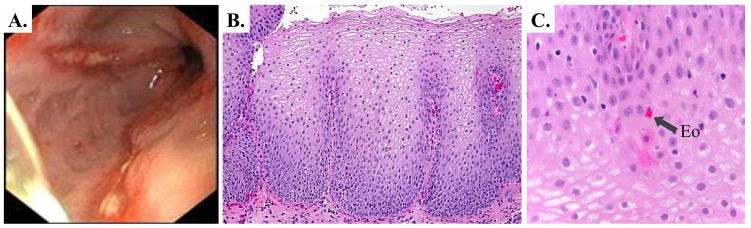
Endoscopic and histologic appearance of gastro-esophageal reflux disease (GERD). A. LA Grade B esophagitis visualized endoscopically, characterized by non-confluent mucosal breaks in the distal esophagus. B. and C. Hematoxylin and Eosin (H&E) stain of GERD esophagitis at low power (B.) and higher power (C.) demonstrating non-specific immune cell infiltrate, including in some rare eosinophils (Eo).
Figure 2.

Endoscopic and histologic appearance of Barrett’s esophagus. A. and B. Endoscopic appearance of Barrett’s esophagus under white-light (A.) and narrow-band imaging (NBI) (B.). NBI uses filters to image using light in the blue and green wavelengths in order to enhance mucosal surface details. C. H&E stain of human Barrett’s esophagus at the squamo-columnar junction (indicated by the green arrow). Normal multilayered squamous epithelium (NSE) is to the left and simple columnar Barrett’s esophagus is to the right (above bracket BE). Within the BE field Goblet-cell (GC) metaplasia and a chronic inflammatory infiltrate are observed.
Development of esophageal inflammatory diseases begins with acid reflux injury followed by the infiltration of immune cells to the site of the injury (Figure 3) (5,8). Secretion of signaling molecules such as growth factors and cytokines orchestrate the activation of immune cells and direct their differentiation. Cytokines bind to naïve CD4+ T cells, upregulating master transcriptional regulators, and inducing differentiation into specialized T helper (TH) subsets (TH1, TH2, TH17, or regulatory T cells). Each of the four lineages then expresses its own distinct set of cytokines and transcription factors, conferring specific functionality (9). In the esophagus, activation of a TH1 pro-inflammatory response, characterized by production of interferon (IFN)-γ, is typical for acid reflux esophagitis (10). Progression to BE is accompanied by a shift in cytokine expression patterns, including increased levels of interleukin (IL)-4, IL-5, IL-10, and IL-13, which are hallmarks of a TH2 humoral immune response (10–12).
Figure 3.

Model for progression from GERD to BE to EAC. Illustration summarizing the transition from normal esophageal epithelium to metaplastic Barrett’s esophagus (BE) to esophageal adenocarcinoma (EAC). Following reflux injury, epithelial cells experience several genetic events that, when combined with microenvironmental signals from stromal fibroblasts and infiltrating immune cells, trigger the development of intestinal columnar cells in place of stratified epithelium. BE is a precursor lesion that will continue to accumulate molecular alterations, ultimately leading to the development of dysplasia and invasive EAC.
Several inflammatory mediators have been linked to BE and EAC pathogenesis and disease progression. Cyclooxygenase-2 (Cox-2) and its product prostaglandin E2 (PGE2) are highly elevated in BE, presumably as result of reflux injury, and aid in the recruitment of immune cells including neutrophils, macrophages, and mast cells (13,14). PGE2 also regulates T cell expansion by activating TH2 effector cells while suppressing the TH1 response, supporting a cytokine profile shift towards BE.
A number of other molecular events have also been implicated in the development of BE and progression to EAC (Table 1). Along the Barrett’s metaplasia to dysplasia sequence (BE → LGD → HGD → EAC), allelic loss or silencing of the tumor suppressor and cyclin dependent kinase (CDK) inhibitor p16 is thought to be one of the earliest occurrences (15). Mutations of the p53 tumor suppressor gene, as well as enhanced expression of Bcl2 and cyclin D1 have also been detected in metaplastic/pre-dysplastic BE (16–18). Progression of BE to dysplasia and EAC is frequently accompanied by p21 upregulation and K-Ras activating mutations (19,20). Altogether, these genes regulate a broad range of cellular functions from cell adhesion to cell cycle checkpoints and apoptosis, illustrating the complexity of this esophageal disease and the challenges faced in modeling them in the research laboratory.
Table 1.
Genes involved in BE onset and progression to EAC
| Gene | Protein | Alteration | Function | Associations | Ref | |
|---|---|---|---|---|---|---|
| Adhesion | APC | Adenomatous polyposis coli | LOH | Tumor suppressor | BE - EAC | (21) |
| CTNNB1 | β-catenin | Nuclear Accumulation | Transcription factor | BE - EAC | (22) | |
| CDH1 | E-cadherin | Decreased Expression | Cell-cell adhesion | BE - EAC | (22) | |
| Apoptosis | BCL2 | B-cell lymphoma 2 | Increased Expression | Apoptosis inhibitor | BE - LGD | (17) |
| FASLG | Fas Ligand | Increased Expression | Apoptosis driver | BE - EAC | (23) | |
| MDM2 | E3 ubiquitin-protein ligase Mdm2 | Increased Expression | Negative p53 regulator | EAC | (24) | |
| TP53 | p53 | Mutation | Tumor suppressor | BE - EAC | (16) | |
| Cell Cycle | CCND1 | Cyclin D1 | Increased Expression | CDK regulation, proto-oncogene | BE - EAC | (18) |
| CDKN2A | p16 | LOH | CDK inhibitor, tumor suppressor | BE - EAC | (15) | |
| CDKN1A | p21 | Increased Expression | CDK inhibitor | LGD - EAC | (19) | |
| CDKN1B | p27 | Cytoplasmic Sequestration | CDK inhibitor, tumor suppressor | HGD - EAC | (25) | |
| Cell Signaling | CDX2 | Caudal type homeobox 2 | Increased Expression | Transcription factor | BE | (26) |
| ERBB2 | Human epidermal growth factor receptor 2 | Increased Expression | Growth factor receptor, proto-oncogene | EAC | (27) | |
| EGFR | Epidermal growth factor receptor | Amplification | Growth factor receptor | BE - EAC | (28) | |
| KRAS | K-Ras | Mutation | MAPK signaling | HGD - EAC | (20) | |
| SRC | Src kinase | Increased Expression | EGF signaling | BE - EAC | (29) | |
| TGFA | Transforming growth factor α | Amplification | EGF signaling | BE - EAC | (28) | |
| Inflammation | PTGS2 | Cyclooxygenase-2 | Increased Expression | Prostaglandin synthesis | BE - EAC | (30) |
| IL1B | Interleukin 1β | Increased Expression | Cytokine | BE - EAC | (10) | |
| NOS2 | Inducible nitric oxide synthase | Increased Expression | Nitric oxide synthesis | BE - EAC | (30) |
Several different approaches have been pursued in the past to study BE and EAC, and while important advances have been made, each has its limitations. We will briefly review past and current models for BE and esophageal metaplasia, discussing the physiological limitations of each and then exploring how novel human cell based organotypic and tissue engineering approaches can be applied to advance our understanding of BE and EAC pathogenesis.
Modeling Esophageal Diseases
Cell Culture Models: Esophagus
The earliest cell culture models for BE studies used human BE tissue explants and esophageal adenocarcinoma cell lines (21–24). More recently, primary normal human esophageal and Barrett’s epithelial cell lines were established with retroviral transduction of human telomerase reverse transcriptase (hTERT) (25,26). Five primary Barrett’s cell lines were developed from various stages of BE progression, with CP-A and BAR-T cell lines originating from metaplastic tissue, while CP-B, CP-C, and CP-D were all derived from Barrett’s with HGD. While the CP-A, B, C, and D cell lines have documented chromosomal abnormalities (27), the BAR-T cell line maintains a diploid status, displays normal morphology, and responds to contact inhibition (28), making them a particularly useful cell line for studies of disease progression to cancer. Many of the studies using these cell lines in 2-dimensional (2D) culture environments have explored the effects of various Barrett’s etiologic agents upon esophageal keratinocytes, including chronic acid and bile acid treatment (24,29), BMP-4 (30), retinoic acid (31,32), and the intestine-specific transcription factors Cdx1 and Cdx2 (33–35). While these treatments did yield increased expression of intestine-specific genes and changes in cell morphology, none succeeded at inducing a complete BE phenotype in keratinocytes.
Animal Models: Esophagus
Animal models have become the cornerstone for biomedical research, as they allow for the evaluation of genetic alterations and/or environmental variables in a controlled manner. However, the development of a murine model that accurately recapitulates human BE continues to be a challenge. Major anatomical differences exist in mice and humans and, as a result, mice do not naturally experience GERD. Other anatomic differences include the absence of submucosal glands, which have been hypothesized as the cell of origin for BE (36), and the mouse squamous epithelium-lined forestomach. Due to this forestomach, the transition from squamous to columnar epithelial cells in rodents occurs in the stomach as opposed to the gastroesophageal junction where the squamo-columnar junction occurs in humans. Since rodents do not typically have GERD, surgical manipulation is necessary to introduce bile and gastric acid to the esophagus in order to mimic GERD and elicit an inflammatory response that progresses to metaplasia, dysplasia, and adenocarcinoma (37). Such procedures are technically challenging to perform and do not guarantee production of the desired phenotype. The utilization of transgenic mice has therefore been adopted as an alternative approach.
To study BE pathogenesis, our laboratory previously generated a transgenic mouse in which an intestine-specific transcription factor, Cdx2, was ectopically expressed in the mouse esophagus using a Keratin-14 promoter (38). Cdx2 is required for intestine-specific gene expression, columnar cell shape and cell-cell adhesion, and is absolutely required for the normal differentiation of the intestinal epithelium (39–47). In human BE tissues, CDX2 mRNA and protein are uniformly expressed in biopsy samples (48, 49). Moreover, Cdx2 expression can be induced in cultures of esophageal keratinocytes treated with short pulses of acid and bile (24, 29). In addition, ectopic gastric expression of Cdx2 induced a gastric intestinal metaplasia (GIM) similar to human GIM . CDX2 is therefore expected to play a similar role in the pathogenesis of BE (33, 50). These K14-Cdx2 mice were found to have significantly reduced esophageal cell proliferation, as well as diminished barrier function. While a complete BE phenotype was not achieved, cells that exhibited ultrastructural features of both squamous and columnar cells were observed. These hybrid cells indicate that Cdx2 promotes the appearance of a transitional phenotype that, under a supportive microenvironment, may adopt a metaplastic columnar phenotype.
More recently, a transgenic mouse has been created that over-expresses the cytokine IL-1β in the esophagus, mimicking chronic esophagitis (51). Expression of IL-1β was specifically targeted to the oral cavity, esophagus, and squamous forestomach using an Epstein-Barr virus L2 promoter (52). By six months of age the IL-1β mice displayed moderate inflammation, followed by metaplasia at 12–15 months. Furthermore, the addition of bile acids in their drinking water resulted in significant acceleration of metaplasia, dysplasia, and cancer in the IL-1β mice but not their wild-type littermates. These data suggest a synergy between cytokine expression and bile acid exposure in the induction of metaplasia at the squamo-columnar junction. Future studies will explore the role of other genetic factors in disease progression using this model, however, inherent differences between murine and human biology persists. Perhaps a better way to accurately model the in vivo events occurring throughout the Barrett’s metaplasia disease sequence will be to use novel human cell-based tissue-engineered culture systems.
Human Engineered Tissue Systems: Esophagus
To be clinically relevant, studies must strive to best mimic human in vivo physiology. However, animal models have significant limitations as we have seen, and traditional 2D cell culture using human cells can only represent the effects of a single cell type and fail to capture the significant contributions of the microenvironment. The development of the organotypic culture (OTC) system that allows for the co-culture of immortalized human epithelial cell lines together with primary fibroblasts in 3-dimensional (3D) tissue reconstructions represents a novel means by which to perform in vitro experiments that are still physiologically relevant (53). Under 3D OTC conditions, esophageal fibroblasts are embedded in a collagen matrix, overtop of which, after a period of growth and matrix modification, keratinocytes are seeded (Figure 4). Exposure of the keratinocytes to the air-liquid interface then triggers differentiation and stratification producing a fully mature epithelium (54). This physiologically relevant model thus provides a novel platform for the study of many human esophageal diseases.
Figure 4.

OTC systems to model normal esophageal growth and inflammation. A. Illustration identifying the main components of the standard OTC method, as well as how to adapt this culture system for the inclusion of immune cells to invoke inflammation. B. H&E stain of normal human esophageal keratinocytes grown under OTC conditions.
When evaluated under OTC conditions, the Barrett’s cell lines CP-A, CP-B, CP-C, and CP-D show surprising variability. The HGD-derived CP-B, CP-C, and CP-D lines displayed little to no stromal invasion, however, the metaplastic CP-A cells not only yielded mucin-producing goblet cells, but also were unexpectedly highly invasive (55). These major phenotypic differences were not apparent previously and were only observed as a result of the more physiological 3D OTC system. This study also underscores the fact that a high level of diversity exists not only between BE patients, but likely within their own BE segments, as well. As a result, it will be imperative in future studies to include a wide panel of cell lines and tissues in every stage of disease.
Our laboratory ventured to examine the early genetic and molecular events that precede the development of BE to gain further insight into its pathogenesis. We demonstrated that epithelial expression of the inflammation-associated enzyme Cox-2 in the hTERT-immortalized human esophageal keratinocytes produced elevated levels of PGE2 and was sufficient to induce a metaplasia with features of BE (56). In addition, using this same OTC system, we determined through a genetic approach that the onset of a true BE is a multistep process that requires increased proliferation, senescence inhibition, and epigenetic alterations (34).
Future innovations will be directed toward increasing the complexity of the OTC cultures by including other relevant cell types including immune, endothelial, smooth muscle, and neural cells (Figure 4). In addition, investigators have recently identified conditions by which primary human Barrett’s organoids can be maintained and expanded in a 3-dimensional culture matrix (57). Together, the novel OTC culture system and new primary human Barrett’s cell culture methods will lead to engineered human esophageal and Barrett’s tissues that better model physiologic responses and therefore, will be far superior research platforms than any that are presently available.
Graft versus Host Disease and Inflammatory Bowel Disease
Graft versus Host Disease (GvHD)
GvHD is an unfortunately common complication from allogeneic bone marrow transplantation (ABMT), the treatment of choice for a number of human malignancies, both hematologic and solid organ. It is a fatal complication to 15% of ABMT recipients, and can be a rare complication of solid organ transplantation as well (58, 59). It is caused by the donor cells responding to and attacking the recipient host alloantigens (60). There are two types of GvHD, acute and chronic, originally defined based on whether they occurred within 100 days of the transplantation (acute) or after (chronic), but are now recognized to have distinctly different clinical presentations and underlying molecular pathogenesis.
Acute GvHD occurs in a third to half of all ABMT recipients, and is marked by strong inflammatory features including dermatitis, cutaneous blisters, crampy abdominal pain with or without diarrhea, nausea and vomiting, and hepatitis (61). The endoscopic and histologic appearance of small intestine GvHD can be quite varied, depending upon the disease severity. In very mild disease, the small intestine mucosa can appear normal, with only focal crypt epithelial apoptosis and mild lymphocytic infiltrate characteristic of GVHD visible histologically. Moderately severe disease can present with small intestinal mucosal edema, increased friability, villus blunting, and small ulcerations (Figure5) (62). The histology in these cases often demonstrates apoptosis with focal dropout of crypt epithelial cells, small ulcerations, and lymphocytic infiltrates in the lamina propria are more numerous. In its most severe form, small intestine GVHD presents with a hemorrhagic epithelium from sloughing of substantial regions of mucosa and the presence of large ulcers. Histologically there is a dense lymphocytic infiltration, epithelial and crypt loss and ulcerations.
Figure 5.

Endoscopic and histologic views of acute GvHD. A. Clinically normal duodenum. B. Moderate acute GvHD diagnosed by mild mucosal edema, hyperemia, villous blunting, and small erosions. C. H&E stain of normal human duodenum demonstrating normal crypt/villous architecture. D. H&E stain of acute GvHD showing architectural distortion, loss of crypts, lymphocytic infiltration with small hemorrhage (H), and crypt cell apoptosis.
Molecularly, it is thought to be largely, though not exclusively, a TH1 and TH17 immune response, whereas chronic GvHD follows more typically a TH2 pathway (60, 63, 64). Treatment of acute GvHD is directed toward optimizing immunosuppressive therapy, and is typically effective, however about half of these patients will go on to develop chronic GvHD associated with greater long-term morbidity and mortality.
Chronic GvHD is the more serious and long-term complication of ABMT, occurring in a one to two-thirds of ABMT transplant recipients surviving more than 100 days (65). Unlike acute GvHD which is mediated by alloreactive cytotoxic effects, chronic GvHD is more indolent, resembling autoimmune vascular diseases (60). Patients can sometimes present with distinctive findings that establish the diagnosis, including sclerosis, esophageal webs, and bronchiolitis obliterans, however more typically they present with less diagnostic features (65). In these cases the diagnosis is confirmed by a biopsy of involved tissues which most commonly are the skin, oral cavity, liver, and intestine/gastrointestinal tract (including esophagus). As in acute GvHD, treatment typically requires immunosuppressive medications for a median of 2 to 3 years, and in some cases for 7 or more years. The mortality associated with chronic GvHD, which is as high as 30% to 50% at 5 years, is caused by two mechanisms, organ dysfunction and failure due to damage by the chronic GvHD, and infectious complications from chronic immune suppressive medications (60, 65). In summary, the significant morbidity and mortality from acute and chronic GvHD after ABMT significantly limits the effectiveness of ABMT for hematologic and solid malignancies and remains an important focus of research.
Inflammatory Bowel Disease (IBD)
Inflammatory bowel diseases such as Crohn’s disease (CD) and ulcerative colitis (UC) are chronic disorders of the intestine and/or colon, in which patients suffer from rectal bleeding, severe diarrhea, abdominal pain, fever, and weight loss (66–68). In North America, 1.4 million people are affected by IBD (69). Our current understanding of the pathogenesis of IBD is that they are a result of abnormal host immune responses brought on by diet and lifestyle in genetically susceptible individuals (70). Host genetic susceptibility plays an important role in the risk of development of IBD. Several genes have been identified including those involved in innate immune signaling pathways (ITLN1) (71), autophagy (IRGM) (72), ER stress responses (ORMDL3) (73), and negative regulators of interferon-gamma (INF-γ) signaling (PTPN2) (74). The cornerstone therapies for IBD are immunosuppressive drugs and steroids, both of which provide variable benefit to the IBD population (75).
Clinical and histopathological observation of tissue from patients with IBD reveals gastric erosions and ulcerations, the presence of a large inflammatory cell infiltrate concurrent with extensive mucosal and transmural injury including edema, loss of goblet cells, decreased mucous production and crypt cell hyperplasia (Figure 6) (66). CD is characterized by an excessive recruitment of leukocytes from the blood circulation into the inflamed gut wall and massive infiltration of neutrophil granulocytes and macrophages into the affected mucosa (76–78). During acute flares of CD, neutrophil granulocytes are found in the stool and have served as a clinical and research tool to quantify levels of disease activity (76). Although it was previously thought that CD was a TH1-mediated disease while UC was a TH2-mediated disease, it has recently been suggested that a TH17 immune response is also involved in the pathogenesis of IBDs; TH17 cytokines are highly expressed in the intestinal mucosa of both CD and UC patients (79).
Figure 6.

IBD visualized endoscopically and histologically. A. Clinically normal small intestine. B. Crohn’s disease diagnosis presented as edematous, erythemous small intestine mucosa with nearly circumferential small bowel ulceration and over-lying inflammatory exudate. C. H&E stain of normal human small intestine, D. H&E stain of Crohn’s disease marked by loss of villi, chronic inflammatory cell infiltrate extending into the submucosa, and an active inflammation in the epithelium.
Modeling Intestinal Diseases
Cell Culture Models: Intestine
The intestinal mucosa contains a large variety of cell types in close proximity to the epithelium and the complexity of this microenvironment is further increased during an inflammatory response with additional immune cells infiltrating the lamina propria. Immune and epithelial cells release a variety of signaling factors and peptides to regulate the inflammatory response (cytokines), recruit phagocytes (chemokines) and facilitate epithelial repair (growth factors) (75) (Figure 7). To better understand these key components in IBD, in vitro models have focused on a variety of culture systems utilizing one or more cell types.
Figure 7.

Illustration of key mediators involved in the development of acute intestinal GvHD (aGvHD) and IBD. aGvHD is caused by donor T cells responding to and attacking recipient host alloantigens with cytotoxic products. Irradiation and BMT conditioning prior to the BMT injure the intestinal epithelium, leading to intestinal barrier disruption and exposure of the mucosal immune compartment to bacterial products. These bacterial components are potent activators of an innate immune response, which trigger a “cytokine storm”, amplifying the immune response and favoring the development of acute GvHD. IBD, such as Crohn’s disease, develops due to the presence of a large inflammatory cell infiltrate with extensive mucosal and transmural injury including edema, loss of goblet cells, decreased mucous production and crypt cell hyperplasia. Factors secreted by the intestinal microbiota and immune cells can activate epithelial cells to produce cytokines that regulate inflammation, chemokines that signal recruitment of phagocytes, and growth factors with autocrine actions that facilitate epithelial repair and/or damage.
Historically, there have been four different cell culture approaches to model and study human small intestine epithelium, 1) primary cell cultures, 2) cell lines established from colon cancers, 3) spontaneously immortalized cell lines from normal intestine, and 4) normal intestinal cell lines immortalized by oncogenes and other genetic approaches. Each approach has important strengths and weaknesses. Cell lines established from human colon cancers have been most widely used experimentally. This is because they are human cells, easy to grow and manipulate in the lab, relatively inexpensive to maintain, and widely available. Several colon cancer cell lines exhibit interesting phenotypes. Three cell lines Caco-2, HT 29 and T84 express morphological and functional characteristics of differentiated intestinal epithelial cells including polarized, columnar morphology, vectoral transport, and tight junction-dependent barrier (80–83). They can be grown on transwell membranes and other structural supports readily, facilitating studies of these properties and functions by cell biologists and physiologists. Bioengineers fabricating devices to model intestinal epithelium likewise favor them for their ease of use and functional epithelial barrier.
However, while individual cell lines may be suitable for certain limited purposes (study of tight-junction formation and barrier disruption, or transporters and channels, for instance), they fall far short of physiological models of normal human small intestine. All are colonic in origin, though one cell line, CaCo-2, express small intestine hydrolases (sucrase-isomaltase, lactase, aminopeptidase N and dipeptidylpeptidase IV) when they spontaneously differentiate after reaching confluency. Moreover, all of these cells have considerable gene mutations and epigenetic changes in gene expression, resulting in many abnormal cellular signaling pathways, stress responses, and cellular metabolism. Perhaps most importantly, they lack normal intestinal stem cells, and therefore are unsuitable for toxicology studies exploring the effects of toxins on the vulnerable intestinal stem cell compartment.
Given these disadvantages, investigators have employed other strategies, including isolating from primary intestinal epithelial cultures spontaneously immortalized cell lines, or immortalizing primary intestinal epithelial cells using oncogenic proteins like SV40 Large-T antigen, or more recently telomerase. Fetal rat intestinal epithelium yielded a half-dozen spontaneously immortalized cell lines (IEC and FRIC cell lines), whereas attempts using fetal human intestine yielded a single cell line (HIEC cells) (84, 85). Another approach was to immortalize cell lines from a primary intestinal epithelial cell culture using a temperature-sensitive SV40-virus Large-T antigen, an established oncogene, or, more recently, a combination of non-oncogenic Cdk4 with telomerase (86). In most cases, these cells were homogeneous, passaged readily in culture, and were typically near-normal genetically. However they typically exhibited characteristics of poorly differentiated intestinal epithelium, more like crypt cells than differentiated cells in the villi. Moreover, except for human colonic HCEC cells (86), they do not form a mature polarized epithelial barrier with tight junctions.
In vitro research efforts in IBD have been directed towards understanding the contribution of the epithelium in the inflammatory response using single cells treated with specific molecules. Several studies using colon cancer cell lines observed that common IBD epithelial responses, including injury and repair, apoptosis, and necrosis, can be induced by microbes or microbial products and immune activation (75). Another common approach to studying these responses is by co-culturing human colon crypt-like T84 epithelial cells with bacterial species or bacterial products and/or immune cells (or conditioned media from such cells). This approach has been particularly effective for studies exploring perturbations in epithelial permeability (i.e. transepithelial electrical resistance) brought out by inflammatory responses (75, 87, 88).
Primary human intestinal epithelial cell cultures in theory should be the most physiologic model, however, the isolation of the epithelial cells from the underlying supportive mesenchyme leads to rapid loss of their differentiated characteristics, loss of stem cell capacity, and limited viability in culture. In addition, the simple culture of cell monolayers in 2D plastic systems does not permit exploration of how cell-to-cell interactions and a physiologic extracellular matrix can influence mucosal immune cell responses. Recent advances in techniques to maintain human small intestinal epithelial cells in long-term culture employ a 3-dimensional matrix and physiologically relevant growth factors (57, 89, 90). This innovative culture system is a significant advance, permitting the long-term growth of intestinal stem cells while permitting normal differentiation along four intestinal lineages, and the formation of a polarized epithelium with normal vectoral transport and cell-cell adhesion including tight junctions. This novel culture system has the potential to significantly advance current GvHD and IBD research practices
Animal Models: Acute GvHD
Mice are far and away the main preclinical animal model for GvHD and studies in mice have made important contributions to our understanding of the pathogenesis of this disorder, though other large animal models have contributed to our improved understanding as well (60). Mouse models for GvHD involve transplantation of donor bone marrow along with different types of donor lymphocytes into irradiated allogeneic recipients. These recipients typically differ from the donor with regard to their MHC class I and/or II molecules. The use of these animal models has led to novel insights regarding disease pathogenesis, including the central role of gastrointestinal injury as an initiating event (91).
Irradiation and other conditioning regimens to prepare the recipient host for the donor BMT also injures the intestinal epithelium, leading to intestinal barrier disruption and exposure of the mucosal immune compartment to bacterial products including lipopolysaccharides (LPS) and DNA (91). These bacterial components are potent activators of an innate immune response, which triggers a “cytokine storm”, amplifying the immune response and favoring the development of acute GvHD (Figure 7). This current model for acute GvHD is supported by a broad array of observations. Early animal and clinical studies found that gut decontamination significantly reduced death rates and acute GvHD incidence (91). Moreover, depletion of T-cells from the graft can abrogate acute GvHD in experimental models (48,69)(60, 91). Additionally, mutation of Toll-like receptor 4 (TLR4), an innate immune receptor for bacterial LPS, can reduce GvHD risk whereas activation of TLR9 by bacterial DNA can enhance the risk for GvHD (92, 93). Based on these observations, strategies that manipulate the gut flora, deplete T-cells, or suppress innate immune responses are an important focus of ongoing research efforts.
The strength of the mouse as a research model for human GvHD lies in our ability to manipulate its genome and generate transgenic mice with strategic genes “knocked-out” by mutation. The generation of transgenic mice with inactivating mutations in a host of cytokines and TLRs has enabled our dissection of their role in the pathogenesis of this disorder (94, 95). For this reason, mouse models of GvHD will likely remain an important tool for the foreseeable future. However, these important mouse models do have significant limitations as well. Fundamentally, while these models can explore the general pathogenesis of GvHD in the setting of MHC incompatibilities, they cannot test specifically human MHC alloreactive responses. An equally important consideration is the effect of mouse strain and genetic drift on GvHD research. It was recently reported that differences in mouse strains which naturally occur over time appear to be altering alloreactive immune responses and GvHD severity (96). Finally, genomic studies of GvHD have identified an increasing number of human polymorphisms that may alter the risk for the development of GvHD (97), however demonstrating this association beyond observational studies in human BMT recipients would require the generation of transgenic mice bearing the polymorphism, a considerable expense. Together these limitations argue for the development of human engineered tissues and systems in which acute and, possibly, chronic GvHD can be credibly modeled using human cells, possibly from BMT patient’s themselves.
Animal Models: IBD
A major contribution to our current understanding of the pathogenic mechanisms responsible for the induction and progression of IBD has been the availability of animal models that recapitulate some aspects of the human disease (66, 98). While not ideal, these rat and mouse models (25–30 models total) of intestinal inflammation have yielded considerable insights and served as platforms for hypothesis testing and IBD disease modeling for more than 20 years.
Based on the nature of the inflammation and the method of disease induction, rodent models of intestinal inflammation are grouped into five major categories: 1) chemically or biologically induced, self-limiting models of colitis (TNBS, DSS, C. rodentium, for example), 2) genetically engineered traditional transgenic/knock-out/knock-in mice (i.e. IL-7 transgenic, IL-10 knock-out, mutant gp130 knock-in), 3) disruption of T cell homeostasis by adoptive transfer (CD45RB, CD8 transfer models), 4) congenital, spontaneous mutations (C3H/HeJBir), and 5) spontaneous (cotton-top tamarin) (66, 98, 99). Two of the more common IBD mouse models currently used in many laboratories around the world are the IL-10-deficient (IL-10−/−) mouse model and the T cell transfer model.
Disruption of the IL-10 gene in mice leads to spontaneous pancolitis and cecal inflammation by 2–4 months of age and histopathologically the mouse colon shows many of the same characteristics as those observed in human IBD (66, 100). The advantage of the IL-10−/− mice is that it is a well-established TH1-mediated model of transmural colitis. Alternatively, the T cell transfer model utilizes the adaptive transfer of CD4+CD45RBhigh T cells (naive T cells) from healthy wild-type (WT) mice into syngeneic recipients that lack T and B cells. Following T cell transfer, pancolitis and small bowel inflammation develop at 5–8 weeks (66, 101, 102). The major advantage of the T cell transfer model is that it shows the earliest immunological events associated with induction of gut inflammation and perpetuation of the IBD. Furthermore, data suggests that transfer of naive T cells into recombinase activating gene-1-deficient (RAG−/−) mice will provide a model similar to CD development with induction of colitis and small bowel inflammation (66, 103, 104). Recent evidence also supports that epithelial cell stress and abnormalities can lead to intestinal inflammation in mice following deletion in XBP1 (105), inositol requiring enzyme 1β (IRE1β) (106), caspase-8 (107, 108), anterior gradient 2 (AGR2) (109), the RNA editing enzyme ADAR1(110), or missense mutations in MUC2 (111).
Although these various rodent models lack the complete recapitulation of the clinical and histopathological characteristics of human IBD, three underlying principles and recurring themes have evolved. First, the reason that suggests the T cell-dependent models of IBD are significantly more relevant than the other models, is that chronic gut inflammation is largely mediated by T lymphocytes. Second, this inflammation is initiated and perpetuated by commensal enteric bacteria. And third, the genetic background of the mouse is an important but poorly understood modulator/modifier of disease onset and severity (66, 67, 99).
Human Engineered Tissue Systems: Intestine
The intestinal and colonic mucosa are both complex and dynamic tissues with epithelial cells arranged in crypts and villi in the small intestine and crypts in the colon (112–115). In 2011, several groups independently published descriptions of methods by which human intestinal and colonic stem cells can be maintained and expanded in culture to mimic in vivo microenvironments (57, 89, 116, 117), building upon an older method of culturing mouse small intestine stem cells (90). In this culture method, human intestinal and colonic crypts or purified stem cells are embedded in Matrigel (Figure 8). Over several days they form spherical or elongated oval structures with a crypt-like lumen, referred to as enterospheres (118). Small intestine and colon spheres can expand into multilobulated enteroids and colonoids that mimic the ordered structure of the epithelium complete with crypts containing multipotent columnar base stem cells and Paneth cells. Other intestinal epithelial cell types, including enterocytes, goblet cells, and enteroendocrine cells (57, 118) can be observed in differentiated cell regions away from the stem cell compartment. While the majority of studies conducted with human enteroids and colonoids thus far have focused on stem cell characterization and regenerative medicine, there is immense potential for this culture system in GI research, particularly to model GvHD and IBD. However, they have not been widely adopted as yet to model these and other human disease conditions.
Figure 8.

Engineered human tissue culture system to model intestinal inflammatory-related diseases. A. Human intestinal enteroids grown from stem cell-rich crypts cultured with immune cells in Matrigel and media. B. Phase contrast microscopic imaging of a multilobular human intestinal enteroid in culture. C. Alcian Blue staining of a human intestinal enteroid identifying mucin-producing Goblet cells.
Ideally, it would seem to be then that the next step in the advancement of these micophysiological intestinal culture systems would be the inclusion of additional intestinal cell types such as fibroblasts, smooth muscle cells, and immune cells to better mimic normal and disease environments. The inclusion of immune cells is of particular interest to our laboratory as a way to induce inflammatory responses that model GvHD and IBD diseases (Figure 8). Human peripheral blood mononuclear cells can be easily and safely isolated from human volunteers and patients; these cells directly, or fractionated subpopulations, can be added to the intestinal cultures and treated with cytokines to induce TH1 or TH2 type responses. The effects of these inflammatory responses on intestinal epithelial stem cell viability and proliferation would be of interest, as would the effects of TReg and other suppressor populations on the inflammatory response. Furthermore, tissues and immune cells from individuals with specific genetic polymorphisms could be tested to determine the effect of these polymorphisms on the in vitro induction of a GvHD or IBD inflammatory response. Interestingly, genetic polymorphisms in E-cadherin and hepatocyte nuclear factor 4 α are thought to contribute to UC risk (119) and would be ideal candidate genes to test in vitro for effects on tissue structure and function in the intestine.
In terms of personalized medical applications, growing intestinal epithelial cells from GvHD or IBD patients, and combining them with their own isolated immune cells could serve as a novel microphysiological platform to screen the effectiveness of various therapies for that particular patient. In addition, the inclusion of other microphysiological systems representing the liver, kidneys, and other organ systems onto the platform would also serve to identify potential unexpected toxic and idiosyncratic responses thereby maximizing therapeutic efficacy and minimizing unwanted side-effects associated with treatment and therapy. One could, for instance, examine the role for the cytotoxic cytokine IL-13 in UC pathogenesis, in particular its effect on epithelial apoptosis. Moreover, one could potentially screen for agents to suppress this IL-13 cytotoxicity in patient-derived organoid systems as a novel approach to drug discovery (120).
Future directions: integrating complex biological systems into innovative platforms
The OTC and enteroid culture systems discussed here for modeling gastrointestinal inflammatory conditions have several distinct advantages. First and foremost, they utilize normal, non-transformed human cells grown under physiological conditions. Second, these innovative culture environments allow for the maintenance of a stem cell population, which until recently was not possible for intestinal epithelial cell cultures (57, 89, 90). Third, and of equal importance, the culture conditions are permissive for full cellular and morphologic differentiation, including the formation of the multilayered squamous epithelium of the esophagus, or the development of crypt structures and multilineage daughter cells (enterocytes, enteroendocrine, Paneth, and goblet). Lastly, these culture systems can be easily modified to include other cell types of interest, such as fibroblasts, immune cells, and smooth muscle cells, for example. Therefore the complexities of the actual human tissue can be more completely modeled.
However, these novel culture systems do have two important limitations which will restrict their application to research labs. The complex conditions that yield these biologically relevant models are not easily adapted to a clinical lab for diagnostic purposes. Nor are they suitable for high-throughput approaches favored by industry and the FDA for screening of novel compounds for therapeutic or toxic effects. Moreover, while it is possible to study vectoral nutrient transport, channel and transporter activity, and epithelial barrier function in the enteroids (121, 122), it is more difficult than when cells are cultured on Transwell or similar platforms.
Innovative bioengineered platforms are under development that, if modified, may resolve these limitations. The effort to microfabricate human organs on small culture devices has yielded a number of successful designs(123–127). These devices can mimic peristalsis, incorporate a microbiome, and absorb and metabolize drugs in a physiologically relevant manner. However, all these devices were tested using human colon cancer CaCo-2 cells because of their ease of culture and their ability to differentiate into polarized, columnar epithelial cells with functional tight junctions. Modifications to these platforms, such that they can support intestinal enteroid growth, including the maintenance of a crypt compartment with viable stem cells, would yield a device with relevant biology and excellent functionality.
In conclusion, novel engineered human gastrointestinal tissue systems that recapitulate normal physiology provide an innovative and attractive approach to model inflammatory diseases of the gastrointestinal track including GvHD and IBD. Adapting these research platforms to the new proposed uses has the potential to yield new insights into human disease pathogenesis. It will also likely advance the development of novel therapeutics and provide tools to better personalize medical therapies.
Acknowledgments
We would like to thank the University of Pennsylvania Morphology and Pathology Imaging Core facility for their technical expertise and assistance. Tissue samples were kindly provided by the Cooperative Human Tissue Network, which is funded by the National Cancer Institute. This work is supported as part of the Integrated Microphysiological Systems program and the Intestinal Stem Cell Consortium with funding to JPL (TR 000536 and DK 085551), JY (DK085570), and MGM (DK 085535). This work was also supported by an NCI Program Project PO1 CA098101 (AKR) and the Morphology, Cell Culture, and Molecular Biology Core Facilities of the Center for Molecular Studies in Digestive and Liver Disease at the University of Pennsylvania (P30-DK050306 and PO1 CA098101)(AKR).
List of Abbreviations
- GI
gastrointestinal
- IBD
inflammatory bowel disease
- GERD
gastroesophageal reflux disease
- EOE
esosinophilic esophagitis
- BE
Barrett’s esophagus
- EAC
esophageal adenocarcinoma
- LGD
low-grad dysplasia
- HGD
high-grade dysplasia
- TH
T-helper
- IFN
interferon
- IL
interleukin
- Cox-2
cyclooxygenase-2
- PGE2
prostaglandin E2
- hTERT
human telomerase reverse transcriptase
- 2D
2-dimensional
- OTC
organotypic culture
- 3D
3-dimensional
- ABMT
allogeneic bone marrow transplantation
- GvHD
graft-vs-host disease
- IBD
inflammatory bowel disease
- CD
Crohn’s disease
- UC
ulcerative colitis
- TLR
Toll-like receptor
- IRE1β
inositol requiring enzyme 1β
- AGR2
anterior gradient 2
Footnotes
Competing Interests
The authors declare that they have no competing interests.
Authors’ Contributions
KGH and JDB were responsible for drafting the manuscript, diagram design, and image acquisition. GWF, GGG, and NJ were responsible for image acquisition and for the concept and critical manuscript revision. JY, MGM, and AKR were responsible for the concept and critical manuscript revision. JPL was responsible for the concept and experimental design, critical manuscript revision and final approval. All authors read and approved the final manuscript.
References
- 1.Kappelman MD, Rifas-Shiman SL, Porter CQ, Ollendorf DA, Sandler RS, Galanko JA, Finkelstein JA. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology. 2008 Dec;135(6):1907–1913. doi: 10.1053/j.gastro.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001 Oct;1(1):46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker BM, Chen CS. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci. 2012 Jul 1;125(Pt 13):3015–3024. doi: 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007 Dec 1;121(11):2381–2386. doi: 10.1002/ijc.23192. [DOI] [PubMed] [Google Scholar]
- 5.Falk GW. Barrett’s esophagus. Gastroenterology. 2002 May;122(6):1569–1591. doi: 10.1053/gast.2002.33427. [DOI] [PubMed] [Google Scholar]
- 6.Sharma P, McQuaid K, Dent J, Fennerty MB, Sampliner R, Spechler S, Cameron A, Corley D, Falk G, Goldblum J, Hunter J, Jankowski J, Lundell L, Reid B, Shaheen NJ, Sonnenberg A, Wang K, Weinstein W AGA Chicago Workshop. A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology. 2004 Jul;127(1):310–330. doi: 10.1053/j.gastro.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Hvid-Jensen F, Pedersen L, Drewes AM, Sorensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med. 2011 Oct 13;365(15):1375–1383. doi: 10.1056/NEJMoa1103042. [DOI] [PubMed] [Google Scholar]
- 8.Picardo SL, Maher SG, O’Sullivan JN, Reynolds JV. Barrett’s to oesophageal cancer sequence: a model of inflammatory-driven upper gastrointestinal cancer. Dig Surg. 2012;29(3):251–260. doi: 10.1159/000341498. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fitzgerald RC, Onwuegbusi BA, Bajaj-Elliott M, Saeed IT, Burnham WR, Farthing MJ. Diversity in the oesophageal phenotypic response to gastro-oesophageal reflux: immunological determinants. Gut. 2002 Apr;50(4):451–459. doi: 10.1136/gut.50.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohata Y, Fujiwara Y, Machida H, Okazaki H, Yamagami H, Tanigawa T, Watanabe K, Watanabe T, Tominaga K, Wei M, Wanibuchi H, Arakawa T. Role of Th-2 cytokines in the development of Barrett’s esophagus in rats. J Gastroenterol. 2011 Jul;46(7):883–893. doi: 10.1007/s00535-011-0405-y. [DOI] [PubMed] [Google Scholar]
- 12.Zhong YQ, Lin Y, Xu Z. Expression of IFN-gamma and IL-4 in the esophageal mucosa of patients with reflux esophagitis and Barrett’s esophagus and their relationship with endoscopic and histologic grading. Dig Dis Sci. 2011 Oct;56(10):2865–2870. doi: 10.1007/s10620-011-1696-9. [DOI] [PubMed] [Google Scholar]
- 13.Morris CD, Armstrong GR, Bigley G, Green H, Attwood SE. Cyclooxygenase-2 expression in the Barrett’s metaplasia-dysplasia-adenocarcinoma sequence. Am J Gastroenterol. 2001 Apr;96(4):990–996. doi: 10.1111/j.1572-0241.2001.03599.x. [DOI] [PubMed] [Google Scholar]
- 14.Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol. 2013 Mar;35(2):123–137. doi: 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett MT, Sanchez CA, Galipeau PC, Neshat K, Emond M, Reid BJ. Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progression in Barrett’s esophagus. Oncogene. 1996 Nov 7;13(9):1867–1873. [PubMed] [Google Scholar]
- 16.Hamelin R, Flejou JF, Muzeau F, Potet F, Laurent-Puig P, Fekete F, Thomas G. TP53 gene mutations and p53 protein immunoreactivity in malignant and premalignant Barrett’s esophagus. Gastroenterology. 1994 Oct;107(4):1012–1018. doi: 10.1016/0016-5085(94)90225-9. [DOI] [PubMed] [Google Scholar]
- 17.Katada N, Hinder RA, Smyrk TC, Hirabayashi N, Perdikis G, Lund RJ, Woodward T, Klingler PJ. Apoptosis is inhibited early in the dysplasia-carcinoma sequence of Barrett esophagus. Arch Surg. 1997 Jul;132(7):728–733. doi: 10.1001/archsurg.1997.01430310042007. [DOI] [PubMed] [Google Scholar]
- 18.Arber N, Lightdale C, Rotterdam H, Han KH, Sgambato A, Yap E, Ahsan H, Finegold J, Stevens PD, Green PH, Hibshoosh H, Neugut AI, Holt PR, Weinstein IB. Increased expression of the cyclin D1 gene in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 1996 Jun;5(6):457–459. [PubMed] [Google Scholar]
- 19.Hanas JS, Lerner MR, Lightfoot SA, Raczkowski C, Kastens DJ, Brackett DJ, Postier RG. Expression of the cyclin-dependent kinase inhibitor p21(WAF1/CIP1) and p53 tumor suppressor in dysplastic progression and adenocarcinoma in Barrett esophagus. Cancer. 1999 Sep 1;86(5):756–763. doi: 10.1002/(sici)1097-0142(19990901)86:5<756::aid-cncr9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 20.Lord RV, O’Grady R, Sheehan C, Field AF, Ward RL. K-ras codon 12 mutations in Barrett’s oesophagus and adenocarcinomas of the oesophagus and oesophagogastric junction. J Gastroenterol Hepatol. 2000 Jul;15(7):730–736. doi: 10.1046/j.1440-1746.2000.02163.x. [DOI] [PubMed] [Google Scholar]
- 21.Fitzgerald RC, Omary MB, Triadafilopoulos G. Acid modulation of HT29 cell growth and differentiation. An in vitro model for Barrett’s esophagus. J Cell Sci. 1997 Mar;110(Pt 5):663–671. doi: 10.1242/jcs.110.5.663. [DOI] [PubMed] [Google Scholar]
- 22.Ouatu-Lascar R, Fitzgerald RC, Triadafilopoulos G. Differentiation and proliferation in Barrett’s esophagus and the effects of acid suppression. Gastroenterology. 1999 Aug;117(2):327–335. doi: 10.1053/gast.1999.0029900327. [DOI] [PubMed] [Google Scholar]
- 23.Souza RF, Shewmake K, Terada LS, Spechler SJ. Acid exposure activates the mitogen-activated protein kinase pathways in Barrett’s esophagus. Gastroenterology. 2002 Feb;122(2):299–307. doi: 10.1053/gast.2002.30993. [DOI] [PubMed] [Google Scholar]
- 24.Marchetti M, Caliot E, Pringault E. Chronic acid exposure leads to activation of the cdx2 intestinal homeobox gene in a long-term culture of mouse esophageal keratinocytes. J Cell Sci. 2003 Apr 15;116(Pt 8):1429–1436. doi: 10.1242/jcs.00338. [DOI] [PubMed] [Google Scholar]
- 25.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, von Werder A, Enders GH, Opitz OG, Rustgi AK. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res. 2003 Aug;1(10):729–738. [PubMed] [Google Scholar]
- 26.Morales CP, Gandia KG, Ramirez RD, Wright WE, Shay JW, Spechler SJ. Characterisation of telomerase immortalised normal human oesophageal squamous cells. Gut. 2003 Mar;52(3):327–333. doi: 10.1136/gut.52.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palanca-Wessels MC, Klingelhutz A, Reid BJ, Norwood TH, Opheim KE, Paulson TG, Feng Z, Rabinovitch PS. Extended lifespan of Barrett’s esophagus epithelium transduced with the human telomerase catalytic subunit: a useful in vitro model. Carcinogenesis. 2003 Jul;24(7):1183–1190. doi: 10.1093/carcin/bgg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaiswal KR, Morales CP, Feagins LA, Gandia KG, Zhang X, Zhang HY, Hormi-Carver K, Shen Y, Elder F, Ramirez RD, Sarosi GA, Jr, Spechler SJ, Souza RF. Characterization of telomerase-immortalized, non-neoplastic, human Barrett’s cell line (BAR-T) Dis Esophagus. 2007;20(3):256–264. doi: 10.1111/j.1442-2050.2007.00683.x. [DOI] [PubMed] [Google Scholar]
- 29.Kazumori H, Ishihara S, Rumi MA, Kadowaki Y, Kinoshita Y. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in Barrett’s epithelium. Gut. 2006 Jan;55(1):16–25. doi: 10.1136/gut.2005.066209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milano F, van Baal JW, Buttar NS, Rygiel AM, de Kort F, DeMars CJ, Rosmolen WD, Bergman JJ, Van Marle J, Wang KK, Peppelenbosch MP, Krishnadath KK. Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology. 2007 Jun;132(7):2412–2421. doi: 10.1053/j.gastro.2007.03.026. [DOI] [PubMed] [Google Scholar]
- 31.Chang CL, Lao-Sirieix P, Save V, De La Cueva Mendez G, Laskey R, Fitzgerald RC. Retinoic acid-induced glandular differentiation of the oesophagus. Gut. 2007 Jul;56(7):906–917. doi: 10.1136/gut.2006.097915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooke G, Blanco-Fernandez A, Seery JP. The effect of retinoic acid and deoxycholic acid on the differentiation of primary human esophageal keratinocytes. Dig Dis Sci. 2008 Nov;53(11):2851–2857. doi: 10.1007/s10620-008-0240-z. [DOI] [PubMed] [Google Scholar]
- 33.Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, Lynch JP, Rustgi AK. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett’s esophagus. PLoS One. 2008;3(10):e3534. doi: 10.1371/journal.pone.0003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong J, Nakagawa H, Isariyawongse BK, Funakoshi S, Silberg DG, Rustgi AK, Lynch JP. Induction of intestinalization in human esophageal keratinocytes is a multistep process. Carcinogenesis. 2009 Jan;30(1):122–130. doi: 10.1093/carcin/bgn227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu T, Zhang X, So CK, Wang S, Wang P, Yan L, Myers R, Chen Z, Patterson AP, Yang CS, Chen X. Regulation of Cdx2 expression by promoter methylation, and effects of Cdx2 transfection on morphology and gene expression of human esophageal epithelial cells. Carcinogenesis. 2007 Feb;28(2):488–496. doi: 10.1093/carcin/bgl176. [DOI] [PubMed] [Google Scholar]
- 36.Nicholson AM, Graham TA, Simpson A, Humphries A, Burch N, Rodriguez-Justo M, Novelli M, Harrison R, Wright NA, McDonald SA, Jankowski JA. Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut. 2012 Oct;61(10):1380–1389. doi: 10.1136/gutjnl-2011-301174. [DOI] [PubMed] [Google Scholar]
- 37.Fein M, Peters JH, Chandrasoma P, Ireland AP, Oberg S, Ritter MP, Bremner CG, Hagen JA, DeMeester TR. Duodenoesophageal reflux induces esophageal adenocarcinoma without exogenous carcinogen. J Gastrointest Surg. 1998 May-Jun;2(3):260–268. doi: 10.1016/s1091-255x(98)80021-8. [DOI] [PubMed] [Google Scholar]
- 38.Kong J, Crissey MA, Funakoshi S, Kreindler JL, Lynch JP. Ectopic Cdx2 expression in murine esophagus models an intermediate stage in the emergence of Barrett’s esophagus. PLoS One. 2011 Apr 6;6(4):e18280. doi: 10.1371/journal.pone.0018280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suh E, Chen L, Taylor J, Traber PG. A homeodomain protein related to caudal regulates intestine-specific gene transcription. Molecular & Cellular Biology. 1994;14(11):7340–51. doi: 10.1128/mcb.14.11.7340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Troelsen JT, Mitchelmore C, Spodsberg N, Jensen AM, Noren O, Sjostrom H. Regulation of lactase-phlorizin hydrolase gene expression by the caudal-related homoeodomain protein Cdx-2. Biochemical Journal. 1997;322(Pt 3):833–8. doi: 10.1042/bj3220833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao N, White P, Kaestner KH. Establishment of Intestinal Identity and Epithelial-Mesenchymal Signaling by Cdx2. Dev Cell. 2009;16(4):588–99. doi: 10.1016/j.devcel.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo R, Funakoshi S, Lee HH, Kong J, Lynch JP. The intestine-specific transcription factor Cdx2 inhibits beta-catenin/TCF transcriptional activity by disrupting the beta-catenin/ TCF protein complex. Carcinogenesis. 2010;31(2):159–66. doi: 10.1093/carcin/bgp213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keller MS, Ezaki T, Guo RJ, Lynch JP. Cdx1 or Cdx2 Expression Activates E-Cadherin-mediated Cell-cell Adhesion and Compaction in Human Colo 205 cells. Am J Physiol Gastrointest Liver Physiol. 2004;287(1):G104–14. doi: 10.1152/ajpgi.00484.2003. [DOI] [PubMed] [Google Scholar]
- 44.Guo RJ, Suh ER, Lynch JP. The Role of Cdx Proteins in Intestinal Development and Cancer. Cancer Biol Ther. 2004;3(7):593–601. doi: 10.4161/cbt.3.7.913. [DOI] [PubMed] [Google Scholar]
- 45.Guo RJ, Huang E, Ezaki T, patel N, Sinclair K, Wu J, et al. Cdx1 inhibits human colon cancer cell proliferation by reducing b-catenin/TCF transcriptional activity. J Biol Chem. 2004;279(35):36865–75. doi: 10.1074/jbc.M405213200. [DOI] [PubMed] [Google Scholar]
- 46.Funakoshi S, Ezaki T, Kong J, Guo RJ, Lynch JP. Repression of the Desmocollin 2 gene in colorectal cancer cells is relieved by the homeodomain transcription factors Cdx1 and Cdx2. Molecular Cancer Research. 2008;6(9):1478–90. doi: 10.1158/1541-7786.MCR-07-2161. [DOI] [PubMed] [Google Scholar]
- 47.Funakoshi S, Kong J, Crissey MA, Dang L, Dang D, Lynch JP. Cdx2 promotes E-cadherin function and cell-cell adhesion in colon cancer cells by enhancing E-cadherin trafficking to the cell membrane. Am J Physiol Gastrointest Liver Physiol. 2010 doi: 10.1152/ajpgi.00297.2010. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phillips RW, Frierson HF, Jr, Moskaluk CA. Cdx2 as a marker of epithelial intestinal differentiation in the esophagus. Am J Surg Pathol. 2003;27(11):1442–7. doi: 10.1097/00000478-200311000-00006. [DOI] [PubMed] [Google Scholar]
- 49.Groisman GM, Amar M, Meir A. Expression of the intestinal marker Cdx2 in the columnar-lined esophagus with and without intestinal (Barrett’s) metaplasia. Mod Pathol. 2004;17(10):1282–8. doi: 10.1038/modpathol.3800182. [DOI] [PubMed] [Google Scholar]
- 50.Colleypriest BJ, Palmer RM, Ward SG, Tosh D. Cdx genes, inflammation and the pathogenesis of Barrett’s metaplasia. Trends Mol Med. 2009;15(7):313–22. doi: 10.1016/j.molmed.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Quante M, Bhagat G, Abrams JA, Marache F, Good P, Lee MD, Lee Y, Friedman R, Asfaha S, Dubeykovskaya Z, Mahmood U, Figueiredo JL, Kitajewski J, Shawber C, Lightdale CJ, Rustgi AK, Wang TC. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012 Jan 17;21(1):36–51. doi: 10.1016/j.ccr.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakagawa H, Wang TC, Zukerberg L, Odze R, Togawa K, May GH, Wilson J, Rustgi AK. The targeting of the cyclin D1 oncogene by an Epstein-Barr virus promoter in transgenic mice causes dysplasia in the tongue, esophagus and forestomach. Oncogene. 1997 Mar 13;14(10):1185–1190. doi: 10.1038/sj.onc.1200937. [DOI] [PubMed] [Google Scholar]
- 53.Kalabis J, Patterson MJ, Enders GH, Marian B, Iozzo RV, Rogler G, Gimotty PA, Herlyn M. Stimulation of human colonic epithelial cells by leukemia inhibitory factor is dependent on collagen-embedded fibroblasts in organotypic culture. FASEB J. 2003 Jun;17(9):1115–1117. doi: 10.1096/fj.02-0852fje. [DOI] [PubMed] [Google Scholar]
- 54.Kalabis J, Wong GS, Vega ME, Natsuizaka M, Robertson ES, Herlyn M, Nakagawa H, Rustgi AK. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc. 2012 Jan 12;7(2):235–246. doi: 10.1038/nprot.2011.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kosoff RE, Gardiner KL, Merlo LM, Pavlov K, Rustgi AK, Maley CC. Development and characterization of an organotypic model of Barrett’s esophagus. J Cell Physiol. 2012 Jun;227(6):2654–2659. doi: 10.1002/jcp.23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kong J, Crissey MA, Stairs DB, Sepulveda AR, Lynch JP. Cox2 and beta-catenin/T-cell factor signaling intestinalize human esophageal keratinocytes when cultured under organotypic conditions. Neoplasia. 2011 Sep;13(9):792–805. doi: 10.1593/neo.11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011 Nov;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 58.Pasquini MC, Wang Z, Horowitz MM, Gale RP. 2010 report from the Center for International Blood and Marrow Transplant Research (CIBMTR): current uses and outcomes of hematopoietic cell transplants for blood and bone marrow disorders. Clin Transpl. 2010:87–105. [PubMed] [Google Scholar]
- 59.Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012 Oct 7;18(37):5240–5248. doi: 10.3748/wjg.v18.i37.5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012 May 11;12(6):443–458. doi: 10.1038/nri3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jacobsohn DA, Vogelsang GB. Acute graft versus host disease. Orphanet J Rare Dis. 2007 Sep 4;2:35. doi: 10.1186/1750-1172-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ponec RJ, Hackman RC, McDonald GB. Endoscopic and histologic diagnosis of intestinal graft-versus-host disease after marrow transplantation. Gastrointest Endosc. 1999;49(5):612–21. doi: 10.1016/s0016-5107(99)70390-1. [DOI] [PubMed] [Google Scholar]
- 63.Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J Clin Invest. 2000 May;105(9):1289–1298. doi: 10.1172/JCI7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood. 2009 Feb 5;113(6):1365–1374. doi: 10.1182/blood-2008-06-162420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee SJ, Flowers ME. Recognizing and managing chronic graft-versus-host disease. Hematology Am Soc Hematol Educ Program. 2008:134–141. doi: 10.1182/asheducation-2008.1.134. [DOI] [PubMed] [Google Scholar]
- 66.Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, Price VH, Grisham MB. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol. 2009 Feb;296(2):G135–46. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006 Jul;3(7):390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 68.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007 Mar;117(3):514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004 May;126(6):1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 70.Hisamatsu T, Kanai T, Mikami Y, Yoneno K, Matsuoka K, Hibi T. Immune aspects of the pathogenesis of inflammatory bowel disease. Pharmacol Ther. 2013 Mar;137(3):283–297. doi: 10.1016/j.pharmthera.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 71.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ NIDDK IBD Genetics Consortium, Belgian-French IBD Consortium, Wellcome Trust Case Control Consortium. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008 Aug;40(8):955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, Drummond H, Lees CW, Khawaja SA, Bagnall R, Burke DA, Todhunter CE, Ahmad T, Onnie CM, McArdle W, Strachan D, Bethel G, Bryan C, Lewis CM, Deloukas P, Forbes A, Sanderson J, Jewell DP, Satsangi J, Mansfield JC, Cardon L, Mathew CG Wellcome Trust Case Control Consortium. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007 Jul;39(7):830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, Neale BM, Ong RT, Lagace C, Li C, Green T, Stevens CR, Beauchamp C, Fleshner PR, Carlson M, D’Amato M, Halfvarson J, Hibberd ML, Lordal M, Padyukov L, Andriulli A, Colombo E, Latiano A, Palmieri O, Bernard EJ, Deslandres C, Hommes DW, de Jong DJ, Stokkers PC, Weersma RK, Sharma Y, Silverberg MS, Cho JH, Wu J, Roeder K, Brant SR, Schumm LP, Duerr RH, Dubinsky MC, Glazer NL, Haritunians T, Ippoliti A, Melmed GY, Siscovick DS, Vasiliauskas EA, Targan SR, Annese V, Wijmenga C, Pettersson S, Rotter JI, Xavier RJ, Daly MJ, Rioux JD, Seielstad M NIDDK IBD Genetics Consortium. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010 Apr;42(4):332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, Tremblay ML, Russell SM, Godfrey DI, Tiganis T. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest. 2011 Dec;121(12):4758–4774. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McKay DM, Philpott DJ, Perdue MH. Review article: In vitro models in inflammatory bowel disease research--a critical review. Aliment Pharmacol Ther. 1997 Dec;11(Suppl 3):70–80. doi: 10.1111/j.1365-2036.1997.tb00811.x. [DOI] [PubMed] [Google Scholar]
- 76.Amanzada A, Malik IA, Blaschke M, Khan S, Rahman H, Ramadori G, Moriconi F. Identification of CD68(+) neutrophil granulocytes in in vitro model of acute inflammation and inflammatory bowel disease. Int J Clin Exp Pathol. 2013;6(4):561–570. [PMC free article] [PubMed] [Google Scholar]
- 77.Bataille F, Klebl F, Rummele P, Schroeder J, Farkas S, Wild PJ, Furst A, Hofstadter F, Scholmerich J, Herfarth H, Rogler G. Morphological characterisation of Crohn’s disease fistulae. Gut. 2004 Sep;53(9):1314–1321. doi: 10.1136/gut.2003.038208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perminow G, Reikvam DH, Lyckander LG, Brandtzaeg P, Vatn MH, Carlsen HS. Increased number and activation of colonic macrophages in pediatric patients with untreated Crohn’s disease. Inflamm Bowel Dis. 2009 Sep;15(9):1368–1378. doi: 10.1002/ibd.20916. [DOI] [PubMed] [Google Scholar]
- 79.Monteleone I, Pallone F, Monteleone G. Th17-related cytokines: new players in the control of chronic intestinal inflammation. BMC Med. 2011 Nov 15;9 doi: 10.1186/1741-7015-9-122. 122-7015-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Phillips TE, Huet C, Bilbo PR, Podolsky DK, Louvard D, Neutra MR. Human intestinal goblet cells in monolayer culture: characterization of a mucus-secreting subclone derived from the HT29 colon adenocarcinoma cell line. Gastroenterology. 1988;94(6):1390–403. doi: 10.1016/0016-5085(88)90678-6. [DOI] [PubMed] [Google Scholar]
- 81.Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst. 1977;59(1):221–6. doi: 10.1093/jnci/59.1.221. [DOI] [PubMed] [Google Scholar]
- 82.Zweibaum A, Pinto M, Chevalier G, Dussaulx E, Triadou N, Lacroix B, et al. Enterocytic differentiation of a subpopulation of the human colon tumor cell line HT-29 selected for growth in sugar-free medium and its inhibition by glucose. J Cell Physiol. 1985;122(1):21–9. doi: 10.1002/jcp.1041220105. [DOI] [PubMed] [Google Scholar]
- 83.Zweibaum A, Triadou N, Kedinger M, Augeron C, Robine-Leon S, Pinto M, et al. Sucrase-isomaltase: a marker of foetal and malignant epithelial cells of the human colon. Int J Cancer. 1983;32(4):407–12. doi: 10.1002/ijc.2910320403. [DOI] [PubMed] [Google Scholar]
- 84.Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80(2):248–65. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brubaker PL, Drucker DJ, Greenberg GR. Synthesis and secretion of somatostatin-28 and -14 by fetal rat intestinal cells in culture. Am J Physiol. 1990;258(6 Pt 1):G974–81. doi: 10.1152/ajpgi.1990.258.6.G974. [DOI] [PubMed] [Google Scholar]
- 86.Roig AI, Eskiocak U, Hight SK, Kim SB, Delgado O, Souza RF, et al. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology. 2010;138(3):1012–21. e1–5. doi: 10.1053/j.gastro.2009.11.052. [DOI] [PubMed] [Google Scholar]
- 87.Leonard F, Collnot EM, Lehr CM. A three-dimensional coculture of enterocytes, monocytes and dendritic cells to model inflamed intestinal mucosa in vitro. Mol Pharm. 2010 Dec 6;7(6):2103–2119. doi: 10.1021/mp1000795. [DOI] [PubMed] [Google Scholar]
- 88.Leonard F, Ali H, Collnot EM, Crielaard BJ, Lammers T, Storm G, Lehr CM. Screening of budesonide nanoformulations for treatment of inflammatory bowel disease in an inflamed 3D cell-culture model. ALTEX. 2012;29(3):275–285. doi: 10.14573/altex.2012.3.275. [DOI] [PubMed] [Google Scholar]
- 89.Jung P, Sato T, Merlos-Suarez A, Barriga FM, Iglesias M, Rossell D, et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 2011;17(10):1225–7. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 90.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 91.Hill GR, Ferrara JL. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood. 2000 May 1;95(9):2754–2759. [PubMed] [Google Scholar]
- 92.Heimesaat MM, Nogai A, Bereswill S, Plickert R, Fischer A, Loddenkemper C, Steinhoff U, Tchaptchet S, Thiel E, Freudenberg MA, Gobel UB, Uharek L. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut. 2010 Aug;59(8):1079–1087. doi: 10.1136/gut.2009.197434. [DOI] [PubMed] [Google Scholar]
- 93.Imado T, Iwasaki T, Kitano S, Satake A, Kuroiwa T, Tsunemi S, Sano H. The protective role of host Toll-like receptor-4 in acute graft-versus-host disease. Transplantation. 2010 Nov 27;90(10):1063–1070. doi: 10.1097/TP.0b013e3181f86947. [DOI] [PubMed] [Google Scholar]
- 94.Tawara I, Maeda Y, Sun Y, Lowler KP, Liu C, Toubai T, McKenzie AN, Reddy P. Combined Th2 cytokine deficiency in donor T cells aggravates experimental acute graft-vs-host disease. Exp Hematol. 2008 Aug;36(8):988–996. doi: 10.1016/j.exphem.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yi T, Zhao D, Lin CL, Zhang C, Chen Y, Todorov I, LeBon T, Kandeel F, Forman S, Zeng D. Absence of donor Th17 leads to augmented Th1 differentiation and exacerbated acute graft-versus-host disease. Blood. 2008 Sep 1;112(5):2101–2110. doi: 10.1182/blood-2007-12-126987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fanning SL, Appel MY, Berger SA, Korngold R, Friedman TM. The immunological impact of genetic drift in the B10.BR congenic inbred mouse strain. J Immunol. 2009 Oct 1;183(7):4261–4272. doi: 10.4049/jimmunol.0900971. [DOI] [PubMed] [Google Scholar]
- 97.Ting C, Alterovitz G, Merlob A, Abdi R. Genomic studies of GVHD-lessons learned thus far. Bone Marrow Transplant. 2013 Jan;48(1):4–9. doi: 10.1038/bmt.2012.9. [DOI] [PubMed] [Google Scholar]
- 98.Mizoguchi A. Animal models of inflammatory bowel disease. Prog Mol Biol Transl Sci. 2012;105:263–320. doi: 10.1016/B978-0-12-394596-9.00009-3. [DOI] [PubMed] [Google Scholar]
- 99.Elson CO, Cong Y, McCracken VJ, Dimmitt RA, Lorenz RG, Weaver CT. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol Rev. 2005 Aug;206:260–276. doi: 10.1111/j.0105-2896.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- 100.Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996 Aug 15;98(4):1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Powrie F. T cells in inflammatory bowel disease: protective and pathogenic roles. Immunity. 1995 Aug;3(2):171–174. doi: 10.1016/1074-7613(95)90086-1. [DOI] [PubMed] [Google Scholar]
- 102.Powrie F. Immune regulation in the intestine: a balancing act between effector and regulatory T cell responses. Ann N Y Acad Sci. 2004 Dec;1029:132–141. doi: 10.1196/annals.1309.030. [DOI] [PubMed] [Google Scholar]
- 103.Ostanin DV, Pavlick KP, Bharwani S, D’Souza D, Furr KL, Brown CM, Grisham MB. T cell-induced inflammation of the small and large intestine in immunodeficient mice. Am J Physiol Gastrointest Liver Physiol. 2006 Jan;290(1):G109–19. doi: 10.1152/ajpgi.00214.2005. [DOI] [PubMed] [Google Scholar]
- 104.Laroux FS, Norris HH, Houghton J, Pavlick KP, Bharwani S, Merrill DM, Fuseler J, Chervenak R, Grisham MB. Regulation of chronic colitis in athymic nu/nu (nude) mice. Int Immunol. 2004 Jan;16(1):77–89. doi: 10.1093/intimm/dxh006. [DOI] [PubMed] [Google Scholar]
- 105.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008 Sep 5;134(5):743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB, Ron D. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 2001 Mar;107(5):585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011 Sep 14;477(7364):335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011 Jul 31;477(7364):330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 109.Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, Ouellette AJ, Andersen B, Lipkin SM. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2−/− mice. Dev Biol. 2010 Feb 15;338(2):270–279. doi: 10.1016/j.ydbio.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Qiu W, Wang X, Buchanan M, He K, Sharma R, Zhang L, Wang Q, Yu J. ADAR1 is essential for intestinal homeostasis and stem cell maintenance. Cell Death Dis. 2013 Apr 18;4:e599. doi: 10.1038/cddis.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, Adams R, Kato M, Nelms KA, Hong NA, Florin TH, Goodnow CC, McGuckin MA. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008 Mar 4;5(3):e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer. 2008 Jun;8(6):415–424. doi: 10.1038/nrc2392. [DOI] [PubMed] [Google Scholar]
- 113.Kuratnik A, Giardina C. Intestinal organoids as tissue surrogates for toxicological and pharmacological studies. Biochem Pharmacol. 2013 Jun 15;85(12):1721–1726. doi: 10.1016/j.bcp.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 114.Miyamoto S, Rosenberg DW. Role of Notch signaling in colon homeostasis and carcinogenesis. Cancer Sci. 2011 Nov;102(11):1938–1942. doi: 10.1111/j.1349-7006.2011.02049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Simons BD, Clevers H. Stem cell self-renewal in intestinal crypt. Exp Cell Res. 2011 Nov 15;317(19):2719–2724. doi: 10.1016/j.yexcr.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 116.Lahar N, Lei NY, Wang J, Jabaji Z, Tung SC, Joshi V, Lewis M, Stelzner M, Martin MG, Dunn JC. Intestinal subepithelial myofibroblasts support in vitro and in vivo growth of human small intestinal epithelium. PLoS One. 2011;6(11):e26898. doi: 10.1371/journal.pone.0026898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Shroyer NF, Wells JM. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011 Feb 3;470(7332):105–109. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stelzner M, Helmrath M, Dunn JC, Henning SJ, Houchen CW, Kuo C, Lynch J, Li L, Magness ST, Martin MG, Wong MH, Yu J NIH Intestinal Stem Cell Consortium. A nomenclature for intestinal in vitro cultures. Am J Physiol Gastrointest Liver Physiol. 2012 Jun 15;302(12):G1359–63. doi: 10.1152/ajpgi.00493.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van Sommeren S, Visschedijk MC, Festen EA, de Jong DJ, Ponsioen CY, Wijmenga C, Weersma RK. HNF4alpha and CDH1 are associated with ulcerative colitis in a Dutch cohort. Inflamm Bowel Dis. 2011 Aug;17(8):1714–1718. doi: 10.1002/ibd.21541. [DOI] [PubMed] [Google Scholar]
- 120.Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol. 2013 Jan 24;8:477–512. doi: 10.1146/annurev-pathol-011110-130318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu J, Walker NM, Cook MT, Ootani A, Clarke LL. Functional Cftr in crypt epithelium of organotypic enteroid cultures from murine small intestine. Am J Physiol Cell Physiol. 2012;302(10):C1492–503. doi: 10.1152/ajpcell.00392.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kovbasnjuk O, Zachos NC, et al. Human enteroids: preclinical models of non-inflammatory diarrhea. Stem Cell Research & Therapy. 2013;4(Suppl 1) doi: 10.1186/scrt364. (S3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huh D, Kim HJ, Fraser JP, Shea DE, Khan M, Bahinski A, et al. Microfabrication of human organs-on-chips. Nat Protoc. 2013;8(11):2135–57. doi: 10.1038/nprot.2013.137. [DOI] [PubMed] [Google Scholar]
- 124.Kim HJ, Huh D, Hamilton G, Ingber DE. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip. 2012;12(12):2165–74. doi: 10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- 125.Mahler GJ, Esch MB, Glahn RP, Shuler ML. Characterization of a gastrointestinal tract microscale cell culture analog used to predict drug toxicity. Biotechnol Bioeng. 2009;104(1):193–205. doi: 10.1002/bit.22366. [DOI] [PubMed] [Google Scholar]
- 126.Kimura H, Yamamoto T, Sakai H, Sakai Y, Fujii T. An integrated microfluidic system for long-term perfusion culture and on-line monitoring of intestinal tissue models. Lab Chip. 2008;8(5):741–6. doi: 10.1039/b717091b. [DOI] [PubMed] [Google Scholar]
- 127.Imura Y, Asano Y, Sato K, Yoshimura E. A microfluidic system to evaluate intestinal absorption. Anal Sci. 2009;25(12):1403–7. doi: 10.2116/analsci.25.1403. [DOI] [PubMed] [Google Scholar]