

Abstract
Cancer is one of the leading causes of morbidity and mortality around the world. Despite some success, traditional anti-cancer drugs developed to reduce tumor growth face important limitations primarily due to undesirable bone marrow and cardiovascular toxicity. Many drugs showing promise in preclinical trials fail during clinical development, suggesting that the available in vitro and animal models are poor predictors of drug efficacy and toxicity in humans. Hence, there exists a great need for developing novel platforms that more accurately mimic the biology of human organs, and thus provide a reliable model for high-throughput drug screening. 3D microphysiological systems can utilize induced pluripotent stem (iPS) cell technology, tissue engineering, and microfabrication techniques to develop tissue models of human tumors, cardiac muscle, and bone marrow on the order of 1 mm3 in size. A functional network of human capillaries and microvessels to overcome diffusion limitations in nutrient delivery and waste removal can also nourish the 3D microphysiological tissues. Importantly, the 3D microphysiological tissues are grown on optically clear platforms that offer non-invasive and non-destructive image acquisition with sub-cellular resolution in real time. Such systems offer a new paradigm for high-throughput drug screening, and will significantly improve the efficiency of identifying new drugs for cancer treatment, that minimize cardiac and bone marrow toxicity.
I. Introduction
Current drug screening methods usually rely on two dimensional (2D) systems or animal models for assessment of toxicity, pharmacokinetics, pharmacodynamics, and organ system effects. However, both models have weaknesses. To more accurately simulate the in vivo physiologic human response to pharmacologic challenge, it is highly desirable to replicate the complex three dimensional (3D) arrangements of human cells, including, preferably, multiple organ systems and a vascular supply. The vasculature not only provides the necessary convective transport of nutrients and waste in 3D culture, it also couples and integrates the response of the multiple organ systems. Additionally, most drugs are delivered to the target tissue through the microcirculation, and thus incorporation of a vasculature best mimics in vivo drug delivery.
Drug delivery to a target tissue depends on the function of other organs. To achieve the desired effect of a selected drug on a given tissue, the presence of multiple organ systems may be required. In chemotherapy, for example, the gastrointestinal, circulatory, and urinary systems each contribute to determine the pharmacokinetics of a given drug. If a drug possesses useful activity, it will be further studied for possible adverse effects on major organs. While adverse effects on the multiple organ systems throughout the body are important, current anticancer therapies are mostly limited by their undesirable side effects on the immune system, the cardiovascular system, and the liver. First-pass drug metabolism in the liver prior to entry into the vascular system can markedly influence the toxicity of a wide range of drugs. However, in cancer treatment it is well established that the majority of anti-proliferative agents used in traditional chemotherapy can cause myelosuppression in a dose-dependent manner (e.g., alkylating agents, pyrimidine analogs, anthracyclines, methotrexate, etc.) [1]. The use of hematopoietic growth factors has significantly improved the primary acute myelosuppression observed during anticancer treatments [2]; however, in some patients these growth factors can also mask development of a latent residual bone marrow injury manifested by a decrease in HSC reserves and an impairment in HSC self-renewal [3]. Regarding the cardiovascular system, systemic anticancer therapy can lead to hypertension, thromboembolic events, left ventricular dysfunction, myocardial ischemia, arrhythmias, and pericarditis [4-6]. In this regard, two types of cardiotoxicity have been well established according to the type of damage in the cardiomyocyte: type I-induced cardiotoxicity (for example, induced by anthracycline), which is dose-dependent and associated with myocyte death; and, type II-related cardiotoxicity (for example, induced by trastuzumab), which is less predictably associated with dose, and typically correlated with reversible myocardial dysfunction rather than histological changes or myocyte death. However, with the increase of a wide range of new anti-cancer agents used in molecularly-targeted therapy, an unintended cardiotoxicity, recently classified as “off-target”, has arisen from many of these targeted compounds. This new class of drugs has increased the risk and need for assessment of chemoteraphy-associated cardiotoxicity [1-3, 6].
To address the need for improved pre-clinical drug toxicity models, we are developing a system for high throughput screening of both drug efficacy and organ specific toxicity. Our system features 3D tissues made entirely of human cells. The tissues are connected and perfused by human microvessels. Initial designs incorporate tumor, cardiac, and bone marrow tissue modules that allow assessment of anticancer drug efficacy as well as potential side effects.
II. Pharmacology
Once a new drug target has been identified, a sequence of studies is initiated to characterize the dose-response relationship prior to clinical trials. A variety of assays at the molecular, cellular, organ, and systemic levels are needed to define the pharmacokinetics and pharmacodynamics of the drug. Pharmacokinetics is the study of changes in drug concentration with time due to absorption, distribution, metabolism, and elimination of the drug. In contrast, pharmacodynamics is the study of the drug concentration-dependent tissue or cell response. The target tissue is normally represented by a dose-response curve. The EC50 of a drug is an index of its sensitivity or potency and refers to the concentration required to produce 50% of that drug’s maximal effect (e.g., tumor reduction or anti-proliferative effect in response to an anticancer agent). The maximal efficacy of a drug represents the upper limit of the dose-response relation on the response axis.
In cancer treatment, the same total drug dose can be delivered over different lengths of time (i.e., different dose intensities), which can impact the effect of the drug on other organ systems. The goal of chemotherapeutics is to achieve a desired effect with minimal adverse side effects; therefore, when designing a microphysiological system for drug screening, it is important to recognize that many anti-cancer drugs are limited by their adverse effect on a number of non-cancerous tissues throughout the body. As further introduced below, the proposed system recognizes that the response of cells and tissues to a pharmacologic challenge is greatly influenced by the microenvironment. Table 1 categorizes a wide range of important microenvironmental factors that are present in the 3D architecture of the tissue that influence tissue response to a drug.
Table I.
Drug testing of tumor cells cultured in 3D
| I. 2D vs 3D culture systems - Examples of observed differences in drug response | ||
|---|---|---|
| Ia. Cancer cells cultured in 3D are more resistant to anticancer drugs compared with 2D | ||
| Tumor cell line | Outcome of drug treatment | Ref. |
| HepG2 human liver hepatocellular carcinoma cells | Responses of cells to cisplatin, 5-fluorouracil and doxorubicin (anti-cancer drugs) showed higher EC50 values in 3D multicellular spheroids compared with 2D monolayer. The 3D model conferred differentiated phenotypes including increased cell-cell adhesion, G1 phase cell cycle arrest, and enhanced cellular resistance to apoptosis | [98] |
| SA87 (brain metastases), NCI-H460 and H460M (lung) lines from primary tumors and metastases | Responses of cells to increasing concentrations of anti-cancer drugs such as doxorubicin and 5-fluorouracil showed that 3D models conferred more resistance to the drugs than the usual monolayer culture system. The anti-cancer drugs were efficient after 24h of treatment in the monolayer cultures, whereas they were significantly efficient after only one week of incubation in the 3D systems | [99] |
| TE2 and TTn human esophageal squamous carcinoma lines | Responses of cells to anti-cancer drugs such as cisplatin and doxorubicin showed higher EC50 values in 3D-cultured cells compared with 2D demonstrating a higher increase drug resistance in 3D models | [100] |
| MCF-7 breast cancer cells | The antiproliferative effect of different anti-cancer agents including doxorubicin, paclitaxel and tamoxifen in 3D models was significantly lower than in 2D monolayer by a 12- to 23-fold difference in their EC50 values. The greater synthesis of collagen in 3D suggested that the ECM can act as a barrier to drug diffusion | [18] |
| Ib. Cancer cells cultured in 3D makes them resistant or desensitizes the cancer depending on the cell and/or drug type | ||
| Tumor cell line | Outcome of drug treatment | Ref. |
| C4-2B bone metastatic prostate cancer cells | Responses of cells to camptothecin, docetaxel, and rapamycin (anti-cancer drugs), alone and in combination, differed between the 3D and 2D monolayer systems | [101] |
| HT29, SW620, SW1116 (colon) UM-SCC-22B (head and neck) and A2780 (ovary) solid tumor cell lines | Responses of cells to anti-cancer drugs such as cisplatin and miltefosine showed that multilayers are more resistant to the drugs than the corresponding monolayers. However, there are substantial differences between the drugs depending on culture conditions | [102] |
| A549 and H358 lung cancer cell lines | Potency and efficacy from a set of 10 anticancer drugs including paclitaxel, doxorubicin, vinorelbine, gemcitabine or cisplatin showed significant differences when tested in 3D cultured cells comparing with traditional 2D cultures. The activity of these drugs varied with individual drugs and the cell line used for testing | [103] |
| Ic. Anti-cancer drugs demonstrate different effectiveness in 3D compared with 2D. ECM involvement and selective pathway interaction differences | ||
| Tumor cell line | Outcome of drug treatment | Ref. |
| U87 (glioblastoma), PC3 (prostate), T47D (breast), and HCT116 (colon) human cancer cell lines | Cells exposed to both PX-866 and wortmannin phosphatidylinositol-3-kinase inhibitors, suppressed 3D spheroid growth at low concentrations, unlike in 2D monolayer which failed to inhibit cell growth even in higher concentrations | [21] |
| AU565, SKBR3, HCC1569 and BT549 HER2-amplified breast cancer cell lines | Drug response to trastuzumab and pertuzumab (two anti-HER2 agents) was highly dependent on whether the cells were cultured in 2D monolayer or 3D laminin-rich ECM gels. Inhibition of β1 integrin significantly increased the sensitivity of the HER2-amplified breast cancer cell lines to the drugs when they grow in a 3D environment. | [22] |
| Ishikawa, RL95-2, KLE endometrial cancer cell | 3D multicellular cultures exposed to the anti-cancer agents such as doxorubicin and cisplatin exhibited greater resistance to the drugs than 2D monolayers. Their effects on the intracellular mediators were not similar in 3D and 2D cultures, including selective paradoxical stimulatory effects on VEGF secretion. Differences were also dependent on cancer cell lines. | [20] |
| II. ECM environment - Examples of observed differences in drug response | ||
| IIa. The ECM that surrounds the tumor cells impacts pharmacodynamics | ||
| Tumor cell line | Outcome of drug treatment | Ref. |
| HT1080 human fibrosarcoma cells | The anti-invasive effect of doxorubicin observed in conventional 2D culture was completely abolished in a 3D environment. The 3D collagen type I matrix inhibited the antimigratory effect of the drug | [24] |
| H1299, PC3, HCT116 colon cancer cells | 3D cultured cells treated with 5-fluorouracil resulted in p53 stabilization and angiogenesis inhibition by increasing the production of arresten, a collagen IV-derived fragment that possesses anti-angiogenic activity. The presence of ECM contributed to the anti-angiogenic effect of an anti-cancer drug | [23] |
| MIA PaCa-2, PANC-1, and Capan-1 pancreatic cell lines | Cancer cell lines adhering to ECM proteins (collagen I, collagen IV, fibronectin, and laminin) showed decreased cytotoxicity of anticancer drugs such as doxorubicin, cisplatin and 5-fluorouracil, but not gemcitabine | [104] |
| III. Other cell types in the 3D microenvironment - Examples of observed differences in drug response | ||
| IIIa. Interactions with stromal cells involved in drug sensitivity and development of drug resistance | ||
| Tumor & stromal cell line | Outcome of drug treatment | Ref. |
| T3M4 and PT45-P1 pancreatic carcinoma cell lines & Murine Pancreatic Fibroblasts | Tumor cell lines cultured in presence of fibroblasts became much less sensitive toward treatment with the anti-cancer agent etoposide. Fibroblasts contribute to the development of chemoresistance via increased secretion of NO and release of IL-1 by the tumor cells | [105] |
| Different human cancer & NIH3T human fibroblast | Fibroblast-derived 3D matrix increased β1-integrin-dependent survival of a subset of human cancer cell lines during taxol treatment, while it sensitized or minimally influenced survival of other cells | [106] |
| IIIb. Presence of ECs are necessary for tumor growth and progression, and also influence the optimal or impaired drug delivery to the tumor tissue | ||
| Observation | Ref. | |
| ECs, beside providing the blood supply to the tumor that allow it to grow and progress, are also responsible for carrying therapeutic drugs to reduce it | [25] | |
| Anti-angiogenic drugs used as a therapeutic strategy must be examined further in order to find a balance between therapeutic efficacy and excessive vascular regression preventing adequate drug delivery | [107] | |
| The use of vascular disrupting agents can reduce tumor growth and progression by inducing occlusion of the tumor vasculature, which in turn leads to tumor necrosis | [108] | |
| IV. Other factors in a 3D-complex that might influence drug response | ||
| IVa. Hypoxia and drug transporter expression | ||
| Tumor cell line | Outcome of drug treatment | Ref. |
| MCF7 breast cancer cells | 3D-cultured spheroids, unlike in 2D monolayer, showed an increase in the multi-drug resistance transporter Pgp via activation of hypoxia inducible factor (HIF-1) which contributes to increased doxorubicin drug resistance | [19] |
| WiDr colon cancer cells | Cells cultured as multilayer have a lower uptake of antifolates, including the anti-cancer drug methotrexate, compared with monolayers. This higher resistance may be related to a decrease in the reduced folate carrier drug transporter when cells are cultured in 3D. | [109] |
| HT29 human colon cancer cells | 3D multicellular layers are necessary to predict hypoxia-activated anticancer drugs such as tirapazamine since their diffusion through the extravascular tumor compartment may limit their activity | [110] |
| IVb. Vascular shear stress | ||
| Colo205 colon human cancer cells | 3D spheroids cultured in flow showed a threefold increase in resistance to doxorubicin compared to monolayer cells cultured under static conditions | [111] |
| V. Example of other organs affected by the need of a 30-Complex in response to an optimal drug screening | ||
| Cell line | Outcome of drug treatment | Ref. |
| Primary human hepatocytes (PHH) | For testing toxicity of novel therapeutics, the culture of hepatocytes needs a 30-complex to retain their functions and predict hepatotoxicity, since they quickly stop producing drug metabolizing enzymes when cultured on a 2D monolayer | [10] |
| Mammary epithelial cells | The cell matrix interactions in 3D are critical to recapitulate the structure and function of the mammary gland. Drug sensitivity of mammary epithelial cells cultured in 3D and in 2D showed significant differences | [9, 112] |
Drug behavior in 2D vs 3D systems
Differences in cell morphology, differentiation, proliferation, viability, response to stimuli, metabolism, and gene/protein expression, are observed when cells, previously cultured in 2D, are moved to a 3D environment [7]. This is not surprising considering that human organs, with few exceptions, need 3D-structure to develop their associated functions [8, 9]. A well-known example of the necessity of 3D models for testing toxicity of novel therapeutics is the culture of hepatocytes, which behave quite differently in 2D versus 3D cultures [10].
In cancer research, the idea of mimicking 3D tissue function in vitro is not new. Tumor spheroids, for example, were presented nearly four decades ago [11]. However, given the fact that 2D systems are relatively easy to culture, provide simple assessment of cell function (e.g. protein expression), and are lower in cost, the adoption of 3D systems has been relatively slow. Nevertheless, recent studies have enhanced our understanding of the necessity for testing the efficacy of anticancer drugs in 3D systems. For example, among other differences, tumor cells in 3D adopt a different morphology than in 2D [12], have different cell surface receptor expression and proliferation [13], regulate differentially genes responsible for angiogenesis, cell migration, and invasion [14-16], and have different extracellular matrix synthesis [17].
Importantly, tumor cells in 3D also show differences in anticancer drug sensitivity. Studies have shown that culturing cancer cells in 3D can shift their dose response, sometimes to the point where they become functionally resistant to the drug. For instance, culturing of cancer cells in a 3D system can lead to an increase of 20-fold or more in the EC50 compared with 2D culture when cancer cells are exposed to doxorubicin [18]. Moreover, recent studies suggest that 3D models, unlike 2D culture, more accurately predict acquired drug resistance [19]. Drugs that target molecular pathways have shown differences in activation or inhibition depending on the 3D architecture of the local microenvironment [20-22]. In addition, the composition of the extracellular matrix (ECM) can significantly affect the anti-migratory effect of some drugs [23, 24]. These examples demonstrate that tumor cell biology in 3D culture is significantly different than 2D, leading to differences in drug efficacy. Although in vitro, the results in 3D culture may be more representative of the way cancer cells in vivo respond to chemotherapeutic treatment. Table I summarizes and highlights the role that the ECM, stromal cells (e.g. fibroblasts), vasculature, and other factors present in the tumor microenvironment can play in modulating drug response, sensitivity, and drug resistance.
The drug response differences between 2D and 3D- models suggest that pathways necessary for survival in a 3D environment may not be activated in 2D. As a result, drug screening performed on 2D monolayers can increase the false-positive and false-negative rates of investigational compounds. While false-positive results can increase failure of drugs in clinical trials, false-negatives discard potentially effective drugs. The use of 3D-microphysiological systems is predicted to reduce the rate of false-positive and false-negative outcomes, thereby reducing the need for animal testing and improving the overall efficiency of drug discovery.
Drug panel and strategy for validation
Microphysiological systems need to exhibit drug responses that parallel those seen in vivo. Our strategy for validating tissue responses includes a panel of common drugs (Table 2). The panel distinguishes drugs considered to be traditional chemotherapy from those that can be classified as molecularly-targeted therapy, and considers known and poorly understood cardiac muscle and bone marrow toxicity. The panel also recognizes other important observed effects in order to validate, predict, and better understand the response, toxicity, and effectiveness of the drug.
Table II.
Example drug panel - Strategy for validation
| I. Physiologic validation of cardiac muscle and microvasculature function
| ||
|---|---|---|
| Ia. Cardiac muscle validation. Example of representative drugs for cardiac validation
| ||
| Target | Approved drug | Outcome of drug treatment |
|
| ||
| L-type Ca2+ channel | verapamil; L-type Ca2+ channel blocker (negative inotrope) | |
|
| ||
| β-adrenergic receptor | epinephrine; β-adrenergic receptor agonist (positive inotrope) | β-adrenergic receptors agonists are associated with a proliferative and angiogenic effect [113] |
|
| ||
| propranolol; β-blocker (negative inotrope) | β-blockers potentiate anti-proliferative and anti-angiogenic effects in tumor and ECs [114] | |
|
| ||
| human ether-a-go-go related gene (hERG) channel | dofetilide; hERG channel blocker | Undesirable blockade of hERG channel represents the most common form of drug-induced QT prolongation associated with risk of arrhythmias and possible sudden death [115] hERG channels are expressed in several tumor cells influencing their activity in cell survival and proliferation [116, 117] |
|
| ||
| Ib. Microvasculature validation. Example of representative drugs for vascular validation
| ||
| Target | Approved drug | Outcome of drug treatment |
|
| ||
| Vascular permeability | thrombin; permeabilizing agent VE-cadherin disruption [118] | Vascular permeability is greatly increased in tumor vasculature resulting in leaky and dysfunctional vessels [119] |
| II. Physiologic validation of tumor tissue reduction
| |||
| IIa. Tumor tissue validation. Example of representative drugs used in traditional chemotherapy
| |||
| Target | Approved drug |
Risk of direct cardiotoxicity //Bone marrow toxicity Reviewed by [1, 4, 6, 120-122] |
Outcome of drug treatment |
|
| |||
| DNA intercalator | doxorubicin (anthracycline) | Recognized risk of cardiotoxicity Myelosuppression. Recognized bone marrow injury | Typical cardiotoxicity type I associated with ROS generation and irreversible cell damage. Differently described behavior in 2D vs 3D-complex (Table I) |
|
| |||
| Platinum analog | cisplatin, oxaliplatin | Possible cardiotoxicity Myelosuppression and recognized bone marrow injury | Differently described behavior in 2D vs 3D-complex (Table I) |
|
| |||
| Anti-metabolite | 5-fluoruracil | Possible cardiotoxicity associated with myocardial ischemia and arrhythmia Recognized bone marrow injury | Differently described behavior in 2D vs 3D-complex (Table I) |
|
| |||
| Mitotic disrupter |
vincristine (vinca alkaloid) |
Low recognized cardiotoxicity Myelosuppression and recognized bone marrow injury | Possible cardioprotection by attenuation of doxorubicin-induced cardiac myocyte toxicity [123] |
|
| |||
| IIb. Tumor tissue validation. Example of representative drugs used in molecularly-targeted chemotherapy
| |||
| Approved drug | Known target | Risk of direct cardiotoxicity | Outcome of drug treatment |
| Small molecule tyrosine kinase & multikinase inhibitors | Growth factor receptors, intracellular signaling pathways, possible channel targets, and other cell functions | Cardiomyocyte survival, contraction dysfunction, or QT prolongation Reviewed by [5, 124, 125] |
In general, possible different drug response in 2D vs 3D-complex due to the differentially 3D activated or inhibited pathways compared with 2D |
|
| |||
| trastuzumab (monoclonal antibody) | HER2 | Recognized risk of cardiotoxicity | Typical type II cardiotoxicity and “on-target” effect [126, 127] Differently described drug response in 2D vs 3D-complex (Table I) |
|
| |||
| lapanitib | HER2 | Low risk of cardiotoxicity | No risk of cardiotoxicity as the risk associated with the monoclonal antibody that targets the same receptor. Different signaling pathways are activated in cardiomyocytes by both strategies [124] |
|
| |||
| gefinitib | EGFR | Low risk of cardiotoxicity | |
|
| |||
| sunitinib | VEGFR, c-KIT, PDGFRα/β, RET, CSF-1R, FLT3 (hERG) | Recognized risk of cardiotoxicity | Typical “off-target” effect PDGFR signaling important in cardiomyocyte survival ATP depletion by mTOR increases activity in cardiomyocytes [124] |
|
| |||
| sorafenib | RAF, VEGFR, c-KIT, PDGFRα/β, FLT3 | Recognized risk of cardiotoxicity | |
|
| |||
| imatinib | BCL:ABL, PDGFRα/β, c-KIT, (hERG) | Recognized risk of cardiotoxicity | Strong warning for QT prolongation, risk of arrhythmia, and sudden death Possible hERG blockade by ABL inhibitors [115] |
|
| |||
| everolimus | mTOR | Warning risk of cardiotoxicity (lack of significant published data) |
Warning for QT prolongation Additional possible HIF-1 activity inhibition under hypoxic conditions (3D tumor environment) [128] |
|
| |||
| bortezomib | proteasome inhibitor 26S | Warning risk of cardiotoxicity | Hypotension, warning for QT prolongation. Additional HIF-1 transcription inhibition under hypoxic conditions (3D tumor environment) [129] |
|
| |||
| vorinostat | HDAC1, 2, 3, 6 | Warning risk of cardiotoxicity | Warning for QT prolongation Additional HIF-1 degradation induction under hypoxic conditions by HDAC inhibitors (3D tumor environment) [130] |
|
| |||
| III. Physiologic validation of EC proliferation and migration/angiogenesis
| |||
| IIIa. Angiogenesis validation. Example of representative drugs used in molecularly-targeted chemotherapy
| |||
| Approved drug | Known target | Risk of direct cardiotoxicity | Outcome of drug treatment |
| Small molecule tyrosine kinase & multikinase inhibitors | Growth factor receptors, intracellular signaling pathways, and possible channels targets | Cardiomyocyte survival, contraction dysfunction or QT prolongation Reviewed by [5, 124, 125] |
In general, possible different drug response in 2D vs 3D-complex due to the differentially 3D activated or inhibited pathways compared with 2D |
|
| |||
| bevacizumab (monoclonal antibody) | VEGF (Growth factor) | Warning risk of cardiotoxicity | Disrupting VEGF signaling, most of these drugs exhibit predictable risk of hypertension and thrombosis (cardiovascular indirect side effects) since VEGF doesn’t just help new vessels grow, but also protects existing blood vessels [131, 132] |
|
| |||
| sunitinib | VEGFR, c-KIT, PDGFRα/β, RET, CSF-1R, FLT3, hERG | Recognized risk of cardiotoxicity | |
|
| |||
| vandetamib | VEGFR, EGFR, RET | Warning risk of cardiotoxicity | |
|
| |||
| pazopanib | VEGFR, PDGFRα/β, c-KIT | Warning risk of cardiotoxicity | |
III. Design of in vitro microphysiological systems
To address the numerous potential side effects of anticancer drugs on multiple tissues and organ systems, robust in vitro microphysiological systems have been developed for pre-clinical high throughput screening of candidate drugs. These systems need to mimic critical functions and anatomical features of in vivo tissues to generate an appropriate response to a pharmacologic challenge. It is important to note that because of the complex nature of in vivo tissues, it may be impractical and unnecessary to mimic all the functions and architecture of in vivo tissues. In this view, an optimal level of anatomical complexity that has bearing on drug distribution and response of in vivo tissues needs to be reflected in the microphysiological systems.
Each specific organ system presents unique challenges to correlate in vitro performance with in vivo physiology and pathology associated with anticancer drugs. The following sections discuss the critical features of major organ systems and their anticancer drug-related behavior. Methods for mimicking these features in vitro within the proposed microphysiological platform are also presented.
Vascular System
One of the most prominent features of all human tissues is vasculature, which provides a convective mode of transport for nourishment and waste removal. The vascular transport is particularly necessary for tissues with a diameter greater than 200 μm [25-27], as passive diffusional transport is inefficient. Thus, to replicate the complex 3D arrangement of cells and ECM, human microphysiological systems must include a vasculature made of perfused vessels that possess a physiologic flow. The molecular transport across the vascular wall into normal tissues is largely driven by diffusion, but limited convection also takes place primarily in capillaries. Vascular permeability varies considerably among different tissues, and is modulated by different factors such as blood flow [28]. Vascular permeability undergoes significant changes in pathological conditions such as wound healing, chronic inflammatory diseases, and cancer [25].
Unlike arteries and veins, capillaries consist of little more than a layer of endothelial cells (ECs) and connective tissue, along with a sparse covering of pericytes. They lack the smooth muscle cells that are present around the major vessels, arteries, and veins. The ECs are embedded in a 3D microenvironment that mainly consists of a collagen-based ECM and pericytes, and is influenced by biochemical (i.e., growth factors) and physical (i.e., shear stress) forces. Mimicking this complex microenvironment in vitro is a major challenge in vascular research. ECs are responsible for regulating a variety of functions including vascular tone, inflammation, coagulation, and sprouting of new vessels. In normal physiological conditions, vasculature is vasodilatory, anti-thrombotic, anti-inflammatory, and non-angiogenic. These functions are controlled mainly by secreted factors from the EC [29]. In this context, since ECs are mostly quiescent under physiological conditions, the development of new anti-cancer treatments are also directed toward targeting cell signaling pathways involved in pathological EC proliferation and migration [5]. By targeting endothelium, the vascular supply to tumor tissue can be reduced, thereby inhibiting tumor growth. Anti-angiogenic drugs are mostly targeted to the vascular endothelial factor (VEGF) signaling pathway. These drugs often have VEGF receptors (VEGFRs) as a common target. However, VEGF doesn’t only help new vessels grow, it also protects existing blood vessels. Thus, due to disruption of VEGF signaling, many of these drugs are associated with a predictable risk of hypertension and coagulation that can lead to vascular dysfunction and thrombosis, respectively [4, 7, 30].
Tumor Tissue
Tumor cells have a remarkable ability to evolve in response to communication with the microenvironment. In this view, drug testing merely on isolated cancer cells is insufficient for a reliable estimation of drug efficacy in humans. The quest to develop improved models has progressed from 2D cancer cell culture to 3D tumor spheroids made up of cancer cells, and 3D tumor spheroids composed of a mixture of cancer and stromal cells [31-33]. However, these models lack perfused vasculature and are limited in their ability to mimic the in vivo microenvironment critical for modeling drug delivery. Hence, we believe in vitro tumors with perfused capillaries, embedded in naturally occurring ECM, will produce improved drug screening studies.
It is widely accepted that tumors actively communicate with vasculature to fulfill their growing metabolic demands, and in some cases to metastasize. Such communication brings several changes in vasculature, including angiogenesis mediated by VEGF, increased vascular leakiness, and irregular vascular interconnections [25, 34, 35]. In additions, it is noted that lymphatic vasculature of tumors is generally dysfunctional [34, 36, 37]. Together, these conditions are believed to increase the interstitial fluid pressure of tumors and consequently, increase the interstitial fluid flow from the tumor into surrounding tissue [34, 38, 39]. As a result, in vivo tumors display uneven distribution of cytokines, nutrition, and drugs across the tissue, posing a major challenge for efficient drug delivery [34, 38-40]. Therefore, to simulate a variety flow and pressure conditions, drug screening platforms need to have control over spatiotemporal resolution of interstitial flow and pressure in tumor-on-chip devices.
The inefficient vascular supply to tissues and increased growth rate creates hypoxic conditions in certain regions of tumors. As a result, the microenvironment remains acidic, adversely affecting the action of many chemotherapeutic drugs as reviewed elsewhere [41, 42]. Hypoxia also promotes epithelial-to-mesenchymal transition (EMT), which is marked by increased motility of cancer cells and results into invasion of surrounding tissue [43]. Further, hypoxia is believed to promote chemotherapeutic drug resistance in cancer cells by gene expression products such as P-glycoprotein [44]. Hence, the in vitro tumors need to grow under hypoxic conditions closer to that experienced in vivo. We, and others, have previously developed a protocol for creating normal tissues that are perfused with dynamic vasculature under hypoxic and non-hypoxic conditions [45-47].
Interstitial flow and pressure, cellular behavior, drug distribution, and, in turn, drug efficacy are influenced by ECM, which constitutes a major part of the tumor tissue. Tumors are characterized by stiff ECM, which is typically composed of collagen, fibronectin, glycosaminoglycans, and proteoglycans [48, 49]. The active role of tumor ECM in cell signaling, as well as creating chemical and mechanical cues for cell migration, are reviewed elsewhere [48, 50]. Interestingly, it has been shown that it is possible to extract acellular ECM from in vivo tumors [51, 52], and it is also possible to achieve collagen of varying stiffness by mixing it with other naturally occurring ECM such as fibrin. We believe such strategies could be implemented to create microphysiological tumor tissue.
Apart from the constituents of the tumor microenvironment discussed above, other major components are immune cells and tumor-associated fibroblasts [50, 53, 54]. We and others have shown that fibroblasts secrete factors necessary for creating perfused vasculature [46, 47, 55], and, recognizing this fact, our current model of perfused network includes fibroblasts [45, 47]. However, for the future advancement of the in vitro tumor model, appropriate tumor-associated fibroblasts may be needed. Among the immune cells, tumor-associated macrophages are perhaps the most significant cells as they affect a plethora of cancer processes, including angiogenesis, invasion, and metastasis [50, 53, 56], and, importantly, they also adversely affect the chemotherapy [56]. Hence, we believe that for future advancement of microphysiological tumor tissue, it is important to integrate immune cells, particularly macrophages, on organ-on-chip platforms.
Cardiac Tissue
Cardiotoxicity and altered cardiac function have been identified as significant side effects of anticancer drugs [4, 57-60]. At the cellular level, anticancer drugs can cause DNA damage, ATP depletion, apoptotic protein release, ROS generation, electrolyte imbalance, lipid accumulation, and myocyte death [4, 59-61]. At the tissue level, these effects manifest as vasospasm, changes in force and frequency of contraction, and modified electrophysiology [4, 59]. As a result of these cell and tissue level effects, whole organ pathologies, such as left ventricular dysfunction, heart failure, myocardial ischemia, arrhythmias, and pericardial disease, [4, 57-60, 62] are realized. These cardiac dysfunctions are often not discovered until clinical trials primarily due to limitations in current drug screening models. Similar to the previous discussion, cardiac animal models provide whole heart response to drugs, but many drugs have human specific effects.
The key features of a microphysiological cardiac tissue system are as follows: 1) Use of a cardiac-specific matrix creates a 3D environment with the appropriate tissue-specific architecture to induce the formation of tissue that better mimics in vivo physiology and pathology [63]. 2) All cells are of human origin allowing assessment of human cell-specific drug responses. With further development of lineage-specific differentiation protocols, all cells implanted in the device may be derived from the same iPS cell line allowing for genetic homogeneity, and thus patient-specific responses, throughout the tissue compartments. 3) Tissues exhibit synchronous and rhythmic spontaneous contraction that allows detection of alterations in electrophysiological and contractile properties. Together, these properties represent the most critical parameters for a high-throughput cardiac tissue module for the screening of drug-induced cardiac side effects.
Bone Marrow
Toxicity of pharmacological agents toward hematopoietic stem cells (HSCs) is a major problem for many therapeutic strategies, including cancer chemotherapy [64, 65]. Many anti-neoplastic drugs directly target the machinery of cell proliferation and thus also target HSC, which can be either quiescent, undergoing self-renewal, or differentiating into erythroid, myeloid, or lymphoid progenitors, or they may target more committed progenitors downstream of HSC [66]. This results in immunosuppression, anemia and thrombocytopenia, and many patients develop infections as a result. Blood transfusions can reverse anemia, and neutropenia can be addressed by treatment with granulocyte-colony stimulating factor (G-CSF). However, in severe cases of myelosuppression patients have to undergo bone marrow transplantation – either restoration of autologous HSC removed before the start of chemotherapy, or by allogeneic transplantation. Identifying new anti-cancer drugs that do not target HSC and their descendants is thus a priority.
HSC reside in the bone marrow in a specialized niche composed primarily of extracellular matrix (ECM), osteoblasts, mesenchymal stem cells (MSC), and vascular endothelial cells. A hematopoietic microphysiological system will need to contain each of these cellular elements and the matrix will need to be extracted from bone marrow to ensure the presence of necessary extracellular cues. It is essential that the system will model the following: 1) the maintenance of healthy HSCs that undergo renewal; 2) the generation of lymphoid lineage cells; 3) the generation of erythroid lineage cells; 4) the generation of myeloid lineage cells; and, 5) the release of these cells into a circulatory system – the “blood.” Incorporation of an immune compartment, specifically a hematopoietic compartment, into any platform of integrated microphysiological systems will provide obvious advantages in anti-cancer drug-screening strategies.
Platform design considerations
Individual modules of microphysiological systems mimicking critical organ functions can be developed separately and integrated to produce a platform for drug screening (Figure 1A and B). Such a platform should have the following features: biocompatible, flexible, high-throughput, reproducible design, easy fabrication, affordable, and a small footprint.
Figure 1. A prototype drug screening platform.
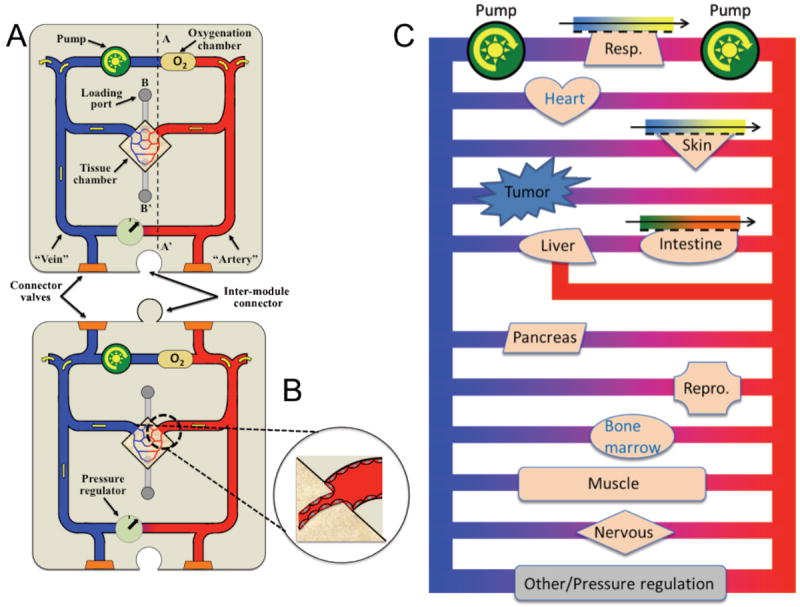
A. The microphysiological systems are developed in a central tissue chamber of individual modules. These microtissues are initially nourished by interstitial flow and later by a perfused capillary network. The medium is pumped around a microfluidic network and is oxygenated through a bubble chamber. A pressure regulator controls “arterial” pressure. Input and output channels on the arterial and venous side, respectively, allow mixing of fresh medium into the system. Individual tissue modules can be connected like jigsaw pieces, and connector valves allow “anastomosis” of microfluidic channels. B. Microfluidic channels are lined by EC and these anastomose with the microvessels in the tissue chamber to form a continuous vascular network linking all of the organs. C. An example how the major organs with a tumor might be placed such that their perfusion is in parallel to each other
Using soft lithography, it is possible to reproducibly and precisely fabricate polydimethylsiloxane (PDMS) devices with micron-range features. In short, the process of fabrication involves the following: a master mold is created by photolithography on silicon wafers that are spin-coated with SU8, and PDMS is poured on the master mold. Features of the mold are impressed on polymerized PDMS, which can be attached to another PDMS sheet or glass in a leak-proof manner by using plasma treatment [45, 47]. PDMS microdevices can be designed to create high throughput platforms and the designs can be easily altered to incorporate tissue-specific requirements, such as absorptive interface for gut tissue. Further, PDMS devices are biocompatible, flexible, affordable, and our current designs can be adapted to create about 100 micro-tissues per 75 cm2 area. Moreover, by manipulating length and/or cross sectional area of microfluidic channels, it is also easy to maintain physiologic pressure drops across the tissue over a time periods exceeding two weeks [67]. Importantly, PDMS is optically clear, offering a non-invasive system of real time tissue imaging with a high degree of spatial resolution.
Our group has recently reported successful creation of perfused vascular networks in PDMS microdevices by allowing self-assembly and growth of endothelial colony forming cell derived-endothelial cells (ECFC-ECs) and stromal fibroblasts in a naturally occurring ECM [45, 68]. These tissues were formed in a PDMS device consisting of 0.1 mm2 tissue chamber, which was fed with microfluidic channels on either side of the tissue chamber, which mimicked arterial and venular supply to the tissue (Figure 1A). Significantly, the tissues were maintained in a medium without exogenous addition of VEGF or bFGF (primary growth factors for ECs and fibroblasts, respectively) and under physiologic oxygen concentration. These studies have shown that using microtechnology, it is possible to achieve an intricate balance of biochemical and mechanical cues, and cell-cell communication to generate anatomical features of inter-connected capillary networks. Importantly, once the microvascular network is developed and anastomosed to the PDMS microfluidic channels, the tissue is fed only with the microvascular flow. This feature of the system is particularly important because it allows control over morphogen delivery to tissues via the vasculature. Also, flow through the vasculature can be controlled by simply altering the hydrostatic pressure gradient [67-69]. Thus, this technology has opened a plethora of opportunities for developing organ systems mimicking in vivo tissues for drug screening platforms.
This technology can be extended to create vascularized tumor, cardiac, reproductive, and other major organ systems incorporating the cells of specific organs that mimic critical functions of the organ. An example of a microphysiological system module with appropriate instrumentation, such as a fluid pressure sensor, pump and oxygenation system, is shown in Fig. 1A and B. To grow micro-tissues in a microdevice, organ specific cells, endothelial cells, and other stromal cells are initially mixed with ECM gels and the mixture is seeded in the microdevice (Figure 1A). The microtissue is initially nourished by interstitial flow controlled by a pressure gradient across the microfluidic channels. Once the microtissue is vascularized, and the vasculature anastomoses with the high and low pressure microfluidic lines (Figure 1A and B), the tissue is completely nourished by the vasculature.
To integrate pre-vascularized organ systems on an integrated drug screening platform, the modules will be equipped with connector valves, which will allow modules to be daisy-chained for system-wide integration (Figure 1A). These modules can be connected so as to broadly mimic the human circulation in which the tissue chambers are connected by the circulation in parallel (Figure 1B). Important considerations for integration are compatible tissue culture media for all tissues on the platform and relative scale of the tissues. Since most primary culture systems need a specialized medium for cell survival, differentiation, and growth, a universal medium supplemented with organ specific growth factors needs to be developed. Previously, it was shown that mixing two or more specialized growth media can be used to facilitate function and growth of cells in co-cultures [70-72].
To simulate the integrated function of various organs on the organ-on-chip platform, it is necessary to appropriately scale these systems, the strategies for which are recently reviewed [73, 74]. The scaling is critical because the vascular blood supply (determined as percent cardiac output of the blood supply) per unit mass of organ varies considerably among various organs, significantly differing the amount of drug received by the organs. For drug screening platforms, the appropriate scaling parameters are the blood supply per unit mass and relative mass of an organ system with respect to other organ systems. These are required to maintain similarity with the in vivo organs. Notably, it may not be possible to maintain the relative mass of some of the organs; particularly skeletal muscle and adipose tissue whose masses are greater than 500 times the mass of testis/ovaries [75]. In such cases, the morphogen or concentrations on the arterial side of the flow of particular organ systems may be modified to accommodate the scaling parameter.
The organ-on-chip platforms can be analyzed at molecular, cellular, tissue, and systemic level functional endpoints. The greatest advantage of the platform material is that it is optically clear and does not interfere with wavelengths of light required for fluorescence or bright field imaging. This feature allows non-invasive image-based measurement of parameters relevant to drug-induced endpoints of pathogenesis, such as fluorescent lifetime imaging (FLIM) of metabolic state [76], assessment of calcium handling using calcium sensitive proteins [77] or calcium dyes [78], electrophysiology monitoring using voltage sensitive dyes [79], force-frequency response to pacing [80], perfusion, and vascular permeability. The organ-on-chip platform also allows easy collection of circulating media to analyze for soluble factors. Importantly, it is also possible to isolate each micro-organ for genomic or proteomic analysis as each micro-tissue is maintained in a separate tissue chamber.
IV. Incorporation of Other Organ Systems
Our device has been designed specifically to address the effects of anticancer drugs in vascular, tumor, cardiac, and bone marrow tissues. However, other organ systems have also demonstrated significant response to anticancer drugs and are important to consider as modules in future iterations of the device. The general approach and methodology of our proposed modular platform is easily adaptable to other tissue systems. The GI tract [81, 82], liver [83], kidneys [84], nervous system [85, 86], skeletal muscle [87, 88], and gonads [89, 90] have all been reported to have anticancer drug related pathologies.
Key features of developing a GI module include analogs of the small intestine (primary site of drug absorption) and the liver (primary site of drug metabolism). Small intestine tissue is comprised of a simple epithelium with absorptive cells and three different secretory lineages. Mimicry of this epithelium will require engineering of a similar epithelium to provide selective barrier function through which circulating media can pass. This will filter drug compounds accordingly and secrete the appropriate factors into the tissue microenvironment. Liver tissue is complex and consists of different zones with specific enzyme activity (Phase I and Phase II enzymes) [91]. A major factor that regulates the enzyme activity in these zones is oxygen tension (i.e. variable distance from the closest blood vessel) [92]. To mimic liver function in a small microphysiological system compartment, an oxygen gradient will need to be established within the compartment to achieve the differential Phase I and Phase II enzyme activity in hepatocytes. Directional flow across the oxygen gradient will be necessary to expose candidate drugs to the appropriate sequence of enzymes to ensure that drugs are metabolized as they are in vivo.
Kidney compartments will need to mimic the main kidney functions of waste removal and metabolite excretion in the urine. Renal tubule epithelial cells are especially susceptible to toxic substances and are the critical cell that coordinates the secretion of waste products (including metabolized drug compounds) and the reabsorption of necessary nutrients [93]. In vivo, both of these objectives are achieved by a counter-current mass exchanger. Thus, an engineered in vitro compartment could mimic this exchanger using microfluidics and a selectively permeable membrane, similar to that used in renal dialysis [94], coated with renal tubule epithelial cells
Nervous tissue modules will require blood-brain barrier (BBB) and blood-cerebrospinal fluid barrier (BCB) analogs to simulate the specialized barrier properties that regulate access to microglia, neurons, oligodendrocytes, and astrocytes. Attempts at BBB reconstitution have been incomplete thus far, but the proper shear stress resulting from blood flow, as provided by our platform, is widely regarded as a key feature [95].
A skeletal muscle module will include contractile tissue and secretion of metabolically active factors into the vascular circuit. Appropriate secretion of metabolically active factors, such as interleukin-6, are necessary for hormonal communication and potential effects on drug metabolism [96]. In addition, the contractile tissue will require exogenous electrical stimulation since certain anticancer drugs affect neuromuscular activity [97].
Gonad compartments will require both a testis and an ovary model. Drug effects on the testis and ovary can be profound and can influence not only immediate reproductive output, but can also have long-term effects on gametes. The main components necessary to mimic are: 1) the maintenance of spermatogonial stem cells in the testis, 2) the development of ovarian follicles in the ovary, 3) recreation of the blood-testis barrier (BTB) in males, and 4) secretion of the sex hormones testosterone and estrogen.
Conclusion
The need for improved methods of screening for anticancer drugs prior to clinical trials is apparent. We have developed a strategy to create a platform that incorporates 3D tissue modules from multiple organ systems to assess the efficacy, as well as the potential side effects, of anticancer drugs. These tissues are comprised entirely of human cells, are perfused by a microvasculature, and mimic the in vivo features of vascular drug delivery and tissue response. The tissues are incorporated into a microfluidic device that allows control of multiple parameters that affect tissue physiology and drug response, as well as non-invasive monitoring of tissue state. The device is currently comprised of vascular, tumor, cardiac, and bone marrow tissues, and is designed to allow for expansion of the system to include additional tissue modules. This in vitro approach represents a significant advance in the ability to identify potential adverse effects of anticancer treatment well before they reach clinical trials.
References
- 1.Marsh JC. The effects of cancer chemotherapeutic agents on normal hematopoietic precursor cells: a review. Cancer Res. 1976;36(6):1853–82. [PubMed] [Google Scholar]
- 2.Dempke W, et al. Human hematopoietic growth factors: old lessons and new perspectives. Anticancer Res. 2000;20(6D):5155–64. [PubMed] [Google Scholar]
- 3.Testa NG, Hendry JH, Molineux G. Long-term bone marrow damage in experimental systems and in patients after radiation or chemotherapy. Anticancer Res. 1985;5(1):101–10. [PubMed] [Google Scholar]
- 4.Albini A, et al. Cardiotoxicity of anticancer drugs: the need for cardio-oncology and cardio-oncological prevention. J Natl Cancer Inst. 2010;102(1):14–25. doi: 10.1093/jnci/djp440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedhli N, Russell KS. Cardiotoxicity of molecularly targeted agents. Curr Cardiol Rev. 2011;7(4):221–33. doi: 10.2174/157340311799960636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Todaro MC, et al. Cardioncology: State of the heart. Int J Cardiol. 2013 doi: 10.1016/j.ijcard.2013.03.133. [DOI] [PubMed] [Google Scholar]
- 7.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7(3):211–24. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 8.Lo AT, et al. Constructing three-dimensional models to study mammary gland branching morphogenesis and functional differentiation. J Mammary Gland Biol Neoplasia. 2012;17(2):103–10. doi: 10.1007/s10911-012-9251-7. [DOI] [PubMed] [Google Scholar]
- 9.Roskelley CD, Desprez PY, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci U S A. 1994;91(26):12378–82. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schyschka L, et al. Hepatic 3D cultures but not 2D cultures preserve specific transporter activity for acetaminophen-induced hepatotoxicity. Arch Toxicol. 2013;87(8):1581–93. doi: 10.1007/s00204-013-1080-y. [DOI] [PubMed] [Google Scholar]
- 11.Dalen H, Burki HJ. Some observations on the three-dimensional growth of L5178Y cell colonies in soft agar culture. Exp Cell Res. 1971;65(2):433–8. doi: 10.1016/0014-4827(71)90023-1. [DOI] [PubMed] [Google Scholar]
- 12.Feder-Mengus C, et al. New dimensions in tumor immunology: what does 3D culture reveal? Trends Mol Med. 2008;14(8):333–40. doi: 10.1016/j.molmed.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Wang F, et al. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci U S A. 1998;95(25):14821–6. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valcarcel M, et al. Three-dimensional growth as multicellular spheroid activates the proangiogenic phenotype of colorectal carcinoma cells via LFA-1-dependent VEGF: implications on hepatic micrometastasis. J Transl Med. 2008;6:57. doi: 10.1186/1479-5876-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wozniak MA, et al. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692(2-3):103–19. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki D, Kurisu S, Takenawa T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene. 2009;28(13):1570–83. doi: 10.1038/onc.2009.2. [DOI] [PubMed] [Google Scholar]
- 17.Beningo KA, Dembo M, Wang YL. Responses of fibroblasts to anchorage of dorsal extracellular matrix receptors. Proc Natl Acad Sci U S A. 2004;101(52):18024–9. doi: 10.1073/pnas.0405747102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horning JL, et al. 3-D tumor model for in vitro evaluation of anticancer drugs. Mol Pharm. 2008;5(5):849–62. doi: 10.1021/mp800047v. [DOI] [PubMed] [Google Scholar]
- 19.Doublier S, et al. HIF-1 activation induces doxorubicin resistance in MCF7 3-D spheroids via P-glycoprotein expression: a potential model of the chemo-resistance of invasive micropapillary carcinoma of the breast. BMC Cancer. 2012;12:4. doi: 10.1186/1471-2407-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chitcholtan K, et al. Differences in growth properties of endometrial cancer in three dimensional (3D) culture and 2D cell monolayer. Exp Cell Res. 2013;319(1):75–87. doi: 10.1016/j.yexcr.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Howes AL, et al. The phosphatidylinositol 3-kinase inhibitor, PX-866, is a potent inhibitor of cancer cell motility and growth in three-dimensional cultures. Mol Cancer Ther. 2007;6(9):2505–14. doi: 10.1158/1535-7163.MCT-06-0698. [DOI] [PubMed] [Google Scholar]
- 22.Weigelt B, et al. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res Treat. 2010;122(1):35–43. doi: 10.1007/s10549-009-0502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Assadian S, et al. p53 inhibits angiogenesis by inducing the production of Arresten. Cancer Res. 2012;72(5):1270–9. doi: 10.1158/0008-5472.CAN-11-2348. [DOI] [PubMed] [Google Scholar]
- 24.Millerot-Serrurot E, et al. 3D collagen type I matrix inhibits the antimigratory effect of doxorubicin. Cancer Cell Int. 2010;10:26. doi: 10.1186/1475-2867-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 26.Helmlinger G, et al. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3(2):177–82. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 27.Muschler GF, Nakamoto C, Griffith LG. Engineering principles of clinical cell-based tissue engineering. J Bone Joint Surg Am. 2004;86-A(7):1541–58. doi: 10.2106/00004623-200407000-00029. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Friedman MH. Adaptive response of vascular endothelial cells to an acute increase in shear stress magnitude. Am J Physiol Heart Circ Physiol. 2012;302(4):H983–91. doi: 10.1152/ajpheart.00168.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cines DB, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91(10):3527–61. [PubMed] [Google Scholar]
- 30.Johnson JI, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84(10):1424–31. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Che ZM, et al. Collagen-based co-culture for invasive study on cancer cells-fibroblasts interaction. Biochemical and Biophysical Research Communications. 2006;346(1):268–275. doi: 10.1016/j.bbrc.2006.05.111. [DOI] [PubMed] [Google Scholar]
- 32.Sutherla Rm, Mccredie JA, Inch WR. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas. Journal of the National Cancer Institute. 1971;46(1):113. [PubMed] [Google Scholar]
- 33.Tanaka R, et al. Three-dimensional coculture of endometrial cancer cells and fibroblasts in human placenta derived collagen sponges and expression matrix metalloproteinases in these cells. Gynecologic Oncology. 2003;90(2):297–304. doi: 10.1016/s0090-8258(03)00335-4. [DOI] [PubMed] [Google Scholar]
- 34.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 35.Jain RK. Antiangiogenic therapy for cancer: current and emerging concepts. Oncology (Williston Park) 2005;19(4 Suppl 3):7–16. [PubMed] [Google Scholar]
- 36.Leu AJ, et al. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Research. 2000;60(16):4324–7. [PubMed] [Google Scholar]
- 37.Padera TP, et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002;296(5574):1883–6. doi: 10.1126/science.1071420. [DOI] [PubMed] [Google Scholar]
- 38.Jain RK. Barriers to Drug-Delivery in Solid Tumors. Scientific American. 1994;271(1):58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 39.Rutkowski JM, Swartz MA. A driving force for change: interstitial flow as a morphoregulator. Trends in Cell Biology. 2007;17(1):44–50. doi: 10.1016/j.tcb.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annual Review of Biomedical Engineering. 1999;1:241–263. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 41.Hockel M, Vaupel P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. Journal of the National Cancer Institute. 2001;93(4):266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 42.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nature Reviews Cancer. 2006;6(8):583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 43.Salnikov AV, et al. Hypoxia Induces EMT in Low and Highly Aggressive Pancreatic Tumor Cells but Only Cells with Cancer Stem Cell Characteristics Acquire Pronounced Migratory Potential. PLoS One. 2012;7(9) doi: 10.1371/journal.pone.0046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Comerford KM, et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Research. 2002;62(12):3387–94. [PubMed] [Google Scholar]
- 45.Hsu YH, et al. Full range physiological mass transport control in 3D tissue cultures. Lab Chip. 2013;13(1):81–9. doi: 10.1039/c2lc40787f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, et al. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab on a Chip. 2013;13(8):1489–1500. doi: 10.1039/c3lc41320a. [DOI] [PubMed] [Google Scholar]
- 47.Moya ML, et al. In Vitro Perfused Human Capillary Networks. Tissue Engineering Part C-Methods. 2013;19(9):730–737. doi: 10.1089/ten.tec.2012.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiig H, Swartz MA. Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiological Reviews. 2012;92(3):1005–60. doi: 10.1152/physrev.00037.2011. [DOI] [PubMed] [Google Scholar]
- 49.Levental KR, et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Egeblad M, Nakasone ES, Werb Z. Tumors as Organs: Complex Tissues that Interface with the Entire Organism. Developmental Cell. 2010;18(6):884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schedin P, et al. Mammary ECM composition and function are altered by reproductive state. Molecular Carcinogenesis. 2004;41(4):207–220. doi: 10.1002/mc.20058. [DOI] [PubMed] [Google Scholar]
- 52.Fleming JM, et al. The normal breast microenvironment of premenopausal women differentially influences the behavior of breast cancer cells in vitro and in vivo. Bmc Medicine. 2010;8 doi: 10.1186/1741-7015-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin EY, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Research. 2006;66(23):11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 54.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature Reviews Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 55.Chen XF, et al. Rapid Anastomosis of Endothelial Progenitor Cell-Derived Vessels with Host Vasculature Is Promoted by a High Density of Cotransplanted Fibroblasts. Tissue Engineering Part A. 2010;16(2):585–594. doi: 10.1089/ten.tea.2009.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Palma M, Lewis CE. CANCER Macrophages limit chemotherapy. Nature. 2011;472(7343):303–304. doi: 10.1038/472303a. [DOI] [PubMed] [Google Scholar]
- 57.Colombo A, et al. Cardiac toxicity of anticancer agents. Curr Cardiol Rep. 2013;15(5):362. doi: 10.1007/s11886-013-0362-6. [DOI] [PubMed] [Google Scholar]
- 58.Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol. 2010;7(10):564–75. doi: 10.1038/nrcardio.2010.121. [DOI] [PubMed] [Google Scholar]
- 59.Raschi E, et al. Anticancer drugs and cardiotoxicity: Insights and perspectives in the era of targeted therapy. Pharmacol Ther. 2010;125(2):196–218. doi: 10.1016/j.pharmthera.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 60.Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol. 2009;53(24):2231–47. doi: 10.1016/j.jacc.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 61.Monsuez JJ, et al. Cardiac side-effects of cancer chemotherapy. Int J Cardiol. 2010;144(1):3–15. doi: 10.1016/j.ijcard.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 62.Bonita R, Pradhan R. Cardiovascular toxicities of cancer chemotherapy. Semin Oncol. 2013;40(2):156–67. doi: 10.1053/j.seminoncol.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 63.French KM, et al. A naturally derived cardiac extracellular matrix enhances cardiac progenitor cell behavior in vitro. Acta Biomater. 2012;8(12):4357–64. doi: 10.1016/j.actbio.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Das B, et al. Squalene selectively protects mouse bone marrow progenitors against cisplatin and carboplatin-induced cytotoxicity in vivo without protecting tumor growth. Neoplasia. 2008;10(10):1105–19. doi: 10.1593/neo.08466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saloustros E, Tryfonidis K, Georgoulias V. Prophylactic and therapeutic strategies in chemotherapy-induced neutropenia. Expert Opin Pharmacother. 2011;12(6):851–63. doi: 10.1517/14656566.2011.541155. [DOI] [PubMed] [Google Scholar]
- 66.Randall TD, Weissman IL. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood. 1997;89(10):3596–606. [PubMed] [Google Scholar]
- 67.Hsu YH, et al. Full range physiological mass transport control in 3D tissue cultures. Lab on a chip. 2013;13(1):81–9. doi: 10.1039/c2lc40787f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moya ML, et al. In Vitro Perfused Human Capillary Networks. Tissue Eng Part C Methods. 2013 doi: 10.1089/ten.tec.2012.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsu YH, et al. A microfluidic platform for generating large-scale nearly identical human microphysiological vascularized tissue arrays. Lab Chip. 2013 doi: 10.1039/c3lc50424g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malavia NK, et al. Airway epithelium stimulates smooth muscle proliferation. American Journal of Respiratory Cell and Molecular Biology. 2009;41(3):297–304. doi: 10.1165/rcmb.2008-0358OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Griffith CK, et al. Diffusion limits of an in vitro thick prevascularized tissue. Tissue Engineering. 2005;11(1-2):257–66. doi: 10.1089/ten.2005.11.257. [DOI] [PubMed] [Google Scholar]
- 72.Thompson HG, et al. Epithelial-derived TGF-beta2 modulates basal and wound-healing subepithelial matrix homeostasis. Am J Physiol Lung Cell Mol Physiol. 2006;291(6):L1277–85. doi: 10.1152/ajplung.00057.2006. [DOI] [PubMed] [Google Scholar]
- 73.Wikswo JP, et al. Engineering challenges for instrumenting and controlling integrated organ-on-chip systems. IEEE Trans Biomed Eng. 2013;60(3):682–90. doi: 10.1109/TBME.2013.2244891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wikswo JP, et al. Scaling and systems biology for integrating multiple organs-on-a-chip. Lab Chip. 2013;13(18):3496–511. doi: 10.1039/c3lc50243k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams LR, Leggett RW. Reference values for resting blood flow to organs of man. Clin Phys Physiol Meas. 1989;10(3):187–217. doi: 10.1088/0143-0815/10/3/001. [DOI] [PubMed] [Google Scholar]
- 76.Stringari C, et al. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc Natl Acad Sci U S A. 2011;108(33):13582–7. doi: 10.1073/pnas.1108161108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499(7458):295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Itzhaki I, et al. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS One. 2011;6(4):e18037. doi: 10.1371/journal.pone.0018037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan P, et al. Palette of fluorinated voltage-sensitive hemicyanine dyes. Proc Natl Acad Sci U S A. 2012;109(50):20443–8. doi: 10.1073/pnas.1214850109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xi J, et al. Comparison of contractile behavior of native murine ventricular tissue and cardiomyocytes derived from embryonic or induced pluripotent stem cells. FASEB J. 2010;24(8):2739–51. doi: 10.1096/fj.09-145177. [DOI] [PubMed] [Google Scholar]
- 81.Gibson RJ, Bowen JM. Biomarkers of regimen-related mucosal injury. Cancer Treat Rev. 2011;37(6):487–93. doi: 10.1016/j.ctrv.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 82.Xue H, et al. Nutrition modulation of gastrointestinal toxicity related to cancer chemotherapy: from preclinical findings to clinical strategy. JPEN J Parenter Enteral Nutr. 2011;35(1):74–90. doi: 10.1177/0148607110377338. [DOI] [PubMed] [Google Scholar]
- 83.Maor Y, Malnick S. Liver injury induced by anticancer chemotherapy and radiation therapy. Int J Hepatol. 2013;2013:815105. doi: 10.1155/2013/815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pabla N, Dong Z. Curtailing side effects in chemotherapy: a tale of PKCdelta in cisplatin treatment. Oncotarget. 2012;3(1):107–11. doi: 10.18632/oncotarget.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Argyriou AA, et al. Chemotherapy-induced peripheral neurotoxicity (CIPN): an update. Crit Rev Oncol Hematol. 2012;82(1):51–77. doi: 10.1016/j.critrevonc.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 86.Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol. 2010;6(12):657–66. doi: 10.1038/nrneurol.2010.160. [DOI] [PubMed] [Google Scholar]
- 87.Gilliam LA, St Clair DK. Chemotherapy-induced weakness and fatigue in skeletal muscle: the role of oxidative stress. Antioxid Redox Signal. 2011;15(9):2543–63. doi: 10.1089/ars.2011.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Norren K, et al. Direct effects of doxorubicin on skeletal muscle contribute to fatigue. Br J Cancer. 2009;100(2):311–4. doi: 10.1038/sj.bjc.6604858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blumenfeld Z. Chemotherapy and fertility. Best Pract Res Clin Obstet Gynaecol. 2012;26(3):379–90. doi: 10.1016/j.bpobgyn.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 90.Ragheb AM, Sabanegh ES., Jr Male fertility-implications of anticancer treatment and strategies to mitigate gonadotoxicity. Anticancer Agents Med Chem. 2010;10(1):92–102. doi: 10.2174/1871520611009010092. [DOI] [PubMed] [Google Scholar]
- 91.Hoekstra R, et al. Phase 1 and phase 2 drug metabolism and bile acid production of HepaRG cells in a bioartificial liver in absence of dimethyl sulfoxide. Drug Metab Dispos. 2013;41(3):562–7. doi: 10.1124/dmd.112.049098. [DOI] [PubMed] [Google Scholar]
- 92.Hilmer SN, Shenfield GM, Le Couteur DG. Clinical implications of changes in hepatic drug metabolism in older people. Ther Clin Risk Manag. 2005;1(2):151–6. doi: 10.2147/tcrm.1.2.151.62914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jang KJ, et al. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr Biol (Camb) 2013;5(9):1119–29. doi: 10.1039/c3ib40049b. [DOI] [PubMed] [Google Scholar]
- 94.Ould-Dris A, et al. Analysis of the mass transfers in an artificial kidney microchip. Journal of Membrane Science. 2010;352(1-2):116–125. [Google Scholar]
- 95.Naik P, Cucullo L. In vitro blood-brain barrier models: current and perspective technologies. J Pharm Sci. 2012;101(4):1337–54. doi: 10.1002/jps.23022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pedersen BK. Muscles and their myokines. J Exp Biol. 2011;214(Pt 2):337–46. doi: 10.1242/jeb.048074. [DOI] [PubMed] [Google Scholar]
- 97.Al Moundhri MS, et al. The effect of curcumin on oxaliplatin and cisplatin neurotoxicity in rats: some behavioral, biochemical, and histopathological studies. J Med Toxicol. 2013;9(1):25–33. doi: 10.1007/s13181-012-0239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li CL, et al. Survival advantages of multicellular spheroids vs. monolayers of HepG2 cells in vitro. Oncol Rep. 2008;20(6):1465–71. [PubMed] [Google Scholar]
- 99.David L, et al. Hyaluronan hydrogel: an appropriate three-dimensional model for evaluation of anticancer drug sensitivity. Acta Biomater. 2008;4(2):256–63. doi: 10.1016/j.actbio.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 100.Fujiwara D, et al. The usefulness of three-dimensional cell culture in induction of cancer stem cells from esophageal squamous cell carcinoma cell lines. Biochem Biophys Res Commun. 2013;434(4):773–8. doi: 10.1016/j.bbrc.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 101.Gurski LA, et al. Hyaluronic acid-based hydrogels as 3D matrices for in vitro evaluation of chemotherapeutic drugs using poorly adherent prostate cancer cells. Biomaterials. 2009;30(30):6076–85. doi: 10.1016/j.biomaterials.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Padron JM, et al. The multilayered postconfluent cell culture as a model for drug screening. Crit Rev Oncol Hematol. 2000;36(2-3):141–57. doi: 10.1016/s1040-8428(00)00083-4. [DOI] [PubMed] [Google Scholar]
- 103.Nirmalanandhan VS, et al. Activity of anticancer agents in a three-dimensional cell culture model. Assay Drug Dev Technol. 2010;8(5):581–90. doi: 10.1089/adt.2010.0276. [DOI] [PubMed] [Google Scholar]
- 104.Miyamoto H, et al. Tumor-stroma interaction of human pancreatic cancer: acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas. 2004;28(1):38–44. doi: 10.1097/00006676-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 105.Muerkoster S, et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004;64(4):1331–7. doi: 10.1158/0008-5472.can-03-1860. [DOI] [PubMed] [Google Scholar]
- 106.Serebriiskii I, et al. Fibroblast-derived 3D matrix differentially regulates the growth and drug-responsiveness of human cancer cells. Matrix Biol. 2008;27(6):573–85. doi: 10.1016/j.matbio.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Goel S, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10(2):415–27. doi: 10.1158/1078-0432.ccr-0642-03. [DOI] [PubMed] [Google Scholar]
- 109.Peters GJ, et al. Determinants of activity of the antifolate thymidylate synthase inhibitors Tomudex (ZD1694) and GW1843U89 against mono- and multilayered colon cancer cell lines under folate-restricted conditions. Cancer Res. 1999;59(21):5529–35. [PubMed] [Google Scholar]
- 110.Hicks KO, et al. Use of three-dimensional tissue cultures to model extravascular transport and predict in vivo activity of hypoxia-targeted anticancer drugs. J Natl Cancer Inst. 2006;98(16):1118–28. doi: 10.1093/jnci/djj306. [DOI] [PubMed] [Google Scholar]
- 111.Agastin S, et al. Continuously perfused microbubble array for 3D tumor spheroid model. Biomicrofluidics. 2011;5(2):24110. doi: 10.1063/1.3596530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen SY, Hung PJ, Lee PJ. Microfluidic array for three-dimensional perfusion culture of human mammary epithelial cells. Biomed Microdevices. 2011;13(4):753–8. doi: 10.1007/s10544-011-9545-3. [DOI] [PubMed] [Google Scholar]
- 113.Masur K, et al. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by beta-blockers. Cancer Res. 2001;61(7):2866–9. [PubMed] [Google Scholar]
- 114.Pasquier E, et al. Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: implication in breast cancer treatment. Oncotarget. 2011;2(10):797–809. doi: 10.18632/oncotarget.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Doherty KR, et al. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol Appl Pharmacol. 2013 doi: 10.1016/j.taap.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 116.Dolderer JH, et al. HERG1 gene expression as a specific tumor marker in colorectal tissues. Eur J Surg Oncol. 2010;36(1):72–7. doi: 10.1016/j.ejso.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 117.Glassmeier G, et al. Inhibition of HERG1 K+ channel protein expression decreases cell proliferation of human small cell lung cancer cells. Pflugers Arch. 2012;463(2):365–76. doi: 10.1007/s00424-011-1045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 119.Ruoslahti E. Specialization of tumour vasculature. Nat Rev Cancer. 2002;2(2):83–90. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- 120.Levesque JP, Winkler IG. It takes nerves to recover from chemotherapy. Nat Med. 2013;19(6):669–71. doi: 10.1038/nm.3231. [DOI] [PubMed] [Google Scholar]
- 121.Schimmel KJ, et al. Cardiotoxicity of cytotoxic drugs. Cancer Treat Rev. 2004;30(2):181–91. doi: 10.1016/j.ctrv.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 122.Wang Y, Probin V, Zhou D. Cancer therapy-induced residual bone marrow injury-Mechanisms of induction and implication for therapy. Curr Cancer Ther Rev. 2006;2(3):271–279. doi: 10.2174/157339406777934717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chatterjee K, et al. Vincristine attenuates doxorubicin cardiotoxicity. Biochem Biophys Res Commun. 2008;373(4):555–60. doi: 10.1016/j.bbrc.2008.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7(5):332–44. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- 125.Raschi E, De Ponti F. Cardiovascular toxicity of anticancer-targeted therapy: emerging issues in the era of cardio-oncology. Intern Emerg Med. 2012;7(2):113–31. doi: 10.1007/s11739-011-0744-y. [DOI] [PubMed] [Google Scholar]
- 126.Ewer MS, Lippman SM. Type II chemotherapy-related cardiac dysfunction: time to recognize a new entity. J Clin Oncol. 2005;23(13):2900–2. doi: 10.1200/JCO.2005.05.827. [DOI] [PubMed] [Google Scholar]
- 127.Telli ML, et al. Trastuzumab-related cardiotoxicity: calling into question the concept of reversibility. J Clin Oncol. 2007;25(23):3525–33. doi: 10.1200/JCO.2007.11.0106. [DOI] [PubMed] [Google Scholar]
- 128.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–64. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 129.Shin DH, et al. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood. 2008;111(6):3131–6. doi: 10.1182/blood-2007-11-120576. [DOI] [PubMed] [Google Scholar]
- 130.Ellis L, Hammers H, Pili R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009;280(2):145–53. doi: 10.1016/j.canlet.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Garber K. Angiogenesis inhibitors suffer new setback. Nat Biotechnol. 2002;20(11):1067–8. doi: 10.1038/nbt1102-1067. [DOI] [PubMed] [Google Scholar]
- 132.Kruzliak P, Kovacova G, Pechanova O. Therapeutic potential of nitric oxide donors in the prevention and treatment of angiogenesis-inhibitor-induced hypertension. Angiogenesis. 2013;16(2):289–95. doi: 10.1007/s10456-012-9327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]