

Abstract
The Ataxia Telangiectasia-mutated (ATM) kinase senses DNA double-strand breaks (DSBs) and facilitates their repair. In humans, ATM deficiency predisposes to B- and T-cell lymphomas, but in mice leads only to thymic lymphomas. We tested the hypothesis that increased DSB frequency at a cellular oncogene could promote B-cell lymphoma by generating ATM-deficient mice with a V(D)J recombination target (“DJβ” cassette) within c-myc intron one (“DA” mice). We also generated ATM-deficient mice carrying an Eμ-Bcl-2 transgene (“AB” mice) to test whether enhanced cellular survival could promote B-cell lymphomas. About 30% of DA or AB mice and nearly 100% of mice harboring the combined genotypes (“DAB” mice) developed mature B-cell lymphomas. In all genotypes, B-cell tumors harbored oncogenic c-myc amplification generated by breakage-fusion-bridge (“BFB”) from dicentric chromosomes formed through fusion of IgH V(D)J recombination-associated DSBs on chromosome 12 to sequences downstream of c-myc on chromosome 15. AB tumors demonstrate that B lineage cells harboring spontaneous DSBs leading to IgH/c-myc dicentrics are blocked from progressing to B cell lymphomas by cellular apoptotic responses. DA and DAB tumor translocations were strictly linked to the cassette, but occurred downstream, frequently in a 6-kb region adjacent to c-myc that harbors multiple cryptic V(D)J recombination targets, suggesting that bona fide V(D)J target sequences may activate linked cryptic targets. Our findings indicate that ATM deficiency allows IgH V(D)J recombination DSBs in developing B cells to generate dicentric translocations that via BFB cycles lead to c-myc-activating oncogenic translocations and amplifications in mature B cells.
Keywords: ATM deficiency, B Cell Lymphoma, V(D)J recombination, c-myc Amplification, Breakage-Fusion-Bridge
INTRODUCTION
The B-cell antigen receptor (BCR) and its secreted antibody form are heterodimers comprising immunoglobulin (Ig) heavy (IgH) and light (IgL) chains. T-cell antigen receptors (TCR) are similarly composed of either αβ or γδ heterodimers. Exons that encode N-terminal variable regions of Ig or TCR chains are assembled in developing bone marrow (BM) B cells and developing thymocytes by V(D)J recombination of germline V, D, and J segments (1). V(D)J recombination is initiated in progenitor (pro) B and T cells by the “RAG” endonuclease, which introduces DSBs at borders of a pair of V, D, or J segments and short conserved recombination signal sequences (RSS) (2). RAG-initiated DSBs at V, D, or J segments occur in the G1 cell-cycle phase, where they are joined by classical non-homologous DNA end-joining (C-NHEJ) (1). IgH V(D)J exons are assembled at the germline JH region just upstream of the Cμ exons, leading to expression of a μ IgH chain (1). Subsequent assembly of an IgL variable region exon and expression of an IgL protein generates IgM, which is expressed on the surface of the resulting mature B cells as a BCR (3).
The C-terminal constant region of IgH chains can be encoded by different sets of exons (CHs) that determine antibody class (e.g. IgM, IgG, IgA) (4). Surface IgM-expressing B cells migrate from the bone marrow (BM) to peripheral lymphoid organs, such as the spleen, where upon antigen stimulation they can undergo IgH class switch recombination (CSR) or Ig variable region exon somatic hypermutation (SHM). CSR changes antibody effector functions by replacing Cμ exons with one of several sets of CH exons that lie 100–200kb downstream (4). CSR employs DSBs initiated by activation-induced cytidine deaminase (AID) within large repetitive switch (S) regions that precede the various sets of CH exons (4). DSBs in the donor Sμ are joined to the DSBs in an acceptor S region by end-joining to complete CSR (1). For SHM, AID introduces lesions into variable region exons that are processed into mutations that contribute to BCR affinity maturation (5, 6).
Chromosomal translocations result from joining of two separate DSBs on the same or different chromosomes (1, 7). Depending on which ends of the DSBs are joined, inter-chromosomal translocations result in the joining of the centromeric portion of one chromosome to the telomeric portion of another or they can be joined to form dicentric chromosomes and/or acentric chromosome fragments (7). The high frequency of RAG-initiated DSBs generated during V(D)J recombination in developing B and T cells provides translocation substrates (8). Indeed, immature B- or T-cell lymphomas in humans and certain mouse models can harbor recurrent translocations that fuse RAG-initiated Ig or TCR locus DSBs to oncogenes and/or join them to other genomic DSBs in a way that deletes tumor suppressors (1, 9). Likewise, in B cells activated for CSR, AID-initiated IgH DSBs can serve as intermediates for chromosomal translocations that contribute to peripheral B-cell lymphomas (1,10). Cryptic RSSs or AID-targeting motifs also can lead to off-target RAG or AID activity, respectively, that contributes to translocations (10–14).
C-NHEJ maintains genomic integrity by re-joining DSBs and, thereby, suppressing chromosome breaks and translocations (1, 15). C-NHEJ is important in G1 when homologous recombination is not available to repair DSBs (15, 16). Despite marked genomic instability, C-NHEJ-deficient mice do not routinely develop cancer, at least in part, due to elimination of cells containing unrepaired DSBs by the G1/S cell cycle checkpoint (8). In this regard, mice with combined deficiency for C-NHEJ and p53, a tumor suppressor that activates the G1 checkpoint, are predisposed to pro-B-cell lymphomas (8) that routinely harbor complex IgH translocations with c-myc oncogene amplification, referred to as “complicons” (17). Such complicons derive from fusion of RAG-initiated IgH DSBs on chromosome 12 to DSBs downstream of c-myc on chromosome 15, generating dicentrics that lead to oncogenic c-myc amplification via a breakage-fusion-bridge (BFB) mechanism (17, 18). A key aspect of complicon formation is persistence of unrepaired RAG-initiated IgH DSBs due to the absence of C-NHEJ and an impaired G1 checkpoint resulting from absence of p53. Consequently, these unrepaired DSBs are replicated and can form dicentrics that promote BFB-mediated c-myc amplification (17, 18).
Unrepaired DSBs in G1, including V(D)J recombination and CSR-associated DSBs, activate the Ataxia Telangiectasia-mutated (ATM) kinase, which phosphorylates downstream factors that then form complexes in chromatin surrounding DSBs, preventing premature DSB separation and promoting joining by C-NHEJ (1, 19, 20). Additionally, this ATM-dependent DSB response activates p53 to enforce the G1 checkpoint (20). Human ATM deficiency leads to ataxia telangiectasia (AT), a condition that includes immunodeficiency and increased predisposition to T- and B-cell cancers (21). ATM inactivation in mice recapitulates some aspects of AT, including predisposition to thymic lymphoma (22). In mice, ATM-deficient thymic lymphomas harbor recurrent translocations involving RAG-initiated DSBs in the TCRδ locus on chromosome 14 that lead to chromosome 14 dicentrics and complicons, as well as joins of TCRδ to sequences on chromosome 12 downstream of IgH to create T(12;14) translocations that delete the Bcl11b haplo-insufficient T-cell tumor suppressor (23, 24). Thus, chromosomal aberrations of ATM-deficient thymic lymphomas appear mechanistically similar to those of C-NHEJ/p53-deficient pro-B-cell lymphomas, potentially because ATM deficiency leads to both a V(D)J joining defect and a G1-checkpoint defect. Notably, ATM deficiency allows unrepaired RAG-initiated IgH DSBs in developing pro-B cells to persist in mature B cells where they can be visualized as large centric chromosome 12 fragments that participate in chromosomal translocations (25). Yet, ATM deficiency in mice has not been reported to predispose to B-cell lymphoma (22).
We now have generated compound mutant mouse models that recurrently develop mature B-cell lymphomas in association with ATM deficiency. Characterization of these ATM-deficient B-cell lymphomas and the recurrent chromosomal translocations that they harbor suggests an unanticipated mechanism for their origin.
MATERIALS and METHODS
Generation of DA, AB, and DAB Mice
To generate the DA cohort, ATM+/− mice (26) were crossed to mice heterozygous or homozygous for the c-mycDJβ allele (27), and the resulting ATM+/−c-mycWT/DJβ offspring intercrossed to obtain experimental and control animals. To generate the AB cohort, mice harboring Eμ-Bcl-2 transgene (28) were bred into the ATM-deficient background. The DAB cohort was generated by crossing ATM+/−c-mycDJβ mice to ATM+/−Eμ-Bcl-2 mice. A productive VHB1-8 knock-in allele (29) was also crossed into the DA and DAB background. All animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital.
Flow-cytometry Analysis
Single-cell suspensions from tumor masses and control organs were stained with three sets of anti-mouse antibodies:αB220-CyChrome (eBiosciences), αCD43-FITC (BD Biosciences), and αIgM-RPE (Southern Biotech); αB220-CyChrome (eBiosciences), αIgκ-FITC (BD Biosciences), and αIgλ-PE (BD Biosciences); αCD3e-PECy5 (eBiosciences), αCD4-PE (eBiosciences), and αCD8a-FITC (eBiosciences). Data acquisition was performed on a FACSCalibur flow cytometer equipped with CellQuest software (Becton Dickinson). Analysis was performed with FlowJo software (Tree Star).
Southern Blotting
Southern blotting was performed with 10–15 μg of genomic DNA isolated from tumor masses or normal control tissues as described (30). The JH4–3 probe is a 1.6-kb HindIII/EcoRI fragment downstream of JH4. The Jκ probe is a 1-kb HindIII/BglII fragment downstream of Jκ5. The Cμ probe is a 869bp XbaI/BamHI fragment spanning Cμ exons 1 and 2. The Iμ probe is a 1-kb XbaI/PstI fragment. The MycA probe is a 1.7-kb XbaI fragment upstream of c-myc exon 1. The MycD probe is a 900bp XhoI/BamHI fragment between c-myc exons 1 and 2. The Myc3’ probe is 2.5 kb XhoI/BamHI fragment comprising the end of c-myc exon 3. A PCR fragment containing MDC1 exon 5 was used as a loading control probe.
c-myc Amplification Quantitation
Intensities of c-mycbands detected by Myc3’ probe were measured by ImageJ software (version 1.48). Fold amplification = (Is-m/Ic-m) / (Is0/Ic0), where Is-m and Ic-m are the intensities of sample band and control band on the Myc3’ Southern Blot; Is0 and Ic0 are the intensities of sample band and control band on the loading control Southern Blot.
Somatic Hypermutation Analysis
The genomic region comprising the rearranged V(D)J exon and the intron downstream of JH4 was PCR-amplified from tumor DNA using degenerate oligonucleotides corresponding to different VH families as forward primers and oligonucleotides downstream of JH4 as reverse primers, as described (31).
Comparative Genomic Hybridization (CGH) Assay
CGH was performed as described (24). Genomic DNA from normal spleen was used as control.
Northern Blotting
Total RNA from tumor and control samples were extracted with TriPure Isolation Reagents following the manufacturer’s instructions (Roche). Northern blotting was preformed according to standard procedures with 10μg of total RNA.
Isolation of translocation junctions
Tumor genomic DNA was digested with appropriate enzymes, (as evaluated by Southern blot analysis of c-myc rearrangements) and phenol/chloroform-purified. Translocation junctions were cloned following a protocol described previously (32), except that the DNA products after Emulsion PCR were further amplified through an additional 30-cycles regular PCR with the same primer. Expected PCR bands were separated on 1% agarose gel, extracted and Sanger-sequenced. Sequencing reads from each PCR products were aligned to mouse reference genome (Mouse July 2007-NCBI Build37/mm9) with the BLAT algorithm.
Cytogenetic Analyses
Metaphase spreads were prepared from lymphoma single-cell suspensions cultured for 3–6 hours in the presence of colcemid (KaryoMAX Colcemid Solution; GIBCO) according to standard protocols (33). Hybridization of metaphases with mouse SKY paints or single-chromosome paints specific for chr12 and chr15 and for two color FISH assays were as performed as described (33).
RESULTS
Mouse Models for ATM-deficient Peripheral B-cell Lymphomas
To test the hypothesis that DSB frequency around oncogenes such as c-myc may be a rate-limiting factor for B-cell lymphoma development in ATM-deficient mice, we generated ATM-deficient mice with a RAG target sequence (“DJβ”cassette) inserted in intron one of c-myc (“c-mycDJβ allele”). This cassette is cut efficiently by RAG during B-cell development (27). We intercrossed ATM+/− mice with c-mycDJβ/DJβ or c-mycDJβ/+ mice to generate ATM−/− c-mycDJβ/DJβ and ATM−/− c-mycDJβ/+ offspring (collectively referred to as “DA” mice), while ATM−/−, ATM+/− c-mycDJβ/DJβ, and ATM+/− c-mycDJβ/+ mice were kept as controls. Of 17 DA mice analyzed, 5 developed IgM+ peripheral B-cell lymphomas between 12 and 20 weeks; while the rest developed T-cell lymphomas with similar onset time and characteristics to those of ATM−/− mice (Fig. 1A; Supplementary Table S1) (24). The c-mycDJβ/DJβ, and ATM+/− c-mycDJβ/+ controls did not succumb to lymphomas or other neoplasias (Fig. 1A; Supplementary Table S1). B-cell lymphomas in the DA cohort presented in peripheral lymph nodes, mesenteric lymph nodes, spleen, and thymus, as well as in liver or kidney (Supplementary Table S2).
Figure 1. DA and AB Mice Develop B-Cell Lymphoma at an Early Age.
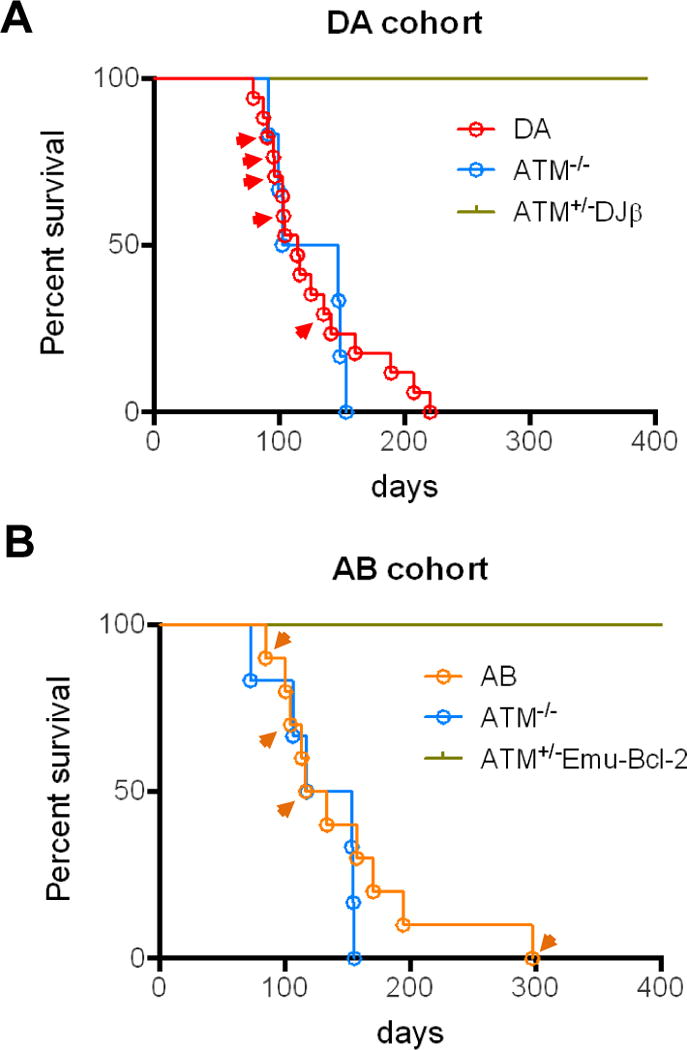
Kaplan-Meier curves showing survival of mice within the DA (A) and AB (B) cohorts. Arrowheads indicate mice that developed B-cell lymphoma.
To test whether enhanced survival of ATM-deficient B cells could promote B-cell lymphoma, we generated ATM-deficient mice that harbored an Eμ-Bcl-2 transgene. Eμ-Bcl-2 transgenic mice develop peripheral B-cell hyperplasia due to decreased apoptotic death of peripheral B cells (28, 34). For this experiment, ATM+/− mice were crossed with Eμ-Bcl-2 transgenic mice and resulting ATM+/−Eμ-Bcl-2 offspring bred to obtain ATM−/− Eμ-Bcl-2 (referred to as “AB”) mice, while ATM−/− mice lacking Eμ-Bcl-2 and ATM+/− Eμ-Bcl-2 mice were kept as controls. Within a cohort of ten AB mice, two developed IgM+ peripheral B-cell lymphomas and one developed an IgM− peripheral B-cell lymphoma between 12 and 17 weeks of age, each with a similar presentation as DA tumors. An older AB mouse developed what appeared to be an unrelated, IgM− B lineage tumor (AB36) in the gastrointestinal tract and spleen; while the remainder of the cohort developed standard ATM−/− T-cell lymphomas (Fig. 1B; Supplementary Tables S3 and S4). ATM+/− Eμ-Bcl-2 littermate controls did not succumb to lymphomas or other neoplasias (Fig. 1B; Supplementary Table S3).
We also generated ATM−/− mice that were either heterozygous or homozygous for the c-mycDJβ allele and which also carried the Eμ-Bcl-2 transgene (referred to as “DAB” mice). Of 9 DAB mice analyzed, 7 developed IgM+ B-cell lymphomas and another developed a related IgM− B-cell lymphoma between 7 and 14 weeks of age, all of which had similar characteristics as the predominant DA and AB B-cell tumors (Supplementary Tables S5 and S6; see below). In these cohorts, the ATM+/− c-mycDJβ Eμ-Bcl-2 or ATM+/− c-mycDJβ control mice again did not develop lymphomas or other neoplasias (Supplementary Table S5). Five DA mice were generated as a by-product of this cohort, and one (DA473) developed an IgM+ B-cell lymphoma (Supplementary Table S5). Finally, three of the DAB mice and one DA mouse (DA473) also carried a productive V(D)J exon knocked into one of their JH alleles to drive early B cell development (29); these four still developed IgM+ peripheral B-cell lymphomas that appeared very similar in characteristics to the others (Supplementary Table S5).
Most ATM-deficient B-Cell Lymphomas Arise Clonally from IgM+ Peripheral B Cells
Of the 17 related DA, AB and DABB-cell lymphomas, 15 expressed surface IgM and either Igκ (13 tumors) or Igλ (2 tumors), while the other two (AB225 and DAB361) did not express surface Ig (Supplementary Tables S2, 4, 6). Southern blotting assays of, respectively, EcoRI- or HindIII-digested tumor DNA for rearrangement of the IgH JH region or the Igκ Jκ region revealed distinct, clonal rearrangements of both JH loci and one or more clonal rearrangements of Jκ in most tumors, confirming monoclonal origin (Fig. 2 and Supplementary Fig. S1). Lack of two distinct rearranged JH bands in some tumors resulted from aberrant V(D)J joins in the absence of ATM that remove the downstream sequences recognized by the probe (see below). These assays also revealed amplification of the JH region and adjacent Cμ exons in most DA, AB, and DAB B-cell tumors (Fig. 2 and Supplementary Fig. S1). The two tumors that lacked surface IgM still had JH rearrangements and amplifications, as well as IgL rearrangements and deletions (Fig. 2, Supplementary Fig. S1, Supplementary Tables S4 and S6), suggesting loss of IgH expression subsequent to a translocation (e.g. ref 35). Sequencing of the region downstream of JH4 from 11 of the ATM-deficient B-cell lymphomas revealed no mutations, consistent with origin from peripheral B cells that had not undergone SHM or CSR (Supplementary Tables S2, 4, 6).
Figure 2. DA, AB, and DAB Tumors have Clonal IgH Rearrangements and c-myc Amplification.
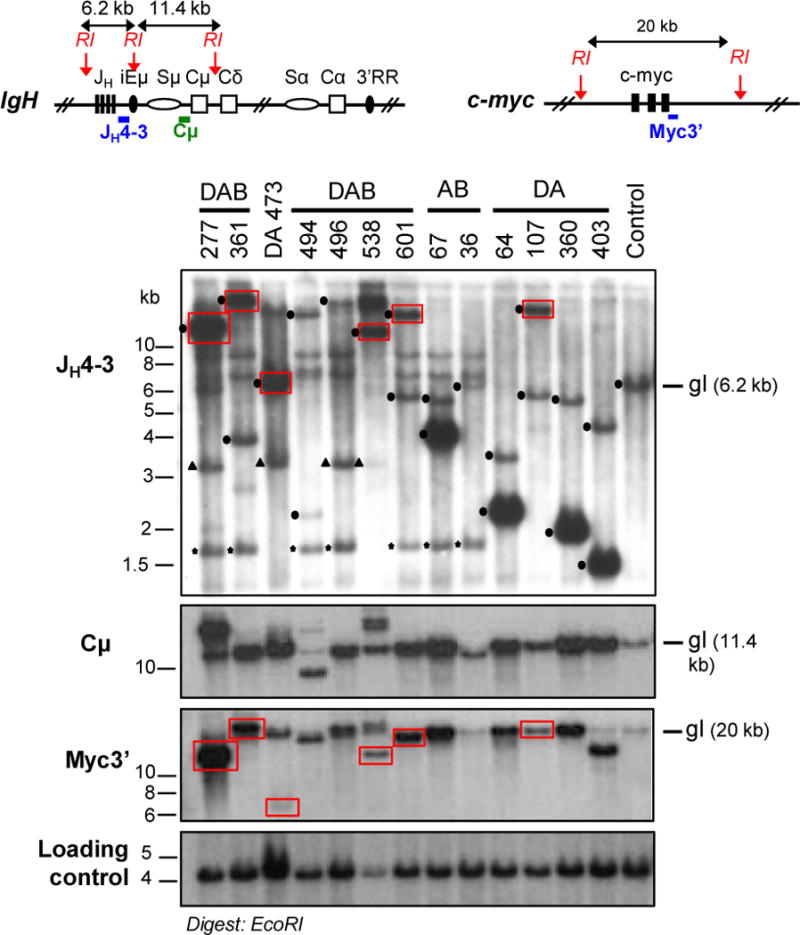
Southern blot analysis on EcoRI-digested genomic DNA of tumor samples from DA, AB, and DAB cohorts. Upper diagrams represent IgH (left) and c-myc (right) loci with position of EcoRI (RI) sites and probes used. For each Southern blot panel, molecular weight markers and probes are indicated on the left, and the running position of the germ-line (gl) band is indicated on the right. In the JH4–3 panel, dots indicate clonal V(D)J rearrangements, triangles indicate bands corresponding to the pre-rearranged VHB1–8 IgH locus present in some of the mice, and stars indicate the cross-reacting band from the Eμ-Bcl-2 transgene in DAB and AB mice. Red boxes indicate co-migrating rearranged c-myc and JH bands. Lack of amplified JH or c-myc band in some of the tumors is due of deletion of the region recognized by the probes upon translocation (details in Supplementary Tables S2, 4, 6).
ATM-deficient B-Cell Lymphomas Harbor Complex Chromosome 12 to 15 Translocations
Amplification of the JH/Cμ portion of IgH in conjunction with co-amplification of c-myc in the context of chromosome 12;15 complicons was previously observed in pro-B-cell lymphomas from C-NHEJ plus p53-deficient mice (17, 18). Southern blot analysis of EcoRI-digested DNA with a probe downstream of c-myc exon 3 (Myc3’) and/or with other probes internal to or upstream of c-myc (probes MycD and MycA, respectively) revealed c-myc amplification in all tumors with JH and/or Cμ amplification (Fig. 2 and Supplementary Fig. S1). Notably, some tumors had amplified rearranged c-myc-hybridizing EcoRI fragments, which in 6 of them co-migrated with JH4-3-hybridizing EcoRI fragment (Fig. 2, red boxes), suggesting that these translocations closely juxtapose c-myc to IgH. Additional Southern blotting studies with MycA and D probes also revealed rearrangements in the upstream portion of c-myc in several tumors (Supplementary Fig. S1). Consistent with c-myc amplification, Northern blot analyses revealed over-expression of c-myc transcripts in tumors that showed amplification (Supplementary Fig. S2A).
To characterize translocations cytogenetically, we performed fluorescent in situ hybridization (FISH) experiments in which several DA, AB, and DAB tumor metaphases were hybridized with paints specific for chromosome 12 (IgH) and 15 (c-myc), respectively. After image acquisition, paint signals were stripped and metaphases re-hybridized with BAC probes specific for the 3′ region of IgHor for c-myc. Translocations juxtaposing these two loci cytogenetically were present in all analyzed tumor metaphases, in the form of complicons that retained either the centromeric portion of chromosome 12 or that of 15, with the latter metaphases also routinely harboring a T(12;15) (Fig. 3A and B; Supplementary Tables S2, 4, 6). One or the other of these two types of IgH/c-myc complicons were found in C-NHEJ/p53-deficient pro-B-cell lymphomas and models for their origins have been discussed (17, 18).
Figure 3. ATM-deficient B-cell lymphomas harbor Clonal IgH/c-myc Complicons.
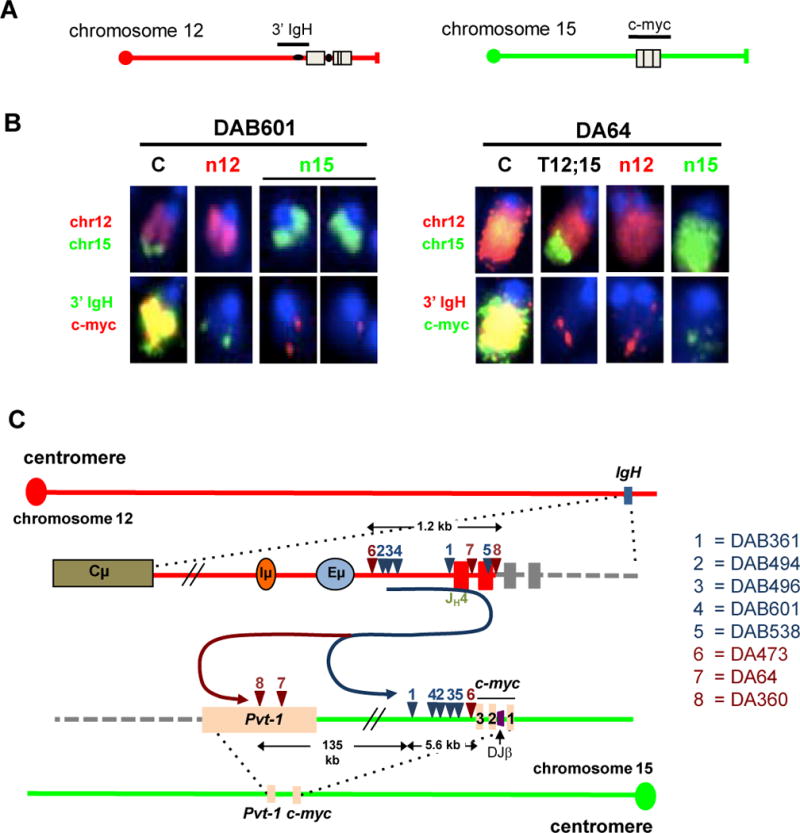
(A) Diagrams represent normal chromosomes 12 (left panel) and 15 (right panel) with position of either IgH or c-myc BAC probes. (B) FISH analysis showing two types of complicons (“C”); only relevant chromosomes (chrs) are shown. Tumors with chr12-based complicon (e.g. DAB601, left) always have one normal chromosome 12 (n12) and two normal chromosome 15s (n15); tumors with chr15-based complicons (e.g. DA64, right) also contain a T(12;15) juxtaposing IgH to sequences downstream of c-myc (details in Supplementary Tables S2, 4, 6). The same metaphase was sequentially hybridized with whole chromosome paints (top) and locus-specific probes (bottom). Probes used are indicated on the left. (C) Schematic showing location of sequenced chromosome 12 and chromosome 15 translocation breakpoints in DAB (blue) and DA (red) tumor samples. Corresponding numbers on the chromosomes diagrams denote the locations of the two break-points joined in a translocation in given tumors (listed on the right). Black arrows show the length of the indicated regions. On chromosome 15, c-myc exons 1, 2, and 3 are indicated. The DJβ cassette is indicated in purple between exon1 and exon 2 of c-myc.
Amplification of c-myc via a Breakage-fusion-bridge Mechanism
We employed CGH to examine amplification around the c-myc and IgH loci in selected tumor DNA samples from each cohort. This analysis showed that amplification at IgH began downstream of the D gene segments in all samples analyzed along with amplification of c-myc, which in some tumors ended immediately downstream of c-myc (DAB494, DAB496, DAB601), but in others (e.g. DAB64) extended 100kb downstream into the Pvt-1 locus (Supplementary Fig. S2B). In one tumor (DAB496) amplification on chromosome 15 was focused rather precisely on c-myc (Supplementary Fig. S2B). Translocation junctions from 8 tumors (DAB361, DAB494, DAB496, DAB601 and DAB538 and DA473, DA64, DA360) were cloned by a PCR approach and sequenced. Individual translocation break-points for each of the tumors involved a sequence lying within or just downstream of JH segments on chromosome 12, consistent with derivation from an attempted V(D)J recombination event, fused in “dicentric” orientation to a sequence on chromosome 15 lying within a region spanning from 99 bp to 140 kb downstream of c-myc (Fig. 3C; Supplementary Tables S2, 4, 6). Consistent with Southern blotting and CGH analyses data, chromosome 15 junctions in six of 8 tumors (DAB261, DAB494, DAB496, DAB601, and DAB538 and DA473) involved sequences lying within a 6-kb region just downstream of c-myc exon 3; whereas two other junctions were further downstream in the Pvt-1 locus (Fig. 3C; Supplementary Tables S2, 6). Southern blotting also revealed tumor DA107 to have an amplified junction just downstream of c-myc exon 3; whereas four additional c-myc-amplified tumors (DA403, AB67, AB143, and DAB277) had co-amplified downstream Pvt-1 sequences, suggesting dicentric translocation junctions likely occurred at least 100 kb downstream of c-myc (Supplementary Fig. S2C; Supplementary Tables S2, 4, 6).
Downstream Translocation Junctions are Promoted by the Upstream DJβ Cassette
Southern blot analyses with various digest and probe combinations revealed that, in most DA and DAB tumors, the DJβ cassette had undergone normal or aberrant rearrangements consistent with expected V(D)J recombination-associated events (27) in the absence of ATM (Fig. 4A and B, Supplementary Tables S2, 4, 6). However, as the major dicentric translocation junctions that result in c-myc amplification in DA and DAB tumors lie downstream of c-myc, the DJβ cassette within c-myc first intron was not directly involved. To determine whether the DJβ cassette indirectly influenced translocations linked in cis, we performed Southern blotting with Myc3’ probe on KpnI-digested DNA from the nine DA or DAB tumors that were heterozygous for the DJβ allele. This blotting strategy distinguishes wild-type (WT) from c-mycDJβ alleles. Strikingly, all 9 of these c-mycDJβ/WT tumors had undergone the downstream dicentric translocation on the chromosome 15 containing the c-mycDJβ allele (Fig. 4C). In this regard, it is notable that a substantial fraction of the DAB and DA translocation breakpoints clustered within the 6-kb region just downstream of c-myc in the vicinity of a series of strong cryptic RSSs (Fig. 4D; Supplementary Table 7). Notably, no strong cryptic RSSs were found within the 6.4-kb region containing the c-myc gene that lies just upstream (Fig. 4D; ref 36).
Figure 4. Chromosome 15 Translocations in DAB and DA Tumors Involve the c-mycDJβ Allele.
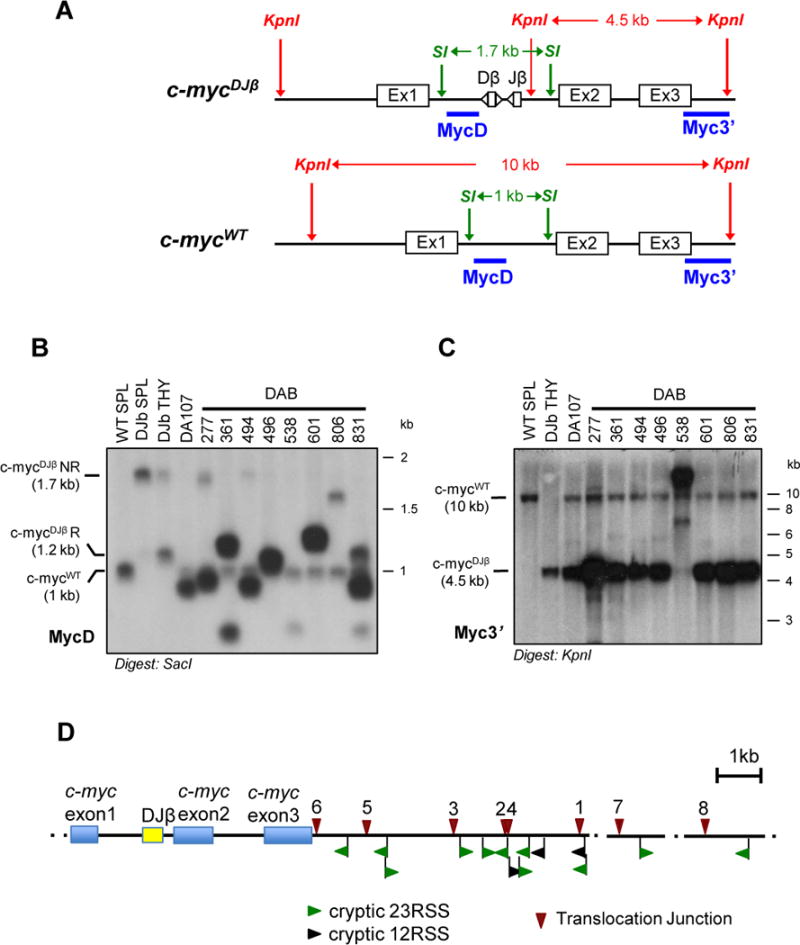
(A) Map of c-mycWT and c-mycDJβ alleles showing SacI (SI) and KpnI (KI) restriction sites and location of probes. (B and C) Southern blot analysis of SacI- (B) or KpnI-digested (C) genomic DNA from DA and DAB tumors with indicated probes. Molecular weight markers and running position of WT c-myc (c-mycWT), non-rearranged c-mycDJβ (c-mycDJβNR) and rearranged c-mycDJβ (c-mycDJβR) alleles are indicated. DNA from 129/Sve spleen (WT SPL) and c-mycDJβ/DJβ spleen (DJβ SPL)and thymus (DJβ THY) were used as controls. In the MycD blot (B), rearrangements of the c-mycDJβ allele are compatible with precise (496) or aberrant (other tumors) V(D)J recombination joins within the DJβ cassette in the absence of ATM. In the Myc3’ blot (C), tumor 538 shows an amplified and rearranged KpnI fragment derived from the c-mycDJβ allele, as expected from the cloned translocation breakpoint (Fig. 4 and Supplementary Table S6). Tumors 538 and 806 shows amplification with Myc3’ probe but not MycD probe, consistent with data shown in Fig. 2 and Supplementary Fig. S1. (D) Schematic showing 1he positions of tumor translocation junctions (top) and cryptic RSSs (bottom) in the region from c-myc exon one to 6 kb downstream and two other short regions more than 100 kb downstream of c-myc. Prediction of cryptic RSSs within this 6kb downstream region was generated with the program described at: http://www.itb.cnr.it/rss/index.html. Note that no strong cryptic RSS was predicted to lie in the 6.4 kb region, spanning the c-myc gene, upstream of the first left cryptic RSS. Blue boxes represent c-myc exons; yellow box represents the DJβ cassette. Red triangles indicate junctions from 1: DAB361, 2: DAB494, 3: DAB496, 4: DAB601, 5: DAB538, 6: DA473, 7: DA64, 8: DA360 tumors. Cryptic 23RSSs are showed in green, while 12RSSs in black; arrowheads indicate RSSs in (−) orientation if pointing to left, RSSs in (+) orientation if pointing to the right.
DISCUSSION
We have developed the DA, AB, and DAB compound mutant mouse models, which, in the context of ATM deficiency, all develop spontaneous IgM+ peripheral B-cell lymphomas that harbor clonal translocations from the IgH JH region to sequences downstream of c-myc to form dicentric derivatives and IgH/c-myc amplifications in the context of complicons. The general structure of the IgH/c-myc complicons in these tumors is strikingly similar to that of RAG-dependent complicons found in pro-B lymphomas from C-NHEJ/p53-deficient mice (17, 18), with the clustering of IgH translocation breakpoints in or near the JH region, strongly indicating that the IgH complicon break-point in ATM-deficient tumors is also RAG-initiated. In the absence of C-NHEJ and p53, pro-B cells replicate persistent RAG-initiated JH DSBs, cycling them into dicentrics and complicons (17, 18). ATM-deficiency results in both V(D)J recombination defects and impaired G1 checkpoints (20, 25, 37); but, unlike C-NHEJ deficiency, does not block V(D)J joining (37). Thus, we propose that some ATM-deficient pro-B cells generate dicentric chromosomes from replication of persistent RAG-initiated DSBs on one IgH allele, but that productive V(D)J rearrangements on the other allele allows such B cells to develop to the IgM+ B cell stage, where they contribute to B-cell lymphomagenesis. Our finding that incorporating a preassembled IgH variable region into the DAB model does not alter the tumor outcome supports this interpretation. Overall, our findings lead us to propose that ATM suppresses mature B-cell lymphomas in susceptible backgrounds by preventing the conversion of unrepaired RAG-initiated IgH breaks in pro-B cells into dicentric chromosomes that, through BFB cycles, give rise to new DNA breaks, translocations and gene amplifications that contribute to mature B-cell tumors (Fig. 5). We now have provided direct support for this model in parallel studies that employed our high throughput genome-wide translocation sequencing (HTGTS) method to study translocations of c-myc DSBs in WT and ATM-deficient B cells (38).
Figure 5. A Proposed BFB Mechanism for the Generation of c-myc Complicons Found in ATM-deficient B-cell Lymphomas.
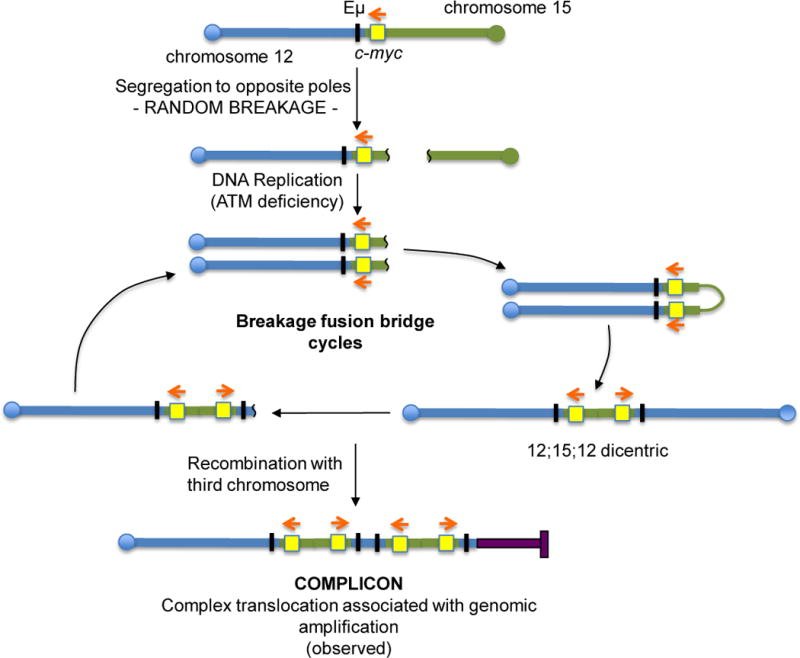
In ATM-deficient pro-B cells, RAG-initiated DSBs generated during DH-to-JH recombination at IgH, are not properly repaired and can be jointed to DSB downstream of c-myc locus, leading to the generation of 12;15 dicentric chromosomes. Dicentrics are unstable and can be broken again during chromosome segregation. In the absence of ATM, these unrepaired chromosomes can persist and due to lack of a G1 checkpoint can be replicated to generate dicentrics. Repeated breakage-fusion-bridge (BFB) cycles result in further amplification of c-myc. Finally, a complicon is formed when the dicentric is stabilized by recombination with a third chromosome via telomere capture. More details of this general model as it applies to NHEJ/p53 double deficient pro-B-cell lymphomas can be found in ref 17. Chr12 is showed in blue, chr15 in green, and the third chromosome participating to the complicon in purple. The black bar represents Eμ enhancer and the yellow box represents c-myc. The red arrows indicate c-myc transcriptional orientation.
The AB B-cell lymphoma model confirmed that the Eμ-Bcl-2 transgene promotes survival of ATM-deficient developing or mature B cells with capacity to contribute to mature B-cell lymphomas. On the other hand, the DJβ cassette in the DA and DAB models did not lead to B-cell lymphoma by increasing DSBs and translocations within c-myc intron one. In human B-cell lymphomas, c-myc intron one is a major translocation hotspot (39). However, peripheral B-cell lymphomas that arise in a different mouse model routinely harbor spontaneous, standard IgH/c-myc translocation junctions upstream of the c-myc promoter (30, 40). Together, these findings are consistent with the possibility that translocations into mouse c-myc intron one may incapacitate the gene by disrupting the alternative promoter upstream of c-myc exon two (41, 42). Yet, the strict linkage of the downstream dicentric translocations in DAB tumors to the c-mycDJβ allele confirms that the cassette contributes to their appearance. Moreover, the clustering of a substantial fraction of the DAB and DA dicentric junctions within 6-kb region just downstream of c-myc near strong cryptic RSSs suggests a potential mechanism (Fig. 4D). We propose that the DJβ cassette may contribute to downstream DSBs by providing a bona fide set of proximal 12/23 RSSs for pairing with closely linked downstream cryptic RSSs; thereby, enhancing RAG cutting at these sites and potentially others within so-called megabase “proximity” domains (1, 43). In the absence of ATM, such RAG-initiated DSBs might be aberrantly repaired (44, 45), with downstream breaks and translocations arising in cells in which cassette DJβ DSBs are resolved within the cassette, thereby preserving c-myc integrity. Notably, dicentric translocations junctions in pro-B lymphomas from C-NHEJ/p53-deficient mice, which do not harbor the DJβ cassette, did not occur within this 6-kb c-myc proximal region, but rather occurred much further downstream (17, 18). While this finding is consistent with the enhanced RSS cleavage model we propose, we note that differences in the effects of C-NHEJ deficiency versus ATM deficiency with respect to repair of particular types of DSBs also could theoretically contribute.
Why do AB, DA, and DAB tumors all arise in association with IgH/c-myc translocations/amplifications generated by the same basic BFB mechanism? The occurrence of AB tumors indicate B lineage cells harboring spontaneous DSBs that lead to IgH/c-myc dicentrics normally arise in the ATM-deficient background at low frequency; but that their progression into B-cell lymphomas is dampened by cellular responses that lead to apoptotic cell death. On the other hand, introduction of the DJβ cassette into intron one of c-myc in the ATM-deficient background appears to greatly increase DSB frequency, perhaps at cryptic RSSs downstream of c-myc, thereby potentially increasing dicentric translocations and providing sufficient numbers of cells with dicentric intermediates that some escape cell death. In this scenario, the compound DAB mutant would both increase translocation frequency and allow cells with such translocations to survive, further increasing tumor penetrance to nearly 100%, as observed.
The peripheral DA, AB, and DAB B-cell lymphomas arise from peripheral IgM+ B cells that had not undergone either CSR or SHM. The lack of B-cell lymphomas derived from germinal center (GC) B cells in ATM-deficient mice is surprising given that activated ATM−/− B cells in culture form AID-initiated dicentrics and other translocations at a high frequency (33). Thus, the lack of such tumors in ATM-deficient mice might result from more strict checkpoints in GC B lymphocytes, which eliminate cells carrying off-target AID DNA damage. In this context, perhaps the susceptible ATM-deficient newly generated IgM+ B-cell population is more tolerant of DNA damage or oncogenic stress than ATM-deficient GC B cells. In addition, progenitors for ATM−/− IgM+ B-cell lymphomas may arrive in the periphery with activated c-myc genes or c-myc genes predisposed to amplification that allows them to more readily achieve full oncogenic transformation. These findings could be relevant to certain pre-GC human B-cell lymphomas, for example, a subset of mantle cell lymphomas (MCLs) derive from pre-GC B cells with ATM mutations (46). MCLs also frequently harbor translocations between chromosome 14 and chromosome 11 that lead to deregulation of cyclinD1 gene through translocation to the IgH JH region and a number of MCLs have complex chromosomal rearrangements with co-amplified IgH/cyclinD1 genes (47, 48). The potential relevance of our findings for mechanisms of B-cell lymphomagenesis in AT patients awaits a more extensive and in depth characterization of these human ATM-deficient B-cell tumors.
Supplementary Material
Acknowledgments
Grant support: This work was supported by grant P01CA109901 from the National Cancer Institute of the National Institutes of Health and a Leukemia and Lymphoma Society of America (LLS) SCOR grant to F.W.A. F.W.A. is an Investigator of the Howard Hughes Medical Institute. M.G. is supported by an Eleanor and Miles Shore/Boston Children’s Hospital Career Development Fellowship Award. J.H. is supported by the Robertson Foundation/Cancer Research Institute Irvington Fellowship.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
S.T., J.H., M.G, and F.W.A. designed the study, analyzed the data and wrote the manuscript; S.T., J.H., and M.G. performed research.
References
- 1.Alt FW, Zhang Y, Meng F-L, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–29. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet. 2010;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- 3.Cobb RM, Oestreich KJ, Osipovich OA, Oltz EM. Accessibility control of V(D)J recombination. Adv Immunol. 2006;91:45–109. doi: 10.1016/S0065-2776(06)91002-5. [DOI] [PubMed] [Google Scholar]
- 4.Matthews AJ, Zheng S, DiMenna LJ, Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol. 2014;122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 6.Storb U. Why does somatic hypermutation by AID require transcription of its target genes? Adv Immunol. 2014;122:253–77. doi: 10.1016/B978-0-12-800267-4.00007-9. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Gostissa M, Hildebrand DG, Becker MS, Boboila C, Chiarle R, et al. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv Immunol. 2010;106:93–133. doi: 10.1016/S0065-2776(10)06004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gostissa M, Alt FW, Chiarle R. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annual Rev Immunol. 2011;29:319–50. doi: 10.1146/annurev-immunol-031210-101329. [DOI] [PubMed] [Google Scholar]
- 9.Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141:27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol. 2013;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 11.Larmonie NS, Dik WA, Meijerink JP, Homminga I, van Dongen JJ, Langerak AW. Breakpoint sites disclose the role of the V(D)J recombination machinery in the formation of T-cell receptor (TCR) and non-TCR associated aberrations in T-cell acute lymphoblastic leukemia. Haematologica. 2013;98:1173–84. doi: 10.3324/haematol.2012.082156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greisman HA, Lu Z, Tsai AG, Greiner TC, Yi HS, Lieber MR. IgH partner breakpoint sequences provide evidence that AID initiates t(11;14) and t(8;14) chromosomal breaks in mantle cell and Burkitt lymphomas. Blood. 2012;120:2864–7. doi: 10.1182/blood-2012-02-412791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Z, Tsai AG, Akasaka T, Ohno H, Jiang Y, Melnick AM, et al. BCL6 breaks occur at different AID sequence motifs in Ig-BCL6 and non-Ig-BCL6 rearrangements. Blood. 2013;121:4551–4. doi: 10.1182/blood-2012-10-464958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai AG, Lieber MR. Mechanisms of chromosomal rearrangement in the human genome. BMC Genomics. 2010;11(Suppl 1):S1. doi: 10.1186/1471-2164-11-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 17.Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–21. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 18.Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, et al. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med. 2002;196:469–80. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bednarski JJ, Sleckman BP. Lymphocyte development: integration of DNA damage response signaling. Adv Immunol. 2012;116:175–204. doi: 10.1016/B978-0-12-394300-2.00006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 21.Perlman S, Becker-Catania S, Gatti RA. Ataxia-telangiectasia: diagnosis and treatment. Semin Pediatr Neurol. 2003;10:173–82. doi: 10.1016/s1071-9091(03)00026-3. [DOI] [PubMed] [Google Scholar]
- 22.Lavin MF. The appropriateness of the mouse model for ataxia-telangiectasia: neurological defects but no neurodegeneration. DNA Repair. 2013;12:612–19. doi: 10.1016/j.dnarep.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood. 2011;118:4169–73. doi: 10.1182/blood-2010-11-318873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zha S, Bassing CH, Sanda T, Brush JW, Patel H, Goff PH, et al. ATM-deficient thymic lymphoma is associated with aberrant tcrd rearrangement and gene amplification. J Exp Med. 2010;207:1369–80. doi: 10.1084/jem.20100285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Callen E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A, et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007;130:63–75. doi: 10.1016/j.cell.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 26.Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, et al. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci U S A. 2000;97:3336–41. doi: 10.1073/pnas.050584897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranganath S, Carpenter AC, Gleason M, Shaw AC, Bassing CH, Alt FW. Productive coupling of accessible Vbeta14 segments and DJbeta complexes determines the frequency of Vbeta14 rearrangement. J Immunol. 2008;180:2339–46. doi: 10.4049/jimmunol.180.4.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A. 1991;88:8661–5. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sonoda E, Pewzner-Jung Y, Schwers S, Taki S, Jung S, Eilat D, et al. B cell development under the condition of allelic inclusion. Immunity. 1997;6:225–33. doi: 10.1016/s1074-7613(00)80325-8. [DOI] [PubMed] [Google Scholar]
- 30.Gostissa M, Yan CT, Bianco JM, Cogné M, Pinaud E, Alt FW. Long-range oncogenic activation of Igh-c-myc translocations by the Igh 3′ regulatory region. Nature. 2009;462:803–7. doi: 10.1038/nature08633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ehlich A, Martin V, Muller W, Rajewsky K. Analysis of the B-cell progenitorcompartment at the level of single cells. Curr Biol. 1994;4:573–83. doi: 10.1016/s0960-9822(00)00129-9. [DOI] [PubMed] [Google Scholar]
- 32.Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho Y-JJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–19. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–14. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 34.McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, et al. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 35.Wang JH, Alt FW, Gostissa M, Datta A, Murphy M, Alimzhanov MB, et al. Oncogenic transformation in the absence of Xrcc4 targets peripheral B cells that have undergone editing and switching. J Exp Med. 2008;205:3079–90. doi: 10.1084/jem.20082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merelli I, Guffanti A, Fabbri M, Cocito A, Furia L, Grazini U, et al. RSSsite: a reference database and prediction tool for the identification of cryptic Recombination Signal Sequences in human and murine genomes. Nucleic Acids Res. 2010;38:W262–7. doi: 10.1093/nar/gkq391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol. 2011;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu J, Tepsuporn S, Meyers RM, Gostissa M, Alt FW. Developmental Propagation of V(D)J Recombination-Associated DSBs and Translocations in Mature B cells Via Dicentric Chromosomes. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1410112111. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janz S. Myc translocations in B cell and plasma cell neoplasms. DNA Repair. 2006;5:1213–24. doi: 10.1016/j.dnarep.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 40.Wang JH, Gostissa M, Yan CT, Goff P, Hickernell T, Hansen E, et al. Mechanisms promoting translocations in editing and switching peripheral B cells. Nature. 2009;460:231–6. doi: 10.1038/nature08159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernard O, Cory S, Gerondakis S, Webb E, Adams JM. Sequence of the murine and human cellular myc oncogenes and two modes of myc transcription resulting from chromosome translocation in B lymphoid tumours. EMBO J. 1982;2:2375–83. doi: 10.1002/j.1460-2075.1983.tb01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saito H, Hayday AC, Wiman K, Hayward WS, Tonegawa S. Activation of the c-myc gene by translocation: a model for translational control. Proc Natl Acad Sci U S A. 1983;80:7476–80. doi: 10.1073/pnas.80.24.7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gostissa M, Schwer B, Chang A, Dong J, Meyers RM, Marecki GT, et al. IgH class switching exploits a general property of two DNA breaks to be joined in cis over long chromosomal distances. Proc Natl Acad Sci U S A. 2014;111:2644–9. doi: 10.1073/pnas.1324176111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–70. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- 45.Huang CY, Sharma GG, Walker LM, Bassing CH, Pandita TK, Sleckman BP. Defects in coding joint formation in vivo in developing ATM-deficient B and T lymphocytes. J Exp Med. 2007;204:1371–81. doi: 10.1084/jem.20061460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stankovic T, Stewart GS, Fegan C, Biggs P, Last J, Byrd PJ, et al. Ataxia telangiectasia mutated-deficient B-cell chronic lymphocytic leukemia occurs in pregerminal center cells and results in defective damage response and unrepaired chromosome damage. Blood. 2002;99:300–9. doi: 10.1182/blood.v99.1.300. [DOI] [PubMed] [Google Scholar]
- 47.Menanteau MR, Martinez-Climent JA. Genomic profiling of mantle cell lymphoma. Methods Mol Biol. 2013;973:147–63. doi: 10.1007/978-1-62703-281-0_9. [DOI] [PubMed] [Google Scholar]
- 48.Setoodeh R, Schwartz S, Papenhausen P, Zhang L, Sagatys EM, Moscinski LC, et al. Double-hit mantle cell lymphoma with MYC gene rearrangement or amplification: a report of four cases and review of the literature. Int J Clin Exp Pathol. 2013;6:155–67. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.