

Abstract
To enhance the water miscibility and increase the mechanical properties of dentin adhesives, a new glycerol-based monomer with vinyl and carboxylic acid, 4-((1,3-bis(-methacryloyloxy)propan-2-yl)oxy)-2-methylene-4-oxobutanoic acid (BMPMOB), was synthesized and characterized. Dentin adhesive formulations containing 2-hydroxyethyl methacrylate (HEMA), 2,2-bis[4-(2-hydroxy-3-methacryloxypropoxy) phenyl]propane (BisGMA), and BMPMOB were characterized with regard to real-time photopolymerization behavior, water sorption, dynamic mechanical analysis, and microscale three-dimensional internal morphologies and compared with HEMA/BisGMA controls. The experimental adhesive copolymers showed higher glass transition temperature and rubbery moduli, as well as improved water miscibility compared to the controls. The enhanced properties of the adhesive copolymers indicated that BMPMOB is a promising comonomer for dental restorative materials.
Keywords: water miscibility, dentin adhesive, dynamical mechanical property, photopolymerization, crosslink polymer
INTRODUCTION
Dentin adhesive is a blend of hydrophobic and hydrophilic monomeric compounds used to obtain amphiphilic materials that bond the hydrophobic restorative composite to the tooth. Acid-etching provides effective mechanical bonding between enamel and adhesive, but bonding to dentin has been fraught with problems. The two fundamental processes involved in bonding adhesives to dentin are: removal of the mineral phase from the dentin substrate without altering the collagen matrix and filling the voids left by the mineral with adhesive that undergoes complete in situ polymerization, that is, the hybrid layer.1 The ideal hybrid layer would be characterized as a three-dimensional (3D) polymer/collagen network that provides both a continuous and stable link between the bulk adhesive and dentin substrate. Numerous studies indicate that this ideal objective is not achieved.2–13
The monomers used in dentin adhesives are critical components because polymerization of the resin monomers produces a crosslinked matrix that provides chemical/physical stability to the hybrid layer.14–19 Monomer selection exerts considerable influence on the properties, durability, and operation of dentin adhesive in the wet, oral environment. Although numerous monomers have been studied, the effectiveness and durability of adhesive copolymers is still a major problem.16,20–23
Under in vivo conditions, it is nearly impossible to control the amount of water left on the tooth and the adhesive undergoes physical separation (hydrophobic-rich and hydrophilic-rich phases) at the wet surface.24 It is well known that the hydrophobic monomers bond poorly to wet dentin.25,26 An increase in water miscibility of the adhesive formulation can reduce the detrimental impact of phase separation, but hydrophilic monomers are plagued by problems such as increased water sorption and decreased mechanical properties.24,27,28 A balance between hydrophobicity and hydrophilicity of adhesive formulations is crucial to promote the durability of dentin adhesives.
Previously our group synthesized a urethane-linked trimethacrylate, 1,1,1-tri-[4-(methacryloxyethlaminocarbonyloxy)-phenyl]ethane (MPE). With the increase in MPE concentration, the adhesives showed less water sorption and higher crosslink density than the 2-hydroxyethyl methacrylate (HEMA)/2,2-bis[4-(2-hydroxy-3-methacryloxypropoxy) phenyl]propane (BisGMA) control.29 Then we synthesized another dimethacrylate with a branched aromatic carboxylic acid group, 2-((1,3-bis(methacryloyloxy)-propan-2-yloxy)carbonyl)benzoic acid (BMPB) to enhance the water miscibility of the adhesive formulations.30 With the addition of BMPB, the formulation can tolerate higher water concentration to suppress detrimental adhesive phase separation. However, the adhesive copolymers may experience reduced mechanical properties under wet conditions when compared with the HEMA/BisGMA control. In combination, these results suggested that the trimethacrylate monomer MPE can increase the crosslink density of the copolymers, and the carboxylic acid containing dimethacrylate monomer BMPB is miscible with and can tolerate higher water content.
In this work, a new glycerol-based dimethacrylate monomer with a vinyl and a carboxylic acid group is proposed as a dental comonomer that will provide improved water miscibility and dynamic mechanical properties in the wet environment. The objective of this work was to synthesize and characterize a new multifunctional monomer and to evaluate the properties of a dentin adhesive formulated using this new monomer under conditions that simulate the wet, oral environment.
MATERIALS AND METHODS
Materials
BisGMA (Polysciences, Warrington, PA) and HEMA (Acros Organics, NJ) were used as received without further purification as monomers in dentin adhesives. BMPB was synthesized as reported previously in our laboratory.30 4-((1,3-Bis(methacryloyloxy)propan-2-yl)oxy)– 2-methylene-4-oxo-butanoic acid (BMPMOB) was synthesized in our laboratory and used as a comonomer (Figure 1). Itaconic anhydride (IAA) and glycerol dimethacrylate (GDMA) were obtained from Sigma-Aldrich (St. Louis, MO). Camphoroquinone (CQ), ethyl-4-(dimethylamino) benzoate (EDMAB), and diphenyliodonium hexafluorophosphate (DPIHP) were obtained from Sigma-Aldrich. All other chemicals were reagent grade and used without further purification.
FIGURE 1.

Reaction scheme for synthesis of BMPMOB.
Synthesis of monomer BMPMOB
The new monomer, BMPMOB, was synthesized based on the following procedures. IAA (0.08 mol) was dissolved in tetrahydrofuran (20 mL) and toluene (60 mL) at room temperature (RT, 25°C). Sulfuric acid was added to the mixture, followed by dropwise addition of GDMA (0.07 mol). The reaction was stirred at 60°C for 3 h. Then the temperature was raised to 80°C and maintained at this temperature for another 3 h. The reaction was monitored by thin layer chromatography (mobile phase: CH2Cl2:MeOH = 5:1). After the reaction was completed, the solvent was removed by rotary evaporation. Diethyl ether was added, and the unreacted IAA was removed by filtration. The organic phase was extracted with 0.5 M NaCHO3. The aqueous basic layer was acidified to a pH of 5–6 with 1 M HCl and then extracted with ethyl acetate. After drying over anhydrous MgSO4, the solvent was removed with a rotary evaporator at 35–40°C to obtain BMPMOB as colorless oil. The scheme for the BMPMOB synthesis is shown in Figure 1. The structure of this synthesized compound (BMPMOB) was confirmed using Fourier transform infrared spectrophotometry (FTIR, Spectrum 400; Perkin-Elmer, Waltham, MA), proton nuclear magnetic resonance (1H–NMR), and carbon nuclear magnetic resonance (13C-NMR; FT-400 MHz Bruker Spectrometer, CDCl3 as solvent) spectroscopy.
Preparation of adhesive formulations
HEMA/BisGMA (45/55, mass ratio) and HEMA/BisGMA/BMPB (45/30/25, mass ratio) were used as control-0 (C0) and control-1 (C1), respectively.29,30 It was found that when BMPB content was 25% (C1) in the adhesive formulation, the polymer showed optimum mechanical properties.29,30 The experimental adhesive formulations (designated E1) consisting of HEMA, BisGMA, and BMPMOB are listed in Table I, in which BMPMOB is used as the third comonomer. CQ (0.5 wt %), EDMAB (0.5 wt %), and DPIHP (0.5 wt %) were used as a three-component photoinitiator system,31,32 with respect to the total amount of monomers. Mixtures of monomers/photoinitiators are prepared in a brown glass vial under an amber light. The preparation of adhesive formulations and their polymer beams have been reported previously.30,33 The solutions containing the monomers/photoinitiators were mixed overnight at 25°C to promote complete dissolution and formation of a homogeneous solution. The prepared resins were injected into a glass-tubing mold (Fiber Optic Center, part no.: ST8100; New Bedford, MA) and light-cured for 40 s at RT with a LED light curing unit (LED Curebox, 200 mW cm−2 irradiance; Proto-tech, Portland, OR). The polymerized samples were stored in dark at 25°C for at least 48 h before being used. The resultant cylindrical beam specimens (1.0 –mm diameter × 15 mm length) were used to determine water sorption, dynamic mechanical properties, and microscale morphologies.
Mixtures for compatibility with water
About 0.5 g of each neat resin was weighed into a brown vial, and water was added in increments of approximately 0.005 g until the mixture was visually observed to be turbid. The percentage of water in the mixture was noted (w1). The mixture was then back titrated using the neat resin until the turbidity disappeared, and the percentage of water in the mixture was noted (w2). Then the water miscibility (Wwm, %) of liquid formulation was calculated as the average of w1 and w2. Three specimens of each material were measured.
Determination of log P
The log P values (ratio of solubility in octanol to solubility in water) for each of the monomers and the model adhesive formulations were predicted using ChemBioDraw Ultra 12.0 (Cambridge Soft from Perkin Elmer).33 The log P value for each adhesive formulation was determined using the mole fraction-average of individual monomer values as seen in the following equation:
| (1) |
where xHEMA, xBisGMA, and xcomonomer are the mole fractions of HEMA, BisGMA, and BMPB/BMPMOB comonomer, respectively.
Water sorption and solubility of adhesive copolymer
The experimental protocol for the water sorption analyses has been reported previously.33 In brief, water sorption and solubility were measured using cylindrical beam specimens (1 mm × 15 mm). Five specimens were prepared for each adhesive formulation. The polymerized samples were stored at 25°C for 2 days in a dark room. Samples were weighed (m1) with a calibrated electronic balance (resolution of 0.01 mg, Mettler Toledo, XS 205 Dual range, Columbus, OH) and were immersed in distilled water to prewash for 7 days. Next, the specimens were dried in a vacuum oven in the presence of freshly dried silica gel at 45°C. The samples were removed every 8 h to determine the weight. This process was continued until a constant mass (m2) was recorded for each beam specimen. After prewashing, the dried specimens were immersed in distilled water and at fixed time intervals (3 h, 6 h, 24 h, 2 days, 3 days, 6 days, and 9 days), they were removed, blotted to remove excess water, weighed (m3) and returned to the water until a constant weight was obtained. All the specimens were then removed from the water and placed in a vacuum oven at 45°C until a constant weight was achieved (m4). The values (%) for water sorption (Wsp) and solubility (Wsu) were calculated by the following equations:
| (2) |
Real-time double bond conversion and maximal polymerization rate
The degree of conversion (DC) and polymerization behavior were determined by FTIR as described previously by our group.32,34 Real-time in situ monitoring of the photopolymerization behavior of the different adhesive formulations was performed using an infrared (IR) spectrometer (Spectrum 400 Fourier transform infrared spectrophotometer; Perkin-Elmer) at a resolution of 4 cm−1. One drop of adhesive solution was placed on the diamond crystal top plate of an attenuated total reflectance (ATR) accessory (PIKE Technologies Gladi-ATR, Madison, WI) and covered with a mylar film to prevent oxygen inhibition of polymerization. A 40-s exposure to the commercial visible light-polymerization unit (Spectrum® 800; Dentsply, Milford, DE) at an intensity of 550 mW cm−2 was initiated after 50 IR spectra had been recorded. Real-time IR spectra were continuously recorded for 600 s after light activation began. A time-based spectrum collector (Spectrum TimeBase; Perkin-Elmer) was used for continuous and automatic collection of spectra during polymerization. A minimum of three replicates were obtained for each adhesive formulation. The change in the band ratio profile-1637 cm−1 (C=C)/1608 cm−1 (phenyl) was monitored, and DC was calculated using the following equation based on the decrease in the absorption intensity band ratio before and after light curing.
| (3) |
The average of the last 50 values of the time-based spectra is reported as the DC value. The maximum polymerization rate was determined using the maximum slope of the linear region of the DC–time plots.31 For all experimental groups, the differences between DC or polymerization rate were analyzed using one-way analysis of variance (ANOVA), together with Tukey’s test at α = 0.05 (Microcal Origin Version 8.0; Microcal Software, Northamp-ton, MA) to identify significant differences in the means.
Dynamic mechanical analysis
Dynamic mechanical analysis (DMA) is a thermal analysis technique that measures the properties of materials as they are deformed under periodic stress. In this study, DMA tests were performed using a TA instruments Q800 DMA (TA Instruments) with a three-point bending clamp. The dynamic mechanical properties of methacrylate-based dentin adhesives have been described by our group.34 A sinusoidal stress is applied, and the resultant strain is measured. The properties measured under this oscillating loading are storage modulus, loss modulus, and tan δ. The storage modulus (E′) represents the stiffness of a visco-elastic material and is proportional to the energy stored during a loading cycle. The loss modulus (E″) is related to the amount of energy lost due to viscous flow. The ratio of loss (E″) to storage modulus (E′) is referred to as the mechanical damping, or tan δ. The frequency used to measure the storage modulus is 1 Hz with an amplitude of 15 μm and a preload of 0.01 N.33,35 The storage modulus is measured from 10 to 250°C with a ramping rate of 3°C min−1. The glass transition temperature (Tg) is determined as the position of the maximum on the derivate storage modulus versus temperature plots. Cylindrical beam specimens (1.0 mm diameter × 15 mm length) prepared as described previously are used for DMA measurements. A minimum four specimens of each material are measured.
Wet-condition DMA tests are operated using the three-point submersion clamp.29 Cylindrical beam specimens (1.0 mm × 15 mm) are immersed in water at 25°C for 5 days to be fully hydrated. The test temperature is varied from 5 to 80°C with a ramping rate of 1.5°C min−1. The results are analyzed statistically using one-way ANOVA, together with Tukey’s test at α = 0.05 (Microcal Origin Version 8.0, Microcal Software).
X-ray microcomputer tomography
To simulate the moist environment of the mouth and compare the copolymer’s microscale structure after polymerization with water, the adhesives C0 and E1-25 were formulated with 13 wt % water. The beams were prepared as mentioned above. The microscale morphologies of cylindrical beam specimens cured in the presence of 13 wt % water were observed using 3D Micro X-ray Computer Tomography (MicroXCT-400; Xradia, Pleasanton, CA). Computer tomography (CT) facilitates viewing of an object in 3D and allows selection of virtual slices spaced, thus illustrating the bulk structure of heterogeneous materials. As described previously,30 the transmission X-ray imaging of the samples was performed using an X-ray tube with a tungsten anode setting of 50 kV at 8 W with an optical magnification of 20X. The 3D images were constructed with the help of software “XM Reconstructor 8.0” (Xradia) using 1600 images acquired with a 10-s exposure time per image.
RESULTS
The structure of the newly synthesized monomer BMPMOB is characterized by FTIR, 1H-NMR, and 13C-NMR spectroscopy (see Figure 2). The characteristic FTIR peaks for BMPMOB are 1716 cm−1 (C=O stretching), 1637 cm−1 (C=C stretching), 1293, 1238, and 1154 cm−1 (C–O stretching), and 813 cm−1 (C=C bending). Disappearance of the anhydride peak at 1843 and 1764 cm−1 confirmed the formation of the new monomer. In the 1H-NMR spectrum, the chemical shifts of BMPMOB are (ppm): (a) 1.93 (6H, –CH3), (b) 3.37 (2H, –CH2), (c) 4.27–4.42 (4H, –CH2O), (d) 5.37 (1H, –CH), (e) 5.60, 6.12 (4H, =CH2), and (f) 5.84, 6.46 (2H, =CH2). In the 13C-NMR spectrum, the chemical shifts of BMPMOB are (ppm): (a) 18.1 (C=C–CH3), (b) 37.0 (CH2–COO), (c) 62.3 (O–CH(CH2)2), (d) 69.1 (O–CH(CH2)2), (e) 126.0 (CH2=C–CH3), (f) 130.9 (CH2=C), (g) 131.8 (CH2=C), (h) 135.6 (CH2=C–CH3), (i) 166.2 (CH2–COO), (j) 166.7 (–COOH), and (k) 169.9 (=C(CH3)COO).
FIGURE 2.

FTIR (A), 1H-NMR (B), and 13C-NMR (C) spectra in CDCl3 of BMPMOB. EA: ethyl acetate. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
As can be seen in Figure 3, the water compatibility of the adhesive formulations is enhanced with the increase in BMPMOB concentration. Compared with BMPB, the water miscibility of the formulation increased from 11.2 to 13.7% when the third comonomer concentration was 25 wt % (Table I). At the same time, the difference (ΔW) between water miscibility of the liquid formulation and water sorption of the solid copolymer showed a maximum value when the BMPMOB concentration is 25%. At this concentration, the water miscibility of the liquid formulation was 13.7%, and the water sorption of the E1–25 copolymer was 10.8%. Compared to the difference value of C0 (ΔW =0.8%), the ΔW of E1-25 was 2.9%, which was threefold larger than the value of the control.
FIGURE 3.
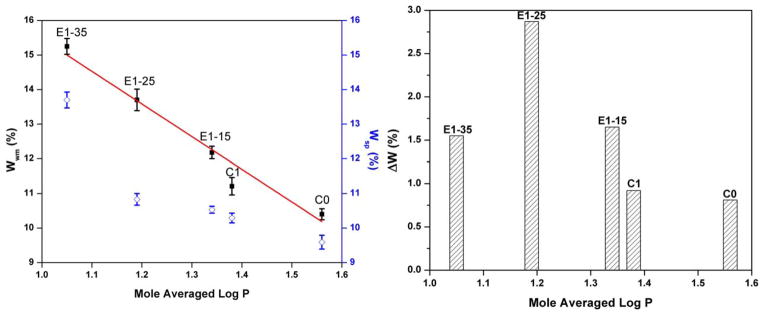
Relation between mole averaged log P and water miscibility of adhesive formulations, water sorption of copolymers contained BMPMOB. Wwm stands for water miscibility, and Wsp stands for water sorption. (■ represents the water miscibility value of liquid formulation; ▨ represents the water sorption of copolymer beam specimen, ⋄ represents the difference between water miscibility of liquid formulation and water sorption of copolymer, ΔW = Wwm − Wsp). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
TABLE I.
Adhesive Formulations of Control and Experimental and Results of Water Miscibility
| Sample | C0 | C1 | E1-15 | E1-25 | E1-35 |
|---|---|---|---|---|---|
| HEMA | 45 | 45 | 45 | 45 | 45 |
| BisGMA | 55 | 30 | 40 | 30 | 20 |
| BMPB | – | 25 | – | – | – |
| BMPMOB | – | – | 15 | 25 | 35 |
| C=C (mmol g−1) | 5.60 | 5.96 | 6.34 | 6.83 | 7.32 |
| Converted C=C (mmol g−1)a | 3.86 | 4.24 | 3.96 | 3.93 | 3.66 |
| Log P (mol avg.) | 1.56 | 1.38 | 1.34 | 1.19 | 1.05 |
| Wwm (%) | 10.40 ± 0.16 | 11.21 ± 0.25 | 12.18 ± 0.18 | 13.70 ± 0.31 | 15.25 ± 0.23 |
The converted C=C was calculated based on the total C=C concentration multiplied by the degree conversion of double bond obtained by real-time FTIR method.
The internal morphology of the control and experimental adhesive copolymers are shown in Figure 4. The voids in the control (A) indicate microphase separation when the control is polymerized in the presence of 13 wt %. In comparison, no voids are observed in the experimental adhesive (B) when it is polymerized in the presence of 13 wt % water.
FIGURE 4.

The microscale morphologies of the control (A1: CT slice at the x–y plane from the 3D image, A2: 3D image) and experimental E1-25 formulation (B1: CT slice at x–y plane from 3D image, B2: 3D image) adhesive copolymer prepared in the presence of 13 wt % water. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Real-time photopolymerization kinetic behavior of the controls and experimental adhesives are shown in Figure 5. With the increase in BMPMOB concentration from 0 to 35, the DC decreased from about 68 to 50%, and the maximum polymerization rate decreased from 0.24 to 0.05 s−1. The control (C0) reached its maximum polymerization rate at about 23% conversion, whereas BMPMOB containing adhesives (E1–15, E1–25, and E1–35) reached their maximum at about 20, 10, and 5%, respectively.
FIGURE 5.
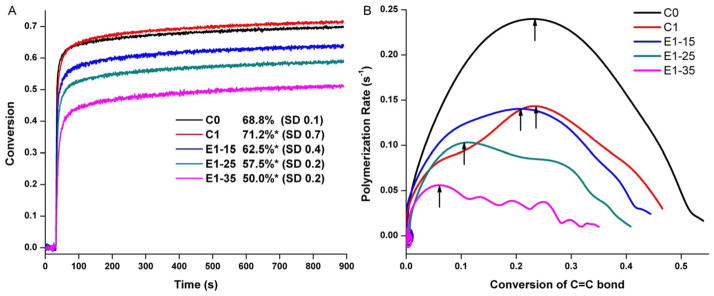
Real-time conversion (A) and comparison of polymerization rates (B) of control (C0 and C1) and experimental adhesives (E1-15, E1-25, and E1-35). The adhesives were light-cured for 40 s at RT using a commercial visible light lamp (Spectrum® 800; Dentsply, Milford, DE. Intensity is 550 mW cm−2). *Significantly (p < 0.05) different from the control (C0). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 6 shows, with the increase in BMPMOB concentration, the final water sorption and solubility of the synthetic copolymers are significantly (p < 0.05) higher than the control (C0). The final water sorption percentage of controls (C0 and C1) and experimental (E1-15, E1-25, and E1-35) are 9.89 ± 0.20, 10.29 ± 0.15, 10.53 ± 0.10, 10.79 ± 0.15, and 13.70 ± 0.23, respectively. The solubility of controls (C0, C1) and experimental (E1-15, E1-25, and E1-35) are 0.98 ± 0.17, 1.45 ± 0.11, 1.71 ± 0.13, 2.77 ± 0.19, and 3.61 ± 0.58, respectively.
FIGURE 6.
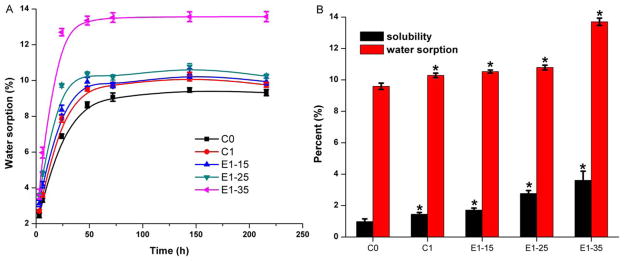
Water sorption of adhesive copolymers with time (A) and the final solubility and water sorption after storage in water at 25°C for 9 days (B). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The dynamic mechanical property of adhesive copolymers in the dry condition is measured by a three-point bending clamp, and in the wet condition it is determined by a three-point water submersion clamp method at various temperatures. These results are summarized in Table II. In dry conditions, the storage modulus values at 37°C, rubbery modulus values and height of tan δ peak for the experimental adhesives were significantly different from control (p < 0.05). The first glass transition temperature (Tg1) obtained from the derivate storage modulus versus temperature profile is significantly different at the level 0.05; however, the second glass transition temperature (Tg2) is not significantly different (p < 0.05). In wet condition, for all samples, the storage modulus decreased slowly at low temperatures (25, 37°C). As the temperature to the transition region was reached (~60°C), the storage modulus decreased markedly [see Figure 7(B)]. Particularly at 75°C, the modulus of E1–25 sample was significantly (p < 0.05) greater and others were significantly (p < 0.05) lower than that of the control.
TABLE II.
DMA Data for Control and Experimental Adhesive Copolymers in the Dry and Wet Conditions
| Sample | Dry Condition
|
Wet Condition
|
||||||
|---|---|---|---|---|---|---|---|---|
| Modulus at 37°C (MPa) | Rubbery Modulus (MPa) | Tg1 (°C) | Tg2 (°C) | ΔTg (°C) | Modulus at (MPa)
|
Tg a(°C) | ||
| 37°C | 75°C | |||||||
| C0 | 4129 ± 149 | 39 ± 1 | 69.1 ± 3.3 | 139.0 ± 1.3 | 69.9 ± 3.8 | 2123 ± 126 | 550 ± 41 | 61.2 ± 0.6 |
| C1 | 4665b ± 148 | 61b ± 1 | 65.7 ± 2.7 | 133.2 ± 2.6 | 67.5 ± 5.2 | 2112c ± 14 | 370b,c ± 24 | 59.6b,c ± 0.5 |
| E1-15 | 4497b ± 73 | 77b ± 2 | 63.0b ± 1.8 | 135.4 ± 2.8 | 72.4 ± 4.5 | 2294c ± 76 | 387b,c ± 19 | 59.7b,c ± 0.1 |
| E1-25 | 4585b ± 88 | 98b ± 2 | 62.8b ± 0.1 | 145.0 ± 2.9 | 82.2b ± 2.9 | 2109c ± 102 | 639b,d ± 32 | 60.9c,d± 0.1 |
| E1-35 | 4783b ± 77 | 95b ± 6 | 60.9b ± 2.6 | 143.5 ± 3.3 | 82.6b ± 3.4 | 1696b,d ± 65 | 447b,e ± 19 | 58.5b,e ± 0.0 |
Tg is obtained from the derivate storage modulus–temperature profile.
Significantly (p < 0.05) different from control C0.
Significantly (p < 0.05) different from each other.
FIGURE 7.

Representative storage modulus (A and B), derivative storage modulus (C and D), and tan δ (E and F) versus temperature curves of the control and experimental adhesive copolymers in dry (A, C, and E) and wet (B, D, and F) condition. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
With the increase in BMPMOB concentration, the storage modulus of dry samples at 37°C is significantly (p < 0.05) higher than C0. However, the storage modulus of samples in wet condition shows no difference (p < 0.05) with C0, except E1–35. From the derivative storage modulus profile in dry condition, the lower transition temperatures decrease from about 69.1 to 60.9°C, and the higher glass transition temperature increase from about 139.0 to 145.0°C (for details see Table II). The ΔT values of E1–25 and E1–35 are significantly higher than the control at the level of 0.05. In wet condition, all of the transition temperatures are close to 60°C. In Figure 7(C), with an increase in BMPMOB concentration, the widths of the tan δ curves become broad. However, in wet condition, the full tan δ curves cannot be obtained due to the limitation in the DMA instrument that the water medium temperature could not be over 80°C.
DISCUSSION
The chemistry of monomers controls the structure and properties of the polymer network.23,36–40 This relationship is critical in determining the durability and stability when the crosslinked polymers are used in an aqueous environment. Monomers containing polar groups such as carboxylic acid, hydroxyl, ester, amine, and/or ether moieties are hydrophilic, and are helpful to increasing the water miscibility of adhesive formulations. Usually, the mechanical properties of adhesives in wet condition are poor compared with those in dry condition. This is due to the adsorbed water acting as a plasticizer in the adhesive copolymers. First, we introduced the carboxylic acid functional group into this new monomer, which would be helpful to a dentin adhesive formulation because of improved water miscibility. Second, the copolymerizable vinyl functional group and lower molecular weight of the new monomer (Mw of BMPMOB and BisGMA is 340.33 and 512.59 g mol−1, respectively; when BMPMOB was used to partially replace BisGMA, the C=C double bond content in the experimental formulations was higher than that of the control) may lead to the copolymer achieving a higher cross-link density and may enhance the mechanical performance in the oral environment (wet condition). In Table I, the higher converted C=C mole number of the E1–25 formulation can be viewed as indirect evidence of a more highly crosslinked structure. Third, the carboxylic acid group may be beneficial to promote the interaction between the adhesive and the dental substrate, such as collagen or the mineral hydroxyapatite, via hydrogen bond or electrostatic attraction.41,42
Figure 3 shows the water miscibility of the liquid formulations (including control and experimental) and mole averaged log P. A negative correlation with R2 =0.96 is found for a linear fit to the data. The negative correlation shows that the contribution of the hydrophilicity of liquid formulations contributes to higher water miscibility. With the increase in BMPMOB concentration, the water sorption of copolymers increased accordingly. Compared to the control, increasing the BMPMOB concentration from 15, 25, and even to 35 wt %, caused the water miscibility of the experimental formulation to increase by about 17, 32, and 50%, respectively. Comparing the difference between Wwm and Wsp of the same formulation, E1–25 shows an inflection point [Figure 3(B)]. This result shows that the Wsp value is lower than the theoretical predication, which indicates that the water sorption behavior is affected by the hydrophilicity of monomers as well as the structure of the copolymer.
With the introduction of the BMPMOB monomer, the water miscibility of the adhesive formulation increased. A homogenous solution was obtained by mixing the control adhesive (C0) and experimental adhesives (E1–25) with 10.4 and 13.7 wt % water, respectively. When the water concentration was greater than the miscibility of the liquid formulation, voids were observed from the micro-CT images [Figure 4(A)]. The results indicate that the experimental formulation tolerated higher water concentration during polymerization.
The polymerization behavior (reactivity and DC) plays an important role in determining the quality of the interfacial hybrid layer. Many studies have reported that conversion of the double bond in the multifunctional methacrylates is rarely complete because the mobility of the propagating free radicals species is reduced as the reaction progresses.43 It has been observed that DC decreased with an increase in the concentration of the new monomer BMPMOB. This is consistent with our previous work.29,44 The difference in DC of controls (C0 and C1) and experimental adhesives may be due to the difference in monomer functionality. In our previous work,30 when BMPB was used to replace part of BisGMA, the viscosity of the formulation decreased, and the DC increased accordingly. BMPB is a dimethacrylate and has the same functionality as that of the BisGMA. In this work, the control contains mono- and difunctional methacrylates, while the experimental sample contains mono-, di-, and trifunctional monomers. The multi-functional methacrylate system will most likely vitrify more quickly, trapping unreacted pendant methacrylate groups. With the increase in BMPMOB concentration, the C=C bond concentration increases accordingly in the formulations (Table I). However, due to the lower DC of C=C obtained from the FTIR spectrum, the converted C=C bond concentration of the experimental decreased. Although, the physical and mechanical properties of dentin adhesive copolymer would be optimal when the C=C DC can reach 100%, the reported values for the DC usually range from 40 to 85%.45 However, it is not easy to clarify the relationship between DC and mechanical properties,46 which are mainly affected by the chemical structure of the monomer.44 In the present work, the DC of C1 was higher than that of E1–25. However, the mechanical properties of C1 were poor compared to E1–25. BMPMOB contains a vinyl group, and it is difficult to distinguish the true DC of vinyl groups and methacrylate groups from the real-time FTIR spectra. The reactivity of the vinyl group is lower than that of methacrylate group. Therefore, it is possible that the C=C bond with lower conversion was mainly affected by the vinyl groups.
The maximum polymerization rates decrease and appear early with the increase in BMPMOB concentration. The control (C0) reached its maximum polymerization rate at about 23% conversion, whereas BMPMOB-containing E1-15, E1-25, and E1-35 adhesives reached their maximum at about 20, 10, and 5%, respectively. Usually, with the increase in functionality, the earlier and steeper autoacceleration can be observed at a lower double bond DC.47 This is due to the localized increases in viscosity of the polymerizing system, which slows termination reactions. The original viscosity of the formulation and the functionality of the monomer determine the maximum polymerization rate and its appearance time, respectively.47 The viscosities of the experimental adhesive formulations are less than the control (unpublished data), which is probably due to the lower molecular weight of BMPMOB and the decreased concentration of BisGMA in the experimental formulations. When the BMPMOB weight percent is over 25%, the bimodal polymerization rate can be observed from the Rp-conversion curves in Figure 5.
During photopolymerization of a dental adhesive/composite, the concept of gel point refers to the transformation of viscous fluid to an elastic gel status. Vitrification occurs after the gel point when the elastic gel transforms to a glass.48 Usually, the maximum polymerization rate will appear due to the autoacceleration (gel effect) in bulk free radical polymerization. According to convention, the steep acceleration of the reaction in the crosslinking polymerization is considered to be the gel point.49 However, the dentin adhesive can form a network structure in just a few seconds of light irradiation, and it is difficult to determine the gel point. When the polymer changes from a viscous state to glassy state, the polymerization rate will usually undergo a significant decrease as the reaction becomes controlled by the diffusion of reactants. It is common for complete vitrification to result in a decrease in the rate of polymerization by 2–3 orders of magnitude. Therefore, the vitrification point can be determined as the conversion at the maximum rate of polymerization.50 In our experiments, the maximum polymerization rates appear at lower degrees of conversion in the experimental formulations (E1-15, E1-25, and E1-35) compared with C0. This indicates that the gel and vitrification point come early in the experimental formulations due to the BMPMOB’s higher functionality. After the gel points are reached, the formed microgels connect with each other, forming the complete polymer network. The sooner the gel/vitrification point occurs, the more heterogeneous and the weaker the structure in some regions of the copolymer. This result can be indirectly verified by the lower storage modulus of E1–35 sample in wet condition.
Since water or saliva is present in the mouth during the dentin bonding, the hygroscopic effect can influence the polymer network’s dimensional stability, mechanical properties, and chemical stability. When the adhesive formulation is photopolymerized in the mouth, the copolymer can be saturated by water, and the polymer matrix can change over time. Therefore, the water sorption and solubility of the adhesive copolymer can be indirectly used to characterize the stability of the network. From Figure 5, the water sorption of the control or experimental adhesive copolymers can reach equilibrium at about 48 h. At the same time, with the increase in BMPMOB concentration the curves became steeper at the initial 24 h mark. This indicates water penetrates more quickly into the experimental copolymers than that of the control. With the increase in BMPMOB concentration from 25 to 35%, there was an obvious increase in the water sorption of the copolymers. This is due to the higher polarity of the carboxylic acid group.
The degree of crosslink density is another important determinant of mechanical properties as well as structural durability. The importance of structural durability is implied because the higher the crosslinking of the adhesive copolymer the less it swells and degrades in the oral environment. To our knowledge, there is currently no method to directly measure the degree of crosslinking. Usually, molecular mobility is reduced with the increase in crosslink density, which is inferred from an increased glass transition temperature (Tg) or decrease in tan δ peak maximum intensity. However, because Tg depends on the DC and other factors such as monomer viscosity and functionality, it is difficult to distinguish between the effects of each of the different factors that contribute to the crosslink density. In our group, we have used the inverse ratio (ζ)29 of the modulus in rubbery region to the temperature and ve33 (ve = E′/3RTg, here E′ is the apparent rubbery modulus, Tg is the glass transition temperature obtained from the tan δ versus temperature profile obtained by the DMA test) to present the relative crosslink density of an adhesive copolymer. The values of ζ or ve all suggested higher relative crosslink density when part of the BisGMA was replaced by BMPMOB in the formulation. Usually, with the increase in crosslink density, the water sorption and solubility of the copolymer will decrease. However, there is a contradiction between the results of the crosslink density and water sorption experiment. Although the crosslink density values of the copolymer were enhanced with the addition of BMPMOB, the water sorption and solubility increased accordingly. The factors contributing to these results could be: (1) BMPMOB is more hydrophilic than BisGMA, so the copolymer will show more hydrophilicity; (2) the crosslink structure becomes heterogeneous (higher crosslink region and lower crosslink region) with an increase in BMPMOB amount; and (3) the photoinitiator system might not be optimized for the experimental formulation.
The DMA temperature scan is particularly well suited to characterize the viscocelastic behavior and has been suggested as a valuable method to obtain network information, such as the crosslink density and heterogeneity. The intensity of maximum tan δ peak reflects the extent of the mobility of the copolymer chain segments at the transition temperature. It is reported that the heterogeneity will increase as the crosslink agent concentration increases within a copolymer system.47 In the current work, BMPMOB can be considered to be a trifunctional comonomer. The broad glass transition range contributes to heterogeneous network containing both highly crosslinked and less densely crosslinked regions. The span of the tan δ peak, or the difference between Tg1 and Tg2, indicates that the glass transition occurs over a wide temperature range. When compared with the data of the maximum rate of polymerization shown in Figure 5, it can be determined that the gel effect came earlier, and the structure of the copolymer became more heterogeneous. All of the experimental copolymers showed lower tan δ peak height than the control, indicating increased elastic behavior and greater crosslink density. The modulus at the rubbery plateau is related to the crosslink density of the polymer. It has been reported that with the increase in a monomer’s functionality, a crosslink structure with a higher glass transition temperature, larger storage modulus and smaller average molecular weight between crosslinks will be formed.47 In the dry condition, the rubber moduli reach a maximum when BMPMOB concentration is 25%. Due to the higher functionality of BMPMOB, the maximum polymerization rate comes early, which indicates that the heterogeneity increases. According to the literature, highly crosslinked structures are very heterogeneous due to the ready formation of a gel region (which is region that has above average crosslink density localized around a center of initiation)51–53 and are considered to have the potential for weaknesses. Therefore, these regions should be carefully evaluated in different adhesive formulations.
In Figure 7, the lower transition temperature may be related to the loosely crosslinked domain of the polymer network; whereas the higher transition temperature could correlate to the highly crosslinked network.54,55 With the increase in BMPMOB concentration, the distance between the lower and higher transition temperatures increased gradually. This result indicated that the structure of the copolymers showed more heterogeneity with an increase in BMPMOB.
The three-point bending water-submersion clamp method used in this work is expected to simulate the wet environment of the mouth. The modulus of the control and experimental samples measured by the water-submersion method was significantly lower than that of the dry samples measured by a standard clamp. The difference is contributed to the plasticization of the copolymer in the water. Water can be attracted to the polar functional groups (such as carboxylic, hydroxyl, and ester) of the copolymer to form hydrogen bonds and decrease the intermolecular interaction of the copolymers. When the BMPMOB concentration was 25%, the modulus of the copolymer showed no significant difference at 37°C and was significantly higher than that of the control at 75°C. This means with the increase in the functionality of the comonomer, the highly crosslinked structure can counteract the effect of water plasticization. However, the storage modulus decreased with the continued increase in BMPMOB concentration at 35%. The poor mechanical property of the E1–35 sample in wet conditions may be attributed to the formulation being too hydrophilic, which causes it to absorb more water. Previously, it is been seen that with the increase in functionality or polarity of a comonomer, a significantly weaker structure may be formed in some regions, potentially causing premature failure.47,56 Importantly, by introducing the polarity group and vinyl group into the new monomer BMPMOB, we demonstrate the mechanical properties of the resulting polymers in the wet condition can be balanced by controlling the water sorption and the crosslink density of the copolymer. The experimental monomer, BMPMOB, shows promise as a hydrophilic crosslinker for dentin adhesive formulations. Meanwhile, when BMPMOB was used to partially replace BisGMA in the adhesive formulation, the lower maximum polymerization rate and decreased C=C bond conversions were mainly attributed to the higher functionality of new monomer. This may lead to a more heterogeneous cross-linked network structure compared to the control.44
CONCLUSION
A new monomer with a branched carboxylic acid group (BMPMOB) was synthesized by the reaction of IAA and GDMA, and it was used as a comonomer in dentin adhesive formulations. BMPMOB containing formulations showed better water compatibility than the control. With increased BMPMOB concentration, the maximum rate of polymerization was decreased and showed a lower DC. Dynamic mechanical analyses in dry condition showed that the experimental copolymers, with a larger storage moduli and higher glass transition temperatures, are significantly more heterogeneous than the controls. In wet conditions, the copolymer with 25% BMPMOB showed the optimal mechanical properties. By increasing the functionality of the comonomer, the balance between water sorption and mechanical properties can be controlled. The experimental adhesive copolymers improved mechanical properties in both dry and wet conditions. These results indicated that the BMPMOB is a promising comonomer for improving dentin adhesives.
Acknowledgments
Contract grant sponsor: National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892; contract grant numbers: R01DE14392; R01DE02202514392-08S1; R01 DE022054
We thank Dr. Ranganathan Parthasarathy for his helpful discussion on the crosslink structures.
References
- 1.Tjaderhane L, Nascimento FD, Breschi L, Mazzoni A, Tersariol ILS, Geraldeli S, Tezvergil-Mutluay A, Carrilho M, Carvalho RM, Tay FR, Pashley DH. Strategies to prevent hydrolytic degradation of the hybrid layer—A review. Dent Mater. 2013;29(10):999–1011. doi: 10.1016/j.dental.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burrow MF, Satoh M, Tagami J. Dentin bond durability after three years using a dentin bonding agent with and without priming. Dent Mater. 1996;12(5–6):302–307. doi: 10.1016/s0109-5641(96)80038-8. [DOI] [PubMed] [Google Scholar]
- 3.Hashimoto M, Ohno H, Kaga M, Endo K, Sano H, Oguchi H. In vivo degradation of resin-dentin bonds in humans over 1 to 3 years. J Dent Res. 2000;79(6):1385–1391. doi: 10.1177/00220345000790060601. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto M, Ohno H, Sano H, Tay FR, Kaga M, Kudou Y, Oguchi H, Araki Y, Kubota M. Micromorphological changes in resin–dentin bonds after 1 year of water storage. J Biomed Mater Res. 2002;63(3):306–311. doi: 10.1002/jbm.10208. [DOI] [PubMed] [Google Scholar]
- 5.Sano H, Yoshikawa T, Pereira PNR, Kanemura N, Morigami M, Tagami J, Pashley DH. Long-term durability of dentin bonds made with a self-etching primer, in vivo. J Dent Res. 1999;78(4):906–911. doi: 10.1177/00220345990780041101. [DOI] [PubMed] [Google Scholar]
- 6.Spencer P, Swafford JR. Unprotected protein at the dentin–adhesive interface. Quintessence Int. 1999;30(7):501–507. [PubMed] [Google Scholar]
- 7.Spencer P, Wang Y, Bohaty B. Interfacial chemistry of moisture-aged class II composite restorations. J Biomed Mater Res Part B: Appl Biomater. 2006;77B(2):234–240. doi: 10.1002/jbm.b.30434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spencer P, Wang Y, Walker MP, Wieliczka DM, Swafford JR. Interfacial chemistry of the dentin/adhesive bond. J Dent Res. 2000;79(7):1458–1463. doi: 10.1177/00220345000790070501. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Spencer P. Hybridization efficiency of the adhesive/dentin interface with wet bonding. J Dent Res. 2003;82(2):141–145. doi: 10.1177/154405910308200213. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Spencer P. Physicochemical interactions at the interfaces between self-etch adhesive systems and dentine. J Dent. 2004;32(7):567–579. doi: 10.1016/j.jdent.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Spencer P. Overestimating hybrid layer quality in polished adhesive/dentin interfaces. J Biomed Mater Res Part A. 2004;68A(4):735–746. doi: 10.1002/jbm.a.20105. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Spencer P. Continuing etching of an all-in-one adhesive in wet dentin tubules. J Dent Res. 2005;84(4):350–354. doi: 10.1177/154405910508400411. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Spencer P, Yao XM. Micro-Raman imaging analysis of monomer/mineral distribution in intertubular region of adhesive/dentin interfaces. J Biomed Opt. 2006;11(2) doi: 10.1117/1.2187992. [DOI] [PubMed] [Google Scholar]
- 14.Moszner N, Hirt T. New polymer-chemical developments in clinical dental polymer materials: Enamel–dentin adhesives and restorative composites. J Polym Sci Part A: Polym Chem. 2012;50(21):4369–4402. [Google Scholar]
- 15.Moszner N, Salz U. New developments of polymeric dental composites. Prog Polym Sci. 2001;26(4):535–576. [Google Scholar]
- 16.Moszner N, Salz U. Recent developments of new components for dental adhesives and composites. Macromol Mater Eng. 2007;292(3):245–271. [Google Scholar]
- 17.Moszner N, Salz U, Zimmermann J. Chemical aspects of self-etching enamel–dentin adhesives: A systematic review. Dent Mater. 2005;21(10):895–910. doi: 10.1016/j.dental.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz RS, Fransman R. Adhesive dentistry and endodontics: Materials, clinical strategies and procedures for restoration of access cavities: A review. J Endod. 2005;31(3):151–165. doi: 10.1097/01.don.0000155222.49442.a1. [DOI] [PubMed] [Google Scholar]
- 19.Vaidyanathan TK, Vaidyanathan J. Recent advances in the theory and mechanism of adhesive resin bonding to dentin: A critical review. J Biomed Mater Res Part B: Appl Biomater. 2009;88B(2):558–578. doi: 10.1002/jbm.b.31253. [DOI] [PubMed] [Google Scholar]
- 20.Park JG, Ye Q, Topp EM, Spencer P. Enzyme-catalyzed hydrolysis of dentin adhesives containing a new urethane-based trimethacrylate monomer. J Biomed Mater Res Part B: Appl Biomater. 2009;91B(2):562–571. doi: 10.1002/jbm.b.31430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park JG, Ye Q, Topp EM, Kostoryz EL, Wang Y, Kieweg SL, Spencer P. Preparation and properties of novel dentin adhesives with esterase resistance. J Appl Polym Sci. 2008;107(6):3588–3597. doi: 10.1002/app.27512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowman CN, Lovell LG, Syrpes DS, Stansbury JW. Dimethacrylate dental resins: The effect of comonomer on the polymerization kinetics. Abstracts of Papers of the American Chemical Society. 1998;246:U125–U125. [Google Scholar]
- 23.Moszner N. New monomers for dental application. Macromol Symp. 2004;217:63–75. [Google Scholar]
- 24.Spencer P, Wang Y. Adhesive phase separation at the dentin interface under wet bonding conditions. J Biomed Mater Res. 2002;62(3):447–456. doi: 10.1002/jbm.10364. [DOI] [PubMed] [Google Scholar]
- 25.Tay FR, Pashley DH, Kapur RR, Carrilho MRO, Hur YB, Garrett LV, Tay KCY. Bonding BisGMA to dentin—A proof of concept for hydrophobic dentin bonding. J Dent Res. 2007;86(11):1034–1039. doi: 10.1177/154405910708601103. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Tjaderhane L, Breschi L, Mazzoni A, Li N, Mao J, Pashley DH, Tay FR. Limitations in bonding to dentin and experimental strategies to prevent bond degradation. J Dent Res. 2011;90(8):953–968. doi: 10.1177/0022034510391799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kostoryz EL, Dharmala K, Ye Q, Wang Y, Huber J, Park JG, Snider G, Katz JL, Spencer P. Enzymatic biodegradation of HEMA/BisGMA adhesives formulated with different water content. J Biomed Mater Res Part B: Appl Biomater. 2009;88B(2):394–401. doi: 10.1002/jbm.b.31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishitani Y, Yoshiyama M, Donnelly AM, Agee KA, Sword J, Tay FR, Pashley DH. Effects of resin hydrophilicity on dentin bond strength. J Dent Res. 2006;85(11):1016–1021. doi: 10.1177/154405910608501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park JG, Ye Q, Topp EM, Misra A, Spencer P. Water sorption and dynamic mechanical properties of dentin adhesives with a urethane-based multifunctional methacrylate monomer. Dent Mater. 2009;25(12):1569–1575. doi: 10.1016/j.dental.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park J, Ye Q, Singh V, Kieweg SL, Misra A, Spencer P. Synthesis and evaluation of novel dental monomer with branched aromatic carboxylic acid group. J Biomed Mater Res Part B: Appl Biomater. 2012;100B(2):569–576. doi: 10.1002/jbm.b.31987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo X, Wang Y, Spencer P, Ye Q, Yao X. Effects of water content and initiator composition on photopolymerization of a model BisGMA/HEMA resin. Dent Mater. 2008;24(6):824–831. doi: 10.1016/j.dental.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Q, Park J, Topp E, Spencer P. Effect of photoinitiators on the in vitro performance of a dentin adhesive exposed to simulated oral environment. Dent Mater. 2009;25(4):452–458. doi: 10.1016/j.dental.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parthasarathy R, Misra A, Park J, Ye Q, Spencer P. Diffusion coefficients of water and leachables in methacrylate-based crosslinked polymers using absorption experiments. J Mater Sci: Mater Med. 2012;23(5):1157–1172. doi: 10.1007/s10856-012-4595-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park J, Ye Q, Topp EM, Misra A, Kieweg SL, Spencer P. Effect of photoinitiator system and water content on dynamic mechanical properties of a light-cured bisGMA/HEMA dental resin. J Biomed Mater Res Part A. 2010;93A(4):1245–1251. doi: 10.1002/jbm.a.32617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JG, Ye Q, Topp EM, Lee CH, Kostoryz EL, Misra A, Spencer P. Dynamic mechanical analysis and esterase degradation of dentin adhesives containing a branched methacrylate. J Biomed Mater Res Part B: Appl Biomater. 2009;91B(1):61–70. doi: 10.1002/jbm.b.31374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anseth KS, Goodner MD, Reill MA, Kannurpatti AR, Newman SM, Bowman CN. The influence of comonomer composition on dimethacrylate resin properties for dental composites. J Dent Res. 1996;75(8):1607–1612. doi: 10.1177/00220345960750081301. [DOI] [PubMed] [Google Scholar]
- 37.Moszner N, Volkel T, Zeuner F, Rheinberger V. Radical ring-opening monomers for dental composites. Abstracts of Papers of the American Chemical Society. 1997;214:10-POLY. [Google Scholar]
- 38.Salz U, Burtscher P, Vogel K, Moszner N, Rheinberger V. New adhesive monomers for dental application. Abstracts of Papers of the American Chemical Society. 1997;214:175-POLY. [Google Scholar]
- 39.Pavlinec J, Zeuner F, Angermann J, Moszner N. Monomers for adhesive polymers, 5-synthesis and radical polymerization behavior of 2,4,6-trimethylphenyl 2,4-(dihydroxyphosphoryl)-2-oxa-butyl acrylate. Macromol Chem Phys. 2005;206(18):1878–1886. [Google Scholar]
- 40.El-Deeb HA, Al Sherbiney HH, Mobarak EH. Bond durability of adhesives containing modified-monomer with/without-fluoride after aging in artificial saliva and under intrapulpal pressure simulation. Oper Dent. 2013;38(1):48–56. doi: 10.2341/11-389-L. [DOI] [PubMed] [Google Scholar]
- 41.Atai M, Nekoomanesh M, Hashemi SA, Yeganeh H. Synthesis and characterization of BTDA-based dimethacrylate dental adhesive monomer and its interaction with Ca2+ ions. J Appl Polym Sci. 2002;86(13):3246–3249. [Google Scholar]
- 42.Solhi L, Atai M, Nodehi A, Imani M, Ghaemi A, Khosravi K. Poly(-acrylic acid) grafted montmorillonite as novel fillers for dental adhesives: Synthesis, characterization and properties of the adhesive. Dent Mater. 2012;28(4):369–377. doi: 10.1016/j.dental.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Sideridou I, Tserki V, Papanastasiou G. Effect of chemical structure on degree of conversion in light-cured dimethacrylate-based dental resins. Biomaterials. 2002;23(8):1819–1829. doi: 10.1016/s0142-9612(01)00308-8. [DOI] [PubMed] [Google Scholar]
- 44.Park J, Eslick J, Ye Q, Misra A, Spencer P. The influence of chemical structure on the properties in methacrylate-based dentin adhesives. Dent Mater. 2011;27(11):1086–1093. doi: 10.1016/j.dental.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santerre JP, Shajii L, Leung BW. Relation of dental composite formulations to their degradation and the release of hydrolyzed polymeric-resin-derived products. Crit Rev Oral Biol Med. 2001;12(2):136–151. doi: 10.1177/10454411010120020401. [DOI] [PubMed] [Google Scholar]
- 46.Ye Q, Spencer P, Wang Y, Misra A. Relationship of solvent to the photopolymerization process, properties, and structure in model dentin adhesives. J Biomed Mater Res Part A. 2007;80A(2):342–350. doi: 10.1002/jbm.a.30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Young JS, Kannurpatti AR, Bowman CN. Effect of comonomer concentration and functionality on photopolymerization rates, mechanical properties and heterogeneity of the polymer. Macromol Chem Phys. 1998;199(6):1043–1049. [Google Scholar]
- 48.Gao BT, Lin H, Zheng G, Xu YX, Yang JL. Comparison between a silorane-based composite and methacrylate-based composites: Shrinkage characteristics, thermal properties, gel point and vitrification point. Dent Mater J. 2012;31(1):76–85. doi: 10.4012/dmj.2011-147. [DOI] [PubMed] [Google Scholar]
- 49.Pavlinec J, Moszner N. Monomers for adhesive polymers, 8(a) crosslinking polymerization of selected N-substituted bis(acrylamide)s for dental filling materials. J Appl Polym Sci. 2009;113(5):3137–3145. [Google Scholar]
- 50.Odian G. Principles of Polymerization. 4. Hoboken, NJ: Wiley; 2004. [Google Scholar]
- 51.Allen PEM, Simon GP, Williams DRG, Williams EH. Dynamic–mechanical properties and cross-polarized, proton-enhanced, magic angle spinning C-13 NMR time constants of polyoligo(ethylene glycol) dimethacrylates. Macromolecules. 1989;22(2):809–816. [Google Scholar]
- 52.Simon GP, Allen PEM, Bennett DJ, Williams DRG, Williams EH. Nature of residual unsaturation during cure of dimethacrylates examined by cppemas C-13 NMR and simulation using a kinetic gelation model. Macromolecules. 1989;22(9):3555–3561. [Google Scholar]
- 53.Simon GP, Allen PEM, Williams DRG. Properties of dimethacrylate copolymers of varying cross-link density. Polymer. 1991;32(14):2577–2587. [Google Scholar]
- 54.Ye Q, Wang Y, Spencer P. Nanophase separation of polymers exposed to simulated bonding conditions. J Biomed Mater Res Part B: Appl Biomater. 2009;88B(2):339–348. doi: 10.1002/jbm.b.31047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye Q, Park JG, Topp E, Wang Y, Misra A, Spencer P. In vitro performance of nano-heterogeneous dentin adhesive. J Dent Res. 2008;87(9):829–833. doi: 10.1177/154405910808700911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ito S, Hashimoto M, Wadgaonkar B, Svizero N, Carvalho RM, Yiu C, Rueggeberg FA, Foulger S, Saito T, Nishitani Y, et al. Effects of resin hydrophilicity on water sorption and changes in modulus of elasticity. Biomaterials. 2005;26(33):6449–6459. doi: 10.1016/j.biomaterials.2005.04.052. [DOI] [PubMed] [Google Scholar]