

Abstract
We report the case of a 27-year-old man with a history of previously undiagnosed renal disease that presented with multiple cerebrovascular infarctions. Workup for traditional causes of cerebrovascular infarction including cardiac telemetry, multiple echocardiograms, and hypercoagulative workup was negative. However, a transcranial Doppler detected circulating microemboli at the rate of 14 per hour. A serum oxalate level greater than the supersaturation point of calcium oxalate was detected, providing a potential source of the microemboli. Furthermore, serial imaging recorded rapid mineralization of the infarcted territories. In the absence of any proximal vessel irregularities, atherosclerosis, valvular abnormalities, arrhythmias, or systemic shunt as potential stroke etiology in this patient, we propose that circulating oxalate precipitate may be a potential mechanism for stroke in patients with primary oxalosis.
Keywords: Stroke, hyperoxaluria, primary oxalosis, neuroimaging, genetics, transcranial Doppler
Introduction
Primary hyperoxalurias are rare genetic conditions in which errors in the metabolism of glycolate result in the increased endogenous production of oxalate leading to progressive renal dysfunction and ultimately systemic deposition of calcium oxalate.1, 2 Stroke in patients with primary oxalosis has been rarely described—to our knowledge, there are only two prior cases reported.3, 4 These two case reports describe patients with profound intravascular deposits of calcium oxalate, with vessel occlusion and embolization of these deposits as potential etiology. Despite multiple imaging modalities, no such deposits were seen in our patient. Due to circulating levels of serum oxalate greater than the supersaturation point, and evidence of microemboli by transcranial Doppler, we propose that serum precipitation of calcium oxalate may also provide a potential etiology of stroke in patients with primary oxalosis.
Case Report
A 27-year-old Indian man with a long history of renal disease was transferred to our institution after two thromboembolic cerebrovascular infarctions. The patient’s medical history began with his first episode of kidney stones at 8 months of age. At the age of two, he required surgery for a bladder stone. He had recurrent kidney stones, up to several per year, requiring multiple attempts at lithotripsy. He underwent multiple 24-hour urine collections and hypercalciuria, hyperoxaluria, and cystinuria were suspected etiologies. A kidney stone analysis at age 16 showed the nephroliths to be composed calcium oxalate. Due to worsening renal dysfunction, the patient began peritoneal dialysis at age 23. At age 24, he underwent a right nephrectomy after a kidney infection. There was no history of kidney stones or renal failure in either side of his family.
His first stroke was at age 27 when he presented with acute onset left face, arm, and leg hemiparesis. Magnetic resonance imaging (MRI) showed an infarction in the right frontoparietal region, in the territory of the right middle cerebral artery. Furthermore, there were multiple other small areas of restricted diffusion in the left frontal lobe, the left occipital lobe, and the right caudate head. MRA showed an abrupt termination of the right distal M1 segment suspicious for occlusion. Workup including transthoracic echocardiogram, transesophageal echocardiogram, hypercoagulative laboratory panel, and cardiac telemetry was all negative or unremarkable. He was discharged on aspirin and recovered well from this infarction with mild residual hemiparesis.
One month later, he returned with acute worsening of left face, arm, and leg hemiparesis. MRI showed a new right anterior cerebral artery infarction with an A1 occlusion. Additionally, this repeat MRI showed early encephalomalacia and mineralization of tissue in the prior middle cerebral artery infarction (Fig 2). On transfer to our facility, he underwent a diagnostic cerebral angiogram that showed multiple filling defects in the right anterior cerebral and middle cerebral artery with slow contrast filling in the M4 branches of the left middle cerebral artery, consistent with emboli in multiple arterial distributions.
Fig. 2.

MRI on admission. (A) and (C) Initial MRI with DWI showing acute right middle cerebral artery infarction and smaller left sided infarction. (B) and (D) FLAIR shows older left frontal infarction as well (large arrow).
His hospital course was complicated by a hemorrhagic transformation of the right anterior cerebral artery stroke. He also suffered a pulmonary embolus and imaging at that time also revealed multiple splenic infarctions. Workup was again negative for hypercoagulative, rheumatologic, or infectious disorder. A second transesophageal echocardiogram showed normal cardiac function with no evidence of intracardiac masses or shunts, and no valvular or aortic abnormalities. Cardiac telemetry was unremarkable. Transcranial Doppler with bubble study did not show any evidence of systemic shunt; however, there were spontaneous asymptomatic microemboli detected at the rate of 8 per hour in the right MCA and 6 per hour in the left MCA territory.
Concomitantly, his underlying metabolic disorder was investigated. A plasma oxalate level was checked and revealed an elevated level of 100 μmol/liter. A genetic test for primary oxalosis was requested and returned positive for Primary Hyperoxaluria Type I with AGXT Mutation 1: c.1049G>A,p.G350D and AGXT Mutation 2: c.1049G>A,p.G350D.5
Discussion
Primary oxalosis is a rare genetic disorder caused by the deficiency of liver-specific peroxisomal alanine-glyoxalate aminotransferase (AGT). This enzyme is needed for the detoxification of glycoxylate to glycine as part of glycolate metabolism. Decreased activity of AGT results in the overproduction of oxalate via glycoxylate metabolism though alternate pathways (Fig 1).2, 6 Oxalate is excreted by the kidneys, and thus excess production by the liver results in calcium oxalate nephrolithiasis and progressive renal dysfunction. With a falling glomerular filtration rate and continued overproduction of oxalate by the liver, plasma oxalate levels may reach the supersaturation point (Pox > 30 μmol/liter). As oxalate is a poorly soluble substance, this leads to the systemic deposition of its precipitate, calcium oxalate.1 Deposition may occur in multiple organ including the heart, blood vessels, and in the brain itself.1 Stroke in the setting of primary oxalosis has rarely been described—to our knowledge, there are only two cases in the literature. Lammie et al describe a case of bilateral carotid territory infarction and moyamoya syndrome in a patient with this disorder. Pathologic analysis of this patient showed deposition of calcium oxalate in the tunica media and progressive fibrosis of the proximal cerebral blood vessels ultimately leading to thrombotic occlusion.3 Di Pasquale et al describe a patient with cardioembolic cerebral infarction due to embolization of cardiac calcium oxalate deposits.4 Furthermore, oxalosis may lead to cardiac conduction abnormalities and arrhythmias; however, despite prolonged cardiac monitoring, our patient did not display any abnormal cardiac rhythms.7–9
Fig. 1.
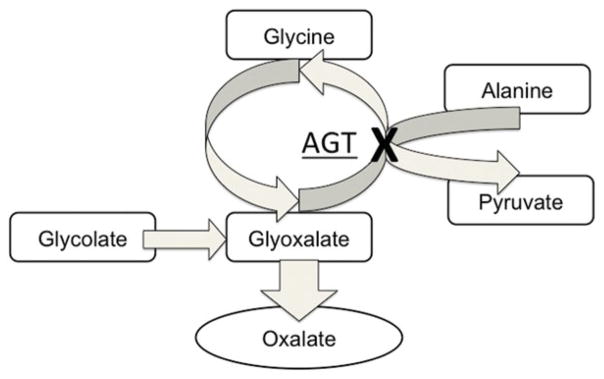
Simplified schema of glycoxalate metabolism. With AGT deficiency, glyoxalate is preferentially metabolized to oxalate, resulting in the overproduction of this metabolite.
Our patient had multiple echocardiograms that did not demonstrate any cardiac abnormalities, thrombus, or cardiac deposits. There was no evidence by CT or conventional angiogram to suggest stenosing vasculopathy from intravascular deposits. Yet, on prolonged transcranial Doppler monitoring, multiple high-intensity transient signals recorded in the bilateral middle cerebral arteries. In the absence of any proximal vessel irregularities, atherosclerosis, valvular abnormalities, arrhythmias, or systemic shunt as potential causes, we postulate that circulating oxalate precipitate may be the source of these microemboli. Further evidence of possible oxalate precipitation can be seen on follow-up imaging taken in the absence of clinical change, which shows rapid laminar necrosis of the infarcted tissue (Figs 2 and 3). The CT shows areas of hyperdensity, which are not typically associated with laminar necrosis.10, 11 These areas of hyperdensity do not correspond with evidence of hemorrhage on the MRI. Thus, given the patient’s underlying disorder, which has been shown to cause deposits of calcium oxalate in tissues such as retina, skin, and heart,1 we posit that disruption of the blood brain barrier seen in laminar necrosis may have allowed these deposits to form in the damaged brain tissue as well.10
Fig. 3.

Follow-up imaging. MRI 1 month after initial presentation showing acute right anterior cerebral artery infarction on DWI (A, small arrow). Progression of laminar necrosis of infarcted cortex seen by CTH seen 9 days (B) and 54 days (C) after the ACA infarction. The areas of hyperdensity on the 54 day CTH may represent calcification due to calcium oxalate deposition as there was no evidence of acute hemorrhage on MRI with FLAIR (D) and T1 (E) 50 days after the ACA infarction. (F) Cerebral angiogram showing filling defect in the right ACA territory (large arrow) and multiple branch occlusions (small arrows). There is no other evidence of vasculopathy.
Although we could not fully exclude microscopic proximal vessel or cardiac deposits below the resolution of our diagnostic testing, it is possible that due to the high serum level of calcium oxalate exceeding the supersaturation point, direct precipitation in the bloodstream may provide a nidus for the formation of calcium oxalate thromboemboli. Direct occlusion of the cerebral vasculature by circulating calcium oxalate thromboemboli may be another potential etiology of stroke in patients with primary oxalosis.
For this patient, we recommended switch of his peritoneal dialysis to conventional hemodialysis for improved oxalate clearance. He was then placed in consideration for combined liver and renal transplant, which is a potentially curative treatment for this disorder.8, 9
Footnotes
Disclosures: None.
References
- 1.Harambat J, Fargue S, Bacchetta J, et al. Primary hyperoxaluria. Int J Nephrol. 2011;2011:864580. doi: 10.4061/2011/864580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmes RP, Assimos DG. Glyoxylate synthesis, and its modulation and influence on oxalate synthesis. J Urol. 1998;160(5):1617–1624. [PubMed] [Google Scholar]
- 3.Lammie GA, Wardlaw J, Dennis M. Thrombo-embolic stroke, moya-moya phenomenon and primary oxalosis. Cerebrovasc Dis. 1998;8(1):45–50. doi: 10.1159/000015815. [DOI] [PubMed] [Google Scholar]
- 4.Di Pasquale G, Ribani M, Andreoli A, et al. Cardioembolic stroke in primary oxalosis with cardiac involvement. Stroke. 1989;20(10):1403–1406. doi: 10.1161/01.str.20.10.1403. [DOI] [PubMed] [Google Scholar]
- 5.Monico CG, Rossetti S, Schwanz HA, et al. Comprehensive mutation screening in 55 probands with type 1 primary hyperoxaluria shows feasibility of a gene-based diagnosis. J Am Soc Nephrol. 2007;18(6):1905–1914. doi: 10.1681/ASN.2006111230. [DOI] [PubMed] [Google Scholar]
- 6.Cochat P. Primary hyperoxaluria type 1. Kidney Int. 1999;55(6):2533–2547. doi: 10.1046/j.1523-1755.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- 7.Vélez-Roa S, Depierreux M, Nortier J, et al. Cardiac oxalosis: a rare cause of diastolic dysfunction. Eur Heart J. 2006;27(21):2496. doi: 10.1093/eurheartj/ehi871. [DOI] [PubMed] [Google Scholar]
- 8.Detry O, Honoré P, DeRoover A, et al. Reversal of oxalosis cardiomyopathy after combined liver and kidney transplantation. Transpl Int. 2002;15(1):50–52. doi: 10.1007/s00147-001-0364-y. [DOI] [PubMed] [Google Scholar]
- 9.Massie BM, Bharati S, Scheinman MM, et al. Primary oxalosis with pan-conduction cardiac disease: electrophysiologic and anatomic correlation. Circulation. 1981;64(4):845–852. doi: 10.1161/01.cir.64.4.845. [DOI] [PubMed] [Google Scholar]
- 10.Komiyama M, Nishikawa M, Yasui T. Cortical laminar necrosis in brain infarcts: chronological changes on MRI. Neuroradiology. 1997;39(7):474–479. doi: 10.1007/s002340050448. [DOI] [PubMed] [Google Scholar]
- 11.Siskas N, Lefkopoulos A, Ioannidis I, et al. Cortical laminar necrosis in brain infarcts: serial MRI. Neuroradiology. 2003;45(5):283–288. doi: 10.1007/s00234-002-0887-7. [DOI] [PubMed] [Google Scholar]