

Significance
Stroke is the leading cause of disability in the United States and has very limited treatment options. Brain stimulation techniques that promote recovery after stroke are a promising area of research; however, current stimulation techniques nonspecifically activate/inhibit the target area, which not only leads to undesired side effects but also makes it difficult to understand which cell types and mechanisms drive recovery. We used the optogenetic technique to specifically stimulate only neurons after stroke and demonstrate that selective neuronal stimulations can activate beneficial mechanisms and promote recovery. Understanding the cell type and mechanisms driving recovery may identify potential drug targets for stroke treatment, as well as ultimately help develop precise brain stimulation techniques for stroke therapy.
Keywords: stroke recovery, channelrhodopsin
Abstract
Clinical and research efforts have focused on promoting functional recovery after stroke. Brain stimulation strategies are particularly promising because they allow direct manipulation of the target area’s excitability. However, elucidating the cell type and mechanisms mediating recovery has been difficult because existing stimulation techniques nonspecifically target all cell types near the stimulated site. To circumvent these barriers, we used optogenetics to selectively activate neurons that express channelrhodopsin 2 and demonstrated that selective neuronal stimulations in the ipsilesional primary motor cortex (iM1) can promote functional recovery. Stroke mice that received repeated neuronal stimulations exhibited significant improvement in cerebral blood flow and the neurovascular coupling response, as well as increased expression of activity-dependent neurotrophins in the contralesional cortex, including brain-derived neurotrophic factor, nerve growth factor, and neurotrophin 3. Western analysis also indicated that stimulated mice exhibited a significant increase in the expression of a plasticity marker growth-associated protein 43. Moreover, iM1 neuronal stimulations promoted functional recovery, as stimulated stroke mice showed faster weight gain and performed significantly better in sensory-motor behavior tests. Interestingly, stimulations in normal nonstroke mice did not alter motor behavior or neurotrophin expression, suggesting that the prorecovery effect of selective neuronal stimulations is dependent on the poststroke environment. These results demonstrate that stimulation of neurons in the stroke hemisphere is sufficient to promote recovery.
Stroke is a major acute neurological insult that disrupts brain function and causes neuron death. Functional recovery after stroke has been observed and is currently attributed to both brain remodeling and plasticity (1–4). Structural and functional remodeling of areas next to an infarct or remote regions can alter signaling within bilateral neuronal networks and thus contribute to functional recovery (3–7). Rewiring of neural connections is mediated by electrical activity, which can activate a number of plasticity mechanisms, including the release of activity-dependent neurotrophins such as brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) (8–10). Both BDNF and NGF have been shown to improve recovery by enhancing axonal and dendritic sprouting (10–12).
Tremendous effort has been focused on promoting recovery after stroke, including pharmacological treatment, rehabilitation (e.g., constraint-induced therapy), stem cell transplantation, and brain stimulation (1, 4, 13). Brain stimulation is a promising area of research because it allows direct activation and manipulation of the target area’s excitability (14–16). The primary motor cortex (M1) is a commonly stimulated area as it directly innervates the corticospinal tract to initiate movement (1, 7). Although electrical stimulation and transcranial magnetic stimulation show promise in promoting recovery (17, 18), these techniques are limited by imprecision and indiscriminate activation or inhibition of all cell types near the stimulated site; thus, they can produce undesired effects such as psychiatric and motor/speech problems (19–21). In addition, it has been difficult to elucidate the cell type and mechanisms driving recovery, as multiple cell types such as neurons, astrocytes, and oligodendrocytes have been shown to contribute to remodeling and recovery processes after stroke (5, 22–27).
To elucidate whether activation of neurons alone can promote recovery, we used optogenetics to selectively manipulate the excitability of specific cell groups with millisecond-scale temporal precision in a manner more similar to endogenous neuronal firing patterns (21, 28, 29). This technique uses light-activated microbial proteins such as Channelrhodopsin 2 (ChR2), which depolarizes neurons when illuminated with blue light, or Halorhodopsin (NpHR), which hyperpolarizes neurons (21, 28, 29). Optogenetic approaches have been used in rodents to probe neuronal circuits for several neurological/neurodegenerative diseases, including Parkinson disease (30) and epilepsy (31). Recent studies have also used optogenetics to map functional organization after stroke (32–35). The safety and efficacy of using optogenetics in nonhuman primates has also been characterized (29, 36).
In this study, we used optogenetics to selectively stimulate neurons in layer V of the ipsilesional primary motor cortex (iM1) and examine the effects of repeated neuronal stimulations in normal and stroke mice. Sensory-motor behavior tests were used to evaluate functional recovery after stroke, and plasticity-associated mechanisms, such as cerebral blood flow (CBF)/neurovascular coupling responses and activity-dependent neurotrophin expression, were investigated.
Results
iM1 Neuronal Stimulations Can Activate Peri-infarct Areas and Contralesional M1.
We used Thy-1–ChR2–YFP line-18 transgenic mice, which exhibit high levels of ChR2 in layer V of primary motor cortex pyramidal neurons (Fig. 1A). An optical fiber was stereotaxically implanted above layer V of iM1, and a transient middle cerebral artery occlusion model was used to generate infarct in the striatum (Str) and somatosensory cortex (S1). We first examined if reliable neuronal activation could be achieved by our iM1 neuronal stimulation paradigm consisting of three successive 1-min laser stimulations, separated by 3-min rest intervals (Fig. 1 B and C). In vivo electrophysiological optrode recording in iM1 indicated that this stimulation paradigm could generate reliable and consistent firing patterns in all three stimulations (Fig. 1B), with individual spiking shown in Fig. 1C. To demonstrate that iM1 stimulation could activate peri-infarct areas and contralesional M1 (cM1), a dual recording was performed: an optrode in iM1 and a recording electrode in either the ipsilesional striatum (iStr), ipsilesional somatosensory cortex (iS1), or cM1. iM1 stimulation can induce reliable firing in iS1 (Fig. 1D), iStr (Fig. 1E), and cM1 (Fig. 1F), indicating that iM1 neuronal stimulation can activate peri-infarct regions (Str and S1) as well as the cM1.
Fig. 1.

iM1 neuronal stimulation activates peri-infarct areas and contralesional cortex. (A) High expression of Thy-1–ChR2–YFP in layer V pyramidal neurons of M1. (Scale bar, 100 µm.) (B, Upper) Stimulation paradigm with three successive 1-min laser stimulations (blue bars) separated by 3-min rest intervals. (Lower) Representative optrode tracing of neuronal firings that result from the application of this paradigm to iM1. (C) Enlarged image of a stimulation interval in the optrode tracing of B, showing individual spiking from the light pulses (red bracket). (D–F, Left) Ischemic regions (striped) and implantation sites in M1. An optrode (blue) with a recording electrode (black) is placed in iM1 and a second recording electrode (brown) is placed in iS1 (D), iStr (E), or cM1 (F). (Center) Representative optrode tracings for dual simultaneous recordings. (Right) Enlarged images of individual spikes. iM1 stimulation resulted in activation of the ischemic iS1 and iStr, as well as cM1.
Repeated iM1 Neuronal Stimulations Enhanced Cerebral Blood Flow/Neurovascular Coupling in Stroke Mice.
To examine if repeated iM1 neuronal stimulation could activate plasticity-associated mechanisms and recovery, we implemented a repeated neuronal stimulation paradigm at poststroke days 5–14 (6 d/wk, see experimental time line, Fig. 2A). We used five groups of Thy1-ChR2-YFP transgenic mice: sham, normal + nonstim, normal + stim, stroke + nonstim, and stroke + stim (Fig. 2B). During the optogenetic neuronal stimulations, we observed visible forelimb movements in the affected limb when the laser was turned on (Movie S1). This indicates that our iM1 stimulations activate sufficient motor-output neurons to generate movements in the affected forelimb.
Fig. 2.

Experimental design and time line. (A) Mice were handled and pretrained on several motor-sensory behavior tests before collecting baseline. Preimplant baseline was collected 1 d before fiber optic cannula implant surgery. Prestroke baseline was collected 1 d before stroke surgery (30-min suture model). Optogenetic neuronal stimulation began at poststroke day 5. Stimulation continued until poststroke day 14 (6 d/wk). Behavior tests were performed on days 2, 7, 10, and 14. On poststroke day 15, one group of mice was placed under anesthesia for CBF measurements and sacrificed that day. Another group was sacrificed for qPCR studies or histology. At poststroke day 5, another group was used for CBF measurement and sacrificed that day (indicated by dotted line). These mice were not used for behavior studies, as indicated by dotted line. (B) Chart of the five experimental groups used for CBF, qPCR, and behavior studies and the types of surgery for each group.
We first examined whether repeated iM1 neuronal stimulations increased CBF and neurovascular coupling response (Fig. 3). Laser doppler flowmetry (LDF) was used to measure changes in CBF in sham, stroke + nonstim, and stroke + stim mice. The site of stimulation, CBF measurement location, and stroke location are indicated in the diagrams (Fig. 3). In the sham group, CBF increased during the 1-min stimulation, followed by a significantly larger increase during the 3-min poststimulation period in both the contralesional and ipsilesional hemispheres (Fig. 3A). Stroke mice, however, despite significantly increased CBF with contralesional stimulation during the 1-min stimulation period, failed to induce the large increase in CBF poststimulation in both contralesional and ipsilesional hemispheres at poststroke day 5 (Fig. S1). This is consistent with the current concept of an overall depressed excitability and blood flow throughout the brain after stroke (37, 38). Interestingly, at poststroke day 15, stimulated stroke mice exhibited an improved CBF/neurovascular coupling response in the ipsilesional hemisphere (Fig. 3B), whereas nonstimulated stroke mice remained unresponsive to the 1-min laser stimulation and exhibited no significant change in ipsilesional CBF during and after the 1-min stimulation.
Fig. 3.

Repeated iM1 neuronal stimulations improved CBF and the neurovascular coupling response after stroke. Changes in CBF in response to a 1-min stimulation (blue bars) on either cM1 or iM1 were measured in (A) sham mice and (B) stimulated and nonstimulated stroke mice at poststroke day 15. (Left) Illustration of the stimulation site (indicated by fiber), the ischemic area (orange) and the CBF measurement site (green). (Center) Time lapse recordings of percentage change in CBF, consisting of three periods: baseline (1 min), laser-on stimulation (1 min), and a laser off (3 min). (Right) Peak percentage CBF change in each period. (A) Sham mice exhibited a similar neurovascular coupling response in both hemispheres, with an increased CBF during the laser-on period and a larger CBF response after laser was turned off. *P < 0.05, ***P < 0.001; one-way ANOVA with Dunnet's post hoc test. n = 4–6 per group. (B) At poststroke day 15, both stimulated and nonstimulated stroke mice exhibited a similar neurovascular coupling response in the cM1, but only the stimulated stroke mice exhibited significant improvement of the neurovascular coupling response in the iM1. *P < 0.05, **P < 0.01, ***P < 0.001; two-way ANOVA with Bonferroni’s post hoc test. n = 4–6 per group.
Repeated iM1 Neuronal Stimulations Increased Neurotrophin Expressions in Stroke Mice.
Next we investigated the effects of repeated iM1 neuronal stimulation on neurotrophic factor expression, as a number of neurotrophic factors have been demonstrated to promote recovery poststroke (9, 11, 39). We examined the expression of the activity-dependent neurotrophin family (BDNF, NGF, and NTF3) at poststroke day 15. Quantitative PCR (qPCR) was performed on iM1, cM1, iS1, and cS1 in all five groups listed in Fig. 2B. The stroke areas and dissected regions (iM1, cM1, iS1, and cS1) are outlined in Fig. 4A. Stroke induced a significant decrease of BDNF expression in iS1 of both stimulated and nonstimulated stroke mice compared with sham. Interestingly, stimulated stroke mice exhibited a significantly higher level of BDNF in cM1 and cS1 than nonstimulated mice (Fig. 4B). Western blot analysis indicated that BDNF protein levels were also significantly higher in stimulated mice, specifically in iM1, cM1, and iS1 (Fig. S2). NGF expression was also significantly up-regulated in cM1 of stimulated vs. nonstimulated mice, whereas stroke alone caused a decrease in iM1 NGF (Fig. 4C). Similarly, neurotrophin 3 (NTF3) expression was also significantly elevated in cM1 and cS1 (Fig. 4D). In contrast, repeated neuronal stimulations in normal nonstroke mice did not cause significant changes in levels of any neurotrophins (Fig. S3).
Fig. 4.

Repeated iM1 neuronal stimulations increased the expression of neurotrophins after stroke. (A) Neurotrophin mRNA expression in brains of stimulated and nonstimulated stroke mice and sham mice sacrificed on day 15. Diagram illustrates the stimulation site, infarct regions, and iM1, cM1, iS1, and cS1 dissected. qPCR was used to examine the expression of neurotrophins. (B) BDNF was significantly lower in stimulated and nonstimulated stroke mice in iS1, compared with sham. Stimulated stroke mice exhibited significantly higher BDNF expression than nonstimulated stroke mice in cM1 and cS1. Stimulated mice also exhibited significantly higher BDNF than sham mice in cM1 (#P < 0.05). (C) NGF and (D) NTF3 expression were also higher in cM1, and NTF3 was higher in cS1 for stimulated vs. nonstimulated mice. *P < 0.05, significant difference between stimulated and nonstimulated stroke mice, one-way ANOVA with Fisher's LSD. ##P < 0.01, ###P < 0.001; significant difference from sham mice, one-way ANOVA with Fisher's LSD test. n = 6–9 per group.
Repeated iM1 Neuronal Stimulations Increased Growth-Associated Protein 43 Expressions in Stroke Mice.
To examine whether repeated iM1 neuronal stimulation might be involved in poststroke synaptic plasticity, we examined the expression of growth-associated protein 43 (GAP43), a plasticity marker involved in synaptic plasticity and reorganization after stroke (3, 4, 40). The stroke areas and dissected regions (iM1, cM1, iS1, and cS1) are outlined in Fig. 4A. Western blot analysis indicated that stimulated stroke mice exhibited a significant increase in GAP43 protein levels in the iS1 and the cM1 (Fig. 5), suggesting that poststroke neuronal stimulations may enhance synaptic plasticity.
Fig. 5.
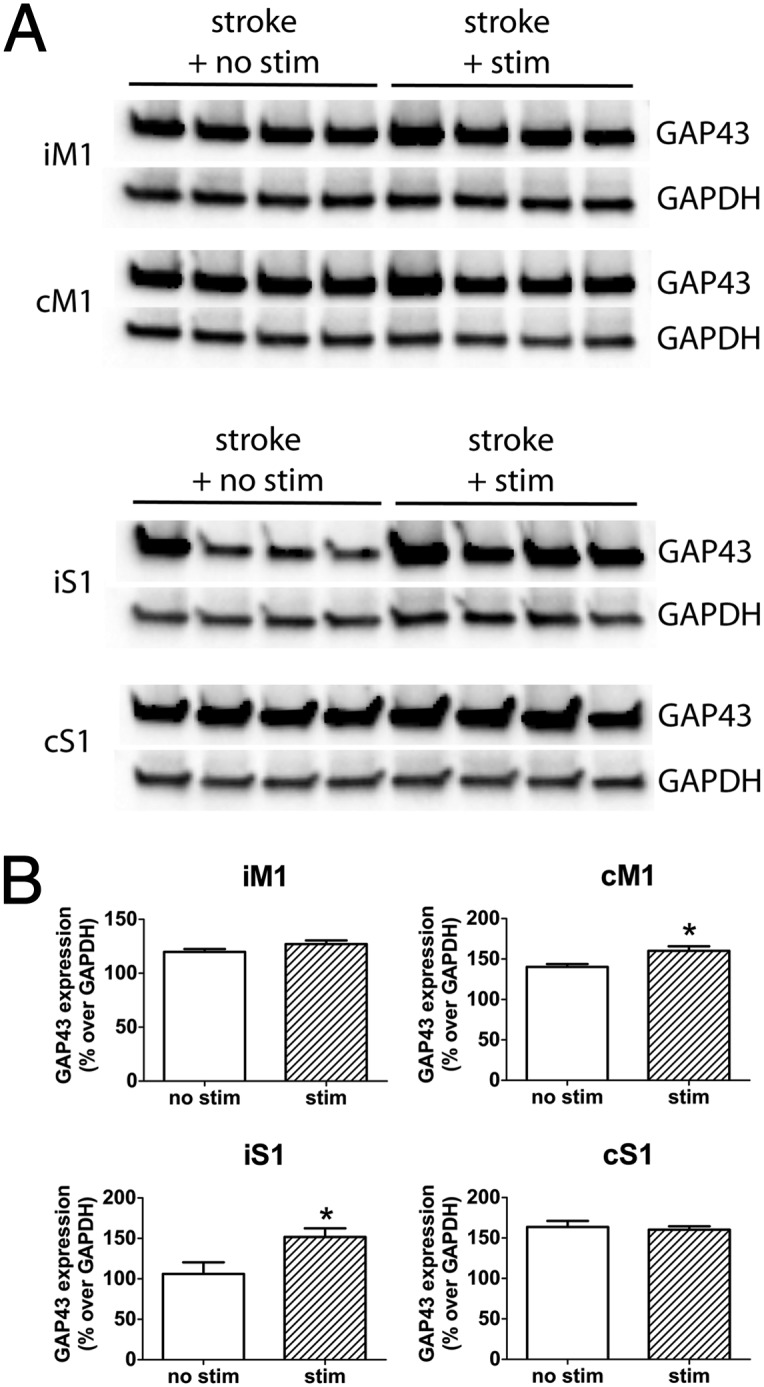
iM1 neuronal stimulations increased GAP43 expression. (A) Western blot of GAP43 and GAPDH expression in iM1, cM1, iS1, and cS1 of nonstimulated and stimulated stroke mice (poststroke day 15). (B) Relative optical density measurements of GAP43 expression expressed as percentage over GAPDH. Stimulated mice exhibit significantly higher GAP43 expression in cM1 and iS1. n = 4 per group. *P < 0.05; significant difference between stim and nonstim group, Student t test.
Repeated iM1 Neuronal Stimulations Promote Functional Behavioral Recovery in Stroke Mice.
To address whether repeated iM1 neuronal stimulations can promote functional recovery, we evaluated the behavioral performance of stroke mice on the rotating beam test, a sensitive and reproducible sensory-motor behavior test used to detect neurological deficit after stroke (41). Baseline performances were evaluated before stroke (day 0) and poststroke days 2, 7, 10, and 14. Body weight changes were also monitored during this period as mice typically lose body weight after stroke. Interestingly, stimulated mice regained their body weight faster than nonstimulated mice at poststroke day 14 (Fig. 6A). Importantly, stimulated stroke mice also exhibited significant improvement in their motor-sensory function, traveling longer distances at day 7 and 10 (Fig. 6B) and faster speeds at day 10 and 14 (Fig. 6C and Movies S2–S4). Infarct analysis indicated that repeated iM1 neuronal stimulations did not alter infarct size (Fig. S4). Interestingly, repeated neuronal stimulations in normal nonstroke mice did not alter or improve motor function (Fig. 6 D and E), suggesting that the effect of neuronal stimulation in stroke mice is dependent on the stroke environment. Stimulation also did not have an effect on body weight in normal nonstroke mice (Fig. S5). Furthermore, stimulation of a control cortical region—the contralesional M1—did not have an effect on functional recovery (Fig. S6), indicating that the prorecovery effect of iM1 stimulation is region dependent and not due to a general stimulation effect.
Fig. 6.

iM1 neuronal stimulations improved functional recovery. (A) Stimulated stroke mice regained their body weight significantly faster than nonstimulated stroke mice at poststroke day 14. (Left) Time course of body weight changes. (Right) Average of percent body weight change during the stimulation period (*P < 0.05, Student t test). Stimulated mice performed significantly better in the rotating beam test, with a longer distance traveled (B) and a faster speed (C). *P < 0.05, **P < 0.01, significant difference between stim and nonstim group, two-way ANOVA repeated measures with Fisher's LSD. Sham, n = 8; nonstim, n = 16; stim, n = 21. Stimulation has no effect on distance traveled (D) or speed (E) in normal mice. n = 6 per group.
Discussion
Our results provide the first demonstration to our knowledge that selective stimulation of neurons can enhance multiple plasticity-associated mechanisms and promote recovery. Specifically, we demonstrate that stimulation of a noninfarct region, iM1, can activate peri-infarct areas and the contralesional cortex (Fig. 1). One form of cortical reorganization involves the balance of interhemispheric interactions between the ipsilesional and the contralesional motor cortex (1, 7). Interestingly, our data show that iM1 neuronal stimulations caused significant increases of multiple neurotrophins in the contralesional cortex in stroke mice (Fig. 4), but not in normal nonstroke mice (Fig. S3), suggesting that stimulation-induced increases in neurotrophins are dependent on the stroke environment. These increases in neurotrophins, possibly resulting from the interactions between the two hemispheres after stimulation, further confirms the involvement of the contralesional cortex in the stroke recovery process (6, 42, 43). It is also possible that the increases in neurotrophins are related to the contralesional limb movement during M1 stimulation (Movie S1), as this movement can send signals back to contralesional and ipsilesional cortices (44, 45). BDNF, the most extensively studied neurotrophin, has been shown to promote poststroke recovery and enhance axonal and dendritic sprouting (10, 11), which are critical repair/plasticity processes for recovery. The enhanced expression of neurotrophins such as BDNF in the contralesional cortex highlights their importance in promoting recovery poststroke. However, it is unclear whether the contralesional increases of neurotrophins are solely responsible for the recovery effect of iM1 neuronal stimulation. Further studies are needed to clarify their role.
Stroke induces a number of repair and rewiring processes that promote axonal sprouting and reorganization (1–4). A number of growth-related molecules such as GAP43, MARCKS, and CAP23 have been associated with directing axonal sprouting and remapping (3, 4, 40). In particular, GAP43 has been used as a plasticity marker and its expression is strongly correlated with poststroke axonal sprouting (3, 4, 40). GAP43 is enriched in growth cones and its expression is up-regulated after stroke in the peri-infarct areas (3, 40). Our stimulated mice exhibited a significant increase of GAP43 expression in both iS1 and cM1 at poststroke day 15 (Fig. 5). These data suggest that poststroke stimulations may enhance synaptic plasticity and reorganization. Interestingly, the enhanced GAP43 expression in cM1 further supports the involvement of the contralesional cortex in recovery. Future studies will fully investigate the effects of poststroke stimulations on other aspects of synaptic plasticity and reorganization, such as axonal sprouting, dendritic branching, and synaptogenesis.
Our data indicate that repeated iM1 neuronal stimulations can restore the temporary depression of the ipsilesional neurovascular coupling response after stroke, with a significant improvement in CBF and neurovascular coupling in stimulated mice at poststroke day 15 (Fig. 3 and Fig. S1). Using LDF, we observed an increase in CBF during the laser-on period, consistent with the view that neuronal activity drives hemodynamic signals. Recent evidence has shown that optogenetic stimulation of cortical excitatory neurons increases blood oxygen level dependent (BOLD) signals (46). Interestingly, we observed a larger CBF increase after the laser was turned off, which has not been previously reported. This larger CBF increase suggests that stimulated stroke mice had a functional neurovascular coupling system similar to sham mice; this effect was completely absent in nonstimulated stroke mice (Fig. 3 and Fig. S1). Moreover, our repeated iM1 stimulation paradigm was able to promote functional recovery at poststroke day 14, with stimulated mice performing significantly better in the rotating beam test (Fig. 6 B and C and Movies S2–S4). However, the relationship between the enhanced neurovascular coupling response and recovery needs to be further investigated. Future studies should also address the effectiveness of the stimulation, such as examining a longer poststroke period (i.e., up to 30 d) to determine if the stimulation effect is transient or persistent.
Our studies demonstrate that stimulating only neurons is sufficient to activate beneficial mechanisms that promote recovery. These specific neuronal stimulations can elicit movements in the affected forelimb and promote functional recovery, possibly through multiple repair/plasticity-associated processes, such as enhanced CBF/neurovascular coupling and increased neurotrophin expression. Understanding the mechanisms driving recovery will help identify potential drug targets for stroke treatment. Our studies also provide to our knowledge the first proof-of-principle use of optogenetics to promote recovery after stroke. Whether optogenetic stimulation can be applied clinically to stroke patients in the future remains to be determined and would require using gene therapy techniques. However, a number of clinical studies are already using intracerebral gene therapy to treat several neurological disorders (47–49). The precision and selectivity of optogenetics might provide an advantage to other stimulation methods, possibly by offering similar or greater therapeutic efficacy with fewer side effects.
Methods
Stereotaxic Surgery.
All studies used Thy-1–ChR2–YFP line-18 transgenic male mice (10–12 wk). Mice were housed under a 12:12 h light:dark cycle with food and water available ad libitum. All experiments were conducted in compliance with animal care laws and institutional guidelines and approved by the Stanford Institutional Animal Care and Use Committee. For stereotaxic surgery, mice were first anesthetized with 5% isoflurane and then maintained on 2–3% isoflurane. Body temperature, heart rate, and respiration were monitored every 15 min and kept in physiological range. Mice were anchored in a digital stereotaxic frame, an incision was made on the top of the scalp, and a small hole was exposed using a drill. The fiber optic cannula (200 µm) was stereotaxically implanted in the iM1 or cM1 using coordinates obtained from the stereotaxic atlas (50) [iM1: anteroposterior (AP) = +0.74 mm, mediolateral (ML) = −1.5 mm, and dorsoventral (DV) = −0.5 mm; cM1: AP = +0.74 mm, ML = +1.5 mm, and DV = −0.5 mm], C&B Metabond (Parkell), and dental cement was applied to secure the fiber optic cannula to the skull. Wounds were closed with suturing and tissue glue, and mice were administered an appropriate amount of buprenorphine and 0.9% saline (s.c.). Mice were monitored for recovery and returned to home cages. Continuous s.c. saline was given for 7 d poststroke to help prevent dehydration due to reduced mobility. Mice were randomized and experimenters were blinded. The stereotaxic implant surgery, stroke surgery, and behavior tests were performed by three different individuals.
Transient Middle Cerebral Artery Occlusion.
Mice were anesthetized with 5% isoflurane and then maintained on 2–3% isoflurane and physiological parameters were kept as mentioned above. An intraluminal suture was inserted into the left internal carotid artery to block the blood flow to the middle cerebral artery. The suture was left in place for 30 min and removed to allow reperfusion. Wounds were closed with suturing and tissue glue, and mice were administered an appropriate amount of buprenorphine and 0.9% saline (s.c.). Mice were monitored for recovery and returned to home cages.
Stimulation Paradigm.
All five mice groups (normal + no stim, normal + stim, sham, stroke + no stim, and stroke + stim) underwent identical handling and behavior procedures with the exception of stimulated mice that received laser pulses (Fig. 2). The difference between normal and sham mice was that sham mice underwent mock stroke procedure, which included anesthesia and skin incision on the chest. Each stimulated mouse received three successive 1-min stimulations daily separated by 3-min rest intervals. A 473-nm blue laser (OEM Laser Systems) was controlled by the Agilent function generator (AGT33210A) and mice were stimulated with the laser set to 10 Hz, 20 ms light pulses with a power range of 0.4–0.8 mW, measured by a power meter (Thorlabs). We used the minimal laser power necessary to elicit movements in the affected forelimb. Stimulations were performed in the morning between 9:00 AM and 12:00 PM and behavior tests were performed between 1:00 PM and 5:00 PM. Thus, each mouse had ∼4 h between stimulation and behavior tests. The effectiveness of iM1 neuronal stimulation was evaluated by visual examination of contralesional forelimb movement during the stimulation period.
In Vivo Optrode Recording.
Optrode recording was performed in isoflurane-anesthetized mice on a stereotaxic frame (KOPF), and recording was carried out as described previously (51). Recordings were performed on stroke mice at poststroke day 5. Briefly, a 500-µm tungsten electrode was bonded to a 200-µm optical fiber ∼0.5–0.7 mm in front of the fiber face to create an optrode. This optrode was installed on one arm of the stereotaxic inserted in iM1 (AP = +0.74 mm, ML = −1.5 mm, DV = −0.5 mm) and used to deliver 473-nm laser pulses at 0.5 mW (10 Hz, 20 ms). Electrophysiological recordings were digitized and then recorded with pCLAMP (Axon). For dual recordings of iM1, iS1, iStr, or cM1, a second recording electrode was used on a separate stereotaxic arm. After obtaining reliable firing in M1, the second electrode was inserted into the follow regions using the coordinates obtained from the stereotaxic atlas (50): iS1 (AP = +0.74 mm, ML = −3.25 mm, DV = −0.5 mm), iStr (AP = +0.74 mm, ML = −2.6 mm, DV = −2.5 mm) or cM1 (AP = +0.74 mm, ML = +1.5 mm, DV = −0.5 mm), and simultaneous optrode and electrode traces were recorded. After the traces were collected, the recording sites were cauterized with a current generator (10 μÅ) attached to the electrode for post hoc electrode localization. Mice were sacrificed and brains were removed, sectioned, and mounted onto slides. Infarct was visualized with cresyl violet and compared with the electrode recording position.
Cerebral Blood Flow Measurements.
Changes in CBF were measured using LDF (PF5001; Perimed). Mice were anesthetized with isoflurane and mounted on a stereotaxic frame. The fiber optic cannula and dental cement were carefully removed without damaging the skull. A small hole was opened on the iM1 and cM1 using a dental drill, and a fiber optic cannula was inserted at a depth of 0.5 mm. A LDF probe was placed adjacent to the fiber optic cannula where the skull is intact (Fig. 3). Once the readings were stable, a 1-min baseline was collected, followed by 1-min laser pulses (10 Hz 20 ms at 0.5 mW) with recording for another 3 min. After measurements were completed, mice were sacrificed and brains were removed for infarct analysis.
RNA Extraction and Quantitative PCR.
At poststroke day 15, mice were sacrificed and perfused with cold sterile 1× PBS and brain regions (iM1, cM1, iS1, and cS1) were dissected on ice with 1× PBS. Dissected regions were kept on ice in RNAlater and frozen at −80 °C. RNA was extracted with the Qiagen RNeasy Plus kit. First-strand cDNA synthesis was performed using SuperScript Reverse Transcriptase II with oligo dT12-18 primer (Invitrogen). qPCR was performed using the CFX96 Real-Time PCR Detection system (Bio-Rad). qPCR reaction mixtures were prepared using Taq polymerase (Life Technologies; 4369016) and Taqman primers (Life Technologies) targeting mouse BDNF (Mm04230607_s1), NGF (Mm00443039_m1), NTF3 (Mm00435413_s1), VEGF (Mm01281449_m1), and GAPDH (Mm99999915_g1). qPCR data were analyzed using the Delta Delta CT method.
Behavior Tests.
We used the rotating beam test to evaluate motor and sensory function on day 0, and poststroke days 2, 7, 10, and 14. The behavior tests were performed by a blinded observer. Three trials were performed and the two closest values were averaged. Mice were handled and pretrained three times before the preimplant baseline. Two baselines were collected: (i) preimplant baseline was collected the day before implant; (ii) prestroke baseline was collected the day before stroke. Prestroke baseline was used as our day 0 data. Exclusion criteria was as follows: Mice that exhibited behavioral deficit after implant surgery were excluded. Mice that did not exhibit behavioral deficit and body weight loss on poststroke day 2 were excluded. Histology with silver staining allowed visualization of the infarct, and mice with striatal-only infarcts were excluded from the study.
Rotating beam test was as follows: This motor/sensory test measures the distance traveled and the speed of mice placed on a rotating white fiberglass beam (length 120 cm, diameter 13 mm, distances marked in centimeters). The beam, attached to a motor that rotates at 3 rpm, is located 60 cm above a table covered with bubble cushions to reduce the mouse’s impact from a fall.
Statistical Tests.
Statistical analyses were performed using Prism 5.0. One-way analysis of variance (ANOVA) with Bonferroni’s post hoc test was used for CBF studies. One-way ANOVA with Fisher’s least significant difference (LSD) was used in qPCR studies in stroke mice. Student t test was used for qPCR studies in normal mice and in Western blot studies. Two-way ANOVA repeated measures with Fisher’s LSD was used for the behavior and body weight studies.
Please see SI Methods for infarct visualization and quantitation and protein extractions and western blots.
Supplementary Material
Acknowledgments
We thank Robin Lemmens, Paul Kalanithi, and Marion Buckwalter for helpful discussions; Cindy H. Samos for scientific editing of the manuscript; Charu Ramakrishnan and Maisie Lo for their helpful technical discussions on optogenetic-related techniques; and Corinne Bart, Alex Bautista, and Aatman Shah for their technical assistance in some of the experiments. This work was supported in part by National Institutes of Health National Institute of Neurological Disorders and Stroke Grant 1R21NS082894, Russell and Elizabeth Siegelman, and Bernard and Ronni Lacroute (G.K.S.). G.K.S. is a member of the Medtronic Neuroscience Strategic Advisory Board.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1404109111/-/DCSupplemental.
References
- 1.Sharma N, Cohen LG. Recovery of motor function after stroke. Dev Psychobiol. 2012;54(3):254–262. doi: 10.1002/dev.20508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward NS. Mechanisms underlying recovery of motor function after stroke. Postgrad Med J. 2005;81(958):510–514. doi: 10.1136/pgmj.2004.030809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmichael ST. Plasticity of cortical projections after stroke. Neuroscientist. 2003;9(1):64–75. doi: 10.1177/1073858402239592. [DOI] [PubMed] [Google Scholar]
- 4.Murphy TH, Corbett D. Plasticity during stroke recovery: From synapse to behaviour. Nat Rev Neurosci. 2009;10(12):861–872. doi: 10.1038/nrn2735. [DOI] [PubMed] [Google Scholar]
- 5.Nudo RJ. Postinfarct cortical plasticity and behavioral recovery. Stroke. 2007;38(2) Suppl:840–845. doi: 10.1161/01.STR.0000247943.12887.d2. [DOI] [PubMed] [Google Scholar]
- 6.van Meer MP, van der Marel K, van der Sprenkel JW, Dijkhuizen RM. MRI of bilateral sensorimotor network activation in response to direct intracortical stimulation in rats after unilateral stroke. J Cereb Blood Flow Metab. 2011;31(7):1583–1587. doi: 10.1038/jcbfm.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calautti C, et al. The relationship between motor deficit and primary motor cortex hemispheric activation balance after stroke: Longitudinal fMRI study. J Neurol Neurosurg Psychiatry. 2010;81(7):788–792. doi: 10.1136/jnnp.2009.190512. [DOI] [PubMed] [Google Scholar]
- 8.Fritsch B, et al. Direct current stimulation promotes BDNF-dependent synaptic plasticity: Potential implications for motor learning. Neuron. 2010;66(2):198–204. doi: 10.1016/j.neuron.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23(12):639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 11.Markus A, Patel TD, Snider WD. Neurotrophic factors and axonal growth. Curr Opin Neurobiol. 2002;12(5):523–531. doi: 10.1016/s0959-4388(02)00372-0. [DOI] [PubMed] [Google Scholar]
- 12.Zhu W, et al. Intranasal nerve growth factor enhances striatal neurogenesis in adult rats with focal cerebral ischemia. Drug Deliv. 2011;18(5):338–343. doi: 10.3109/10717544.2011.557785. [DOI] [PubMed] [Google Scholar]
- 13.Floel A, Cohen LG. Recovery of function in humans: Cortical stimulation and pharmacological treatments after stroke. Neurobiol Dis. 2010;37(2):243–251. doi: 10.1016/j.nbd.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paquette C, Sidel M, Radinska BA, Soucy JP, Thiel A. Bilateral transcranial direct current stimulation modulates activation-induced regional blood flow changes during voluntary movement. J Cereb Blood Flow Metab. 2011;31(10):2086–2095. doi: 10.1038/jcbfm.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown JA, Lutsep HL, Weinand M, Cramer SC. Motor cortex stimulation for the enhancement of recovery from stroke: A prospective, multicenter safety study. Neurosurgery. 2006;58(3):464–473. doi: 10.1227/01.NEU.0000197100.63931.04. [DOI] [PubMed] [Google Scholar]
- 16.Takeuchi N, Izumi S. Noninvasive brain stimulation for motor recovery after stroke: Mechanisms and future views. Stroke Res Treat. 2012;2012:584727. doi: 10.1155/2012/584727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bashir S, Mizrahi I, Weaver K, Fregni F, Pascual-Leone A. Assessment and modulation of neural plasticity in rehabilitation with transcranial magnetic stimulation. PM R. 2010;2(12) Suppl 2:S253–S268. doi: 10.1016/j.pmrj.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fregni F, Pascual-Leone A. Technology insight: Noninvasive brain stimulation in neurology-perspectives on the therapeutic potential of rTMS and tDCS. Nat Clin Pract Neurol. 2007;3(7):383–393. doi: 10.1038/ncpneuro0530. [DOI] [PubMed] [Google Scholar]
- 19.Mandat TS, Hurwitz T, Honey CR. Hypomania as an adverse effect of subthalamic nucleus stimulation: Report of two cases. Acta Neurochir (Wien) 2006;148(8):895–897, discussion 898. doi: 10.1007/s00701-006-0795-4. [DOI] [PubMed] [Google Scholar]
- 20.Blomstedt P, Hariz MI. Are complications less common in deep brain stimulation than in ablative procedures for movement disorders? Stereotact Funct Neurosurg. 2006;84(2–3):72–81. doi: 10.1159/000094035. [DOI] [PubMed] [Google Scholar]
- 21.Aravanis AM, et al. An optical neural interface: In vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. J Neural Eng. 2007;4(3):S143–S156. doi: 10.1088/1741-2560/4/3/S02. [DOI] [PubMed] [Google Scholar]
- 22.Takatsuru Y, et al. Critical role of the astrocyte for functional remodeling in contralateral hemisphere of somatosensory cortex after stroke. J Neurosci. 2013;33(11):4683–4692. doi: 10.1523/JNEUROSCI.2657-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takatsuru Y, Nabekura J, Koibuchi N. Contribution of neuronal and glial circuit in intact hemisphere for functional remodeling after focal ischemia. Neurosci Res. 2014;78:38–44. doi: 10.1016/j.neures.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 24.Takatsuru Y, Nabekura J, Koibuchi N. Activity of the layer II/III neurons in the somatosensory cortex (SSC) plays a critical role on functional recovery after focal stroke in the contralateral SSC. Neurosci Lett. 2013;543:168–171. doi: 10.1016/j.neulet.2013.03.049. [DOI] [PubMed] [Google Scholar]
- 25.Pham L-DD, et al. Crosstalk between oligodendrocytes and cerebral endothelium contributes to vascular remodeling after white matter injury. Glia. 2012;60(6):875–881. doi: 10.1002/glia.22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwai M, et al. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke. 2010;41(5):1032–1037. doi: 10.1161/STROKEAHA.109.570325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y, Rempe DA. Targeting astrocytes for stroke therapy. Neurotherapeutics. 2010;7(4):439–451. doi: 10.1016/j.nurt.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalanithi PS, Henderson JM. Optogenetic neuromodulation. Int Rev Neurobiol. 2012;107:185–205. doi: 10.1016/B978-0-12-404706-8.00010-3. [DOI] [PubMed] [Google Scholar]
- 30.Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fisher RS. Therapeutic devices for epilepsy. Ann Neurol. 2012;71(2):157–168. doi: 10.1002/ana.22621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim DH, et al. In vivo large-scale cortical mapping using channelrhodopsin-2 stimulation in transgenic mice reveals asymmetric and reciprocal relationships between cortical areas. Front Neural Circuits. 2012;6:11. doi: 10.3389/fncir.2012.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S, Mohajerani MH, Xie Y, Murphy TH. Optogenetic analysis of neuronal excitability during global ischemia reveals selective deficits in sensory processing following reperfusion in mouse cortex. J Neurosci. 2012;32(39):13510–13519. doi: 10.1523/JNEUROSCI.1439-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anenberg E, et al. Ministrokes in channelrhodopsin-2 transgenic mice reveal widespread deficits in motor output despite maintenance of cortical neuronal excitability. J Neurosci. 2014;34(4):1094–1104. doi: 10.1523/JNEUROSCI.1442-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silasi G, Boyd JD, Ledue J, Murphy TH. Improved methods for chronic light-based motor mapping in mice: Automated movement tracking with accelerometers, and chronic EEG recording in a bilateral thin-skull preparation. Front Neural Circuits. 2013;7:123. doi: 10.3389/fncir.2013.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerits A, et al. Optogenetically induced behavioral and functional network changes in primates. Curr Biol. 2012;22(18):1722–1726. doi: 10.1016/j.cub.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lauritzen M, et al. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31(1):17–35. doi: 10.1038/jcbfm.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol (1985) 2006;100(1):328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 39.Schäbitz W-R, et al. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke. 2004;35(4):992–997. doi: 10.1161/01.STR.0000119754.85848.0D. [DOI] [PubMed] [Google Scholar]
- 40.Carmichael ST, et al. Growth-associated gene expression after stroke: Evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol. 2005;193(2):291–311. doi: 10.1016/j.expneurol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Nygren J, Wieloch T. Enriched environment enhances recovery of motor function after focal ischemia in mice, and downregulates the transcription factor NGFI-A. J Cereb Blood Flow Metab. 2005;25(12):1625–1633. doi: 10.1038/sj.jcbfm.9600157. [DOI] [PubMed] [Google Scholar]
- 42.Takatsuru Y, et al. Neuronal circuit remodeling in the contralateral cortical hemisphere during functional recovery from cerebral infarction. J Neurosci. 2009;29(32):10081–10086. doi: 10.1523/JNEUROSCI.1638-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Z, Li Y, Zhang X, Savant-Bhonsale S, Chopp M. Contralesional axonal remodeling of the corticospinal system in adult rats after stroke and bone marrow stromal cell treatment. Stroke. 2008;39(9):2571–2577. doi: 10.1161/STROKEAHA.107.511659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hattox AM, Nelson SB. Layer V neurons in mouse cortex projecting to different targets have distinct physiological properties. J Neurophysiol. 2007;98(6):3330–3340. doi: 10.1152/jn.00397.2007. [DOI] [PubMed] [Google Scholar]
- 45.Molyneaux BJ, Arlotta P, Menezes JRL, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8(6):427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 46.Li N, et al. Optogenetic-guided cortical plasticity after nerve injury. Proc Natl Acad Sci USA. 2011;108(21):8838–8843. doi: 10.1073/pnas.1100815108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaplitt MG, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet. 2007;369(9579):2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- 48.Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11(17):2389–2401. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 49.Mandel RJ, Burger C. Clinical trials in neurological disorders using AAV vectors: Promises and challenges. Curr Opin Mol Ther. 2004;6(5):482–490. [PubMed] [Google Scholar]
- 50. Paxinos and Franklin (2004) The mouse brain in stereotaxic coordinates (Elsevier, San Diego), 2nd Ed.
- 51.Gradinaru V, et al. Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci. 2007;27(52):14231–14238. doi: 10.1523/JNEUROSCI.3578-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.